
 , Xiangyu Zhu 1,2,†, Yuting Sun 1,2, Yakun Yang 1,2, Aodi Fan 1,2, Ke Yang 1,2, Zixuan Gao 1, Chuanrui Ma 2,*
, Xiangyu Zhu 1,2,†, Yuting Sun 1,2, Yakun Yang 1,2, Aodi Fan 1,2, Ke Yang 1,2, Zixuan Gao 1, Chuanrui Ma 2,* , Guanwei Fan 1,2, Lan Li 1,2,*
, Guanwei Fan 1,2, Lan Li 1,2,*1 State Key Laboratory of Modern Chinese Medicine, Key Laboratory of Pharmacology of Traditional Chinese Medical Formulae for the Ministry of Education, Tianjin University of Traditional Chinese Medicine, 301617 Tianjin, China
2 National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, 300193 Tianjin, China
†These authors contributed equally.
Abstract
Cardiovascular diseases (CVDs) are the primary cause of mortality globally. The Global Burden of Disease Study indicates that CVDs account for one-third of all global deaths, with a notable 18.17% increase in their incidence over the last decade. Despite the widespread use and proven effectiveness of therapies like antiplatelet agents, lipid-lowering drugs, anticoagulants, and percutaneous coronary intervention (PCI) in managing risk factors, the long-term prognosis of patients remains suboptimal. The prevailing academic viewpoint asserts that chronic inflammation and dysregulated apoptosis lead to the accumulation of apoptotic cells or fragments, triggering inflammatory injury and oxidative stress. Efferocytosis, encompassing the recognition, engulfment, and degradation of apoptotic cells by phagocytes, is essential for preserving tissue homeostasis and limiting excessive inflammation in this context. Macrophages, being professional phagocytes, demonstrate the highest efficiency and precision in clearing apoptotic cells. Hence, this review will center on macrophage-mediated efferocytosis, systematically investigating its roles and mechanisms in the pathogenesis and progression of CVDs.
Keywords
- macrophage
- CVDs
- efferocytosis
- treatment
Cardiovascular diseases (CVDs) remain the leading cause of mortality worldwide [1]. Although contemporary risk-factor control and revascularization have improved early outcomes, long-term prognosis is still limited by maladaptive repair and chronic inflammation after injury [2]. A consistent trigger of non-resolving inflammation is the insufficient clearance of dying cells and cell debris, which promotes secondary necrosis, oxidative stress, and sustained leukocyte recruitment [3].
Efferocytosis refers to the immunologically silent recognition, engulfment, and degradation of apoptotic cells by phagocytes. Macrophages are the dominant professional efferocytes in cardiovascular tissues; by coupling corpse clearance to pro-digesting signaling, they constrain inflammation and instruct tissue repair [2, 4]. Conversely, defects at specific checkpoints (e.g., reduced phosphatidylserine sensing, excess anti-phagocytic signaling such as CD47, or impaired efferosome maturation) can amplify inflammation and remodeling across multiple CVDs.
Here, we summarize the conserved molecular framework of macrophage efferocytosis with stage-specific regulators; discuss how dysregulated efferocytosis contributes to atherosclerosis, myocardial infarction, heart failure, myocarditis, abdominal aortic aneurysm, atrial fibrillation, and diabetic cardiomyopathy; and highlight emerging therapeutic strategies that restore efferocytosis and promote inflammation resolution.
The term “efferocytosis” was officially coined in 2003, defining the process by which phagocytic cells recognize, engulf, and degrade apoptotic cells before they undergo secondary necrosis, with its core functions being anti-inflammatory and pro-digesting [5]. The introduction of the concept of efferocytosis has deepened the understanding of macrophage functions, shifting the focus from mere immune clearance to active immune regulation. In the human body, billions of cells undergo programmed cell death daily, highlighting the crucial role of efficient macrophage efferocytosis in timely clearance of apoptotic cells and cell debris to prevent their accumulation. This process actively suppresses inflammatory responses and plays a significant role in key physiological functions such as embryonic development, inflammation resolution, and tissue homeostasis maintenance [6]. Efferocytosis can be described as three core stages—Find, Pro-phagocytic, and Digest—with the pro-phagocytic stage often subdivided into tethering/recognition and internalization; accordingly, some studies describe a four-step framework (recognition, binding, engulfment, and post-engulfment processing).
Macrophages are a crucial component of the innate immune system [7] and represent the most abundant immune cell population in the cardiac tissue microenvironment [8]. As integral cells of the innate immune system, their primary functions include phagocytosis and clearance of pathogenic microorganisms, antigen processing and presentation, debris clearance, regulation of inflammatory responses, and promotion of tissue repair [9]. Macrophages exhibit significant diversity and heterogeneity, being able to adopt different functional phenotypes based on microenvironmental signals, such as pro-inflammatory (M1) and anti-inflammatory (M2) polarization states, thereby exerting differential effects on tissue repair processes [10].
Cardiac macrophages originate from two lineages: tissue-resident macrophages of
embryonic origin and monocyte-derived macrophages that differentiate after
infiltrating the heart from the bone marrow [11, 12]. Based on the expression of
the chemokine receptor CCR2, cardiac macrophages can be divided into two main
subtypes: CCR2+ macrophages, continuously replenished by circulating
monocytes, primarily involved in inflammatory responses and immune defense, but
may also have detrimental effects under pathological conditions [13, 14]; while
CCR2– macrophages mainly consist of embryonically derived cardiac-resident
macrophages (CRMs), accounting for approximately 6%–8% of normal adult
myocardial cells, capable of self-renewal through local proliferation, playing a
crucial role in maintaining cardiac homeostasis, electrophysiological function,
and tissue repair [15]. During the acute phase of myocardial infarction,
CCR2+ macrophages rapidly accumulate in the infarct area via the CCR2-CCL2
axis, initiating phagocytosis to clear a large number of necrotic and apoptotic
cell debris [16]. Inadequate or delayed clearance of debris can lead to the
release of cellular components, triggering a stronger inflammatory response and
exacerbating damage. As the repair phase progresses, CCR2– macrophages
become the primary effector cells for debris clearance. These cells exhibit high
expression of key efferocytosis receptors such as TIM-4, MERTK, and CD163,
enabling efficient recognition and engulfment of apoptotic cells [17].
