


 , Cheryl C.H. Yang 3,4,5, Yi-Chao Hsu 1,2,*
, Cheryl C.H. Yang 3,4,5, Yi-Chao Hsu 1,2,*
1 Department of Audiology and Speech-Language Pathology, Mackay Medical University, 252005 New Taipei City, Taiwan
2 Institute of Biomedical Sciences, Mackay Medical University, 252005 New Taipei City, Taiwan
3 Institute of Brain Science, National Yang Ming Chiao Tung University, 112304 Taipei City, Taiwan
4 Sleep Research Center, National Yang Ming Chiao Tung University, 112304 Taipei City, Taiwan
5 Brain Research Center, National Yang Ming Chiao Tung University, 112304 Taipei City, Taiwan
Abstract
Inositol hexaphosphate (IP6), a natural polyphosphorylated carbohydrate widely present in grains, legumes, and mammalian cells, has been increasingly recognized as a key regulator of cellular metabolism. This review examines how IP6 modulates metabolic reprogramming and plasticity in stem cells and disease states and discusses the implications of these mechanisms for translational medicine. IP6 influences central metabolic circuits, including glycolysis, mitochondrial oxidative phosphorylation, and redox balance. In stem cells, IP6 modulates energy utilization, enhances antioxidant defenses, and stabilizes pluripotency networks, thereby helping maintain self-renewal and delay premature differentiation. In pathological settings, such as cancer, IP6 acts as a metabolic checkpoint by attenuating aerobic glycolysis, modulating the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mechanistic target of rapamycin (mTOR) signaling pathway, and restoring mitochondrial integrity, which leads to growth arrest and apoptosis. These cancer-associated metabolic effects provide a framework for interpreting the IP6-mediated regulation of metabolic thresholds in stem and progenitor cells. Emerging evidence further suggests that IP6 can influence epigenetic landscapes through metabolite-dependent chromatin remodeling, thereby establishing a link between metabolic state and the transcriptional regulation of stemness and lineage commitment. In summary, IP6 is a versatile metabolic rheostat that preserves stem cell function while counteracting maladaptive metabolic reprogramming in disease states. Its pleiotropic effects are also implicated in neurodegeneration, hearing loss, and metabolic syndromes. Future studies should focus on defining IP6-regulated metabolic checkpoints, identifying direct molecular targets, and addressing pharmacokinetic and delivery challenges to help translate IP6-based strategies into regenerative and disease-modifying applications.
Keywords
- phytic acid
- metabolic reprogramming
- glycolysis
- apoptosis
- stem cell
- translational science
- biomedical
Inositol hexaphosphate (IP6), also known as phytic acid, is a highly phosphorylated derivative of myoinositol that is widely distributed in nature and particularly abundant in plant seeds, grains, and legumes, where it functions as the primary storage form of phosphorus and mineral ions [1, 2].
IP6 has attracted major attention in nutrition, cancer biology, and metabolism studies. Researchers have established that orally administered IP6 is absorbed, metabolized into lower inositol phosphates (InsPs), and distributed systemically, thereby supporting its biological activity beyond the gastrointestinal tract [3]. IP6 has also been extensively examined for its cytostatic, differentiation-associated, and chemopreventive properties in cancer, with numerous in vitro and in vivo studies reporting growth inhibition and favorable safety profiles across multiple tumor models [3]. These findings position IP6 as a naturally occurring bioactive compound with translational potential, particularly as a dietary supplement or adjunctive agent.
At the cellular level, IP6 is recognized as a central node within the broader InsP and pyrophosphate signaling network. By regulating phosphorylation and dephosphorylation reactions, IP6 influences phosphate homeostasis, cellular energy balance, and signal transduction. This network links IP6 metabolism to fundamental processes such as adenosine triphosphate (ATP) utilization, insulin signaling, and stress responses, which highlights its role as an intracellular integrator of metabolic information [4].
Recently, IP6 has attracted research attention in neurological and neurodegenerative settings. Experimental studies have reported its antioxidant activity and high metal-chelating capacity in neuronal and neurodegenerative models, with indirect evidence supporting its protective effect against oxidative-stress-associated mitochondrial vulnerability [5, 6, 7]. These effects overlap with pathways modulated by InsP metabolism, which suggests a potential relationship between IP6 signaling and stem cell metabolic regulation. Other studies involving peripheral neurons and sensory systems have further supported a broader neuroprotective profile of IP6, confirming its protective effect against toxic insults and age-related degeneration [8, 9]. These findings strongly suggest that IP6 influences both mature neurons and upstream cellular populations involved in tissue maintenance and repair.
Despite this extensive body of work, the potential relevance of IP6 to stem and progenitor cell biology has not been systematically examined [10, 11]. Stem cells are uniquely sensitive to metabolic state, redox balance, and mitochondrial integrity [12]. Although individual studies have suggested the existence of relationships among InsP signaling, cellular stress responses, and cell fate, no integrated framework linking IP6 to stem cell regulation across physiological and disease-related settings has yet been developed [10, 11]. Accumulating evidence suggests that IP6 functions not as a single-pathway effector, but as a metabolic rheostat that integrates nutrient, redox, and phosphate status to coordinate cellular stress responses and fate. Fig. 1 presents a summary of the multimodal protective mechanisms of IP6.
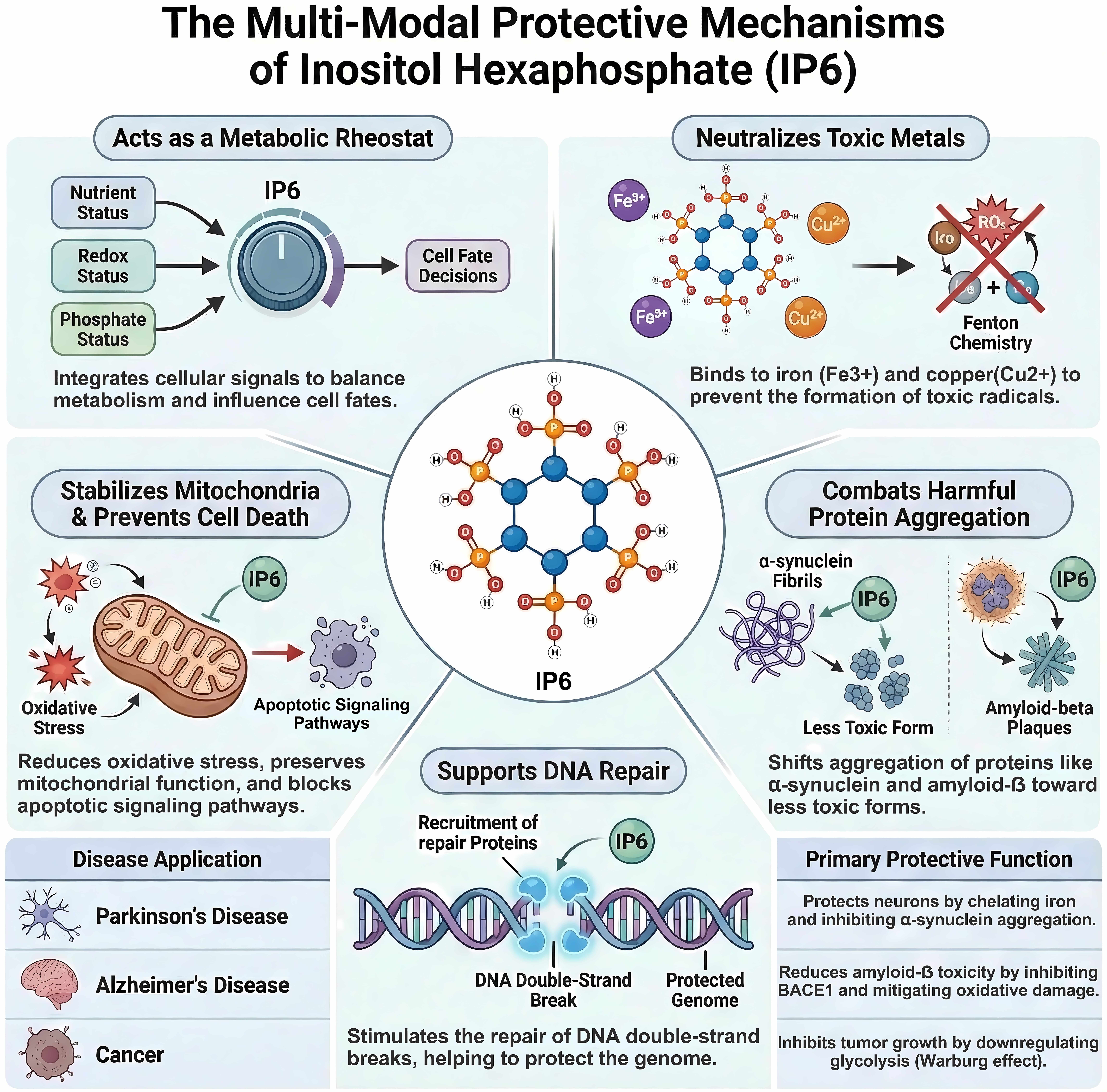 Fig. 1.
