


1 Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, D-52074 Aachen, Germany
The review by Damiano et al. [1] synthesizes a large body of work, arguing that oxidants produced during chronic inflammation can actively reshape deoxyribonucleic acid (DNA) methylation landscapes and thereby contribute to carcinogenesis rather than merely cause random genetic damage. The authors highlight the fact that ~20% of human cancers arise in chronic-inflammation settings and propose that immune cell–derived reactive oxygen species (ROS) are an underappreciated mechanistic bridge between persistent inflammation and the aberrant methylation patterns typical of tumors.
The review starts with a primer on DNA methylation, highlighting how DNA methyltransferase 1 (DNMT1) maintains existing methylation patterns while DNA methyltransferases 3A and 3B (DNMT3A and DNMT3B, respectively) establish new marks and showing how ten-eleven translocation (TET) enzymes oxidize 5-methylcytosine to 5-hydroxymethylcytosine (5-hmC) and beyond as part of active demethylation. The authors emphasize that cytosine-phosphate-guanine (CpG) islands at gene promoters are normally unmethylated and transcriptionally permissive, whereas the cancer-associated hypermethylation of tumor suppressor promoters and global hypomethylation of oncogenes are hallmarks of neoplastic epigenomes.
The novelty of the review lies in connecting these canonical epigenetic pathways to specific inflammatory oxidants, particularly neutrophil-derived species produced via the NADPH oxidase 2 (NOX2)-derived H2O2–myeloperoxidase (MPO; a heme-containing enzyme)–hypochlorous acid axis and secondary chloramine derivatives such as glycine chloramine. Mechanistically, the authors describe how oxidative base lesions such as 8-oxo-2′-deoxyguanosine (8-oxo-dG) can hinder DNMT1 binding at neighboring CpGs and thereby induce local hypomethylation, while the HOCl-derived 5-chlorocytosine can mimic 5-methylcytosine (5-mC) and misdirect methylation-sensitive proteins, promoting aberrant methylation at normally unmethylated CpG sites. The authors also discuss how (i) ROS may indirectly drive hypomethylation by impairing DNMT1 activity and depleting methionine and S-adenosyl methionine (SAM) and (ii) oxidative interference with Fe2+-dependent TET activity can modulate the 5-mC to 5-hmC ratio. In this scenario, blocking the reduction of Fe3+ to Fe2+ through exposure to H2O2 reduces TET activity and hinders the conversion of 5-mC to 5-hmC, which normally protects cells against oxidative stress.
A notable strength of the paper is the integration of genome-wide studies showing that the exposure of proliferating cells to sublethal H2O2 or chloramine levels leads to persistent site-specific methylation changes that are enriched in cancer- and inflammation-associated genes and often occur at chromosomal ends, which has implications for telomere biology and genomic instability. This fact supports the notion that oxidative stress can induce heritable epigenetic heterogeneity within inflamed tissues and thus seed clonal selection and progression toward malignancy.
The review gains further relevance by walking through disease models in which chronic inflammation, inflammatory oxidants, and methylation changes co-occur in a clinically meaningful way. For hepatitis C–associated hepatocellular carcinoma, the authors highlight the fact that H2O2 upregulates the transcription factor Snail and recruits DNMT1 and histone deacetylases to the CDH1/E-cadherin promoter, leading to promoter hypermethylation, gene silencing, and the loss of a key adhesion molecule constraining metastasis. The authors also cite work linking oxidant-induced 8-oxo-dG accumulation in hepatocytes to the hypermethylation of multiple tumor suppressor genes and repressive chromatin states.
For Helicobacter pylori–driven gastric carcinogenesis, the article compiles in vivo and human data showing that infection-associated gastritis correlates with progressive promoter hypermethylation in genes involved in cell adhesion, cell cycle regulation, DNA repair, and inflammation, with some methylation persisting even after bacterial eradication. The authors underscore the experimental results obtained for animal models, where demethylating agents were found to reduce gastritis-induced methylation and prevent gastric cancer, and discuss the nitric oxide–mediated methylation of RUNT-related transcription factor 3 (RUNX3) as a putative pathway by which inflammatory cells can epigenetically silence tumor suppressors in the gastric mucosa.