Concurrently, efferocytosis promotes polarization of macrophages towards a
reparative phenotype characterized by secretion of factors like TGF-
Efferocytosis is organized here into three functional stages—Find, Pro-phagocytic, and Digest (Table 1, Ref. [23, 24, 25, 26, 27, 28, 29, 30, 31, 32]). Find denotes chemotaxis toward apoptotic cells driven by soluble cues; Pro-phagocytic encompasses tethering/recognition and internalization into an efferosome; Digest covers efferosome maturation, lysosomal degradation, and the coupled metabolic/pro-digesting programs that terminate inflammation. This unified framework is used throughout the disease sections to map where each pathology disrupts efferocytosis.
| Phase | Apoptotic cell signals | Macrophage receptors/mechanisms | Reference |
| 1. Find | chemotactic signals: ATP, UTP, S1P, LPC, CX3CL1 | Receptors: | [23, 24, 25, 26] |
| P2Y2, S1PR1/3, G2A, CX3CR1 | |||
| 2. Pro-phagocytic | pro-phagocytic signals: PtdSer (Primary), Calreticulin | Pro-phagocytic Receptors: | [23, 25, 26, 32] |
| Direct: TIM4, BAI1; | |||
| Bridging-related receptors: MERTK; | |||
| Integrins: |
|||
| Inhibitory checkpoint: SIRP |
|||
| 3. Digest | - | Degradation: | [27, 28, 29, 30, 31] |
| LC3-associated phagocytosis (LAP); Rab5/Rab7 | |||
| Metabolic/resolution outputs: | |||
| ABCA1 |
ATP, adenosine triphosphate; UTP, uridine triphosphate; S1P,
sphingosine-1-phosphate; LPC, lysophosphatidylcholine; CX3CL1, chemokine (C-X3-C
motif) ligand 1; P2Y2, purinergic receptor P2Y2; S1PR1/3, sphingosine-1-phosphate
receptor 1/3; G2A, G2 accumulation protein; CX3CR1, chemokine (C-X3-C motif)
receptor 1; PtdSer, phosphatidylserine; TIM4, T cell immunoglobulin and mucin
domain-containing protein 4; BAI1, brain-specific angiogenesis inhibitor 1;
MERTK, MER tyrosine kinase; SIRP
The precise localization of macrophages to apoptotic cells is essential for
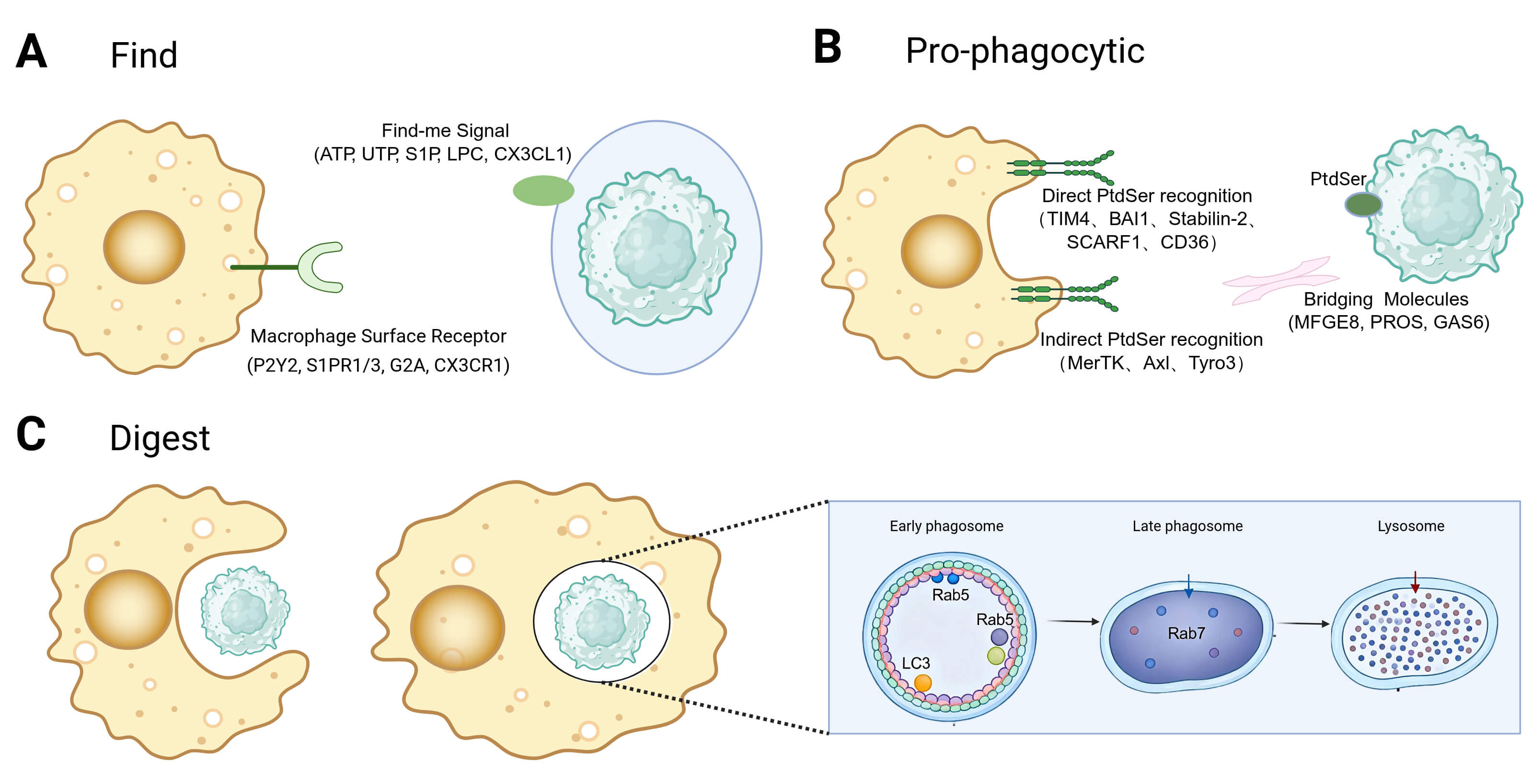
initiating efferocytosis (Fig. 1A). During the find-me stage, apoptotic cells
release various soluble factors, including nucleotides (e.g., ATP, UTP), lipid
derivatives (e.g., S1P, LPC), and chemokines (e.g., CX3CL1), to attract
macrophages [33]. These find-me signals also possess immunomodulatory properties
that promote the polarization of macrophages towards an anti-inflammatory
phenotype. For example, nucleotides activate the P2Y2 receptor, leading to the
suppression of pro-inflammatory cytokines such as TNF-
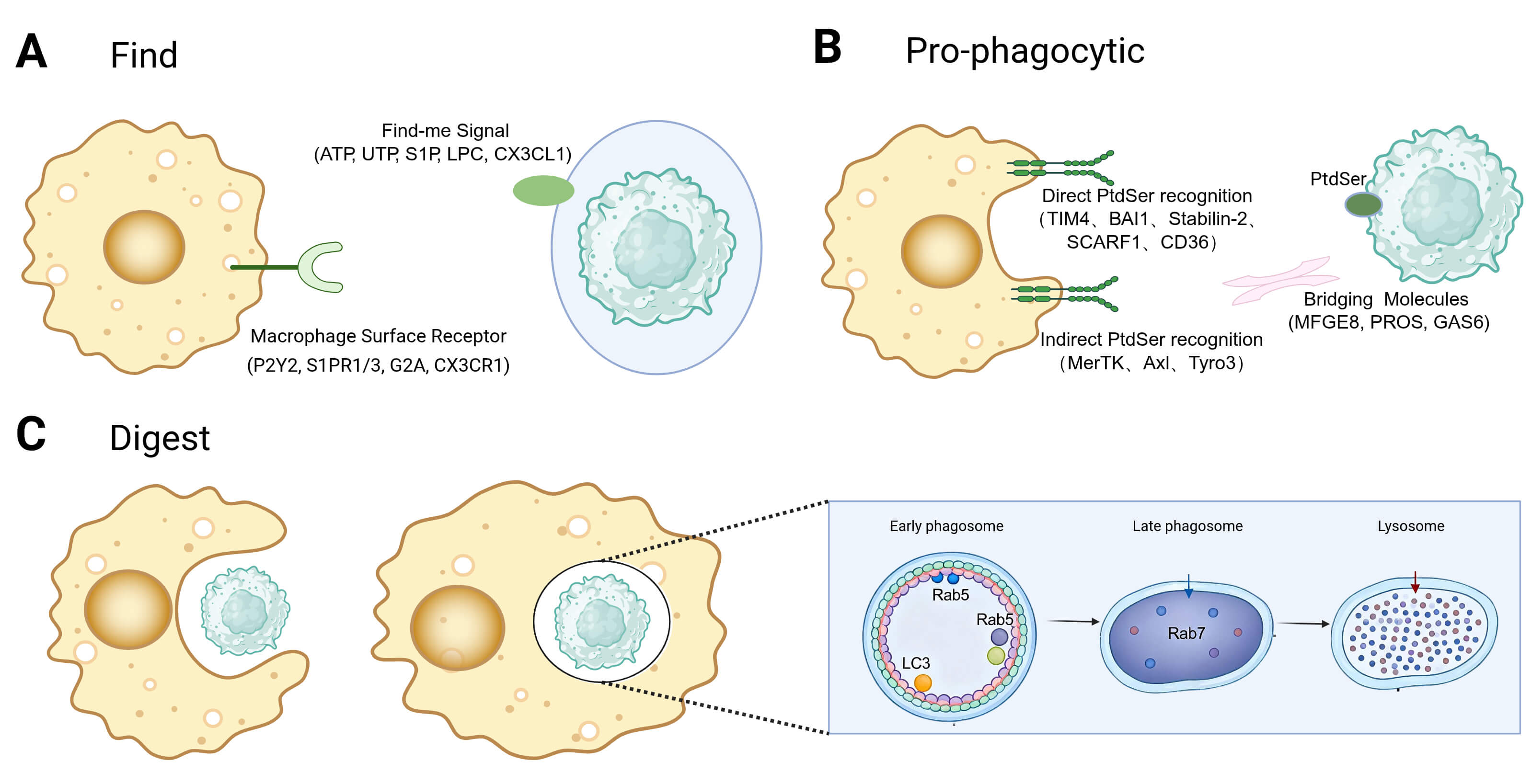 Fig. 1.
Fig. 1.
