


 , Huiye Feng 1,†, Liting Li 1, Jiao Lin 1, Liang Xu 1, Jiayi Zhu 2,*
, Huiye Feng 1,†, Liting Li 1, Jiao Lin 1, Liang Xu 1, Jiayi Zhu 2,* , Yan Liu 1,3,*
, Yan Liu 1,3,*1 MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, 510275 Guangzhou, Guangdong, China
2 Hubei Key Laboratory of Pollutant Analysis and Reuse Technology, College of Chemistry and Chemical Engineering, Hubei Normal University, 435002 Huangshi, Hubei, China
3 Department of Biochemistry and Molecular Biology, School of Basic Medicine, Guangdong Medical University, 524023 Zhanjiang, Guangdong, China
†These authors contributed equally.
Abstract
Synthetic RNA circuits offer powerful tools for reprogramming cellular behavior, but constructing ligand-responsive RNA switches that function reliably inside living cells remains challenging. Existing cis-acting designs often lack modularity and programmability due to tight coupling between sensing and output domains.
We developed a generalizable strategy termed Ligand-Induced Trigger RNA Cleavage (LITC). This approach integrates an aptamer-embedded hammerhead ribozyme (aptazyme) as a trans-acting trigger. The aptazyme sequence is inserted into the spacer region of an RNA trigger strand, separating its toehold and displacement domains. Ligand-induced aptazyme self-cleavage inactivates the trigger, thereby controlling downstream toehold-mediated strand displacement reactions. We validated this system in both prokaryotic (E. coli) and eukaryotic (HEK-293T) cells using translation-controlling toehold switches and gRNA switches within the CRISPR/Cas9 system.
The LITC strategy successfully enabled programmable, dose-dependent regulation of gene expression. A spacer inserted between toehold and displacement domains did not impair trigger function. Embedding self-cleaving ribozymes (HHR, sTRSV) constitutively silenced trigger activity. Using a theophylline-responsive aptazyme, we achieved ligand-controlled regulation of a toehold switch, with different communication modules (CMs) yielding varied regulatory performance and theophylline concentrations up to 4 mM providing graded control. Furthermore, this approach was extended to control CRISPR interference (CRISPRi) in E. coli and CRISPR activation (CRISPRa) of the endogenous ASCL1 gene in HEK-293T cells, demonstrating cross-system portability.
The LITC platform provides a general, modular, and transferable strategy for small-molecule control of toehold-mediated strand displacement reactions. It enables precise conditional regulation of RNA-based devices, including translation switches and CRISPR-Cas9 systems, across both prokaryotic and eukaryotic cells, thereby offering a versatile framework for constructing intelligent genetic circuits.
Keywords
- nucleic acid hybridization
- RNA aptamers
- ribozymes
- RNA cleavage
- CRISPR/Cas9 systems
- gene expression regulation
Genetically expressed RNA gene circuits enable dynamic and long-term regulation of biological processes within living cells through in situ transcription and assembly [1, 2, 3, 4]. Unlike static systems relying on pre-assembled DNA strands, RNA circuits can directly form functional structures during transcription and are inherently integrated into cellular gene expression and signaling networks, thereby supporting more sophisticated logical operations, signal amplification, and conditional responses [5, 6, 7, 8]. Moreover, the intrinsic biological activities of RNA molecules in gene regulation further expand their application potential in cellular manipulation for disease diagnostics and intervention [9, 10]. Thus, RNA gene circuits not only offer high programmability and modular design capabilities but also provide a more flexible and physiologically compatible technological platform for achieving precise and dynamic cellular manipulation.
Precise control over the toehold-mediated strand displacement reaction (TMSDR) is essential for advancing the design and implementation of sophisticated nucleic acid machines [11, 12]. Unlike the conventional control achievable for DNA strand displacement in vitro, all the RNA assemblies are generated co-transcriptionally in the cellular context without pre-assembling [13, 14]. Hence, many strategies involving preformed intermolecular nucleic acid structures applied in strand displacement systems. More than sensing cognate nucleic acid species, integrating aptamers into nucleic acid architectures enables ligand recognition to be coupled with structural rearrangement or functional output, thereby allowing dynamic response to non-nucleic acid ligands [15, 16, 17, 18]. Despite these advances, most RNA-based signaling systems designed for ligand response typically rely on a cis-acting approach, wherein the aptamer domain is embedded within a regulatory RNA structure to control functions such as riboswitches, ribozyme cleavage, translation initiation, or alternative splicing [19, 20, 21, 22, 23, 24]. Although conceptually straightforward, cis designs are often limited in modularity due to the physical and functional coupling between the aptamer and output domains, which frequently require sequence constraints with careful designs. Therefore, this restricts programmability of RNA circuits.
To address these limitations, we previously developed a ligand-triggered trans-acting nucleic acid platform that enables programmable conversion of ligand signals into nucleic acid outputs in test tubes [25]. Moreover, these systems have been successfully implemented in living cells [26]. While they exhibit robust responses to small molecules or protein metabolites, they all operate as switch-on platforms that become activated upon ligand binding. Accordingly, the development of switch-off systems that are inhibited upon ligand recognition is also critical to achieve broader and more scalable systems for enriching the programmability of RNA circuits. On this basis, we integrate aptamers into ribozymes to construct aptazymes to establish a switch-off system. Ligand binding to the aptamer domain triggers a conformational rearrangement, which promotes ribozyme self-cleavage and results in the formation of an active trigger that subsequently suppresses downstream strand displacement reactions. This trans-acting mechanism decouples ligand sensing from direct output execution, allowing aptamer modules to be flexibly integrated into arbitrary sequences without requiring sequence constraints with downstream modules.
