






 , Sixuan Wu 1,†, Lijun Zeng 1, Zhongxiang Fan 1, Yongmei Luo 1, Xinyu Yi 1, Pan Wang 1, Liang Mo 3,*
, Sixuan Wu 1,†, Lijun Zeng 1, Zhongxiang Fan 1, Yongmei Luo 1, Xinyu Yi 1, Pan Wang 1, Liang Mo 3,* , Yuehua Li 1,*
, Yuehua Li 1,*1 Department of Oncology, The First Affiliated Hospital, Hengyang Medical School, University of South China, 421001 Hengyang, Hunan, China
2 Department of Oncology, People’s Hospital of Xiangxi Tujia and Miao Autonomous Prefecture, The First Affiliated Hospital of Jishou University, 416000 Jishou, Hunan, China
3 Department of Thoracic Surgery, The First Affiliated Hospital, Hengyang Medical School, University of South China, 421001 Hengyang, Hunan, China
†These authors contributed equally.
Abstract
Lung adenocarcinoma (LUAD) is recognized as the predominant subtype of non-small cell lung cancer (NSCLC), characterized by its aggressive behavior and notable capacity for metastasis. Recent studies have uncovered the significance of palmitoylation, a vital post-translational modification, in regulating biological functions such as cellular signaling and protein localization, suggesting its potential role in cancer biology. However, the implications of palmitoylation-associated long non-coding RNAs (lncRNAs) in LUAD pathology remain insufficiently characterized.
Utilizing publicly available datasets, we conducted an extensive bioinformatics analysis to evaluate the expression and prognostic significance of palmitoylation-associated lncRNAs in LUAD. Our analysis identified 13 lncRNAs as potential prognostic biomarkers. Specifically, we sought to develop a prognostic model based on these lncRNAs to improve our understanding of their contribution to LUAD prognosis. The robustness of the prognostic model was subsequently verified through survival analysis and receiver operating characteristic (ROC) curve assessments.
This study established a prognostic risk model based on 13 palmitoylation-related lncRNAs, demonstrating excellent predictive performance, with area under the curve (AUC) values of 0.743, 0.724, and 0.748 for 1-year, 3-year, and 5-year overall survival, respectively. Multivariate Cox regression analysis indicated that the model’s risk score serves as an independent prognostic predictor, surpassing traditional clinicopathological features. Differential expression analysis and quantitative reverse transcription polymerase chain reaction (qRT-PCR) validation revealed significant downregulation of AL157895.1 in LUAD tissues and cell lines, whereas AL355472.3, SALRNA1, AL590666.4, AC026355.2, and LINC00862 were significantly upregulated. Importantly, low expression of AL157895.1 was strongly associated with poor patient prognosis.
The findings underscore the potential of palmitoylation-associated lncRNAs as independent prognostic markers for patients with LUAD, paving the way for more personalized treatment approaches. Future studies should further explore their biological mechanisms.
Keywords
- lung adenocarcinoma
- palmitoylation
- lncRNAs
- prognostic model
- survival analysis
Lung adenocarcinoma (LUAD) is the most widespread type of non-small cell lung cancer (NSCLC) [1], and currently represents about 35%–40% of all lung malignancy cases [2]. The prevalence of LUAD is progressively younger, with many patients being diagnosed before the age of 40 [3]. Investigations showed that the survival rate of early-stage LUAD patients is substantially higher than that of advanced-stage patients, highlighting the importance of early diagnosis and treatment [4]. Although targeted therapies and immunotherapies have brought new hope for LUAD treatment, many patients develop resistance after initial targeted therapy [5], and the effectiveness of immunotherapy varies across patient populations [6]. The selection of suitable patients for immunotherapy remains an area requiring further research and exploration [7]. Faced with challenges such as resistance, personalized treatment, and immunotherapy, it is essential to determine additional biomarkers to enhance patient survival and quality of life.
Palmitoylation is a vital post-translational modification that involves the reversible attachment of fatty acid chains, affecting protein stability, localization, and function [8, 9]. Recently, research has revealed a close association between long non-coding RNAs (lncRNAs) and palmitoylation in tumors [10]. For example, the lncRNA lncZBTB10 promotes the androgen receptor (AR) function in prostate cancer by triggering S-palmitoylation, thereby driving tumor progression and resistance [11]. Additionally, the lncRNA PVT1 promotes exosome secretion in pancreatic cancer cells, a process closely associated with the palmitoylation of YKT6, RAB7, and VAMP3 [12]. The lncRNA LOC541471 is significantly correlated with lymph node metastasis and nerve invasion in head and neck squamous cell carcinoma [13]. Lastly, lncRNA HNF1A-AS1 exhibits aberrant expression in multiple cancers, and its palmitoylation status may influence tumor cell proliferation and migration. These outcomes demonstrate that palmitoylation-associated lncRNAs play a vital role in tumor initiation and progression. Future research should further explore their potential as therapeutic targets for LUAD.
Recently, there has been an increasing focus on the function of lncRNAs in LUAD, particularly those associated with protein palmitoylation. For instance, ZDHHC9, a palmitoyltransferase, is significantly upregulated in LUAD, and its deletion suppresses cell aggressiveness while triggering apoptosis, suggesting that ZDHHC9 may facilitate tumorigenesis by regulating PD-L1 palmitoylation [14, 15]. Additionally, the overexpression of lncRNA MALAT1 in LUAD shows significant correlations with TNM stage, tumor size, and lymph node metastasis [16]. MALAT1 controls RhoA expression by competing with miR-429, thereby affecting cell growth and epithelial-mesenchymal transition (EMT) [17]. In studies of lncRNAs, DUXAP8 has been demonstrated to inhibit ferroptosis by inducing SLC7A11 palmitoylation, a process already confirmed in hepatocellular carcinoma [18]. A similar mechanism may exist in LUAD, where DUXAP8 expression correlates with tumor resistance, suggesting its significant role in the tumor microenvironment [18]. Moreover, lncRNA H19 shows a significant overexpression in LUAD tissues and is related to the methylation status of CDH1. In addition, cisplatin resistance in LUAD is mediated by lncRNA H19 through its translocation from the nucleus to the mitochondria and subsequent promotion of mitophagy [19]. Therefore, a comprehensive investigation on the link between these lncRNAs and palmitoylation will help elucidate the molecular mechanisms of LUAD and offer new strategies for clinical treatment. However, the above studies have not integrated a comprehensive bioinformatics analysis with large-scale databases, which presents certain limitations.