Fig. 1.
IP6 as a metabolic rheostat in multimodal protective mechanisms. Inositol hexaphosphate (IP6) acts as a metabolic rheostat integrating nutrient, redox, and phosphate cues to coordinate cellular stress responses. By modulating metabolic signaling, chelating redox-active metals, preserving mitochondrial function, supporting genome stability, and influencing proteostasis, IP6 exerts multi-modal protective effects relevant to neurodegeneration, metabolic dysfunction, and cancer. All figures were created with the assistance of NotebookLM (Google LLC, Mountain View, CA, USA), an AI-based organizational tool, and were subsequently edited and verified by the authors.
In this review, we synthesize evidence from metabolic biology, stem cell research, and disease models to provide a unified view of IP6. Instead of treating IP6 only as a dietary antioxidant or anticancer agent, we explore its emerging role as a metabolic modulator with potential relevance to stem cell plasticity, neurodegeneration, and regenerative medicine [11, 13]. By integrating findings across these traditionally separate domains, we aim to clarify whether IP6 represents not only a biological marker of metabolic state but also a candidate therapeutic or adjunctive factor in cellular stress and impaired regeneration [10].
IP6 functions as a central node in a highly conserved metabolic network that regulates phosphate homeostasis, cellular energy balance, signal transduction, and stress responses [4, 11]. IP6, historically regarded as a structural metabolite or dietary antioxidant, is currently recognized as a precursor, product, and regulator in a broad InsP signaling architecture that functions as an integrated metabolic control system [11, 13]. IP6 is naturally regulated by biosynthetic, degradative, and signaling pathways, which establishes its role as a central integrator of cellular energy metabolism, phosphate balance, mitochondrial activity, and redox homeostasis. Through tightly controlled synthesis, degradation, and conversion into inositol pyrophosphates, IP6 exerts a wide-ranging influence on metabolic flux, stress responses, and developmental programming. In addition to its metabolic and redox-regulatory roles, IP6 directly interacts with specific intracellular proteins across the cytoplasmic, nuclear, and neuronal domains, which provides a mechanistic basis for integrating metabolic state with signal transduction, gene regulation, and cellular fate control. Overall, this biochemical framework helps understand the emerging roles of IP6 in stem cell regulation, neural progenitor biology, and neuroprotection.
IP6 is synthesized in mammalian cells from myoinositol through a stepwise phosphorylation cascade catalyzed primarily by inositol polyphosphate multikinase and inositol-pentakisphosphate 2-kinase [11, 14]. This pathway sequentially converts inositol trisphosphate into inositol tetraphosphate (IP4), inositol pentaphosphate (IP5), and ultimately IP6 [4, 11]. IP6 biosynthesis is tightly coupled to cellular energetic status, which is regulated by ATP availability, nutrient conditions, and opposing phosphatase activity [4, 15]. In addition to its endogenous synthesis, IP6 can be absorbed through the gastrointestinal tract (dietary route), which in turn contributes to systemic and intracellular InsP pools [3]. Rodent and human studies have indicated that orally administered IP6 undergoes partial dephosphorylation during digestion and is subsequently detected in peripheral tissues, which indicates the role of dietary IP6 in overall InsP homeostasis instead of functioning only as a luminal chelator [2, 3, 16].
Multiple inositol polyphosphate phosphatase 1 (MINPP1) is the main enzyme responsible for the catabolism of IP6 in mammalian cells. This enzyme hydrolyzes IP6 into IP5, IP4, and lower InsPs [17]. MINPP1 maintains the levels of intracellular IP6 within a physiological range and prevents the excessive chelation of polyvalent cations. Genetic disruption of MINPP1 leads to the pathological intracellular accumulation of IP6, which in turn results in disturbed calcium homeostasis, impaired mitochondrial function, and severe defects in neural development. Pathogenic MINPP1 mutations have been identified in patients with pontocerebellar hypoplasia, which highlights the necessity of balanced IP6 synthesis and degradation for normal neuronal and cerebellar development. Overall, these findings underscore the importance of maintaining the balance between IP6 synthesis and degradation, particularly in neural tissues [17].
In addition to its role as an endpoint metabolite, IP6 serves as an obligate precursor for the synthesis of inositol pyrophosphates [10, 11]. IP6 kinases (IP6K1, IP6K2, and IP6K3) phosphorylate IP6 to generate 5-diphosphoinositol pentakisphosphate (IP7), whereas diphosphoinositol pentakisphosphate kinases (PPIP5Ks) catalyze the formation of 1-diphosphoinositol pentakisphosphate and bis-diphosphoinositol tetrakisphosphate (IP8) [14, 18]. These molecules contain high-energy phosphoanhydride bonds and function as potent regulators of cellular metabolism and signaling [4, 14, 18].
Genetic and biochemical studies have indicated that IP6K1- and IP6K2-derived diphosphoinositol pentakisphosphate strongly influences glycolytic flux, mitochondrial respiration, and adenosine triphosphate (ATP)-to-adenosine diphosphate (ADP) ratios, partly through protein pyrophosphorylation and key metabolic enzyme modulation [4, 15]. IP6K1 deletion alters systemic energy expenditure, insulin sensitivity, and mitochondrial homeostasis, which underscores the physiological importance of IP6-derived pyrophosphates in whole-body metabolic regulation [4, 19]. In neural settings, IP6K2-dependent IP7 production regulates transcriptional programs in the neurons and glia, which establishes a direct link between PP-InsP metabolism and neural development and function [11, 20].