In this work, we report a generalizable strategy to engineer ligand-responsive switch through programmable RNA circuits in mammalian cells. To enable in situ control of RNA strand displacement reaction (SDR) in living systems, this study integrates the aptazyme framework with the remote toehold strategy. We developed a method termed ligand-induced trigger RNA cleavage (LITC) for the precise regulation of toehold-mediated strand displacement in cells. As illustrated in Fig. 1, LITC involves embedding an aptazyme sequence within the spacer region of an RNA trigger strand, between its toehold and displacement segments. Ligand-dependent activation of the aptazyme cleaves the trigger, thereby controlling its ability to engage in strand displacement. Using this approach, we successfully regulated various toehold-based RNA devices, including translation-regulating riboswitches and gRNA switches within the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system. With the ligand-induced switch-off design, this system may be combined with the aforementioned switch-on platforms to enable more complex logic operations, thereby laying a solid foundation for the implementation of sophisticated RNA circuits in intracellular environments.
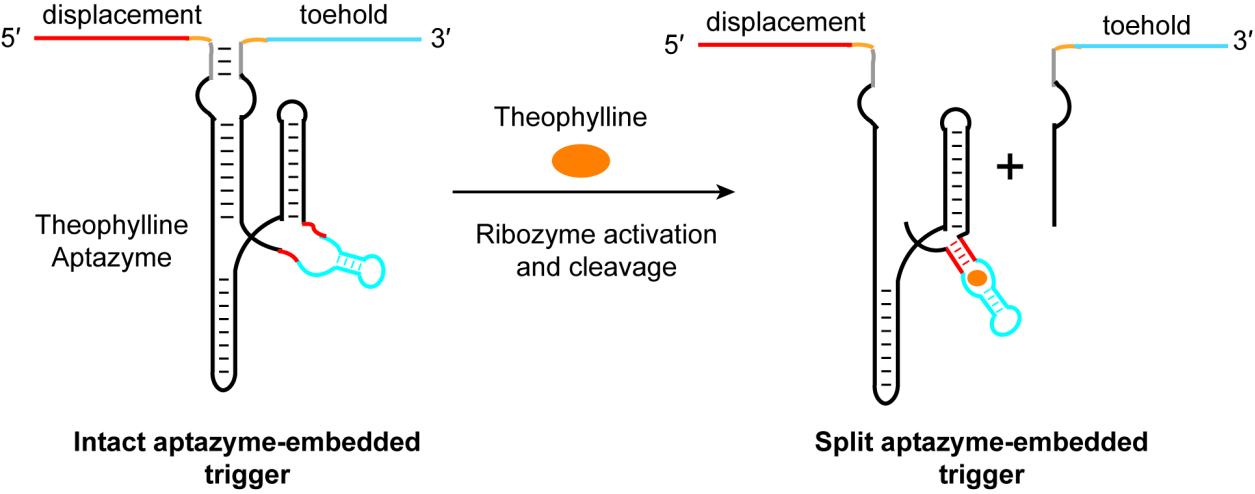
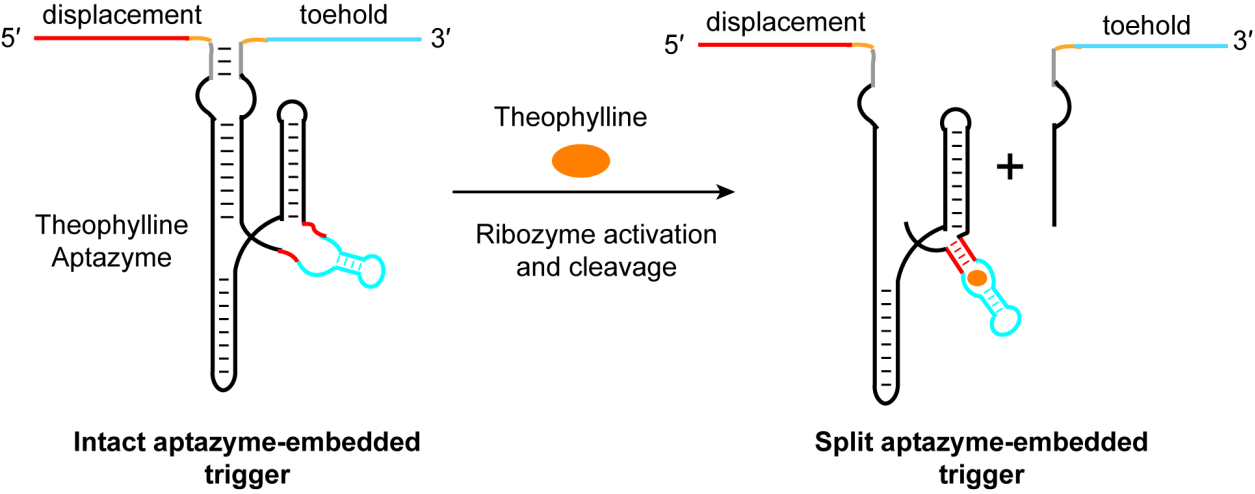 Fig. 1.
Fig. 1.
The principle of ligand-induced trigger RNA cleavage strategy. In the absence of theophylline, the aptamer does not adopt a stable structure, and the aptazyme remains inactive. Under this condition, the 5′- and 3′-sequences of the aptazyme—which also serve as the displacement and toehold regions of the trigger RNA—are brought into proximity through intramolecular base pairing within the aptazyme, allowing the trigger RNA to remain functional. Upon addition of theophylline, ligand binding induces the aptazyme to fold into an active conformation capable of cis-cleavage. This self-cleavage reaction breaks the phosphodiester bond at the cleavage site, physically separating the 5′- and 3′-sequences of the aptazyme and thereby inactivating the trigger RNA.