More broadly, related studies have emphasized the significance of lncRNAs in cancer prognosis from various perspectives. For example, Chaturvedi et al. [20] recently identified the prognostic lncRNA BANCR in LUAD using weighted gene co-expression network analysis (WGCNA). Although this study also investigates LUAD, its analytical approach differs from ours, as we specifically focus on screening lncRNAs associated with the palmitoylation process. Identifying distinct lncRNAs through different analytical strategies highlights the complexity of lncRNA regulatory networks in LUAD. Moreover, constructing prognostic models based on palmitoylation-associated lncRNAs is effective in other cancers [21], as demonstrated by Wang et al. [10] in breast cancer and Zou et al. [22] in hepatocellular carcinoma. These studies provide compelling evidence supporting the potential of “palmitoylation-associated lncRNAs” as an emerging biomarker category while also establishing a strong theoretical foundation for applying this innovative approach to LUAD.
This investigation aims to develop a prognostic model based on palmitoylation-related lncRNAs to offer innovative biomarkers for personalized treatment of individuals with LUAD. By obtaining clinical and RNA sequencing data from individuals with LUAD in the Cancer Genome Atlas (TCGA) database, we identified palmitoylation-associated lncRNAs and developed a LUAD prognostic model using least absolute shrinkage and selection operator (LASSO) regression and Cox regression. Additionally, the study explores the link between these lncRNAs and immune escape mechanisms, immune cell (IC) infiltration, and tumor mutational burden (TMB). Ultimately, the diagnostic, therapeutic, and prognostic utility of these lncRNAs will be evaluated, and the role of a specific lncRNA, AL157895.1, in the disease will be further studied. In summary, focusing on the function of palmitoylation-related lncRNAs in tumor immunity will contribute to a comprehensive investigation of potential therapeutic targets for LUAD and improve the prognosis of LUAD patients.
Clinical data for individuals with LUAD were acquired from the TCGA database (https://portal.gdc.cancer.gov/), including RNA-seq data, patient demographics, and somatic mutation data, covering 522 tumor samples. Because different analyses required different types of available and matched data, the final sample size varied across analyses. Specifically, 507 samples with complete survival information and matched expression data were included in the survival analysis, 496 samples with available somatic mutation data were included in the mutation analysis, and 448 samples with complete subtype-related information were included in the subtype analysis. We excluded the samples missing complete prognostic data or healthy tissue specimens. Gene selection for palmitoylation was based on a recent study [23, 24, 25, 26, 27]. The flowchart and the specific model construction process are illustrated in Fig. 1 and Supplementary Fig. 1.
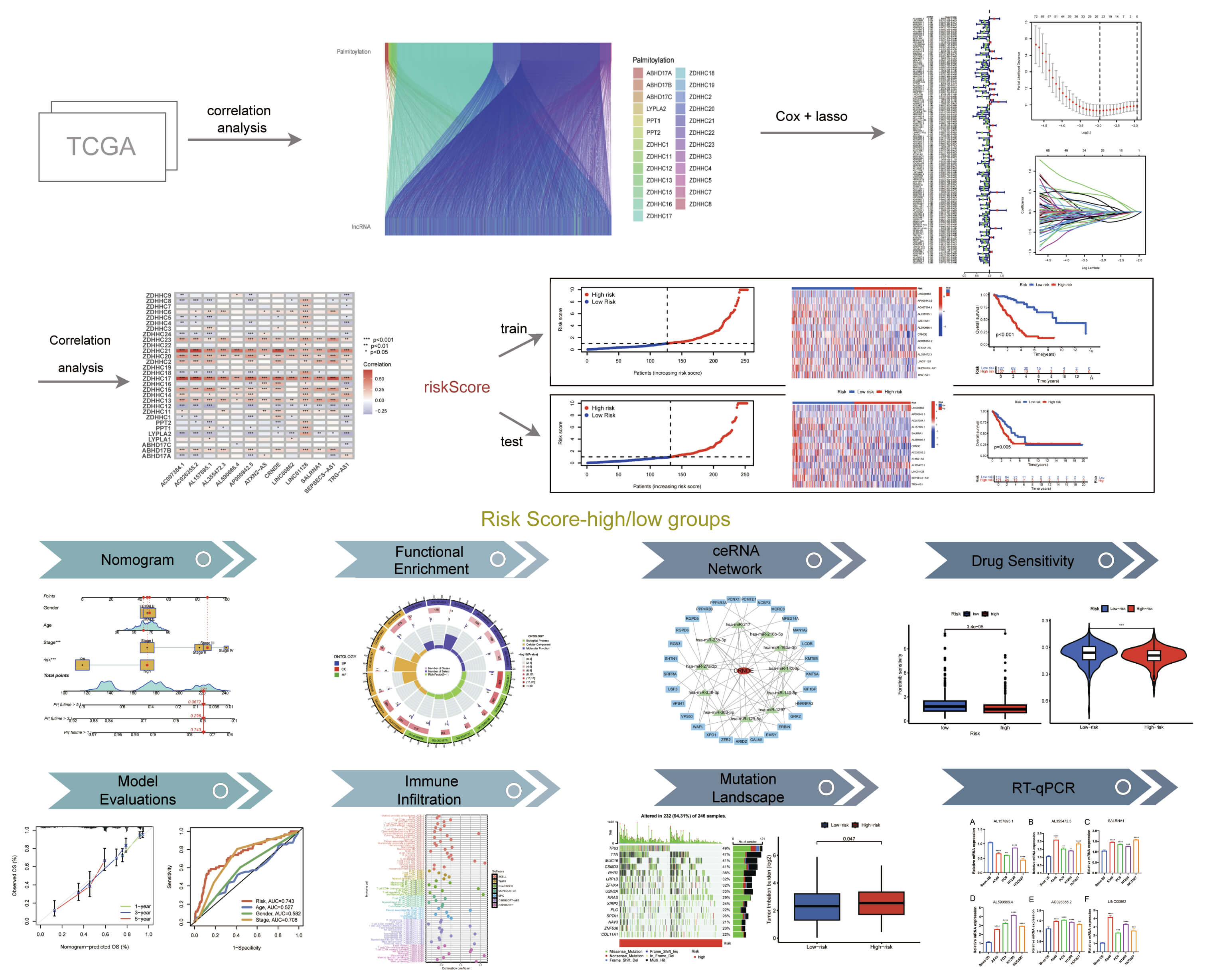 Fig. 1.
Fig. 1.