Through its conversion into inositol pyrophosphates, IP6 integrates multiple core metabolic pathways and stress-responsive signaling axes to coordinate cellular energy balance with nutrient availability. IP6-derived IP7 functions as a metabolic brake under energy-replete conditions by inhibiting protein kinase B (PKB) or serine/threonine-protein kinase (Akt)-dependent insulin and growth-factor signaling, which in turn constrains glycolytic and mitochondrial energy metabolism and limits excessive ATP production [15, 19, 21]. Hence, IP6K1 genetic deletion enhances oxidative phosphorylation efficiency and increases whole-body energy expenditure, highlighting the role of IP6/IP7 signaling in the control of mitochondrial and systemic metabolism [19, 21]. Inositol pyrophosphates directly participate in phosphate and ATP homeostasis by binding SYG1/PHO81/XPR1 (SPX)-domain-containing proteins, which in turn enables the cells to couple intracellular phosphate transport to the energetic state [22]. At the signaling level, IP6K1-generated IP7 modulates insulin and growth-factor pathways by inhibiting Akt phosphorylation, which ensures a balance between anabolic signaling and metabolic capacity [19, 21]. These functions position IP6 and its pyrophosphate derivatives at the core of nutrient sensing, energy demand, and stress adaptation—metabolic parameters that play a central role in the regulation of stem cell quiescence, activation, and lineage commitment.
In addition to exogenous IP6 supplementation, direct pharmacological modulation of endogenous inositol pyrophosphate metabolism provides a mechanistically coherent translational approach. IP7 functions as a physiological inhibitor of Akt signaling, and IP6K-dependent regulation of IP7 levels therefore constitutes a regulatory node linking nutrient sensing to energy homeostasis and stress responsiveness [21]. In stem-cell–relevant settings, dysregulated inositol pyrophosphate signaling has been associated with age-related vulnerability and impaired Akt activation [23], whereas pharmacological or genetic inhibition of IP6Ks has enhanced engraftment efficiency and therapeutic performance in transplantation-based models [24]. Together, these findings indicate that exogenous IP6 administration and targeted manipulation of the IP6K/IP7 axis may operate as complementary strategies for recalibrating metabolic and stress-responsive signaling networks. However, extension of such approaches to neural or regenerative applications will require careful evaluation of cell-type specificity, developmental timing, and long-term systemic consequences [11, 25].
In addition to its phosphorylation-dependent signaling roles, IP6 functions as a
redox buffer and polyvalent metal chelator [7, 13]. By binding ferric iron
(Fe3+) and copper (Cu2+), IP6 limits Fenton chemistry and reduces
hydroxyl radical generation [7]. Biochemical and biophysical research has
indicated that IP6 protects dopamine and ascorbate against Fe3+-catalyzed
oxidation and modulates
In addition to its role as a metabolic intermediate and metal chelator, IP6 directly binds multiple intracellular proteins, supporting its function as an active signaling ligand relevant to stem cell maintenance and neural lineage regulation [26, 27, 28, 29].
A well-defined example of this protein-binding function is adenosine deaminase acting on RNA (ADAR), wherein IP6 is tightly bound in the catalytic core and is required for proper protein folding and RNA-editing activity—a process that contributes to transcriptome diversification and stress adaptation in neural progenitors and differentiating neurons [30]. Consistent with its broader role in gene expression control, IP6 synergistically functions with Gle1 (mRNA export mediator) to activate the DEAD (Asp-Glu-Ala-Asp)-box helicase 5 (Dbp5), which in turn drives nuclear mRNA export and establishes a link between intracellular IP6 levels and posttranscriptional regulation [31].
In the nucleus, IP6 interacts with ATP-dependent chromatin-remodeling complexes such as Switch/Sucrose Non-Fermentable complex (SWI/SNF) for nucleosome remodeling and transcriptional regulation, linking metabolic state to epigenetic programs relevant to stem cell plasticity and neural differentiation [32]. IP6 also acts as a metabolite-dependent regulator of Cullin–RING ubiquitin ligases through the constitutive photomorphogenesis 9 (COP9) signalosome, which establishes a link between InsP metabolism and both protein turnover and signaling robustness in proliferative and differentiating cells [28, 29].
In the cytoplasm, IP6 and its related soluble InsPs interact with pleckstrin homology domains to competitively modulate phosphoinositide binding, with implications for PI3K–Akt signaling, a pathway central to stem cell survival, proliferation, and lineage commitment [33, 34]. IP6 also binds signaling scaffolds such as nonvisual arrestins, regulating their oligomerization and subcellular localization and shaping receptor-proximal signaling dynamics [26]. Moreover, IP6 binds cytoskeletal regulatory proteins such as gelsolin and profilin and modulates actin filament dynamics—processes broadly implicated in the regulation of cell polarity and migration [35, 36]. In hippocampal neurons, IP6 binds the second C2 (C2B) domain of synaptotagmin-1 to suppress excitatory neurotransmission, confirming that IP6–protein interactions extend to activity-dependent neural signaling [27].
These interactions provide a mechanistic framework that links intracellular IP6 availability to phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mechanistic target of rapamycin (mTOR) signaling, ubiquitin-mediated protein turnover, metabolic reprogramming, cytoskeletal remodeling, and chromatin accessibility—key pathways highlighted in this review as central regulators of stem cell fate and neural resilience.
Emerging evidence suggests that IP6 and its downstream pyrophosphate metabolites actively reprogram stem cell metabolism, stress responses, and fate [10, 11, 23, 37, 38]. As presented in Table 1 (Ref. [17, 23, 24, 37, 38, 39, 40]), recent studies involving distinct stem cell systems have indicated that IP6-related interventions modulate stem cell fate and function in a context-dependent manner, shaped by disease-relevant metabolic and stress conditions rather than by uniform, lineage-instructive effects. Instead of acting as a simple antioxidant or static metabolite, IP6 functions within a dynamic InsP network whose impact on stem cells depends on intracellular concentration, enzymatic flux through IP6K/PPIP5K pathways, and cellular metabolic or pathological settings [11, 25, 41]. This section synthesizes evidence obtained from studies involving mesenchymal stem cells and related progenitor systems to explain how IP6/IP7 signaling reshapes stem cell behavior in aging, metabolic stress, and regenerative settings [37, 38].