For inflammatory bowel disease and colitis-associated colorectal cancer, the review notes elevated oxidant markers such as 8-oxo-dG, MPO, and calprotectin in the inflamed colon and stool, alongside distinctive methylation profiles differing from those of sporadic colorectal cancer. A particularly interesting mechanistic thread is the ability of H2O2 to recruit DNMT1 and other repressive complex components (including sirtuin-1 and histone methyltransferases) to 8-oxo-dG–containing CpG island promoters, which leads to the targeted silencing of tumor suppressor genes such as RUNX3.
In the context of ultraviolet (UV)-induced skin cancer, the authors relate
chronic UV exposure to inflammatory signaling via nuclear factor
Beyond organ-specific examples, the paper compiles a table of chronic inflammatory conditions (ranging from chronic pancreatitis and Barrett’s esophagus to periodontal disease, Hashimoto’s thyroiditis, and rheumatoid arthritis) epidemiologically associated with heightened cancer risk, reinforcing the broad relevance of inflammation-associated epigenetic dysregulation.
Inflammatory immunology [2], aberrant redox signaling [3, 4], and epigenetics [5, 6] are often viewed as separate disciplines in cancer research. However, they converge on shared pathways that critically influence cancer risk in chronically inflamed tissues. The persistent activation of immune cells leads to the sustained production of reactive oxygen and nitrogen species, which not only damage DNA but also interfere with the enzymes governing DNA methylation and chromatin organization, thereby reprogramming gene expression in epithelial and stromal cells. Viewing carcinogenesis through this integrative lens underscores how immune cell–derived oxidants function as both mediators of host defense and drivers of maladaptive epigenetic remodeling that can foster malignant transformation.
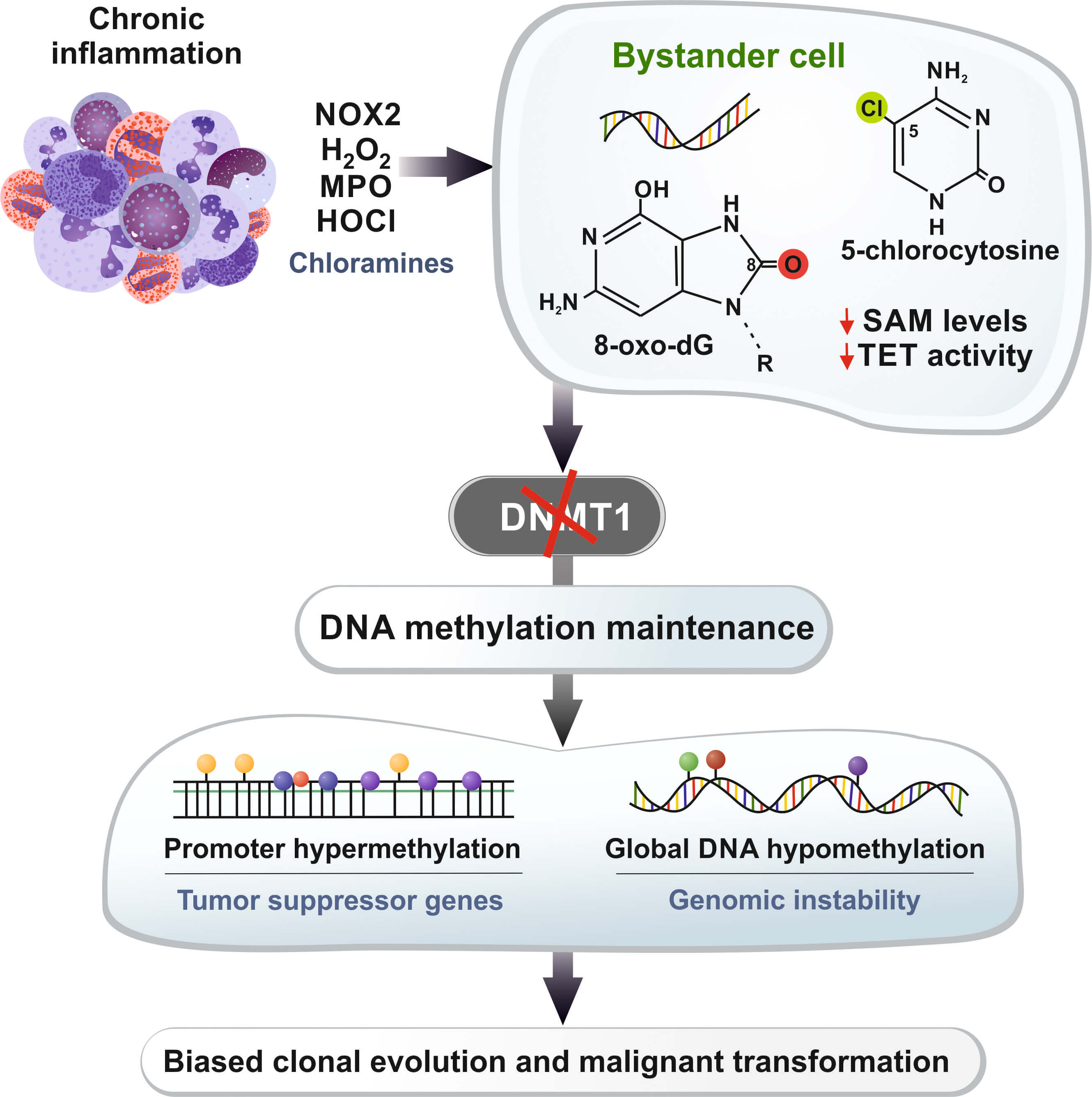