The flowchart of this paper. HRG, high-risk group; LRG,
low-risk group; *p
After an exhaustive review of the literature, 30 genes implicated in
palmitoylation were identified, including ZDHHC1-9, ZDHHC11-24, PPT1, PPT2, ABHD17A,
ABHD17B, ABHD17C, LYPLA1, and LYPLA2. To examine the link between these
palmitoylation-associated genes and lncRNAs, a Pearson correlation analysis was
performed. This analysis used a Pearson correlation coefficient threshold of
Separate training and test sets of LUAD tumor samples were created. To identify
more representative genes, the training cohort underwent LASSO regression
analysis. Then, genes that could be prognostic were filtered using univariate Cox regression [29, 30]. Genes were categorized as potential prognostic indicators if
they showed statistical significance in the Cox regression analysis
(p-value
By performing a correlation analysis between palmitoylation-correlated genes and the modeled lncRNAs, we identified connections between these lncRNAs and palmitoylation genes, aiming to clarify their relationship and deepen our understanding of the pathways involved.
We established a correlation between the modeled lncRNAs and genes associated with palmitoylation using a correlation analysis. This analysis illustrated the interactions between numerous lncRNAs and palmitoylation-associated genes, thereby improving our understanding of how these lncRNAs are interconnected with pathways relevant to palmitoylation.
To evaluate survival disparities between HRG and LRG, we conducted a receiver operating characteristic (ROC) analysis, which provided insights into the prognostic capabilities of the genetic characteristics. Additionally, to corroborate the predictive accuracy of our model, we evaluated its performance across distinct subgroups by classifying the samples and executing separate survival analyses based on gender and disease stage.
By analyzing immune-associated functions, distinct immune-related functions that differed between HRG and LRG were identified, providing a valuable reference for future research.
By integrating the risk values for each sample with the gene expression matrix, we conducted a functional enrichment analysis of the differentially expressed genes identified in HRG and LRG. Using gene ontology (GO), gene set enrichment analysis (GSEA), and Kyoto encyclopedia of genes and genomes (KEGG), we established filtering criteria based on pathways showing differential expression between the two groups. Subsequently, we selected the pathways that showed significant expression differences for further investigation [31, 32, 33].
The ESTIMATE algorithm assessed stromal and immune scores between the two patient groups, and we also examined the relationship between TMB and the two risk groups.
We analyzed variations in TMB and TIDE using the TCGA database mutation load data and correlated them with sample risk values. This helped us compare mutation loads and variations in modeled genes between HRG and LRG, revealing tumor mutation mechanisms. Additionally, using http://tide.dfci.harvard.edu, we assessed the potential for immunotherapy evasion in LUAD samples, comparing HRG and LRG [34].
To explore the relationship between the prognostic risk score developed in this
study and chemotherapy drug sensitivity, the oncoPredict package was used to
predict drug response. The training dataset was sourced from the publicly
available Genomics of Drug Sensitivity in Cancer 2 (GDSC2) database. First, the
gene expression matrix from LUAD tumor samples obtained from the TCGA database
was preprocessed. Duplicate genes were averaged using the avereps function, and
low-expression genes (rowMeans
The ATCC provided the Beas-2B, A549, PC9, H1299, and HCC827 cell lines. After rapid thawing in a 37 °C water bath, the frozen cell stocks were transferred to sterile 15 mL centrifuge tubes containing 10 mL of DMEM/F12 medium and centrifuged to remove the freezing medium. The cell pellets were then resuspended in fresh complete medium and seeded into culture flasks. All cell lines were validated by STR profiling and tested for mycoplasma contamination. The tubes were then maintained in a humidified incubator at 37 °C with 5% CO2. For RNA extraction, total RNA was isolated via TRIzol reagent (Invitrogen, Carlsbad, CA, USA). This total RNA was then utilized alongside a PrimeScript RT reagent kit (Takara) to synthesize complementary DNA (cDNA). qRT-PCR was performed using a CFX-96 apparatus (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with Takara SYBR Green assays. Data normalization was performed using the 2-ΔΔCt method, with GAPDH as the reference gene. Table 1 displays the primer sequences employed in this investigation’s qRT-PCR.
| lncRNA | Sequence (5′-3′) |
| AL157895.1 | F: TGCCTACAACACATTCATCTACAC |
| R: TCCACATCCTCCCTCTATTCAC | |
| AL355472.3 | F: TAGGTTATGGAAGGCTGGAGTC |
| R: CTGGGCAAGTCTTACACAAGT | |
| SALRNA1 | F: GCTAATGCTGTCTTCCTTGTAACT |
| R: TTGGTGGAGTGCTTGTAGAGA | |
| AL590666.4 | F: TGGTGAAATGGGAGAGTGAGA |
| R: CGTCATGGTCAGGATCAGTTC | |
| AC026355.2 | F: CACCTCCAGTGATGGCAAAT |
| R: GTGTGAGACAACCTGAGCATT | |
| LINC00862 | F: AGGAGAAGAGAAGACACGAAGG |
| R: CAGAAGTCCCAAGTCCCAAATC |
lncRNA, long non-coding RNAs; F, forward; R, reverse.
All statistical analyses in this study were conducted usingR software version 4.2.2
(R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism version 9.0
(GraphPad Software, San Diego, CA, USA).. LASSO-Cox regression was used to construct
prognostic models, with the median risk score serving as the cut-off to separate
samples into high- and low-risk groups. Survival comparisons were performed using
Kaplan-Meier curves and log-rank tests. In Cox regression analyses, all variables
satisfied the proportional hazards assumption. Comparisons of clinical and
molecular features between subtypes were conducted using chi-square tests.