| Stem cell category | Model/condition | Context (disease) | IP6 intervention | Key metabolic/molecular effects | Functional outcomes | Ref. |
| Mesenchymal stem cells (BMSC/MSC) | Human BMSCs under high-glucose stress (diabetic-like microenvironment) | Diabetic osteoporosis/impaired bone regeneration under hyperglycemia | IP6 supplementation (in vitro + in vivo) | ↓ ROS; improved mitochondrial function; anti-senescence (↓ p21/p53); ERK pathway activation; circEIF4B–miR-186-5p axis (FOXO1) and IGF2BP3/ITGA5 stabilization | Rescued osteogenesis and mineralization; reduced high-glucose-induced senescence; improved bone regeneration in a diabetic model | [37, 38] |
| Mesenchymal stem cells (BMSC/MSC) | Mouse BMSCs: young vs aged donors (aging-associated decline) | Aging-related MSC dysfunction | IP6K inhibition (↓ IP7/inositol pyrophosphates) | Relief of Akt inhibition (↑ Akt phosphorylation); enhanced mitochondrial survival pathways | ↓ apoptosis; ↑ viability; improved regenerative potential in aged MSCs | [23] |
| Mesenchymal stem cells (BMSC/MSC) | MSC transplantation into the infarcted mouse heart (myocardial infarction) | Cell therapy optimization for MI | IP6K inhibition (↓ IP7) in MSCs prior to transplant | ↑ Akt activation; improved functional survival; enhanced paracrine effect (reported) via down-regulating IP7 | ↑ engraftment and therapeutic efficacy after transplantation | [24] |
| Pluripotent stem cell–derived neural lineage | Patient-derived and genome-edited MINPP1−/− iPSCs and differentiating neurons | Neurodevelopmental disorder/emphasizes the need for IP6 homeostasis | Genetic IP6 accumulation due to impaired degradation (MINPP1 deficiency) | Accumulation of highly phosphorylated inositols (IP6), disturbed Ca2+ homeostasis and ER stress, and altered inositol phosphate balance | Inefficient neuronal differentiation; increased cell death | [17] |
| Neural stem cells (NSCs) + biomaterials | Spinal cord injury (SCI) model with NSC therapy + Ca-phytate nanoparticles | Regenerative medicine: adjunct to NSC transplantation | Ca-phytate (Ca-IP6) nanoparticles to trigger intracellular ion/small-molecule release; combined with NSC therapy | Reported intracellular small molecule/ion storm’ to promote neuroregeneration and immunomodulation | Synergized NSC therapy; improved functional recovery in the SCI model | [39] |
| Dental pulp stem/progenitor cells (DPSC) | Dentine conditioning/regenerative endodontics in vitro | Dental tissue regeneration/scaffold preparation | Phytic acid (IP6) as dentine chelator/conditioner (vs EDTA/HEDP) | Effective TGF- |
Supports DPSC migration/viability for regenerative endodontics workflows | [40] |
Chronic hyperglycemia, as observed in diabetes, strongly impairs the regenerative capacity of bone-marrow-derived mesenchymal stem cells (BMSCs), in turn reducing osteogenic differentiation, diminishing mineralization, and accelerating cellular senescence [42, 43]. These changes contribute to diabetic bone fragility and impaired skeletal repair [43]. Mechanistically, high glucose concentrations increase oxidative stress, disrupt growth-factor signaling, and skew cellular metabolism toward senescence-associated states [43].
Recent studies have indicated that supplementation with phytic acid can reverse
these pathological changes [37, 38]. As presented in Table 1, IP6 restores the
osteogenic differentiation of BMSCs under high-glucose conditions, as evidenced
by increased alkaline phosphatase activity, enhanced matrix mineralization, and
osteogenic gene upregulation [37, 38]. These findings confirm that IP6 increases
the capacity of bone formation in diabetic or hyperglycemic settings [37, 38]. At
the cellular level, IP6 suppresses high-glucose-induced senescence by reducing
reactive oxygen species (ROS) generation, lowering senescence-associated
These findings indicate that IP6 can reprogram BMSCs’ metabolism under pathological glucose stress conditions, which causes the cells to shift away from senescence and toward a regenerative, osteogenic state. The findings also suggest that phytic acid engages defined stress-responsive signaling (e.g., ERK) alongside redox modulation, instead of acting only as a nonspecific antioxidant (Table 1).
In addition to hyperglycemic stress, aging represents a distinct metabolic challenge for stem cells, characterized by diminished survival, impaired paracrine function, and reduced regenerative potential. Aged BMSCs were shown to produce an increased amount of the inositol pyrophosphate IP7, driven by increased IP6K1 activity. This shift correlates with reduced Akt phosphorylation, heightened susceptibility to apoptosis under hypoxic or serum-deprived conditions, and compromised stem cell function [23].
Pharmacological inhibition of IP6K activity, which lowers IP7 levels without eliminating IP6, restores Akt signaling and substantially extends the survival of aged BMSCs [23]. These effects support a model in which excessive flux from IP6 toward IP7 acts as a metabolic brake on stem cell survival pathways during aging. Overall, these findings indicate that IP6/IP7 balance, not absolute IP6 abundance, is a key determinant of stem cell fate.
In addition to mesenchymal stem cells, the InsP and pyrophosphate network broadly influences cellular processes central to stem cell biology, including proliferation, cytoskeletal dynamics, migration, transcriptional regulation, and metabolic homeostasis [10, 25, 41]. Through these mechanisms, IP6K activity and downstream PP-InsPs act as molecular switches that bias cells toward quiescence, proliferation, differentiation, or apoptosis, depending on intracellular metabolic state and environmental cues [25, 41].
Notably, the effects of IP6/IP7 signaling are highly dependent on context and cell type [11, 25]. Distinct IP6K isoforms (IP6K1, IP6K2, and IP6K3) and PPIP5Ks exhibit tissue-specific expression, subcellular localization, and regulatory interactions, which lead to the differential control of IP7 and IP8 pools [25, 44]. This specificity provides a biochemical basis for the selective modulation of stem cell fate across diverse progenitor populations and developmental stages [11, 25].
IP6 functions as a metabolic rheostat in stem cells [37, 38]. Under metabolic stress conditions, such as hyperglycemia or aging, altered InsP or pyrophosphate signaling, including elevated IP7 in aged BMSCs, suppresses prosurvival signaling pathways, reduces differentiation capacity, and promotes senescence or apoptosis (Table 1) [21, 23, 37, 38]. By contrast, supplementation with IP6 or modulation of IP6K activity rebalances InsP flux, restores metabolic homeostasis, and promotes survival and differentiation [23, 25, 37, 38]. This rheostat model emphasizes that the biological effects of IP6 are governed not only by its presence but also by its controlled conversion in the InsP network [11, 15, 25, 41]. This context-dependent regulatory pattern provides a biochemical explanation for various observations across stem cell systems, including diabetic osteogenesis rescue, aging-associated dysfunction mitigation, and potential extension to neural progenitor protection and regenerative applications [11, 23, 25, 37, 38].
Emerging evidence suggests that the functional relevance of IP6 is not limited to intrinsic metabolic regulation in a single stem cell type. IP6 has also been used in distinct stem cell-based regenerative workflows as a microenvironment-conditioning factor to modulate matrix-associated signaling and support stem cell viability and migration. This application potential underscores the broader role of IP6 in shaping stem cell-permissive niches rather than directly determining lineage fate [40].
As presented in Table 1, the functional consequences of IP6-related interventions are best interpreted in the context of disease- or stress-specific conditions, including metabolic overload, aging, ischemia, and neurodevelopmental vulnerability, rather than as uniform stem cell-intrinsic effects.
Although cancer is not the primary focus of stem cell-based regeneration, tumor models provide a clear framework for examining how IP6 limits abnormal metabolic activity and cell survival. Recent cancer metabolism studies have redefined IP6 from a dietary compound to a low-toxicity adjunct capable of simultaneously modulating growth signaling and metabolic stress [3, 45, 46]. Notably, many cancer cells share metabolic features with stressed or maladapted stem and progenitor cells, including sustained activation of growth-promoting pathways, elevated glycolytic flux, and enhanced resistance to oxidative stress. These features enable cancer systems to serve as mechanistic reference models for IP6-driven metabolic regulation across different cellular contexts [47].