Stage-resolved framework of macrophage efferocytosis. (A) Find: apoptotic cells release chemotactic cues that recruit and position macrophages. (B) Pro-phagocytic: macrophages tether to apoptotic cells through PtdSer-dependent receptors and bridging molecules and internalize them into an efferosome. (C) Digest: efferosomes mature via Rab GTPases and fuse with lysosomes; coupled metabolic programs (e.g., ABCA1-driven lipid handling) and pro-digesting signaling ensure immunologically silent degradation and termination of inflammation. Created with BioRender.com. PtdSer, phosphatidylserine; Rab GTPases, Rab guanosine triphosphatases; ABCA1, ATP-binding cassette transporter A1.
Pro-phagocytic cues on apoptotic cells are essential for their selective recognition and clearance by macrophages [38]. Among these cues, externalized phosphatidylserine (PtdSer; PS) is the best characterized. Under homeostatic conditions, ATP-dependent flippases (e.g., ATP11C) restrict PtdSer to the inner leaflet of the plasma membrane. During apoptosis, caspases inactivate ATP11C and activate scramblases such as Xkr8, disrupting membrane asymmetry and causing sustained PtdSer exposure on the cell surface [39, 40]. Additional surface changes, including calreticulin accumulation, can further enhance uptake efficiency and context-specific signaling [41].
Macrophages recognize pro-phagocytic cues through a variety of surface
receptors, which can be broadly categorized into direct PS-binding receptors and
receptors that signal via soluble bridging molecules (Fig. 1B). Direct receptors
like TIM4, BAI1, Stabilin-2, and CD36 bind directly to exposed PS on apoptotic
cells, initiating intracellular signaling cascades that lead to cytoskeletal
rearrangement and phagosome formation. In contrast, indirect receptors, such as
the TAM receptor family (MERTK, Axl, Tyro3), integrin
Upon internalization of the apoptotic cell to form an efferosome, degradation primarily occurs through the phagosome-lysosome pathway (Fig. 1C). Activation of Rho family GTPases, such as Rac1, initiates actin polymerization for engulfment [48]. The maturation of the efferosome is a tightly regulated process controlled by the Rab GTPase family: Rab5 marks early efferosomes, while Rab7 activation signifies the transition to the late stage [27]. Efferosome maturation involves rapid acidification of the phagocytic compartment (pH 4.5–5.0) mediated by V-ATPase, activating hydrolytic enzymes. In addition to the classical pathway, LC3-associated phagocytosis (LAP) functions as a specialized degradation pathway [28]. Binding of apoptotic cells to receptors like TIM-4 or integrins activates the PI3K complex, leading to LC3 lipidation and LAPosome formation on the efferosome membrane [28, 29, 42]. LAP does not alter phagocytic uptake but is crucial for efferosome-lysosome fusion. Knockout studies reveal that deficiencies in key LAP proteins (e.g., Rubicon and ATG5) do not impede phagocytosis but significantly delay degradation, triggering inflammation. This underscores the essential role of LAP in ensuring efficient and immunologically silent digestion [28, 42].