Multiple group comparisons were analyzed using analysis of variance (ANOVA),
followed by Bonferroni and Dunnett multiple-comparison tests. Two-group
comparisons were assessed using t-tests. A p-value
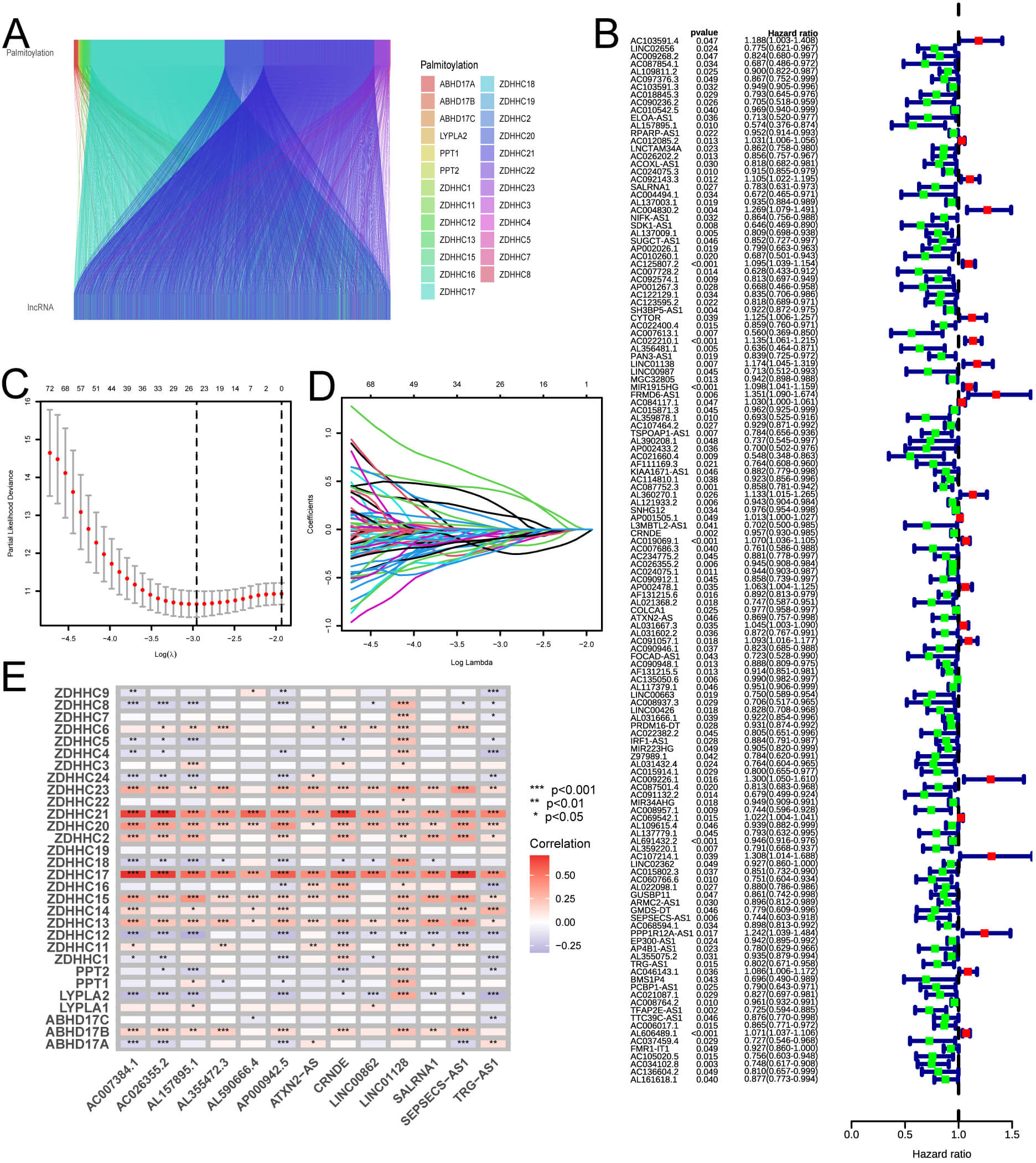
Through the application of co-expression analysis, we determined 1662 lncRNAs related to palmitoylation. The interconnections between palmitoylation-related genes and lncRNAs were illustrated via a Sankey diagram (Fig. 2A). We categorized the 522 LUAD samples retrieved from the TCGA database into training and validating groups. Subsequently, a univariate Cox regression analysis was conducted on the training cohort to determine potential prognostic genes. Fig. 2B depicts the outcomes.
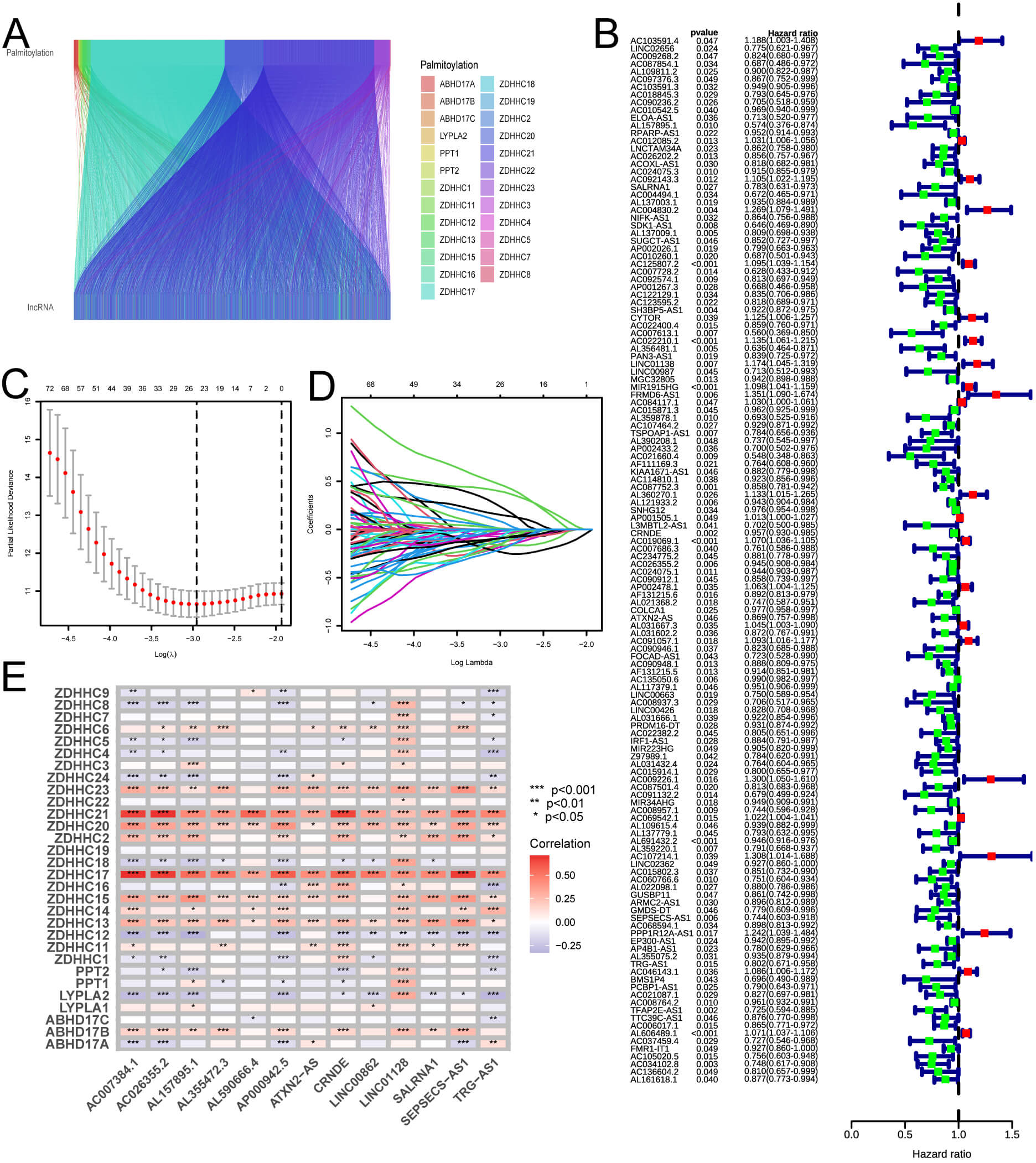 Fig. 2.