IP6 suppresses uncontrolled cellular proliferation in cancer by limiting PI3K/Akt/mTOR signaling and its associated metabolic reprogramming (Fig. 2). IP6 treatment in multiple tumor cell types has also been linked to reduced Akt phosphorylation, downstream mTOR inhibition, and apoptotic and autophagic cell-death program activation, collectively shifting cells away from highly stress-tolerant, prosurvival states [3, 45]. Because this pathway also integrates glucose metabolism, anabolic growth, and redox buffering, its suppression provides a mechanistic bridge between growth inhibition and metabolic state alteration [47, 48].
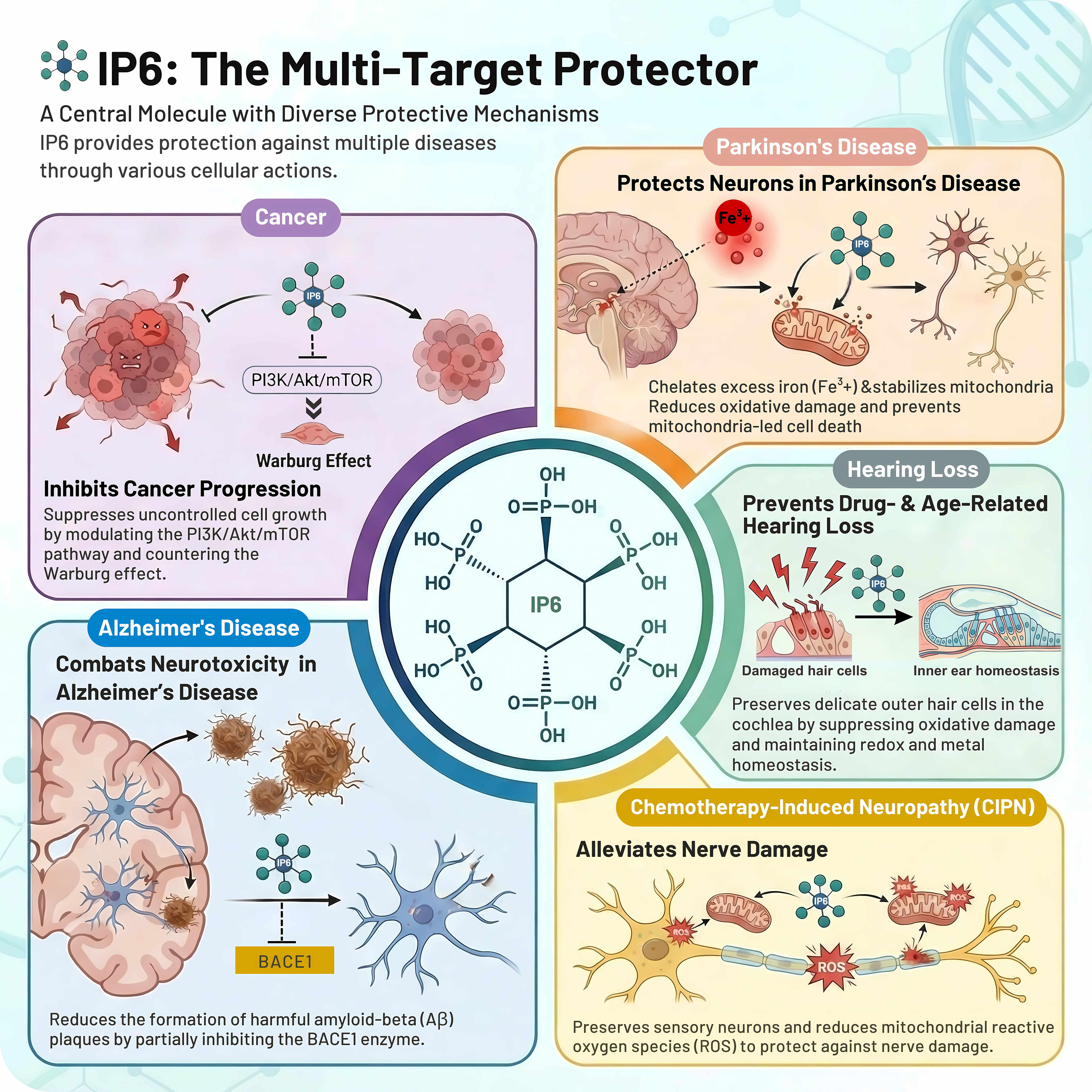 Fig. 2.
Fig. 2.
Diverse protective mechanisms of IP6 in different disease
settings. IP6 acts as a context-dependent modulator of metabolic and stress
responses in various disease models. In cancer, IP6 suppresses uncontrolled
cellular proliferation by inhibiting PI3K/Akt/mTOR signaling and counteracting
Warburg-like metabolic reprogramming, which in turn promotes apoptotic and
autophagic clearance. In neurodegenerative settings, such as in Parkinson’s
disease (PD) and Alzheimer’s disease (AD), IP6 exerts a neuroprotective effect by
chelating excess redox-active metals, stabilizing mitochondrial function,
reducing oxidative stress, and partially inhibiting amyloidogenic processing
through
PI3K/Akt/mTOR activity attenuation by IP6 has been associated with reduced reliance on Warburg-like aerobic glycolysis, which lowers metabolic flexibility and stress-buffering capacity and increases vulnerability to chemotherapeutic or oxidative challenges [45, 49]. Supporting evidence from bioengineering and nanomedicine approaches, including phytate-based metal coordination platforms and nanozyme formulations, suggests that IP6-based strategies can simultaneously influence redox balance, mitochondrial function, and glycolytic flux, with reported reductions in glucose consumption and lactate production in lung cancer and its related models [50].
Clinical translational evidence remains limited and largely supportive rather than curative. A randomized trial involving patients with breast cancer receiving chemotherapy reported an improvement in quality of life and a reduction in treatment-associated side effects with combined IP6 and inositol supplementation, supporting the feasibility of IP6 as a low-toxicity adjunct [51]. Despite these findings, whether IP6 can function as a standalone anticancer intervention remains unclear, and larger-scale trials are required to further validate the dose–exposure relationships, pharmacokinetics, and pathway-specific biomarkers of IP6 [46, 51]. Overall, these findings support the metabolic rheostat model proposed earlier.
Cancer represents an extreme state of metabolic dysregulation, which contrasts with the plasticity and reversibility of stem cells along this continuum [48, 52]. Therefore, the ability of IP6 to limit anabolic and survival signaling in cancer conceptually supports its role in modifying metabolic thresholds in stem and progenitor cells, which in turn shapes survival, differentiation capacity, and stress tolerance indirectly through metabolic control rather than direct lineage instruction. This cross-system perspective provides valuable insights into neural progenitors and neurodegenerative vulnerability.