After phagocytosis, macrophages face significant metabolic challenges and undergo complex reprogramming to maintain homeostasis. They upregulate the reverse cholesterol transporter ABCA1 to manage lipid and cholesterol influx, facilitating cholesterol efflux to ApoA1 and preventing lipid accumulation [30, 31]. Additionally, degradation products, such as arginine from apoptotic cell breakdown, are recycled to sustain clearance functions. Arginine is transported into the cytoplasm via the lysosomal transporter PQLC2 and metabolized by Arg1 and Ornithine decarboxylase (ODC) into polyamines. Disruption of this pathway hinders efferocytosis, while polyamine supplementation restores it, indicating a feedback loop that sustains cellular function [49]. Furthermore, post-efferocytosis, macrophages enhance mitochondrial membrane potential and upregulate UCP2 to optimize energy metabolism, supporting ongoing clearance activities [42].
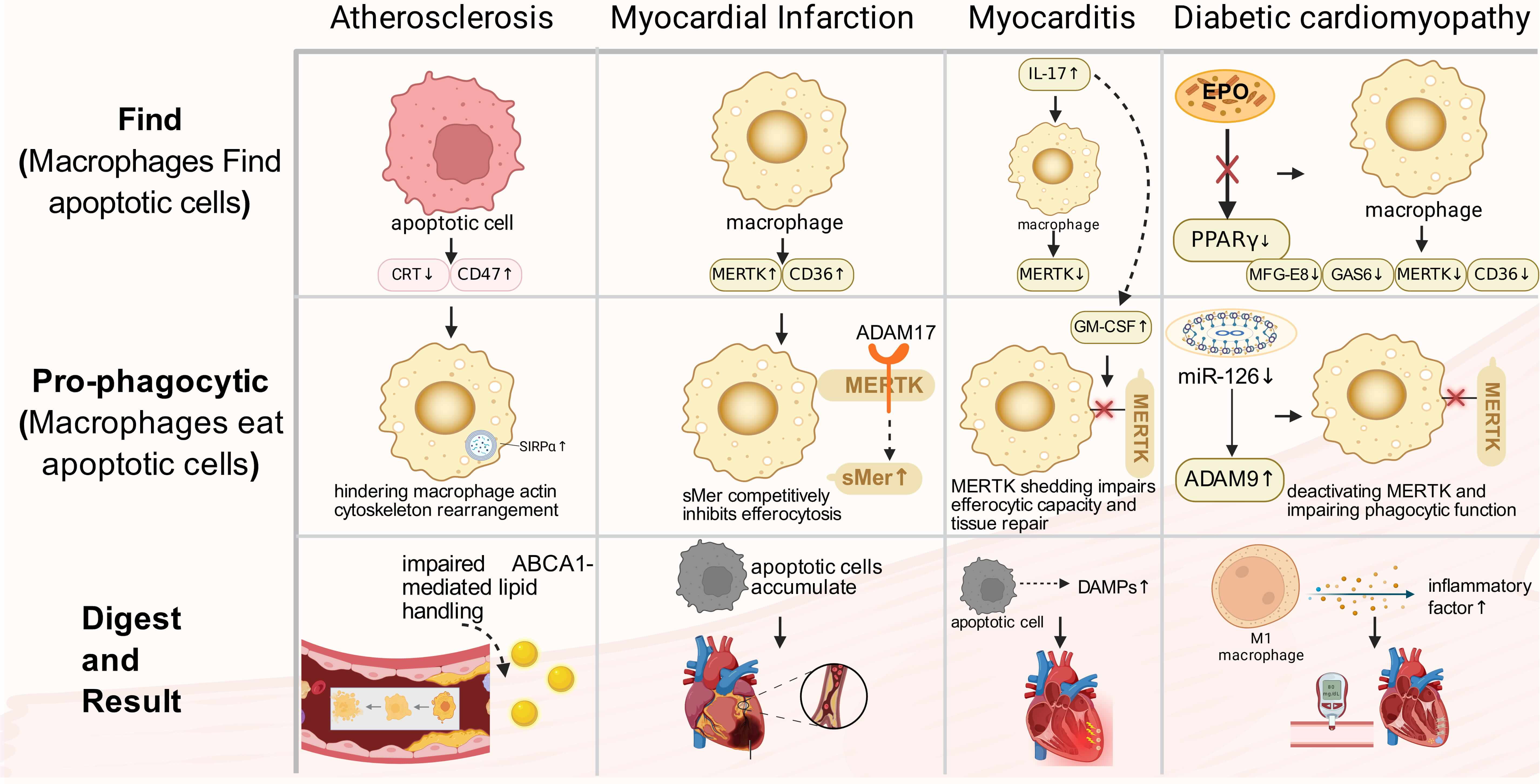
Defective efferocytosis has been implicated in multiple cardiovascular diseases. Fig. 2 provides a stage-mapped schematic of shared efferocytosis checkpoints and representative disease-specific dysregulated nodes, while Table 2 (Ref. [50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67]) serves as a concise, at-a-glance index of the main altered molecules across diseases. We then discuss each disease context in detail and finally compare shared versus disease-tailored mechanisms (Section 4.8).