Fig. 2.
Correlation analysis outcomes. (A) The correlation analysis indicates a relationship between palmitoylation and lncRNAs, revealing connections among various modules, with distinct palmitoylation-associated genes denoted by different colors. (B) The outcomes of Cox regression analysis are shown; red dots indicate an HRG classification, and green dots indicate an LRG. (C,D) LASSO regression analysis results demonstrate that a model incorporating 13 genes offers increased accuracy and reliability. (E) A correlation analysis between lncRNAs included in the model and the palmitoylation-associated genes is presented, with positive (red) and negative (blue) correlations. HRG, high-risk group; LRG, low-risk group; LASSO, least absolute shrinkage and selection operator.
Fig. 2C,D display the results of LASSO regression analysis, which identified genes more representative of the population. By combining 13 lncRNAs related with palmitoylation (AC007384.1, AC026355.2, AL157895.1, AL355472.3, AL590666.4, AP000942.5, ATXN2-AS, CRNDE, LINC00862, LINC01128, SALRNA1, SEPSECS-AS1, TRG-AS1), a predictive model was subsequently produced. The prognostic model was used to determine the risk value for each sample. Individuals were then categorized into two groups, LRG and HRG, with the median risk score serving as the cut-off point. A correlation analysis was performed between palmitoylation-correlated genes and the lncRNAs used to build the model to detect the link between lncRNAs and palmitoylation genes. The outcomes illustrated that the strongest correlation was with 13 lncRNAs associated with palmitoylation, which belonged to the Zinc Finger DHHC-Type Containing protein family (ZDHHC). This helped us understand the relationship between various lncRNAs and palmitoylation genes. Fig. 2E displays the results. The previous studies of 13 lncRNAs are also provided (Table 2, Ref. [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45]).