Neural stem cells (NSCs) reside in metabolically demanding niches, where mitochondrial integrity, redox homeostasis, and genome maintenance considerably influence survival and lineage outcomes [53, 54]. Disrupting these processes, particularly under aging or disease-associated stress conditions, profoundly alters the fate of NSCs [53]. Emerging evidence suggests that IP6 and its metabolic derivatives are involved in these processes, which provides a mechanistic framework for preserving NSC function and enhancing neuroprotective effects under stress, similar to the IP6-driven metabolic reprogramming observed in other stem cell systems [11, 23, 55]. In neural systems, IP6 functions as a multifaceted regulator of mitochondrial integrity, redox homeostasis, metal buffering, DNA repair, and neural progenitor gene expression by integrating into the InsP and pyrophosphate metabolic network [11, 55]. These functions position IP6 as a metabolic rheostat capable of enhancing NSC resilience and influencing lineage outcomes during development, aging, and neurodegeneration [11].
Although NSCs predominantly rely on glycolysis during quiescence, their
activation and neuronal differentiation are accompanied by metabolic remodeling
toward mitochondrial oxidative phosphorylation [53]. Perturbations in
mitochondrial function or redox balance promote the astrocytic differentiation,
suppress the neurogenesis, or trigger the apoptosis of NSCs, which highlights the
role of metabolism as a central regulator of NSC fate [56]. In disease states,
amyloid-
According to the literature, IP6 binds DNA-dependent protein kinase and stimulates nonhomologous end-joining-mediated DNA double-strand break repair in mammalian systems, and it also strongly attenuates oxidative stress in neuronal models [58, 59]. Although studies examining the direct interaction between IP6 and NSCs remain limited, these properties provide a mechanistic rationale for IP6 acting as a metabolic and genome-protective factor in NSCs under stress [13, 55].
In neuronal models of Parkinson’s disease (PD), IP6 reduces ROS production,
preserves mitochondrial membrane potential, and suppresses apoptotic signaling
cascades—features that overlap with the key metabolic vulnerabilities of NSCs
[58, 60]. IP6 also exhibits a high ferric iron chelation capacity, which limits
iron-catalyzed oxidative reactions and modulates
The metabolic conversion of IP6 into inositol pyrophosphates has profound effects on neural development and progenitor regulation [11, 25]. In the enteric nervous system, IP6K2-dependent IP7 production regulates neuronal and glial gene expression, and genetic IP6K2 disruption alters the composition of neural progenitors [44]. IP6K1 has also been implicated in cytoskeletal organization and focal adhesion, which are essential for neuronal migration during cortical development, suggesting that IP6/IP7 signaling influences the spatial patterning of neural progenitors [11, 41]. MINPP1 loss is an example of the pathological consequences of IP6 metabolism disruption. This process leads to intracellular IP6 accumulation, calcium dysregulation, endoplasmic reticulum stress, and severe neurodevelopmental disorders, such as pontocerebellar hypoplasia (Table 1). Together, these findings indicate that both excessive IP6 accumulation and insufficient inositol pyrophosphate signaling impair neural progenitor development, which highlights the need for a balanced IP6–IP7/IP8 metabolic axis [17, 44].
Although direct investigations of IP6 in neural stem or progenitor cells and advanced neural culture systems remain limited, evidence from related neural and sensory cell models supports the translational relevance of IP6-mediated metabolic modulation [11, 55]. IP6 protects peripheral neurons against platinum-induced neurotoxicity by preserving mitochondrial function and reducing oxidative stress [8]. It also protects cochlear hair cells against chemotherapeutic, ototoxic, and age-related degeneration in vivo [9]. Because of the shared reliance of NSCs, peripheral neurons, and sensory cells on mitochondrial stability and redox balance, these findings are consistent with the metabolic rheostat model described earlier and suggest potential applications of IP6 as a preconditioning agent for NSC transplantation, as an additive to brain or otic organoid cultures, or as a metabolic modulator to bias NSC fate toward neuronal lineages [8, 9, 11].
IP6 has been evaluated in diverse neurodegenerative and neurological disease models, predominantly in cellular and preclinical settings [1, 55, 59]. Current evidence supports pleiotropic neuroprotective effects such as redox–metal buffering, mitochondrial stabilization, apoptotic signaling suppression, and pathogenic protein aggregation modulation, with an additional layer of metabolic control through the InsP or pyrophosphate network [7, 11, 58, 59]. Fig. 2 presents a summary of the disease-specific protective effects of IP6 on cancer, neurodegenerative disorders, sensory degeneration, and chemotherapy-associated neuropathy.
PD is characterized by mitochondrial dysfunction, oxidative stress, iron
dyshomeostasis, and pathological
Cellular models of PD commonly involve dopaminergic cell lines such as human neuroblastoma cell line (SH-SY5Y) and 1RB3AN27 rat dopaminergic neuronal cell line (N27) exposed to mitochondrial and oxidative toxins such as 6-hydroxydopamine, which induces dopamine redox cycling, ROS accumulation, mitochondrial membrane depolarization, and intrinsic apoptotic cascade activation. In these systems, IP6 (administered as phytic acid) consistently attenuates toxin-induced neuronal injury. Protective phenotypes include reduced intracellular ROS and lipid peroxidation, mitochondrial membrane potential preservation, reduced cytochrome-c release, Bax/Bcl-2 ratio normalization, and caspase-dependent apoptosis suppression, which together improve dopaminergic cell survival [58, 60].
These effects target the mitochondria–apoptosis axis, which represents a primary vulnerability in PD pathology. Instead of acting only as a radical scavenger, IP6 appears to stabilize mitochondrial function upstream of irreversible apoptotic commitment, which in turn mitigates downstream execution pathways. This pattern is consistent with IP6 modulating stress thresholds in dopaminergic neurons under toxin-induced metabolic overload conditions, which aligns with the broader concepts of metabolic buffering in neurodegenerative settings [58, 59].
Iron accumulation and redox-active metal dysregulation are increasingly recognized as amplifiers of dopaminergic neurodegeneration. Ferric iron (Fe3+) catalyzes the oxidation of dopamine and other cellular metabolites, which generates ROS and promotes oxidative damage to lipids, proteins, and mitochondrial DNA [61]. Because of its highly phosphorylated structure, IP6 exhibits a high Fe3+-chelating capacity and can limit iron-catalyzed redox reactions. Biochemical and biophysical research has indicated that IP6 suppresses the Fe3+-mediated oxidation of dopamine and ascorbate, which in turn mitigates ROS burden at its source [7]. These neuroprotective effects, including iron chelation and mitochondrial stabilization in PD systems, are summarized in Fig. 2.
In addition to redox chemistry, iron availability influences the aggregation
kinetics and conformational landscape of
Integrative reviews of PD-related IP6 research highlight consistent protective
mechanisms observed across cellular, biochemical, and limited in vivo
settings. These mechanisms include the buffering of redox balance and metal
homeostasis, the stabilization of mitochondrial integrity, the attenuation of
apoptotic signaling, the modulation of protein aggregation dynamics, and the
dampening of stress-activated inflammatory pathways, such as NF-
These findings are consistent with the metabolic rheostat framework described earlier, in which IP6-mediated redox–metal buffering and mitochondrial stabilization interact with broader InsP or pyrophosphate signaling to shape stress resilience and degenerative trajectories [7, 11].
AD pathology emerges from the interaction between amyloidogenic processing, metal-dependent oxidative stress, mitochondrial dysfunction, and progressive impairment of neuronal and progenitor cell resilience. Although fewer studies have directly examined the effects of IP6 in AD than in PD, evidence supports several mechanistic entry points through which IP6 may modulate AD-relevant stress pathways.