To evaluate the impact of spacer incorporation on trigger functionality, we first compared the activation of a toehold switch by conventional trigger RNAs versus those containing an intervening spacer. As depicted in Fig. 2, the toehold switch maintains a conformation that represses translation in its default state. Activation occurs via toehold-mediated strand displacement with a complementary trigger RNA, which releases the repression and initiates translation of the downstream reporter. The conventional trigger (Fig. 2a) is composed of a toehold domain and a displacement domain, each complementary to the corresponding regions of the switch. In the engineered construct (Fig. 2b), a spacer sequence was inserted between these two domains. To test whether this modification affects switch activation, we co-expressed plasmids encoding the toehold switch and each trigger variant in E. coli (strain BL21). Cells were quantified by measuring the fluorescence intensity of the downstream superfolder GFP (sfGFP) via flow cytometry. As shown in Fig. 2c (and Supplementary Fig. 1), both the conventional trigger (T) and the spacer-containing trigger (En-T) robustly activated the switch, resulting in significantly higher sfGFP fluorescence than the non-targeting control trigger (Tw). Importantly, En-T and T elicited comparable levels of activation, indicating that the introduction of a spacer does not impair toehold-mediated strand displacement in vivo. These results confirm that the distal toehold strategy remains functional within cellular environments and can be utilized to engineer controllable strand-displacement systems.
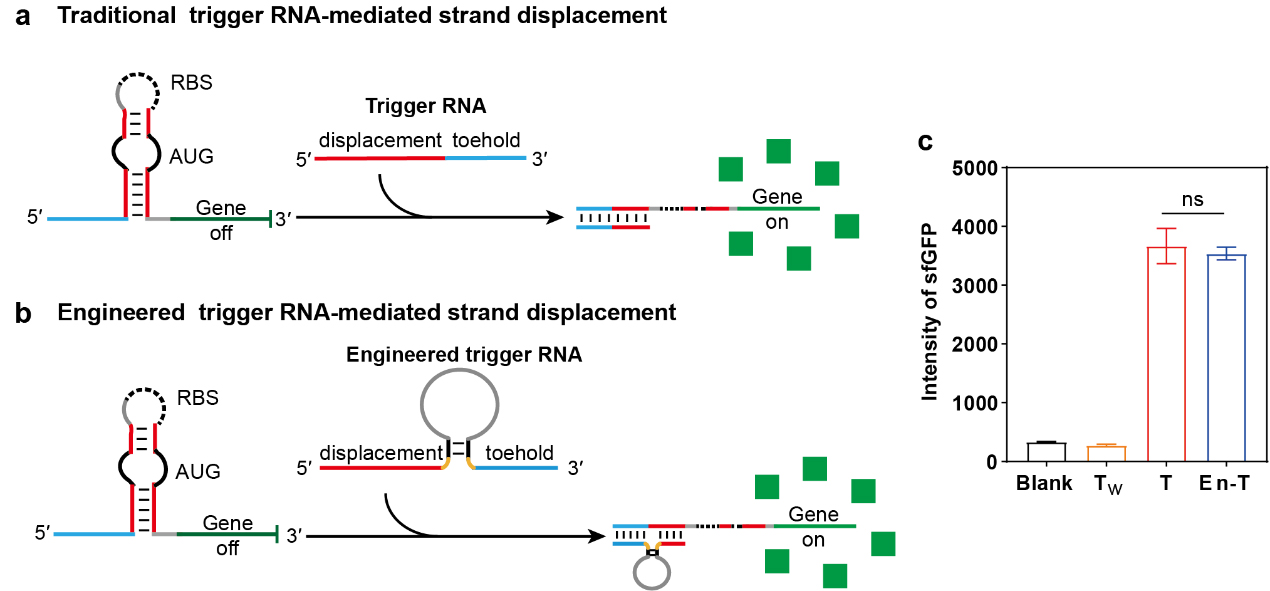 Fig. 2.
Fig. 2.
Comparison of conventional and engineered trigger RNAs in
activating a toehold switch. (a) Schematic of toehold-switch activation by
trigger RNA to control translation. (b) Schematic of toehold-switch activation by
engineered trigger RNA to control translation. Engineered trigger RNA includes an
additional spacer region inserted between the toehold and displacement domains.
(c) Quantitative assessment of toehold-switch activation by different triggers,
based on mean sfGFP fluorescence intensity. Data are presented as mean
Subsequently, we systematically characterized how toehold length influences the regulatory performance of the engineered trigger RNA (En-T) on the toehold switch. A set of seven En-T variants with toehold domains of 4, 6, 8, 9, 10, 12, and 15 nucleotides (nt) was constructed. The activation efficiency of each variant was quantified by measuring the fluorescence intensity of the downstream reporter protein sfGFP using flow cytometry. As shown in Supplementary Fig. 2, sfGFP fluorescence increased progressively with longer toehold lengths, reflecting enhanced restoration of translation. At the shortest toehold (4 nt), En-T exhibited only weak activation, with sfGFP fluorescence slightly above background levels. For toehold lengths between 6 and 12 nt, activation efficiency improved steadily, peaking at 12 nt. Notably, the 12 nt and 15 nt variants produced comparable sfGFP signals, both significantly above background and the fluorescence intensity increased approximately 5-fold. Based on these results, a toehold length of 12 nt was selected as optimal for En-T in all subsequent experiments.
Building on the demonstration that spacer insertion does not impair trigger function, we next engineered trigger RNAs by incorporating enzymatically active self-cleaving ribozymes into the spacer region. First, the hammerhead ribozyme (HHR) sequence was inserted between the toehold and displacement domains, generating the construct HHR-T (Supplementary Fig. 3a). The inherent self-cleavage activity of HHR is expected to sever the linkage between these domains, thereby inactivating the trigger. To test this, we used a trigger containing a catalytically inactive HHR variant (InHHR-T), which differs from the active ribozyme by only a single nucleotide, as a control. Flow cytometry analysis revealed substantially higher sfGFP fluorescence in cells expressing InHHR-T compared to those expressing HHR-T (~7-fold) (Supplementary Fig. 3b,c). This result confirms that the activation of the toehold switch—and thus translation of the downstream reporter—can be effectively silenced by the constitutive self-cleavage activity of the embedded HHR.