The review of Damiano et al. [1] is conceptually valuable because it weaves together these three domains, arguing that the oxidant-mediated disruption of methylation maintenance is a coherent mechanistic axis linking them. The schematic hypothetical model centered on neutrophil-derived oxidants, methionine/SAM depletion, and the failure of DNMT1 to maintain methylation fidelity during replication is particularly useful as a unifying framework for thinking about how chronic inflammation can progressively rewire the epigenome of bystander cells in inflamed tissues. A simplified version of the proposed oxidant–epigenome axis is summarized in Fig. 1, tracing the sequence from chronic inflammation and immune cell–derived oxidant production to stable cancer-relevant changes in DNA methylation patterns in neighboring epithelial cells.
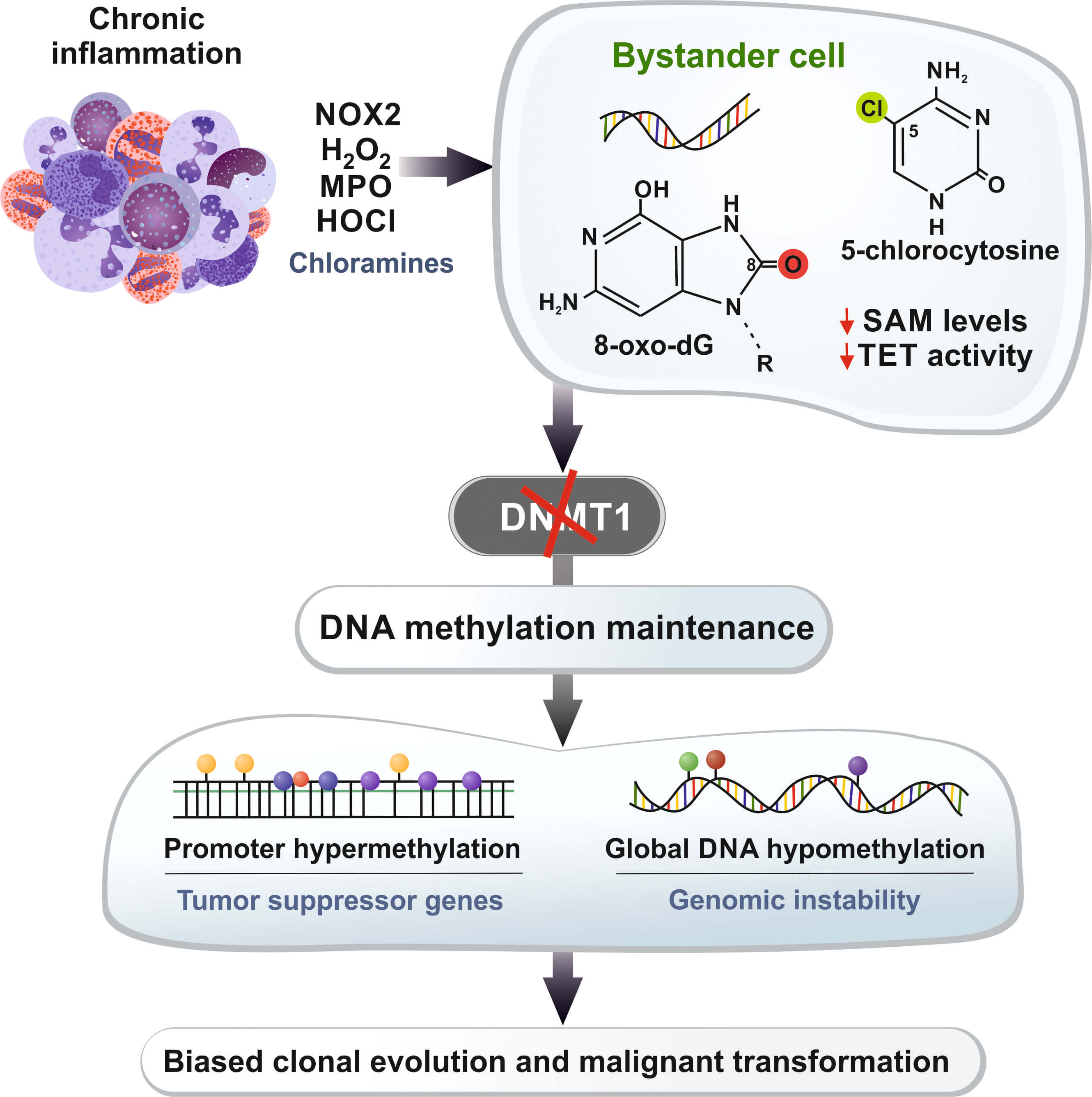 Fig. 1.
Fig. 1.
Simplified model of the oxidant-driven disruption of DNA
methylation in chronic inflammation. Chronic inflammatory infiltrates, which are
rich in neutrophils and other immune cells, produce oxidants through the
NOX2–H2O2–MPO–HOCl pathway, which involves long-lived chloramines
that diffuse into neighboring epithelial or progenitor cells. Within these
bystander cells, oxidative DNA lesions such as 8-oxo-dG and HOCl-derived
5-chlorocytosine, along with methionine/SAM depletion and the inhibition of TET
enzyme activity, disrupt DNMT1-dependent maintenance methylation and normal
demethylation dynamics at CpG-rich promoters. Over multiple cell divisions, this
disruption can lead to the promoter hypermethylation of tumor suppressor genes
and broader hypomethylation at other loci. This process generates persistent
epigenetic heterogeneity within inflamed tissues and potentially drives clonal
evolution toward malignant phenotypes. Moreover, 8-oxo-dG can create mismatches
via pairing with adenine, thus leading to guanine