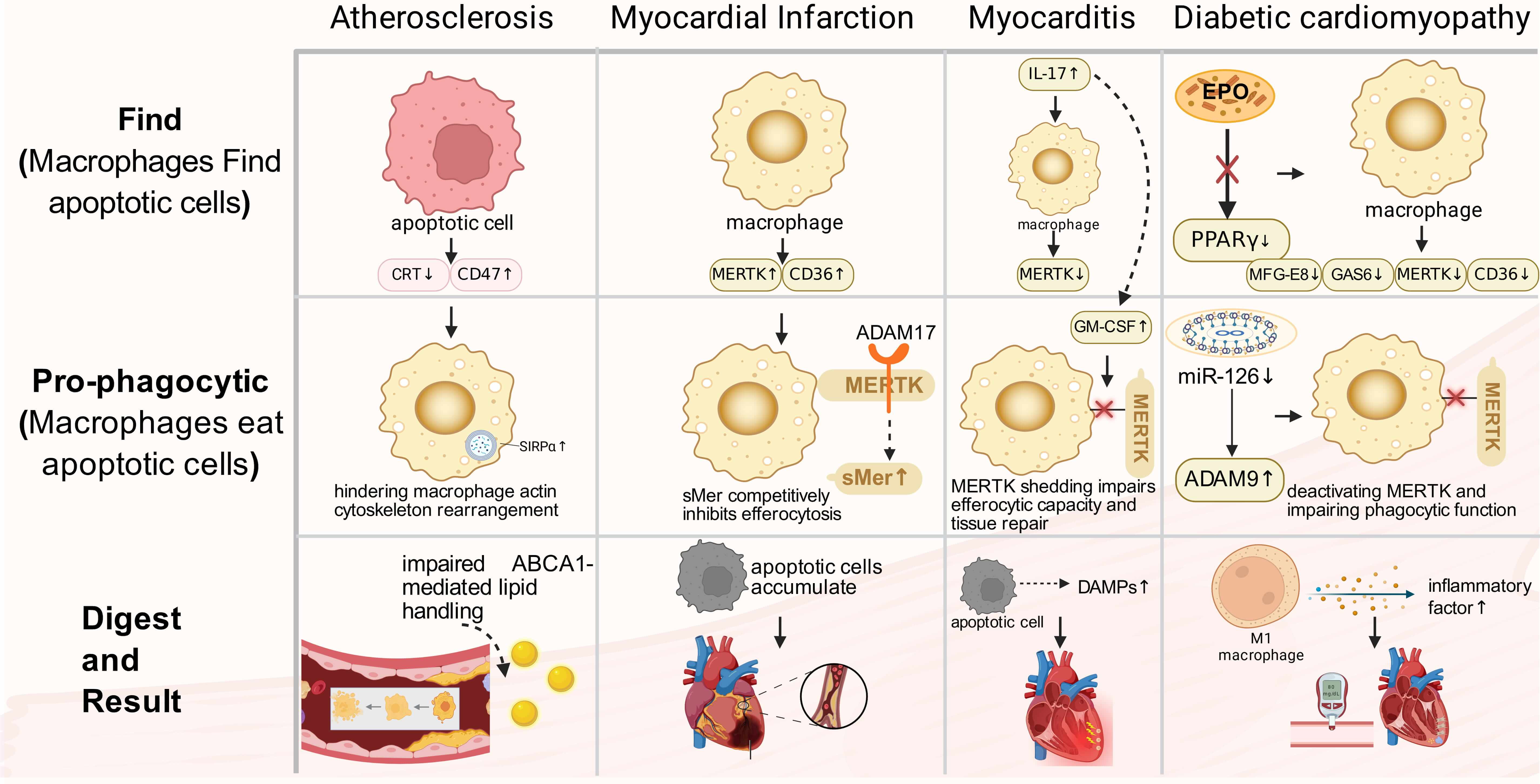 Fig. 2.
Fig. 2.
CVDs related to macrophage efferocytosis. Stage-mapped
dysregulation of macrophage efferocytosis across major cardiovascular diseases.
Rows indicate the predominant disrupted stage (Find, pro-phagocytic, or digest),
and nodes highlight representative molecular alterations reported in each
disease. Shared checkpoints include (i) excessive anti-phagocytic signaling
(e.g., CD47-SIRP
| Disease context | Predominant defective stage | Representative dysregulated nodes | Reference |
| AS | Pro-phagocytic/Digest | ↑CD47, CypA; | [50, 51] |
| ↓MERTK, CRT, ABCA1 | |||
| MI | Pro-phagocytic | ↑ADAM17 (activity); | [64, 66] |
| ↓MERTK (cleaved by ADAM17) | |||
| HF | Digest | ↓Overall Efferocytosis Function | [53, 54, 55] |
| Myocarditis | Pro-phagocytic/Digest | ↑IL-17, GM-CSF (MERTK shedding); | [56] |
| ↓MERTK | |||
| AAA | Pro-phagocytic | ↑CD47, NINJ1, ILF3, IRF5, PI3K |
[57, 58, 59, 60, 61] |
| AF | Digest | ↓Cx43 | [65, 67] |
| Diabetic cardiomyopathy | Pro-phagocytic/Digest | ↑ADAM9, SLC7A11; | [52, 62, 63] |
| ↓MERTK, AXL, TREM2, MFG-E8 |
Atherosclerosis (AS) is a chronic inflammatory vascular condition characterized by lipid accumulation in the arterial intima, infiltration of inflammatory cells, and plaque formation [68]. Within the plaque, macrophages play a crucial role in clearing two types of apoptotic foam cells: those derived from macrophages themselves and those originating from smooth muscle cells that migrate to the intima from the media and become lipid-laden [69, 70]. Efficient clearance of these apoptotic cells by macrophages helps maintain cholesterol homeostasis, limits necrotic core expansion, and enhances plaque stability [28, 71]. Conversely, impaired efferocytosis leads to apoptotic cell accumulation, necrotic core formation, fibrous cap thinning, significantly increasing the risk of plaque rupture, which can ultimately result in myocardial infarction or stroke [72].
Impaired efferocytosis in AS is primarily attributed to the disruption of
apoptotic cell recognition signals. Within atherosclerotic lesions, apoptotic
cells exhibit heightened expression of the anti-phagocytic signal CD47, which
chronically activates SIRP
Furthermore, a detrimental cycle is established within the plaque, where the
perpetually aggravated inflammatory response and compromised efferocytosis
mutually reinforce each other. Inflammatory cytokines induce macrophages to
acquire a pro-inflammatory state, hindering efferocytosis-associated pathways
like ELMO1/Rac1 [76]. Apoptotic cells that remain uncleared release
damage-associated molecular patterns (DAMPs), activating the NLRP3 inflammasome.