To generalize this strategy, we employed another self-cleaving ribozyme, satellite RNA of tobacco ringspot virus (sTRSV). The corresponding trigger, sTRSV-T, was constructed similarly by placing the sTRSV sequence within the spacer (Supplementary Fig. 4a). A control trigger with an inactive sTRSV variant (InsTRSV-T) served as a non-cleaving counterpart. Consistent with the HHR results, the sfGFP signal was significantly stronger in the InsTRSV-T group than in the sTRSV-T group (~4-fold) (Supplementary Fig. 4b,c), indicating that sTRSV self-cleavage also disrupts trigger integrity and abrogates switch activation.
Collectively, these experiments establish that embedding functional ribozymes within the spacer region of a trigger RNA provides a reliable mechanism to control toehold-mediated strand displacement and the consequent regulation of gene expression in E. coli.
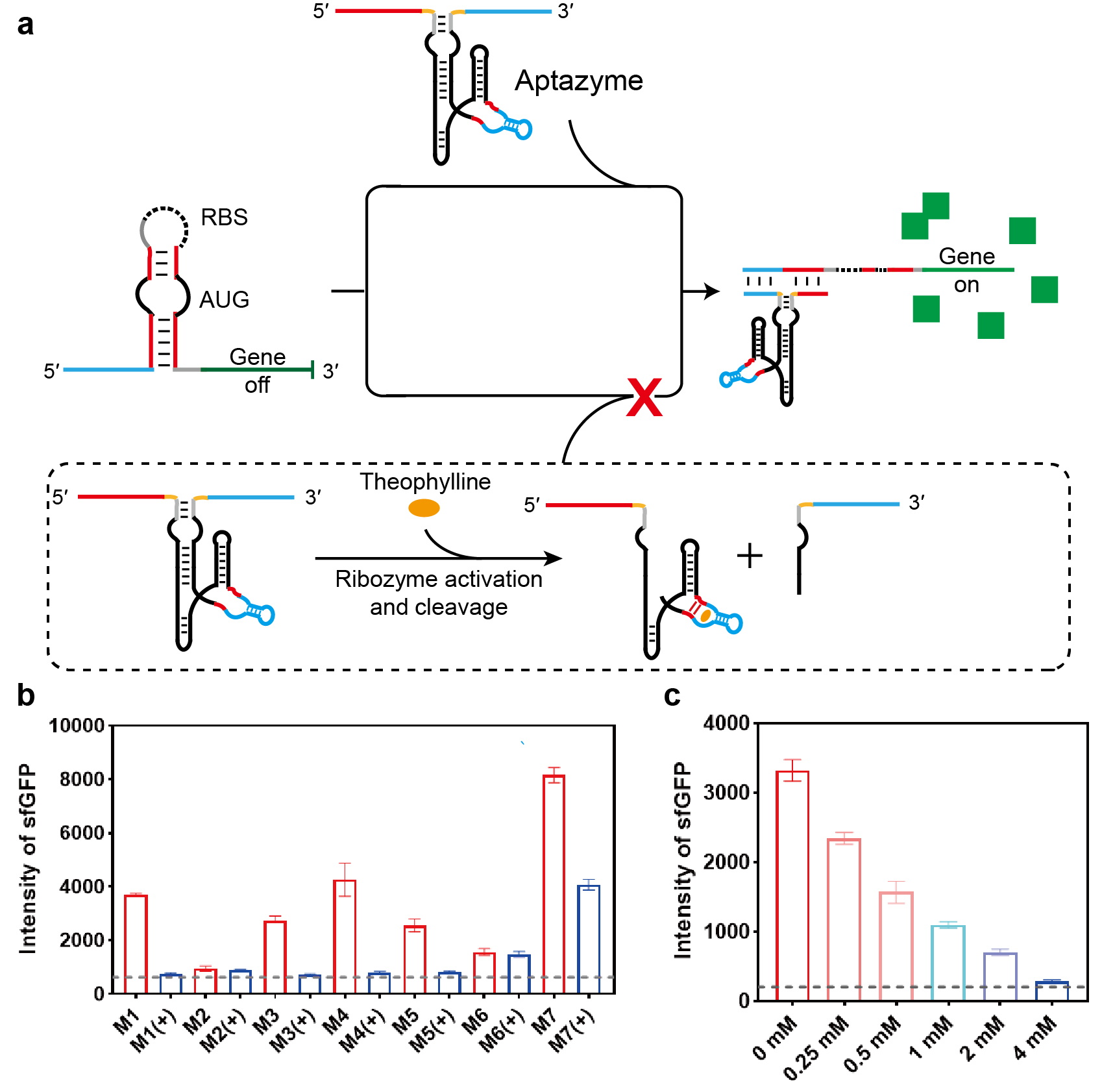
Next, we extended our strategy to achieve ligand-responsive control of the toehold switch by embedding aptazyme sequences into the trigger RNA. An aptazyme is a chimeric RNA molecule in which an aptamer domain is fused to a self-cleaving ribozyme via a designed communication module (CM), allowing small-molecule binding to modulate ribozyme activity [27]. As illustrated in Fig. 3a (and Supplementary Fig. 5), we employed a well-characterized theophylline-responsive aptazyme, in which a theophylline aptamer is connected to a HHR through a CM. By inserting this aptazyme into the spacer region, we constructed a theophylline-regulated trigger, Theo-HHR-T. In the absence of theophylline, Theo-HHR-T remains intact and activates the toehold switch via strand displacement, enabling translation of sfGFP. Upon theophylline addition, aptamer binding activates the ribozyme domain, leading to self-cleavage of the trigger and abolishing switch activation.