Importantly, the authors are appropriately cautious, repeatedly emphasizing that (i) current evidence is limited and often correlative and (ii) the question of whether oxidant-induced methylation changes are the primary drivers of carcinogenesis or secondary consequences of broader cellular stress responses remains unresolved. Much of the mechanistic work still relies on in vitro systems or animal models exposed to defined oxidants, and the field lacks longitudinal human data tracking oxidative lesions, methylation, and clonal evolution in the same tissues over time.
The review could have delved more deeply into the crosstalk between DNA methylation and other epigenetic layers (histone modifications, chromatin architecture, noncoding ribonucleic acids), although it does touch on histone deacetylation and Polycomb recruitment in specific contexts. Likewise, although the authors discuss the decline of 5-hmC and reduced TET expression in certain inflammation-associated cancers, they only briefly address the possibility of 5hmC itself functioning as a stable epigenetic mark rather than solely being an intermediate in demethylation. These choices keep the narrative focused but leave room for future syntheses to integrate the broader epigenomic landscape.
Clinically, the authors argue that defining oxidant-driven methylation signatures could support biomarker development and inform epigenetic or redox-targeted therapies in high-cancer-risk patients with chronic inflammatory diseases, taking advantage of the reversibility of epigenetic modifications. The authors conclude by calling for more work to disentangle causality, delineate precise mechanisms, and explore therapeutic strategies that prevent oxidant-induced epigenetic damage or reprogram established aberrant methylation patterns.
Several points and hypotheses building on the paper’s themes but not explicitly discussed in the review are provided below.