This activation leads to caspase-1 activation and the release of pro-inflammatory
cytokines, such as IL-1
Impaired efferocytosis exacerbates the dysregulation of cholesterol metabolism within the plaque. Macrophages normally manage cholesterol influx after engulfing apoptotic cells by esterification through ACAT1 and efflux via the ABCA1/ABCG1 pathways [79]. However, compromised clearance function disrupts cholesterol homeostasis, leading to excessive accumulation of free cholesterol in the plasma membrane and abnormal lipid raft clustering. Accumulated cholesterol in the membrane facilitates TLR4 aggregation and activation within these lipid rafts. Activated TLR4 triggers a signaling cascade through MYD88, ultimately activating the NADPH NOX2 complex. Subsequently, activated NOX2 generates superoxide anions and reactive reactive oxygen species (ROS), increasing intracellular oxidative stress and eventually causing cell apoptosis [80]. Studies have shown that inhibiting NOX2 activates the MERTK signaling pathway, promoting efferocytosis [81].
The decline in macrophages’ efferocytic ability is a critical factor in AS
progression. Within the inflammatory and cytotoxic environment of atherosclerotic
plaques, macrophages are prone to apoptosis or necrosis, leading to the loss of
their efferocytic function [82]. Moreover, the local inflammatory signaling
network, including IFN-
In conclusion, the targeting of macrophage efferocytosis enhancement shows potential as a therapeutic strategy. This method seeks to promote lipid breakdown and induce M2 polarization to clear apoptotic cells in the plaque and stabilize atherosclerotic lesions [87, 88].
Myocardial Infarction (MI) remains a leading global cause of mortality. Precise modulation of the inflammatory response is crucial for optimal repair outcomes [89]. During the acute phase of MI, a significant influx of macrophages to the necrotic region occurs, playing a key role in clearing dead cells. The MERTK receptor on their surface is essential for the rapid recognition and phagocytosis of apoptotic cells [52, 90, 91]. The functional status of MERTK directly influences inflammatory progression and cardiac prognosis. Studies using MERTK knockout mouse models have shown that impaired apoptotic cell clearance post-MI leads to their accumulation in the heart, resulting in worsened cardiac function and increased infarct size [66]. Conversely, enhancing efferocytosis has been linked to improved cardiac function recovery, suggesting that targeting MERTK could be a promising therapeutic approach. Additionally, other efferocytosis-related receptors, such as CD36 and Stabilin-1, show increased expression post-MI and work together to facilitate the clearance process [92, 93].
The microenvironment of MI plays a crucial role in modulating efferocytosis. Within the infarct area, N2 neutrophils have been shown to stimulate macrophages to upregulate efferocytosis receptors, such as MERTK, CD36, and CX3CR1, as well as bridging molecules like MFGE8 and Gas6, thereby enhancing macrophages’ efferocytic capacity. However, detrimental factors also exist [93]. Pathologically, ADAM17 protease activation leads to the cleavage of MERTK’s extracellular domain, generating soluble sMer. This not only reduces functional MERTK levels on cell membranes but also competitively inhibits efferocytosis, impairing clearance efficiency [64, 66]. Studies have demonstrated that using a cleavage-resistant MERTK mutant can reduce infarct size and improve cardiac function, suggesting targeting ADAM17-mediated MERTK hydrolysis as a potential therapeutic strategy for MI [94]. Furthermore, monocytes/macrophages expressing MERTK and MFGE8 synergistically enhance efferocytic capacity and promote VEGFA secretion, facilitating local repair processes [95].
Heart Failure (HF) is a clinical syndrome resulting from impaired ventricular function, with HF progression being driven by myocardial apoptosis [96]. Persistent inflammation and oxidative stress in HF, combined with an overload of apoptotic cells, hinder macrophage efferocytic capacity. This impaired clearance process leads to the accumulation of dead cells, secondary necrosis, and the release of DAMPs, exacerbating inflammation and injury in a self-perpetuating cycle. Dysregulated efferocytosis is a crucial pathological mechanism in HF myocardial damage [53, 75, 97]. Macrophages not only directly eliminate apoptotic cells but also indirectly influence fibrosis by modulating cardiac fibroblasts. Research indicates that cardiac fibroblasts can engage in efferocytosis and secrete MFG-E8 to enhance phagocytosis [54].
The molecular network involved in efferocytosis also plays a role in regulating
extracellular matrix (ECM) metabolism. For instance, TSP1 participates in
fibroblast differentiation and collagen synthesis [55]. Deficiency in TSP1
hinders TGF-
Myocarditis, an inflammatory disorder of the cardiac muscle, is primarily induced by factors such as viral infections. Pathologically, it is distinguished by infiltration of immune cells, resulting in myocardial damage and necrosis [98, 99]. The condition can rapidly advance from a subclinical phase to HF, posing a significant risk to the well-being of patients [100].
In a CVB3-induced myocarditis model, macrophages infiltrate cardiac tissue early in infection, contributing to the antiviral response through the secretion of inflammatory factors [101]. Impaired clearance of apoptotic cells by these macrophages can result in secondary necrosis, exacerbating inflammation and worsening myocardial injury. Recent studies have highlighted the crucial role of MERTK expression levels in determining apoptotic cell clearance efficiency. Clinical investigations have revealed downregulated MERTK expression in CD11b+ myeloid cells in the blood of myocarditis patients, indicating a potential link to impaired efferocytosis and disease progression. Animal models, such as the experimental autoimmune myocarditis (EAM) model, have shown that the IL-17 signaling pathway induces the secretion of granulocyte–macrophage colony-stimulating factor (GM-CSF) , leading to the shedding of MERTK from macrophages. This shedding impairs efferocytic capacity and tissue repair [56]. Targeting macrophage MERTK expression to enhance efferocytosis represents a novel approach to mitigating myocarditis-induced damage.
The current myocarditis research predominantly focuses on macrophage
polarization, yet the crucial functional effector linking different polarization
states to outcomes, efferocytosis, is integral throughout disease progression. In
viral myocarditis, efficient efferocytosis removes apoptotic cells, limits DAMP
release, and reduces monocyte recruitment (mediated by CCL2 and MIF-
Dysregulation of macrophage efferocytosis in the pathogenesis of Abdominal
Aortic Aneurysm (AAA) is influenced by inflammatory cell infiltration and direct
suppression within a chronic inflammatory microenvironment driven by factors such
as NINJ1, ILF3, and IRF5 [57]. NINJ1 enhances macrophage infiltration and M1
polarization via the TLR4/NF-
In atrial fibrillation (AF), defective efferocytosis may couple electrical instability to persistent atrial inflammation and structural remodeling. Connexin 43 (Cx43), a key gap-junction protein for coordinated conduction, is frequently downregulated or mislocalized in AF. Beyond electrophysiology, altered Cx43 signaling can aggravate mitochondrial stress and bias macrophages toward a pro-inflammatory state with reduced efferocytic capacity. Conversely, restoring Cx43-associated programs has been linked to improved macrophage migration, enhanced corpse clearance, and a shift toward reparative phenotypes, thereby facilitating resolution and limiting fibrosis-driven remodeling [65, 67, 106, 107, 108].