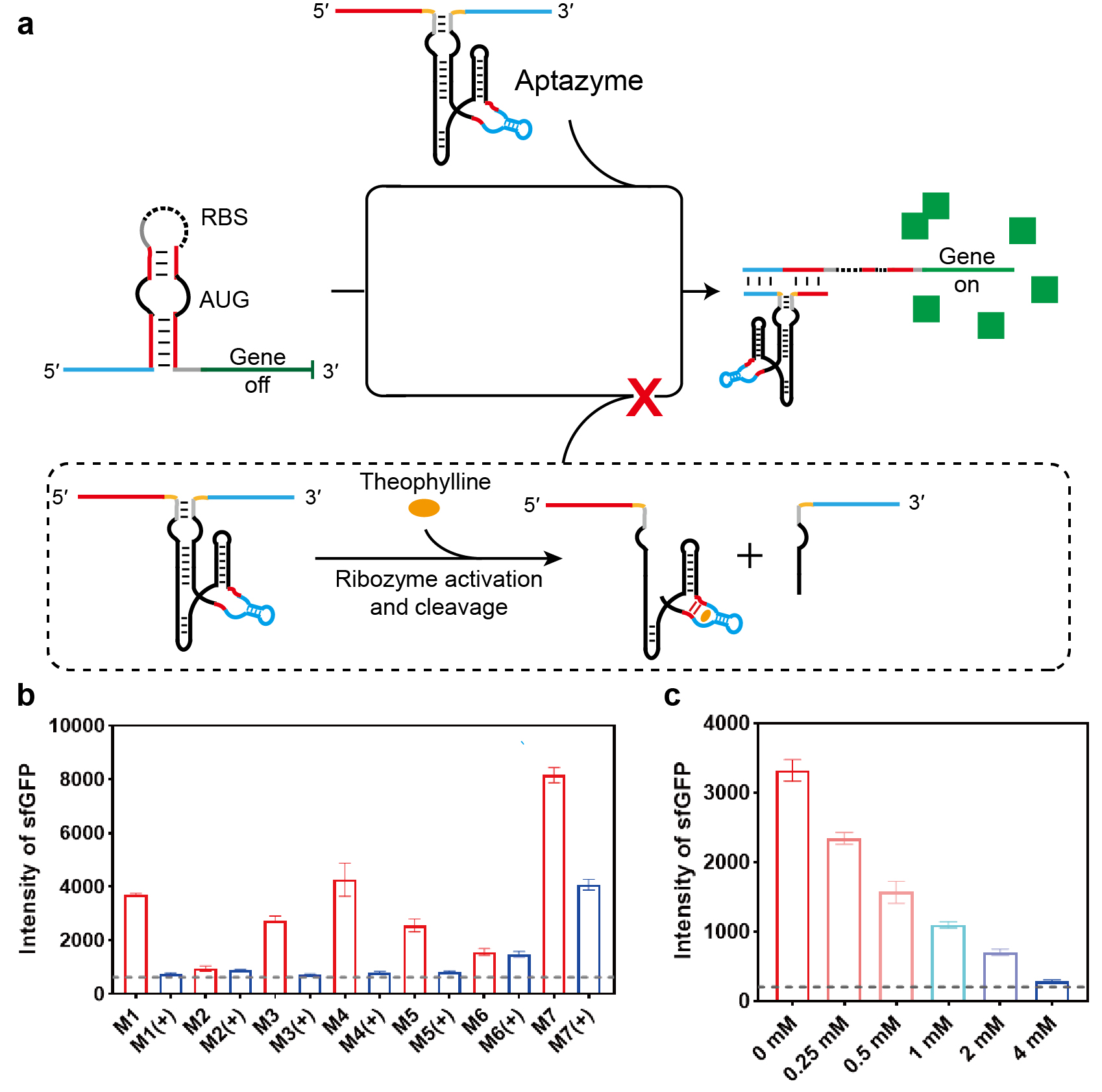 Fig. 3.
Fig. 3.
Ligand-dependent regulation of toehold switch activity using
aptazyme-engineered trigger RNAs with different communication modules. (a)
Schematic of the theophylline-responsive aptazyme trigger RNA (Theo-HHR-T). The
theophylline aptazyme sequence was inserted into the spacer region of the
invading strand, generating Theo-HHR-T. This invader triggers the toehold switch
via strand displacement, activating sfGFP translation. Upon theophylline binding,
the aptazyme cleaves the invader, shutting down translation. (b) Quantitative
analysis of toehold switch activation by Theo-HHR-T variants containing different
CMs, in the presence (+) or absence (–) of theophylline. Switch activity is
reported as mean sfGFP fluorescence intensity measured by flow cytometry. (c)
Dose-dependent regulation of toehold-switch activity by CM‑1 theophylline
aptazyme trigger RNA at varying theophylline concentrations (0, 0.25, 0.5, 1, 2,
and 4 mM). The gray dotted line indicates background fluorescence from a
non-activating control. Data are presented as mean
It has been established that the CM critically influences ligand-dependent regulation of aptazyme activity. We therefore engineered seven variants of Theo-HHR-T, each containing a different CM (M1–M7), and evaluated their ability to regulate the toehold switch in the presence or absence of theophylline. As is known, the stability of a double-stranded structure is positively correlated with the number of base pairs. Fewer base pairs result in lower stability and reduced strand displacement efficiency, whereas an excessive number of base pairs leads to an undesirably high background. Therefore, we kept the total base number of CM constant and adjusted the number of base pairs to achieve a high small-molecule response with a low background [28]. As shown in Fig. 3b, triggers containing the M2 or M6 module exhibited minimal sfGFP fluorescence regardless of theophylline addition, possibly because both structures contain three pairs of completely complementary base pairs. The M2 construct approached background levels, consistent with constitutive ribozyme activity, while M6 remained largely unresponsive to theophylline. In contrast, triggers containing M1, M3, M4, M5, or M7 displayed clear ligand-dependent behavior: sfGFP fluorescence was significantly reduced upon theophylline treatment, with M1, M3, M4, and M5 reducing fluorescence to near-background levels. These results confirm that aptazyme-integrated triggers can confer theophylline-dependent control over toehold-switch activation, with CM selection being crucial for regulatory performance. Appropriate base mismatches can improve the sensitivity of the system to theophylline without increasing the background.