| lncRNA | Research status | Literature evidence |
| AC007384.1 | Research on colon cancer and lung adenocarcinoma | Huang and Pan [35], Li et al. [36] |
| AC026355.2 | Research on lung adenocarcinoma | Lu et al. [37] |
| AL157895.1 | Reported on lung adenocarcinoma | Sun et al. [38] |
| CRNDE | Research on hepatocellular carcinoma | Tang et al. [39], Li et al. [40] |
| LINC00862 | Research on cervical and gastric cancer | Liu et al. [41] |
| LINC01128 | Research on prostate cancer | Zhao et al. [42] |
| SALRNA1 | Research on non-small cell lung cancer | Yi et al. [43] |
| SEPSECS-AS1 | Research on lung adenocarcinoma | Wang et al. [44] |
| TRG-AS1 | Research on breast cancer | Zhu et al. [45] |
| AL355472.3 | Newly reported | No studies available |
| AL590666.4 | Newly reported | No studies available |
| AP000942.5 | Newly reported | No studies available |
| ATXN2-AS | Newly reported | No studies available |
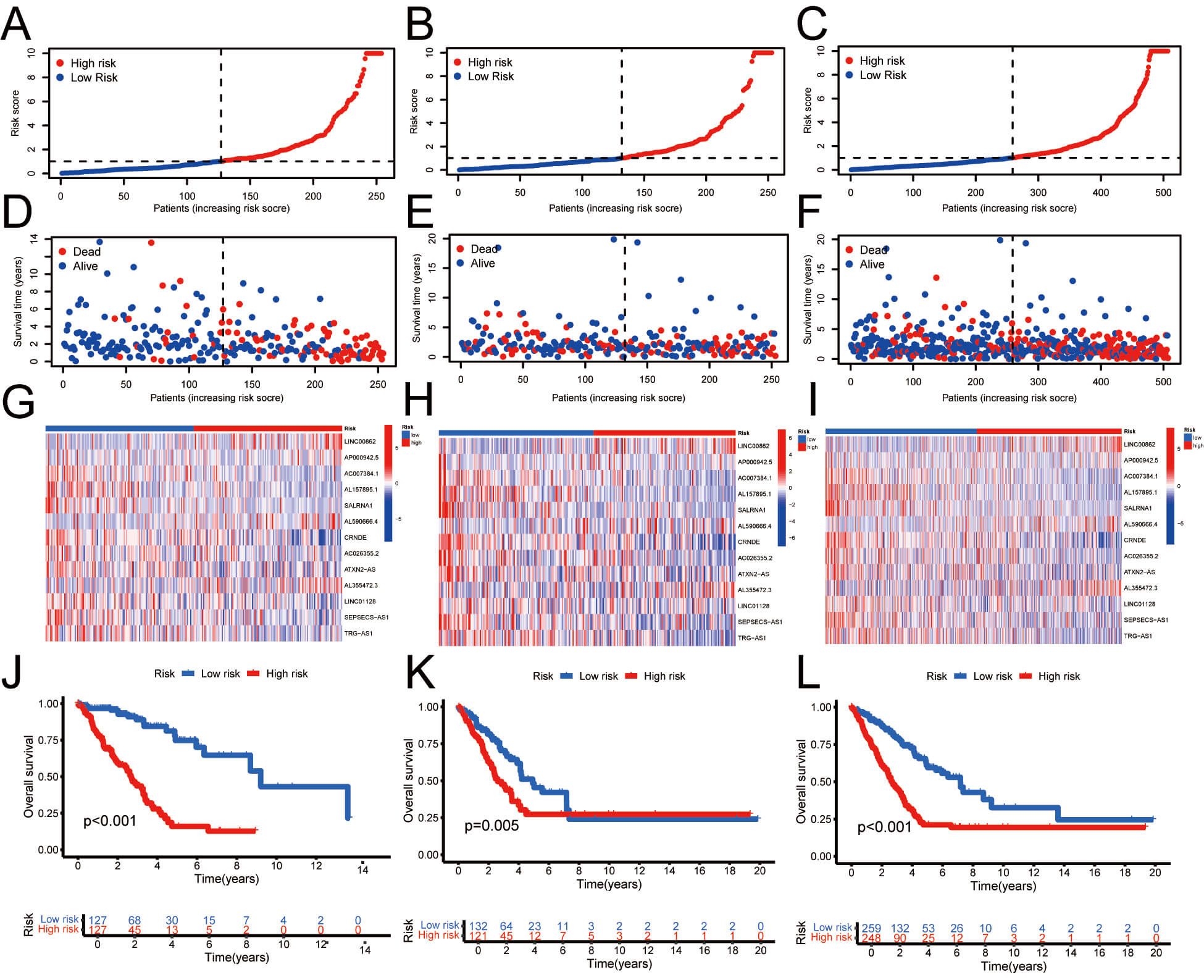
We represented each sample using risk curves, illustrating that, over time, mortality rates among individuals in the HRG exceeded those of the LRG. Fig. 3A–I depict these results. Survival analysis revealed that, across all samples and within both training and validation groups, the survival rates for the LRG exceeded those of the HRG over time (Fig. 3J–L). In conclusion, the predictive model based on risk scores effectively distinguishes survival outcomes between HRG and LRG, with patients classified as LRG generally displaying higher survival rates at all evaluated time points. This underscores the potential utility of risk scores as a predictive tool. The risk score was measured as follows:
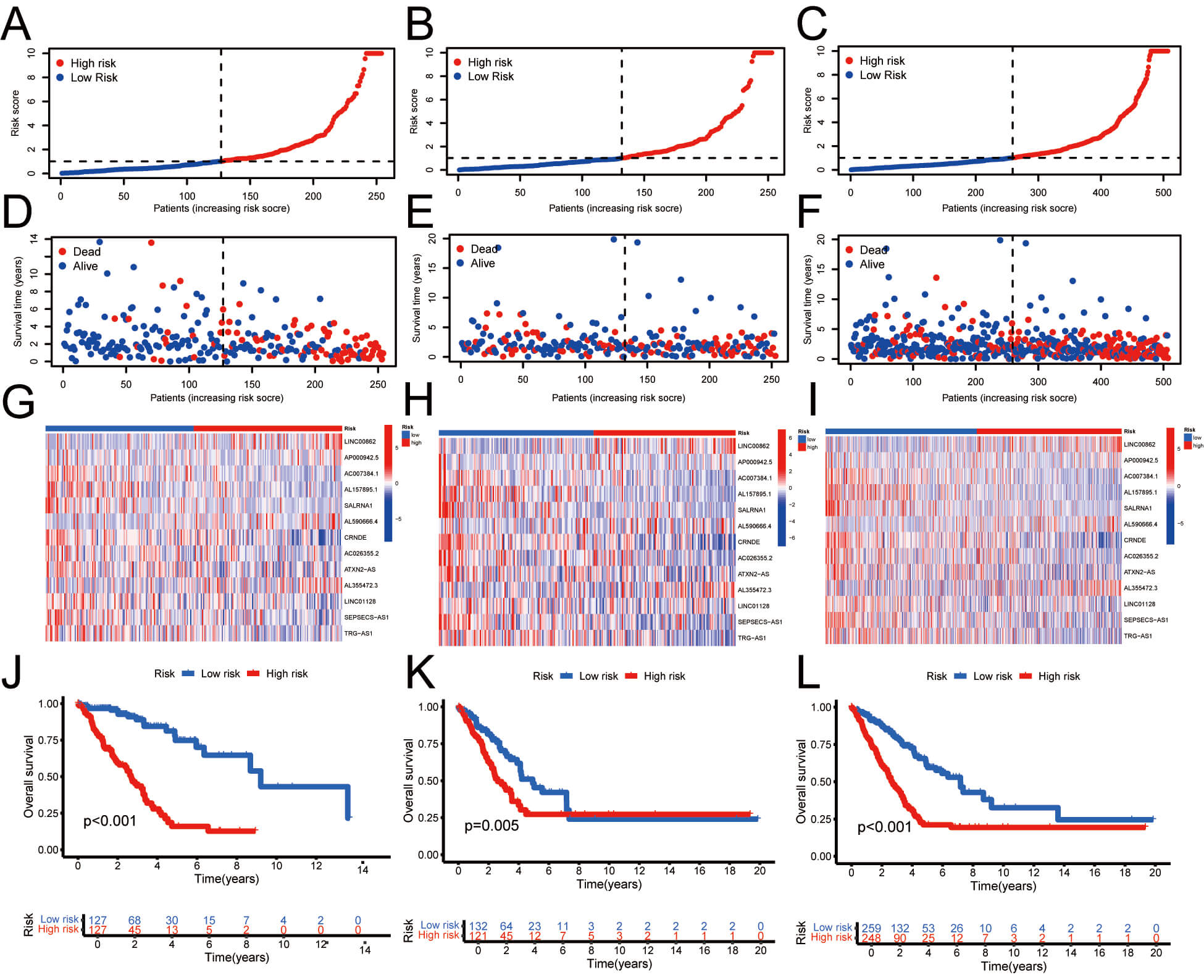 Fig. 3.
Fig. 3.