During amyloid precursor protein processing, IP6 and its related InsPs function
as inhibitors of BACE1, a key enzyme that initiates the amyloidogenic cleavage of
amyloid precursor protein. In cell-based assays, IP6 reduces BACE1 activity and
subsequent A
In addition to its enzymatic inhibitory effect, phytic acid has been explored in
preclinical research as a broader modulator of AD pathology. In animal and
in vitro AD models, supplementation with IP6 attenuates
A
A
Although direct in vivo studies on the interactions between IP6,
A
Various preclinical studies have examined the effects of phytic acid in AD
models beyond enzymatic and cellular assays. Research involving
A
Evidence suggests that IP6 can function as a disease-dependent stress modulator in amyloidogenic processing, metal-dependent oxidative injury, and mitochondrial vulnerability. These mechanisms mirror those implicated in neural progenitor dysfunction under AD-associated stress conditions, which reinforces the conceptual link between neurodegeneration and stem cell metabolic regulation discussed in the previous sections [7, 11, 57, 59, 61].
Although the majority of studies on the neuroprotective effects of IP6 have focused on domains related to the central nervous system (CNS), accumulating evidence suggests that peripheral neurons and sensory cells can help evaluate IP6-mediated metabolic and redox protection (Fig. 2). These cell types share key vulnerabilities with central neurons and neural stem or progenitor cells, including high energetic demand, limited antioxidant reserve, and pronounced sensitivity to mitochondrial and oxidative stress [53, 54, 59]. Hence, peripheral and sensory models provide a crucial translational bridge between cellular mechanisms and system-level neuroprotection.
Chemotherapy-induced peripheral neuropathy (CIPN) is a common and dose-limiting complication of platinum-based chemotherapeutic agents, such as cisplatin and oxaliplatin. Its pathophysiology is characterized by mitochondrial dysfunction, excessive ROS generation, calcium dysregulation, and intrinsic apoptotic pathway activation in dorsal root ganglion (DRG) neurons. In contrast to many types of central neurons, DRG neurons lack robust protective barriers and exhibit a limited capacity to compensate for sustained mitochondrial stress, which renders them particularly vulnerable to metabolic and redox insults [8, 59].
IP6 treatment considerably attenuates platinum-induced neurotoxicity in cultured DRG neuronal systems. Mechanistically, IP6 reduces mitochondrial ROS accumulation, preserves mitochondrial membrane potential, and improves neuronal survival under chemotherapeutic stress conditions. These protective effects are accompanied by the stabilization of mitochondrial bioenergetics and mitigation of oxidative damage, rather than direct interference with chemotherapeutic uptake or cytotoxic efficacy [8]. This distinction is crucial from a translational standpoint because it suggests that IP6 acts by raising neuronal stress tolerance thresholds rather than by antagonizing anticancer mechanisms.
In CIPN models, IP6 targets the conserved mitochondrial redox failure modes shared across neuronal subtypes. Its ability to preserve mitochondrial integrity under acute toxic conditions aligns with its effects in dopaminergic PD models and supports its general role as a metabolic buffer in neurons exposed to high oxidative and energetic stress [8, 11, 59].
Cochlear sensory hair cells represent a highly specialized and metabolically active neuronal cell type characterized by sustained mitochondrial activity, high calcium flux, and vulnerability to oxidative damage. Ototoxic drugs such as cisplatin and aminoglycosides and aging-related stress increase mitochondrial ROS overproduction, impair antioxidant defenses, and activate apoptotic signaling pathways, which lead to irreversible hair cell loss and progressive hearing impairment [9].
In vivo research indicates that IP6 protects cochlear hair cells against drug-induced ototoxicity and age-related degeneration, which helps preserve hair cell integrity and auditory function. These protective effects are associated with reduced oxidative stress markers and improved mitochondrial resilience in the cochlea, consistent with IP6-mediated stabilization of redox and energy homeostasis rather than the regeneration of lost cells [9]. In this context, the efficacy of IP6 underscores its capacity to act within complex tissue environments subject to chronic metabolic stress.
Both sensory hair cells and neural stem or progenitor cells rely on tight mitochondrial regulation and redox balance to maintain their viability and functional identity. Therefore, IP6-mediated protection in cochlear systems confirms the ability of IP6 to modulate stress sensitivity across diverse neural lineages, including progenitor populations exposed to inflammatory, toxic, or age-associated microenvironments [11, 53].
Evidence from peripheral neuropathy and sensory degeneration models confirms that IP6 confers neuroprotective effects by buffering conserved mitochondrial and redox stress pathways. These findings provide a mechanistic rationale for extending IP6-based conditioning strategies to neural stem or progenitor cells and regenerative platforms under comparable stress conditions [8, 9, 11, 53, 59].
IP6 neuroprotection across disease models can be grouped into three related
mechanisms that are also relevant to its translational potential. First, IP6
buffers redox–metal stress by chelating iron and other redox-active metals,
which in turn limits Fenton chemistry, reduces oxidative damage, and exerts a
secondary effect on metal-dependent aggregation processes such as
Because of the multifactorial nature of neurodegenerative disorders and the limited success of single-target interventions, IP6 is positioned as an adjunctive modulator rather than a standalone disease-modifying agent. In the disease models discussed earlier, IP6 consistently lowers oxidative and metal-associated stress and stabilizes mitochondrial function—processes that act upstream of irreversible neuronal injury [7, 58, 59, 60]. In AD settings, IP6 is linked to antiamyloidogenic mechanisms through BACE1 inhibition in cell-based systems [6] and to improved pathological or oxidative indices in preclinical models [5, 59]. In primary DRG neurons, phytic acid reduces mitochondrial ROS and preserves membrane potential under platinum neurotoxicity conditions. Further in vivo research is required to determine whether neuroprotection can be achieved without compromising the anticancer efficacy of phytic acid [8]. In sensory systems, in vivo evidence from auditory pathways demonstrates that IP6 preserves cochlear hair cells and attenuates hearing deficits in mice, highlighting shared mitochondrial and redox vulnerabilities between sensory and central neurons [9].
Despite encouraging preclinical evidence, the optimal dosing, long-term safety, and potential consequences of sustained metal chelation, particularly in the case of essential trace element homeostasis, remain incompletely defined [63, 64]. In addition, much of the currently available evidence is derived from in vitro, biochemical, or short-term preclinical systems, with comparatively limited validation across diverse disease-relevant in vivo settings [13, 55, 59]. Further translational research is required to explore the pleiotropic profile of IP6, particularly during early or presymptomatic disease stages and in contexts such as CIPN, provided that bioavailability and mineral balance are carefully monitored [59, 62]. In the future, developing IP6 derivatives and targeted delivery strategies and facilitating the pharmacological modulation of the endogenous IP6/IP7 axis may enable improved tissue specificity, enhanced brain exposure, and more precise regulation of metabolic and stress-responsive pathways relevant to neurodegeneration [10, 11, 55].