We further characterized the dose–response relationship for the five responsive Theo-HHR-T variants (M1, M3, M4, M5, M7) by treating cells with theophylline concentrations ranging from 0 to 4 mM. As shown in Fig. 3c and Supplementary Fig. 6, sfGFP fluorescence decreased progressively with increasing theophylline concentration, demonstrating concentration-dependent inhibition of switch activation. At 4 mM theophylline, fluorescence in the M1, M3, and M4 groups approached background levels, while the M5 and M7 groups retained slightly or moderately higher signals, respectively. These data establish that embedding a theophylline aptazyme into the trigger RNA enables graded, small-molecule-controlled regulation of strand displacement in living cells, with efficiency tunable by both CM choice and ligand concentration.
To generalize the ligand-responsive strategy across different ribozyme scaffolds, we also engineered a trigger based on a second theophylline-dependent aptazyme, Theo-sTRSV, which combines the theophylline aptamer with the sTRSV self-cleaving ribozyme. The corresponding trigger RNA, Theo-sTRSV-T, was constructed by inserting the Theo-sTRSV sequence into the spacer region between the toehold and displacement domains (Supplementary Fig. 7a). In the absence of theophylline, Theo-sTRSV-T efficiently activated the toehold switch and promoted sfGFP translation. However, upon theophylline addition, ligand binding activated the embedded aptazyme, leading to self-cleavage of the trigger, disruption of its toehold-displacement continuity, and consequent loss of switch activation. As demonstrated in Supplementary Fig. 7b,c, Theo-sTRSV-T produced strong sfGFP fluorescence in the absence of ligand, which was markedly reduced following theophylline treatment (~4-fold). These results further validate that ligand-induced cleavage of engineered trigger RNAs provides a robust and generalizable mechanism to conditionally control toehold-mediated strand displacement in E. coli.
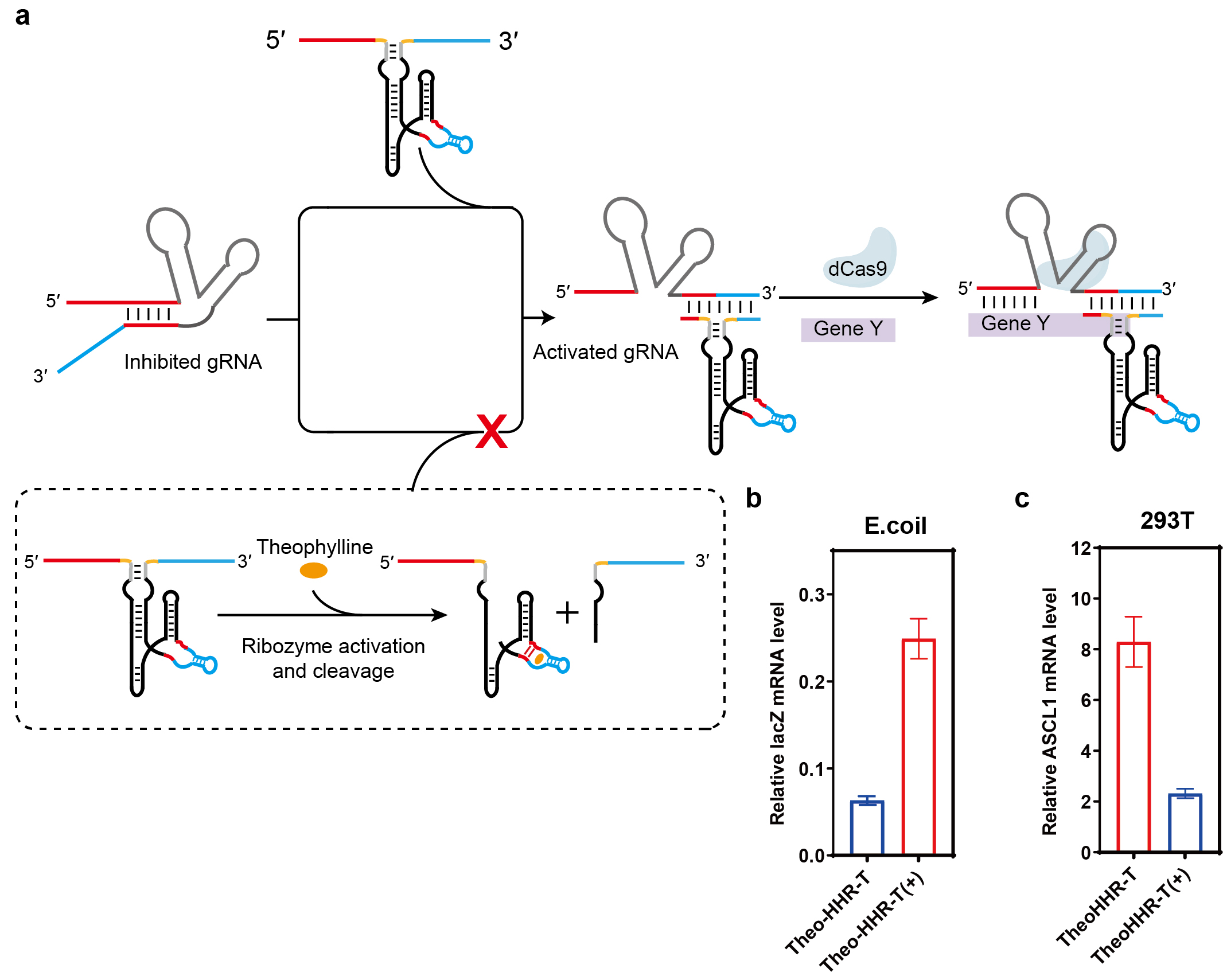
We further extended the ligand-responsive trigger strategy to regulate the CRISPR/Cas9 system via toehold-mediated activation of a gRNA-switch—an RNA device in which the guide sequence of a dead gRNA is masked and can be unmasked through strand displacement with a complementary trigger RNA (Fig. 4a). This allows conditional restoration of CRISPR activity in both interference (CRISPRi) and activation (CRISPRa) modes.