Prognostic assessment of the risk model in different cohorts. (A–C) Risk score distribution in training, testing, and full sets. (D–F) OS
and survival status. (G–I) Heatmap of 13 lncRNA expression. (J–L) Kaplan-Meier
survival curves. T, tumor; N, node; M, metastasis. The risk score = LINC00862
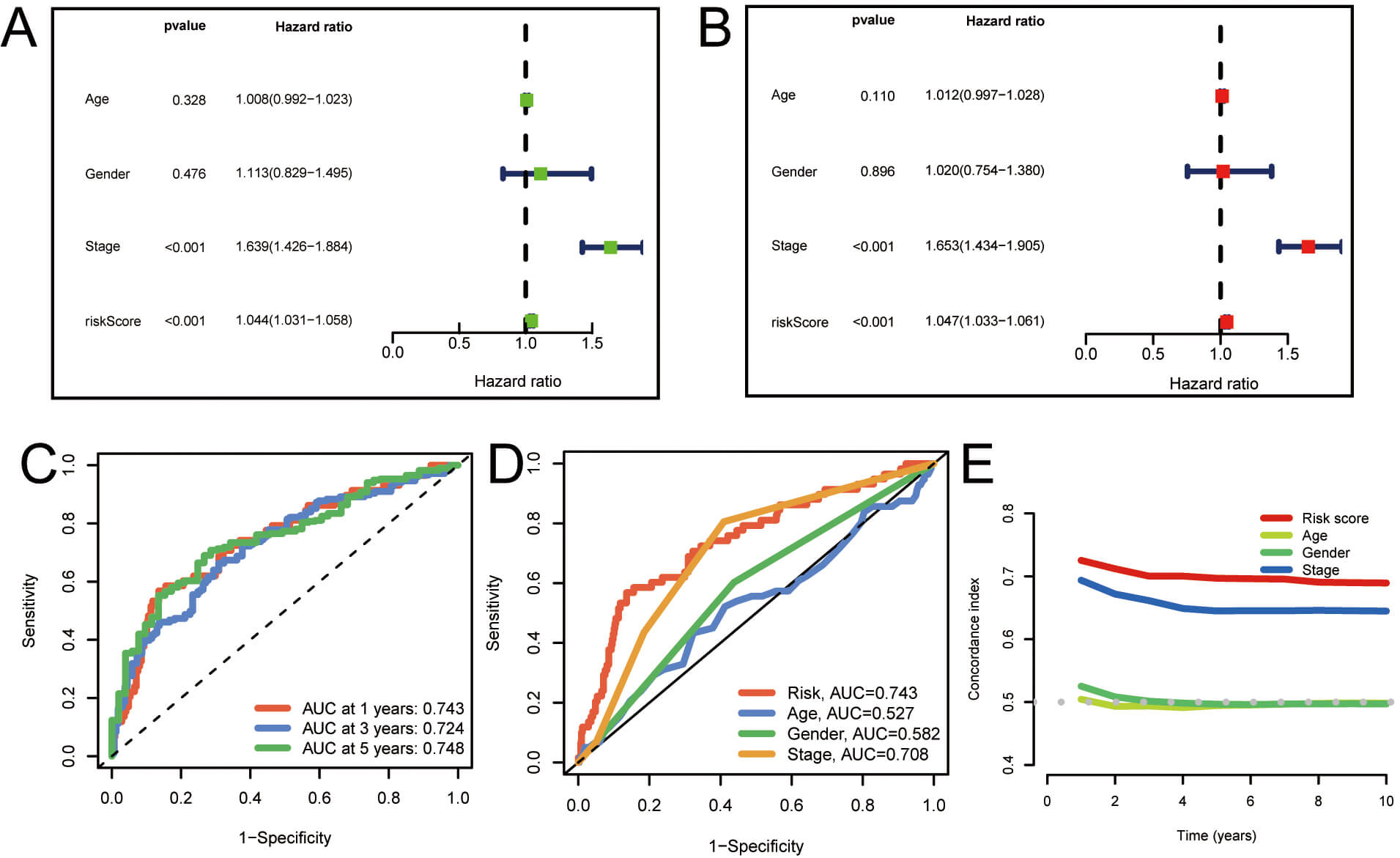
To assess the prognostic significance of palmitoylation-related lncRNAs in individuals with LUAD, univariate and multivariate Cox regression analyses were performed. The findings demonstrated a significant correlation between the risk score derived from palmitoylation-related lncRNAs and overall survival (OS) in LUAD patients, establishing it as an independent prognostic factor (Fig. 4A,B). Moreover, the area under the ROC curve (AUC) for 1-, 3-, and 5-year survival rates was 0.743, 0.724, and 0.748, respectively (Fig. 4C). Furthermore, the predictive capacity of this model’s risk score was superior to that of other clinical parameters (Fig. 4D). Similarly, the concordance index (C-index) for the risk model surpassed that of other clinical features evaluated (Fig. 4E). These analyses show that the risk score model constructed from palmitoylation-associated lncRNAs exhibits high independence and accuracy in prognostic prediction for LUAD patients. Moreover, its predictive ability surpasses that of other clinical features, demonstrating its effectiveness as an independent prognostic factor and as a predictive tool.
 Fig. 4.
Fig. 4.
Evaluation of the prognostic value of the palmitoylation-related lncRNA risk model in LUAD patients. (A,B) Univariate and multivariate analyses demonstrating the model’s independent prognostic significance. (C) AUC values for forecasting 1-, 3-, and 5-year OS. (D) Comparison of the model’s predictive accuracy with clinical and pathological factors (age, gender, and stage). (E) Concordance index curve of the risk model. AUC, Area under the curve; OS, overall survival.