Diabetic cardiomyopathy (DCM) is characterized by chronic metabolic stress, cardiomyocyte injury, and low-grade inflammation, in which macrophage dysfunction contributes to impaired repair. In diabetes, efferocytosis is frequently weakened because key receptors (e.g., MERTK, AXL) and pro-digesting programs are downregulated, while protease activity and metabolic reprogramming further compromise uptake and degradation. These defects favor apoptotic-cell accumulation, secondary necrosis, and sustained inflammatory signaling, accelerating fibrosis and functional decline in the diabetic heart [109].
Chronic hyperglycemia hampers the expression of key efferocytosis-related
molecules. Studies have shown that diabetic individuals or macrophages exposed to
high glucose levels exhibit significant downregulation of crucial receptors like
MERTK and AXL. This downregulation is linked to microRNA dysregulation,
particularly the reduced expression of miR-126 leading to increased ADAM9
protease activity. ADAM9 cleaves MERTK, deactivating it and impairing phagocytic
function [62]. Moreover, hyperglycemia leads to downregulation of TREM2,
hindering macrophage polarization towards a pro-reparative M2 phenotype, further
compromising efferocytic capacity [63]. Additionally, diabetes-induced metabolic
reprogramming directly hinders efferocytosis efficiency by disrupting macrophage
metabolic pathways including glycolysis and oxidative phosphorylation. Activation
of SLC7A11 inhibits aerobic glycolysis, limiting dendritic cells and macrophages’
ability to phagocytose apoptotic cells [110]. Furthermore, crucial reparative
pathways are suppressed under diabetic conditions. Normally, erythropoietin (EPO)
activates PPAR
Dysfunctional efferocytosis exacerbates diabetic complications by impeding critical processes such as wound healing and bone metabolism [113]. Specifically, diabetic macrophages exhibit impaired efferocytosis during wound healing, leading to delayed closure. Efficient efferocytosis is essential for bone healing; however, in diabetic bone defects, persistent dysfunction results in overactivation of M1 macrophages, prolonging inflammation resolution [114, 115]. Moreover, diabetic macrophage-conditioned medium hinders osteogenic differentiation of bone marrow-derived mesenchymal stem cells. In diabetic periodontitis, impaired efferocytosis contributes to intensified alveolar bone resorption [116]. Overall, dysfunctional macrophage efferocytosis emerges as a pivotal mechanism underlying impaired bone repair in diabetes.
Across cardiovascular diseases, efferocytosis failure converges on a limited set
of checkpoints. First, an imbalance between pro-phagocytic and anti-phagocytic
cues reduces corpse recognition (e.g., excessive CD47-SIRP
Superimposed on these shared checkpoints are disease-tailored patterns. In atherosclerosis, lipid loading and chronic anti-phagocytic signaling favor apoptotic foam-cell persistence and necrotic core growth [123]. After myocardial infarction, acute protease activation and cytokine surges can cleave or inactivate engulfment receptors (e.g., ADAM17–MERTK), making the early window of repair particularly sensitive to interventions that preserve receptor integrity [124, 125]. In abdominal aortic aneurysm, adventitial apoptosis and defective clearance appear to contribute to wall degeneration, whereas in atrial fibrillation, electrical/mitochondrial stress pathways (e.g., Cx43-linked remodeling) may indirectly restrict macrophage resolving capacity [126]. Diabetic cardiomyopathy adds a systemic metabolic layer, in which hyperglycemia and dyslipidemia suppress TAM signaling and degrade clearance efficiency, shifting the balance toward persistent inflammation and fibrosis [127].
These comparisons suggest that effective therapies will likely combine a shared ‘core’ module (relieving inhibitory cues and restoring TAM-dependent uptake) with context-specific modules that address the dominant local constraint (protease activity after infarction, lipid overload in plaques, or metabolic stress in diabetes).
Targeting macrophage efferocytosis is an emerging therapeutic strategy for
various diseases, with ongoing clinical trials investigating methods to impede
disease progression. For instance, a clinical trial is currently assessing the
impact of lovastatin on macrophage-mediated phagocytosis and apoptosis in COPD
patients (ClinicalTrials.gov registration number: NCT00700921) [128]. However, in
the context of cancer, enhancing efferocytosis may have adverse effects.
Tumor-associated macrophages (TAMs) exhibit heightened expression of TAM
receptors, leading to excessive engulfment of apoptotic cells and subsequent
secretion of immunosuppressive factors that promote tumor immune evasion and
disease progression [129]. In this scenario, inhibiting efferocytosis emerges as
a potential therapeutic approach. While interventional studies exploring the
modulation of efferocytosis are in early stages, further validation is necessary
to assess their clinical utility [130]. The subsequent discussion will delve into
the mechanisms by which statins and PPAR
| Drug class | Core pathway | Key mechanisms in efferocytosis | Reference |
| Statins | RhoA Pathway (Primary) | 1. ↓ Directly RhoA activity | [22, 131, 132, 133] |
| 2. ↑ PPAR |
|||
| 3. ↓ CD47 (anti-phagocytic signal) | |||
| 4. ↓ MMP-9 secretion | |||
| PPAR |
PPAR |
1. Promotes M1 → M2 polarization | [134, 135] |
| 2. ↑ CD36 and other receptors | |||
| 3. ↓ NF- |
Statins, as first-line therapeutic agents for CVDs, are well-known for their efficacy in lipid-lowering as HMG-CoA reductase inhibitors. Recent studies have further revealed their anti-atherosclerotic properties [136].
Statins enhance efferocytosis primarily by inhibiting the RhoA signaling pathway during the “digest” stage. RhoA activation relies on prenylation for membrane localization, a process hindered by statins through mevalonate synthesis suppression. Statins act as pharmacological RhoA inhibitors, notably boosting efferocytosis in vitro and in vivo [131, 132]. Lovastatin, for instance, improved alveolar macrophage efferocytosis in COPD patients by suppressing RhoA. Additionally, statins’ antioxidant properties mitigate oxidative stress, a precursor to RhoA activation, indirectly fostering a microenvironment conducive to efferocytosis.