IP6 has a unique combination of biochemical properties, including metal chelation, redox buffering, mitochondrial stress modulation, and InsP or pyrophosphate signaling network integration, which collectively support its identification as a translational neuroprotective adjunct [11, 13]. Despite the detailed mechanistic and disease model evidence presented in the previous sections, the therapeutic relevance of IP6 ultimately depends on pharmacokinetics, safety, delivery strategies, and rational positioning within multifactorial treatment paradigms [63]. This section focuses on these translational determinants rather than reiterating mechanistic findings.
A radiotracer study involving rodents established that orally administered IP6 can be absorbed and distributed systemically, which suggests its bioavailability beyond the gastrointestinal tract [65]. A subsequent quantitative analysis involving chromatographic approaches confirmed that the tissue levels of InsPs, including InsP6, respond to dietary phytate intake, with measurable changes detected across multiple organs [16]. These studies have also reported detectable brain InsP6 levels in rodents, indicating that dietary exposure influences cerebral InsP pools [16, 66].
Further research is required to establish pharmacokinetic evidence for blood–brain barrier transport and the neurological effects of dietary IP6 intake. Although rodent data confirm that the concentration of brain InsP6 is consistent with dietary intake, no studies have yet defined the transport kinetics, regional brain distribution, or concentration thresholds required for neuroprotection [16, 63, 66].
Human pharmacokinetic data remain limited and are largely confined to plasma and urinary measurements. A controlled human study reported measurable absorption and renal excretion after oral IP6 exposure, with circulating levels sensitive to dietary restriction and intake [67]. Additional human data further support detectable plasma InsP6 changes after defined oral dosing [68].
In summary, current evidence supports the partial oral absorption and diet-responsive systemic or tissue distribution of IP6/InsP6, including measurable brain InsP6 concentrations in rodents. However, quantitative human CNS exposure remains insufficiently defined and represents a key barrier to clinical translation [63].
IP6 has long been used by humans as a natural dietary component and nutraceutical and is generally regarded as safe to consume within normal intake ranges [13, 63]. However, its strong affinity for multivalent cations, including iron, zinc, and calcium, introduces an inherent translational tradeoff. Redox-active iron chelation may be biologically advantageous by limiting the metal-catalyzed oxidative stress implicated in neurodegeneration and age-related cellular damage [7, 61]. By contrast, excessive phytate exposure can reduce the intestinal absorption of essential trace elements under low dietary diversity or marginal micronutrient conditions, particularly in vulnerable populations [63, 64].
Evidence from nutritional reviews, regulatory summaries, and human metabolic studies indicates that phytate–mineral interactions are strongly dose- and context-dependent [63]. Phytate represents a major organic storage form of dietary phosphorus in plant-based foods. However, its biological effects extend beyond phosphorus-based nutrition and should not be conflated with total phosphorus intake. Dietary surveys involving regulatory assessments indicate that the concentration of phytate in typical mixed diets ranges between hundreds of milligrams and approximately 1 g per day, without identifying mineral deficiency as a public health concern in nutritionally adequate populations [69]. IP6 is widely consumed as a natural dietary component in grains and legumes, with estimated habitual intake ranging from several hundred milligrams to approximately 1–1.5 g per day in mixed diets, without evidence of systemic toxicity in nutritionally adequate populations [13, 63]. In small clinical investigations, oral administration of IP6—often in combination with inositol—has been evaluated as an adjunctive intervention in oncology settings. In a prospective randomized pilot study involving patients undergoing chemotherapy, daily supplementation of IP6 plus inositol (approximately 6 g per day in divided doses) was reported to be well tolerated over several months, with no significant disturbances observed in hepatic enzymes, renal function markers, or routine biochemical parameters [51]. Although limited by small sample size, these findings suggest short- to intermediate-term tolerability of oral IP6-containing formulations in clinical settings. Comparative summaries of international regulatory frameworks further indicate that the permitted phytate intake levels across countries and regions are positioned well below the levels associated with adverse mineral effects. These findings confirm that moderate phytate exposure through balanced diets is compatible with mineral homeostasis [1].
From a translational perspective, IP6 supplementation should be evaluated within defined dosing frameworks that account for nutritional background, exposure duration, and population-specific risk. Short- to intermediate-term supplementation at nutraceutical doses is likely to be well tolerated by individuals with adequate mineral intake [13]. However, prolonged or high-dose regimens, particularly in older adults or individuals with preexisting micronutrient insufficiency, should be accompanied by iron and zinc status monitoring and rational dosing strategies, such as timing relative to meals and individualized assessment of mineral sufficiency [13, 63]. These findings underscore the need for carefully designed pharmacokinetic and safety studies as a prerequisite to large-scale neurological efficacy trials.
Systemic administration is not the only translational route for IP6. Incorporating IP6 into biomaterial platforms provides an alternative strategy to achieve high local concentrations, which in turn addresses the limitations of oral pharmacokinetics. Proof-of-concept research has indicated that IP6-containing hydrogels can exhibit favorable in vitro and in vivo biocompatibility, which supports the feasibility of IP6 local delivery in neural or tissue–interface settings [70].
Calcium-phytate-based nanoparticles have been reported to be internalized by NSCs and microglia and to undergo lysosomal degradation, releasing phytate and calcium to alter intracellular ionic and metabolic states. In a spinal cord injury model, this approach was found to be associated with enhanced neuronal differentiation and immunomodulatory effects functioning synergistically with NSC transplantation (Table 1) [39]. These findings highlight the potential of cell-targeted or microenvironment-focused IP6 delivery in modulating regenerative niches, which in turn complements systemic nutraceutical strategies.
IP6 serves as a modulatory node within the cellular metabolic network that influences redox balance, mitochondrial function, and energy homeostasis under physiological plasticity and stress conditions. Instead of acting through a single dominant pathway, it appears to exert context-dependent, pleiotropic effects that contribute to core metabolic and stress-responsive processes. This view helps integrate observations across experimental systems and highlights how metabolic buffering mechanisms may shape cellular resilience and vulnerability in complex biological settings [10, 11, 55].
From a translational perspective, IP6 has potential as an adjunct or preventive agent, particularly during the early or presymptomatic stages of disease, in which metabolic flexibility remains partially preserved [59, 62]. Future studies should focus on better understanding the dose–response relationships, long-term safety, and context-dependent effects of IP6 on mineral homeostasis, as well as on the development of optimized delivery strategies and pharmacological tools targeting the endogenous IP6/IP7 axis. Addressing these challenges would help determine whether IP6-based metabolic buffering can be modulated to enhance stem cell function and mitigate metabolic stress in neurodegenerative and other complex disease states.
JYL and YCH conceived and designed the review. JYL, CCHY, and YCH conducted the literature search and analysis. JYL and YCH wrote the manuscript. CCHY critically reviewed the manuscript and contributed to the scientific interpretation. All authors have participated sufficiently in the work, read, revised, and approved the final manuscript, and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
We thank the National Science and Technology Council (NSTC) of the Taiwan Government for grant support (MOST 111-2314-B-715-009-MY3), and from Mackay Medical University for their intramural research grants (MMU-RD-114-1B-P005, MMU-RD-114-RM-G001-02).
The authors declare no conflicts of interest.
During the preparation of this work, the authors utilized ChatGPT (OpenAI, San Francisco, CA, USA) to verify spelling and grammar, and NotebookLM (Google LLC, Mountain View, CA, USA) to assist in figure generation. After using these tools, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.