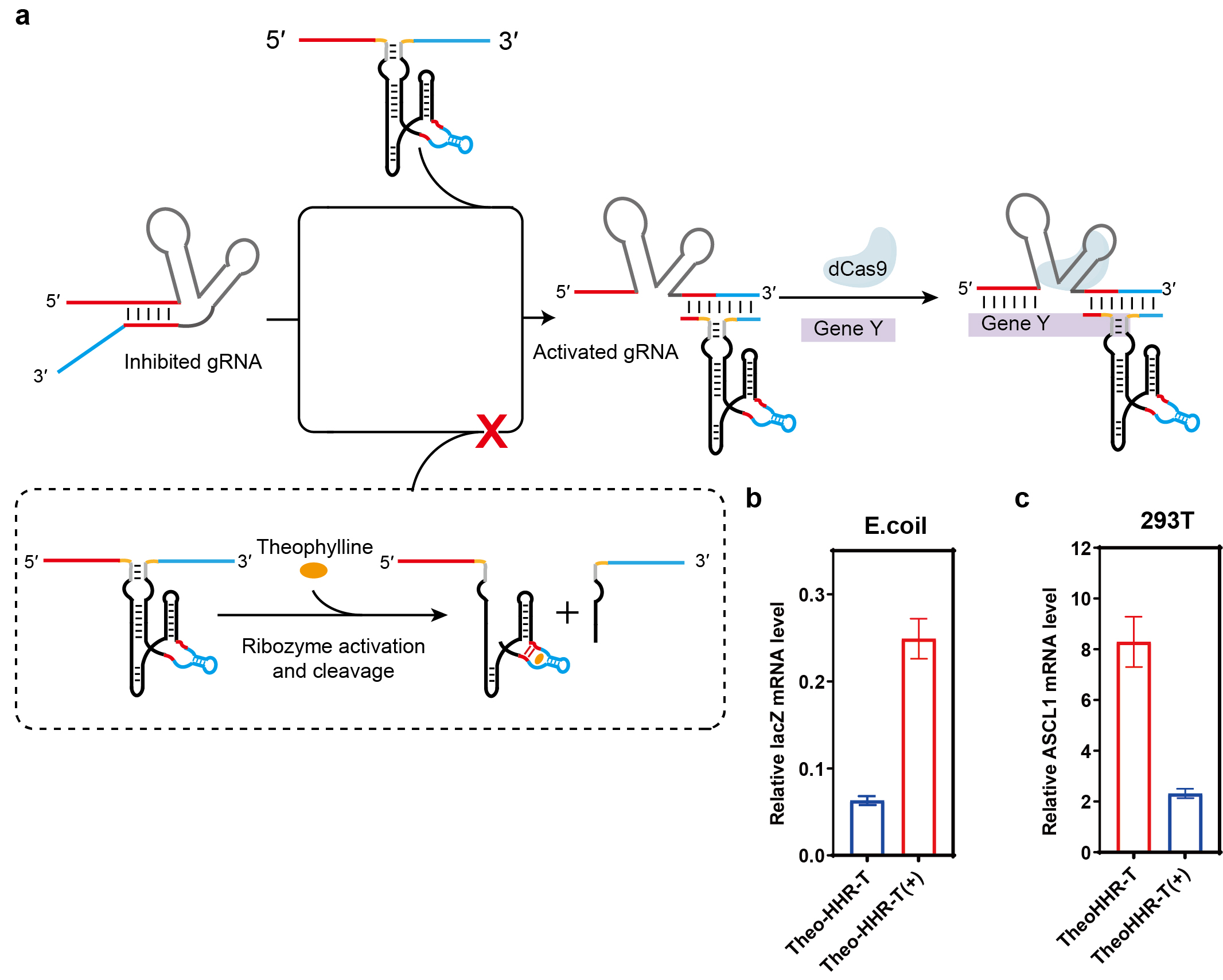 Fig. 4.
Fig. 4.
Ligand-responsive control of CRISPR-Cas9 function via
aptazyme-engineered trigger RNAs in prokaryotic and eukaryotic cells. (a)
Schematic of toehold-mediated gRNA-switch activation in the CRISPR-Cas9 system. A
dead gRNA (guide sequence masked) is converted to an active gRNA through strand
displacement with a complementary trigger RNA, restoring DNA targeting. Based on
the theophylline-responsive aptazyme trigger RNA (Theo-HHR-T). The aptazyme
(theophylline aptamer fused to HHR ribozyme) is embedded in the spacer region of
the trigger; its 5′ and 3′ ends serve as the toehold and displacement
domains for activating the gRNA-switch. (b) Regulation of CRISPR interference
(CRISPRi) in E. coli: Theo-HHR-T controls lacZ repression in a
theophylline-dependent manner. Data show relative lacZ expression normalized to a
constitutively active gRNA control. (c) Regulation of CRISPR activation (CRISPRa)
in HEK-293T cells: Theo-HHR-T controls theophylline-dependent upregulation of the
endogenous gene ASCL1. Fold activation is relative to a non-targeting control.