1. Microbiome–redox–epigenome axis: Chronic inflammatory diseases of barrier organs (gut, skin, lung) arise in the context of complex microbial communities. Microbial metabolites such as short-chain fatty acids can modulate redox status and directly influence host low-grade inflammation, DNA methylation, and histone acetylation. Integrating microbiome profiling with redox and methylome measurements could clarify whether specific microbial configurations predispose tissues to oxidant-driven epigenetic reprogramming [7, 8, 9].
2. Single-cell and spatial multiomics: The review largely discusses methylation changes at the bulk-tissue level; however, clonal evolution in inflamed microenvironments is inherently heterogeneous in space and time. Single-cell multiomic technologies (combining methylome, transcriptome, and chromatin accessibility) and spatially resolved assays could reveal how oxidative lesions and epigenetic modifications (histone acetylation, methylation, phosphorylation, ubiquitination, and DNA methylation) change and propagate through chromatin accessibility alterations across distinct cell types (epithelial, immune, stromal) within a chronically inflamed niche [10, 11].
3. Integration with immunotherapy and immune escape: If oxidant-driven promoter hypermethylation contributes to the silencing of antigen presentation pathways or interferon signaling, it could facilitate immune evasion in inflammation-associated cancers. This suggests that epigenetic drugs aimed at reversing such methylation patterns and capable of reprogramming aberrant transcription networks might synergize with immune checkpoint inhibitors, particularly in patients with chronic inflammatory diseases, and promote immunogenic cell death, modulate inflammatory cytokine profiles, and reverse local immune suppression [12, 13].
4. Metabolic and lifestyle interventions: Many risk factors for chronic inflammation (obesity, Western diet, sedentary behavior) are modifiable and influence systemic redox balance and one-carbon metabolism, which provides substrates for methylation reactions. Carefully designed interventional studies could test whether structured changes in diet, physical activity, or microbiome-directed therapies alter both oxidative stress markers and DNA methylation trajectories in at-risk tissues before overt neoplasia develops [14]. Notably, dietary interventions such as a switch to Mediterranean and Dietary Approaches to Stop Hypertension diets slow epigenetic aging through favorable DNA methylation [15].
5. Selectivity and safety of redox modulation: Although systemic antioxidant supplementation has often failed in cancer prevention trials, the targeted modulation of specific redox pathways (for example, MPO inhibition or selective NOX2 blockade) might reduce harmful inflammation-associated tissue damage and oxidative epigenetic remodeling without broadly suppressing beneficial ROS-dependent signaling [16, 17, 18]. The challenge is maintaining host defense and physiological redox signaling while reducing the chronic low-grade oxidative insults that can gradually reshape the epigenome toward a malignant state.
6. Non-CpG methylation and three-dimensional genome organization: Emerging evidence indicates that non-CpG methylation and higher-order chromatin structures (topologically associated domains, loops) also respond to environmental and inflammatory cues [19, 20]. Hence, one should aim to determine whether oxidant exposure preferentially affects specific three-dimensional genomic domains and thereby coordinates methylation changes across functionally related gene clusters.
Overall, the review succeeds in crystallizing a compelling hypothesis: Immune cell–derived ROS in chronic inflammatory settings do not merely damage DNA in a stochastic fashion but can bias epigenetic programming at defined genomic loci, which helps explain why inflammation-associated cancers have distinctive methylation signatures. Extending this framework using microbiome science, single-cell spatial data, and clinically oriented interventional studies could transform a strong conceptual model into actionable strategies for cancer prevention and early interception in patients with chronic inflammatory diseases.
5-hmC, 5-hydroxymethylcytosine; 8-oxo-dG, 8-oxo-2′-deoxyguanosine; CpG, cytosine-phosphate-guanine; DNA, deoxyribonucleic acid; DNMT1, DNA methyltransferase 1; DNMT3A, DNA methyltransferase 3A; DNMT3B, DNA methyltransferase 3B; MPO, myeloperoxidase; NF-
RW conceived the topic of the commentary, critically evaluated and synthesized the relevant literature, and wrote the manuscript. RW read and approved the final version of the manuscript and agrees to be accountable for all aspects of the work.
Not applicable.
The author gratefully acknowledges the assistance from Sabine Weiskirchen of the Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC) for preparing Fig. 1 of this commentary.
This research received no external funding.
Given his role as the Guest Editor, Ralf Weiskirchen had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
During the preparation of this work, the author used ChatGPT-5.1 in order to check spelling and grammar. After using this tool, the author reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.