Statins not only directly inhibit RhoA but also enhance efferocytosis through
additional mechanisms. Firstly, they increase PPAR
The ability of statins to enhance efferocytosis is closely linked to their physicochemical properties. Studies have shown that hydrophobic statins, such as atorvastatin, lovastatin, and simvastatin, increase efferocytosis in a dose-dependent manner, whereas hydrophilic statins like pravastatin do not have this effect. This suggests that the lipophilic nature of statins enables them to penetrate cell membranes and target intracellular molecules like RhoA [137]. For example, the inflammatory mediator 12(S)-HETE, which is elevated in patients with coronary artery disease, inhibits efferocytosis by activating the RhoA pathway. It has been demonstrated that low-dose atorvastatin effectively reverses the 12(S)-HETE-induced impairment of efferocytosis [138]. In conclusion, the cardiovascular benefits of statins stem from their dual action of lowering lipids and exerting pleiotropic effects, with a significant contribution being the enhancement of efferocytosis through RhoA inhibition.
PPAR
Macrophage efferocytosis is fundamental to cardiovascular health, with its
dysregulation driving disease progression via unstable plaques and poor repair.
Targeting this process (e.g., with statins or PPAR
Several limitations should be acknowledged [86]. First, much of the mechanistic evidence is derived from animal models or reductionist in vitro systems, and the relative contribution of individual checkpoints may differ across species, disease stages, and tissue niches. Cardiovascular pathology is highly heterogeneous in humans, often shaped by age, sex, comorbidities (e.g., diabetes, CKD), medication background, and repeated injury—features that are difficult to recapitulate in single-hit experimental models [117, 121]. Second, efferocytosis is frequently inferred rather than directly measured. Many studies use static surrogates (e.g., receptor expression, apoptotic-cell burden, or macrophage polarization markers) that do not necessarily report efferocytic flux (the rate of corpse uptake and processing). Stage-resolved assays that distinguish “Find” limitations (reduced encounters), “Pro-phagocytic” limitations (recognition/internalization failure), and “Digest” limitations (impaired degradation and resolution outputs) are not yet standardized across laboratories or disease contexts. Moreover, apoptotic targets differ substantially among diseases (foam cells, cardiomyocytes, VSMCs, infiltrating leukocytes), and the composition of the lesion microenvironment can strongly bias which checkpoint becomes rate-limiting [142, 143]. Third, macrophage heterogeneity and temporal dynamics complicate causal attribution. Resident versus recruited macrophages, CCR2+ inflammatory versus CCR2– reparative subsets, and disease-stage–dependent transitions can all alter efferocytic capacity. Without consistent cross-study phenotyping and time-resolved sampling, it remains challenging to assign specific efferocytosis defects to defined macrophage states or to determine whether impaired efferocytosis is a driver of pathology, a consequence of tissue stress, or both [52]. Finally, therapeutic translation faces safety and context-dependence constraints. Strategies that enhance efferocytosis could carry systemic immunological risks (e.g., excessive immunosuppression or off-target phagocytosis) and may have opposite implications in non-cardiovascular settings such as cancer, where efferocytosis can support immune evasion [123, 144]. Therefore, a central limitation of the field is the scarcity of human, lesion-resolved, functional validation and the limited clinical evidence defining optimal timing, dosing, and patient selection for pro-efferocytic interventions [122, 145].
Future work should move beyond describing “impaired efferocytosis” as a broad label and instead build a stage-resolved, cell-state–resolved, and lesion-resolved map of cardiovascular efferocytosis in humans. A key priority is to integrate single-cell/spatial profiling with functional efferocytosis readouts to answer the practical question of “who clears what, when, and where” in human disease. This will require harmonized pipelines that combine (i) spatially anchored macrophage states, (ii) in situ quantification of apoptotic targets and uptake events, and (iii) molecular readouts of post-engulfment processing and resolution programs. Such integration can also enable patient stratification by dominant checkpoint failure (inhibitory signaling [123, 146], receptor shedding [130] or defective digest outputs [121]), which is essential for precision therapeutics.
Mechanistically, several areas deserve focused attention. (i) Receptor availability and stability in vivo: defining how protease activity, inflammatory cytokines, and metabolic stress remodel the TAM axis and bridging pathways across time [147, 148]. (ii) Post-engulfment biology as a therapeutic lever: clarifying how efferosome maturation, lysosomal competence, LAP-associated programs, and immunometabolic rewiring collectively determine whether uptake culminates in inflammation resolution or in persistent stress signaling [121, 122]. (iii) Systems-level crosstalk: elucidating how efferocytosis interfaces with inflammasome activation, oxidative stress, fibrosis programs, and electrophysiological remodeling, particularly in chronic diseases where “Digest” failure may dominate [125]. Addressing these questions will refine which molecular nodes are actionable, which are merely correlates, and which are context-specific liabilities [149].
Clinically, the field is likely to benefit from combination strategies that
reflect the comparative framework summarized in this review: pairing a shared
core module (e.g., relieving inhibitory cues such as CD47–SIRP
Finally, translational progress will depend on biomarker and endpoint development that can report checkpoint-specific engagement in patients. Candidate directions include signatures of receptor shedding versus preserved uptake but defective digest/resolution outputs, lesion imaging approaches that quantify apoptotic burden and clearance dynamics, and circulating markers that reflect pro-resolving mediator tone [130]. Together, these advances can transform efferocytosis-targeted therapy from a compelling mechanistic concept into a clinically testable, precision-guided strategy for cardiovascular repair and remodeling prevention [32].
Author HL and Author XZ contributed equally as co-first authors, leading the conceptualization, literature review, and manuscript drafting. Author YY contributed to the conceptual design of the review and critical interpretation of the literature, while Author AF and KY designed the literature search strategy and synthesized trends. Author ZG contributed to the intellectual framework, and Author YS assisted with literature compilation and editing. Author GF contributed to the design of the review scope and thematic structure, overall supervision, and critical revision of the manuscript. Author LL and CM, as corresponding authors, contributed to the conceptualization of the study, interpretation of findings, supervision of the project, and critical revision of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thank the reviewers for their valuable comments and suggestions.
This work was supported by the National Natural Science Foundation of China (NSFC) (82574792, Li L; 82430121, Fan GW); the National Science and Technology Major Project for Prevention and Treatment Research of Cancer, Cardio-cerebrovascular, Respiratory and Metabolic Diseases (2023ZD0502706, Fan GW); and the Youth Talent Lifting Project of China Association of Chinese Medicine (2024-QNRC2-B22, Li L).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.