Data are presented as mean
First, we tested ribozyme- and aptazyme-integrated triggers for regulating CRISPRi via a gRNA-switch targeting the lacZ gene in E. coli. A trigger containing an active hammerhead ribozyme (HHR-T) was compared to a control with an inactive HHR variant (InHHR-T). As shown in Supplementary Fig. 8a, InHHR-T effectively activated the dead gRNA, repressing lacZ expression to ~7% of the level seen with a constitutively active gRNA (~6%, grey line). In contrast, HHR-T, which undergoes self-cleavage, showed markedly reduced activity, yielding ~28% lacZ expression. We then constructed a theophylline-responsive trigger (Theo-HHR-T) by embedding a theophylline aptazyme into the spacer region. In the absence of theophylline, Theo-HHR-T activated the gRNA-switch, repressing lacZ expression to ~6%. Upon theophylline addition, aptazyme cleavage inactivated the trigger, restoring lacZ expression to ~25% (Fig. 4b). These results confirm that ligand-dependent trigger cleavage can dynamically control CRISPRi activity in prokaryotic cells.
We next applied the same design principle to regulate CRISPRa in HEK-293T cells using a gRNA-switch targeting the endogenous gene ASCL1. A trigger with an active HHR (HHR-T) was again compared to an inactive control (InHHR-T). While InHHR-T robustly activated the dead gRNA, producing a 27.6-fold upregulation of ASCL1, HHR-T showed only 9.9-fold activation due to self-cleavage (Supplementary Fig. 8b). We then evaluated the theophylline-responsive trigger Theo-HHR-T. Without ligand, Theo-HHR-T activated the switch, upregulating ASCL1 by 8.3-fold. In the presence of theophylline, trigger cleavage reduced activation to only 2.3-fold (Fig. 4c), demonstrating efficient ligand-dependent control of CRISPRa in eukaryotic cells. All cell lines were validated by STR profiling and tested negative for mycoplasma.
Together, these experiments establish that embedding aptazymes into trigger RNAs provides a generalizable and transferable strategy for small-molecule regulation of toehold-mediated gRNA-switch activation across both prokaryotic and eukaryotic systems, thereby enabling precise chemical control over CRISPR-based transcriptional modulation.
Despite the successful demonstration of the LITC strategy for ligand-controlled gene expression regulation in both prokaryotic and eukaryotic cells, several limitations should be acknowledged. First, the current study relies exclusively on theophylline as the inducing ligand, a small molecule that is not endogenously present in mammalian or bacterial systems. While theophylline serves as a well-characterized model ligand for proof-of-concept validation, its exogenous administration at millimolar concentrations (up to 4 mM) may raise concerns regarding potential cytotoxicity or off-target effects in certain cellular contexts, although no overt toxicity was observed in our experimental settings. Consequently, the translational applicability of this system to therapeutic settings would require replacement of the theophylline aptamer with aptamers recognizing clinically relevant or endogenous ligands. Second, the aptazyme-based switch described here operates exclusively in a “switch-off” mode, where ligand binding attenuates trigger function. Many synthetic biology applications would benefit from both ON- and OFF-type responses, and although our previous work has established switch-on platforms, the current design does not directly enable orthogonal ON-OFF control within the same circuit without additional engineering. Third, the regulatory performance of different communication modules (CMs) varied substantially, and the sequence determinants that govern optimal ligand-dependent activation remain incompletely understood. While we identified several functional CMs (M1, M3, M4, M5, M7), the inability to predict CM behavior a priori necessitates empirical screening, which may limit the rapid deployment of this strategy for new aptamer-ribozyme combinations. Fourth, although we validated the approach in two representative cell types (E. coli and HEK-293T), the efficiency of aptazyme cleavage and toehold-mediated strand displacement may differ in other cell lines or primary cells due to variations in RNA folding kinetics, intracellular ionic conditions, or nuclease activities. Finally, the potential for unintended cleavage of endogenous RNAs by the expressed aptazyme or off-target strand displacement interactions has not been systematically evaluated. These limitations should be considered when interpreting the quantitative results, particularly the absolute fluorescence levels and fold-changes reported across different conditions. Addressing these challenges in future work—through aptamer engineering, computational CM design, and comprehensive specificity profiling—will further enhance the generalizability and safety of the LITC platform for diverse biological and therapeutic applications.
In summary, this study presents a general and programmable method, termed ligand-induced trigger RNA cleavage (LITC), for achieving precise, small-molecule-controlled strand displacement within living cells. By integrating ligand-responsive aptazymes into the spacer region of an RNA trigger strand, we established that the structural integrity and function of the trigger can be dynamically modulated by external ligands, enabling conditional activation or inactivation of toehold-mediated reactions. We systematically optimized the design, demonstrating robust regulation of two central RNA-based devices—the toehold switch for translation control and the gRNA-switch for CRISPR-Cas9-mediated transcriptional regulation—across both prokaryotic (E. coli) and eukaryotic (HEK-293T) systems.
This platform demonstrates high modular design flexibility, allowing for the customization of regulatory behaviors through the selection of different aptamer and ribozyme scaffolds and the optimization of communication modules. Additionally, it possesses favorable dose–responsiveness, enabling gradient control of gene expression based on ligand concentration. Furthermore, its design exhibits excellent cross-system portability, functioning reliably in diverse cellular environments, including both prokaryotic and eukaryotic systems. This provides a versatile and robust framework for constructing programmable gene-regulatory networks across different biological contexts.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
LX, JL, JZ and YL conceived and designed the research. CZ and HF performed the experiments. JL, LL, LX and JZ analyzed the results and data. JL, CZ and YL wrote, YL and LX revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Dr. Chongxi Yang from La Trobe University and Ziyi Li from Jihua Laboratory for language editing.
This work was supported by Guangdong Basic and Applied Basic Research Foundation (2024B1515040028), the National Natural Science Foundation of China (No. 22477144, 22222706, and 22377034), the National Key R&D Program of China (2020YFA0211200 and 2022YFC2804101), GBRCE for Functional Molecular Engineering, and Open Fund of Hubei Key Laboratory of Pollutant Analysis and Resource Technology (PA250207).
The authors declare no conflicts of interest.
During the preparation of this work the authors used Deepseek-R1 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL51369.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.