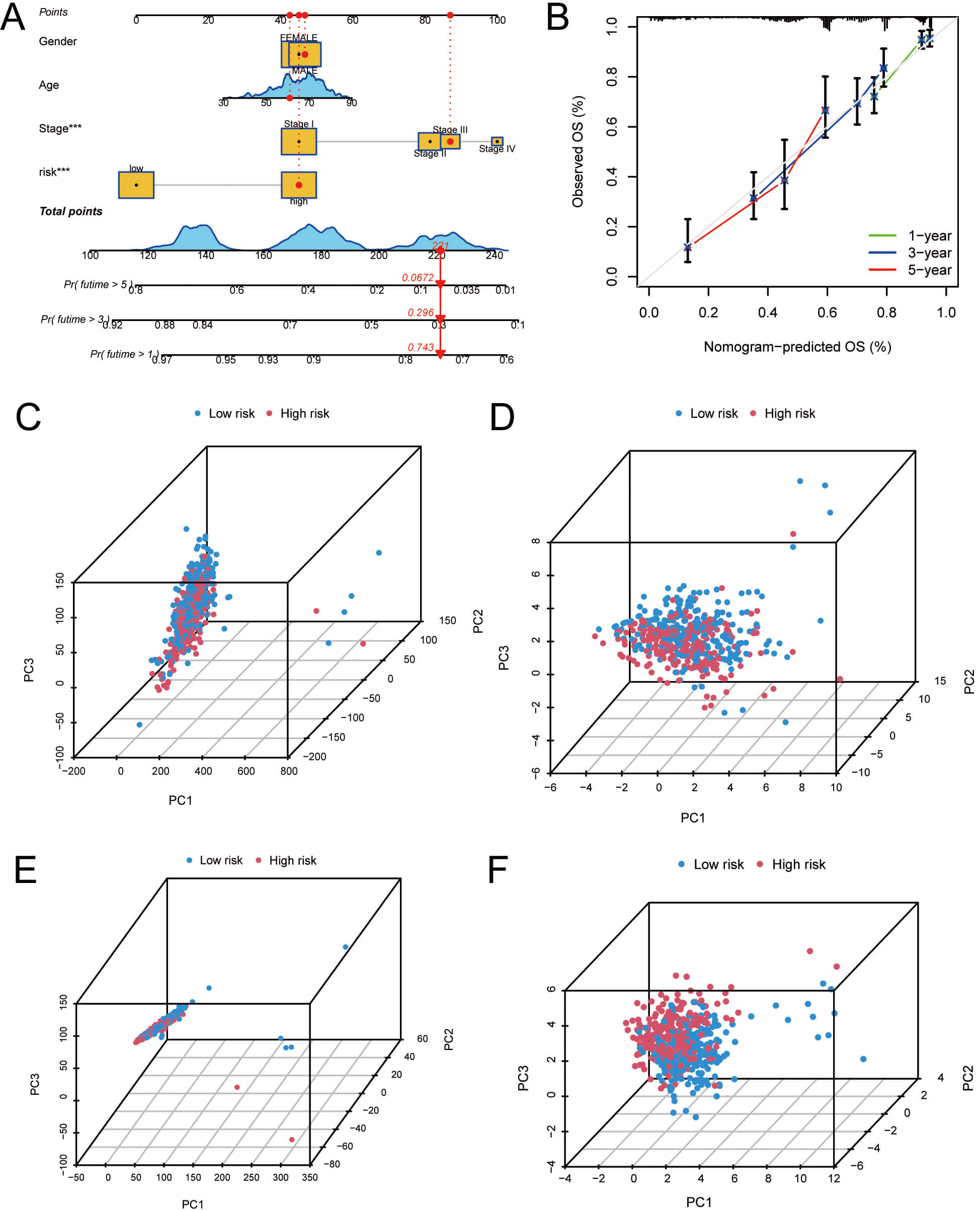
To assess the 1-, 3-, and 5-year OS rates in LUAD patients, a nomogram integrating clinical features and risk scores was developed (Fig. 5A). The calibration curve illustrated that OS rates predicted by the nomogram were in strong agreement with the actual observed rates (Fig. 5B). PCA was performed to examine gene expression differences between HRG and LRG. The results illustrated significant variations between HRG and LRG in the expression profiles of total genes, palmitoylation-associated genes, palmitoylation-related lncRNAs, and the 13 palmitoylation-associated lncRNAs (Fig. 5C–F). Among these, the expression profiles of the 13 palmitoylation-associated lncRNAs showed the most significant variations between HRG and LRG, illustrating that these 13 palmitoylation-associated lncRNAs have the best prognostic capability in distinguishing HRG and LRG for LUAD (Fig. 5F). In summary, these analyses suggest that identifying and analyzing specific palmitoylation-related lncRNAs can provide more accurate risk stratification and prognostic prediction for LUAD patients.
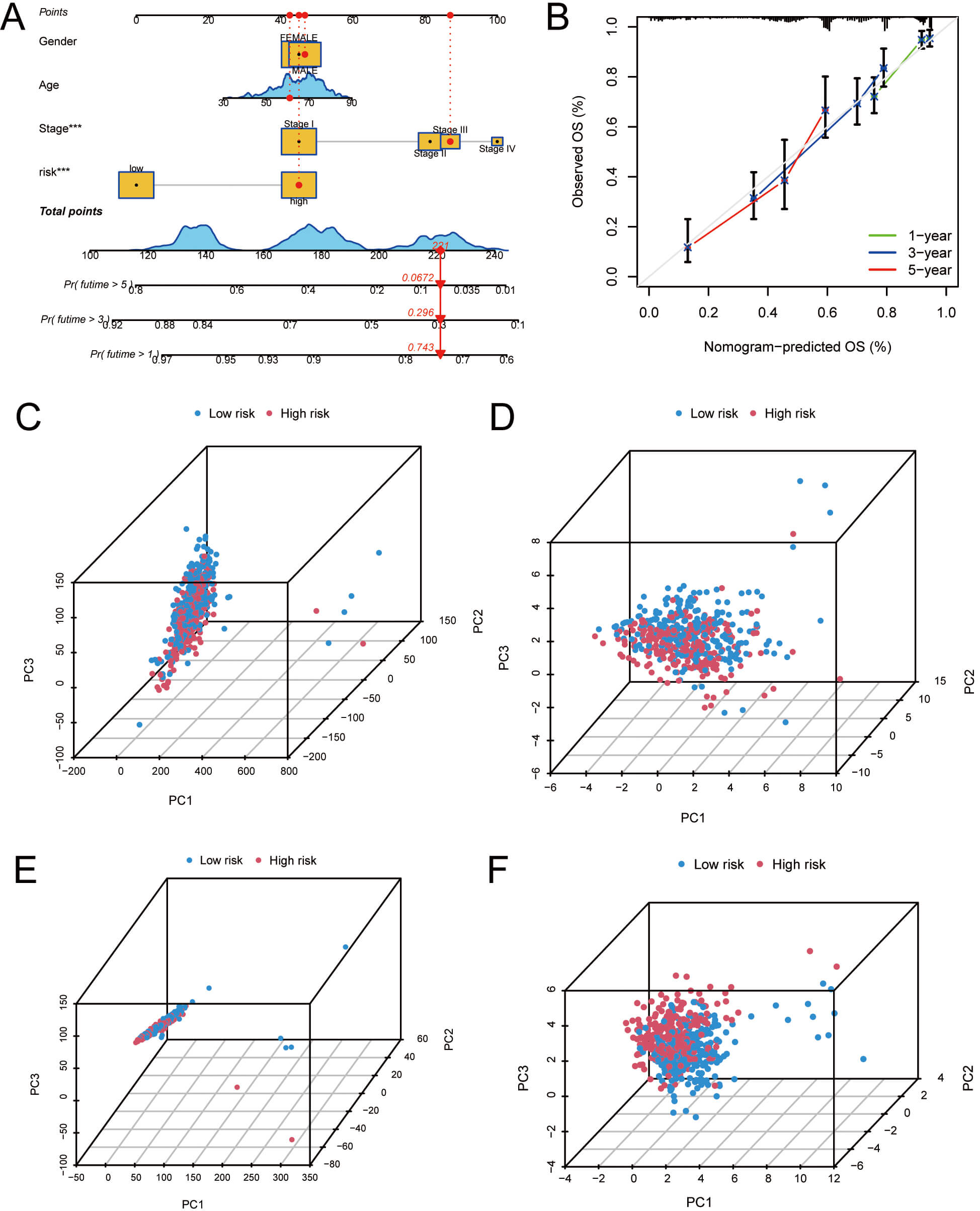 Fig. 5.
Fig. 5.
Nomogram development and PCA analysis. (A) Nomogram predicting
1-, 3-, and 5-year OS for LUAD patients. (B) Calibration curves assessing
consistency between predicted and actual OS. (C) PCA according to all genes. (D)
PCA according to palmitoylation-related genes. (E) PCA based on
palmitoylation-associated lncRNAs. (F) PCA based on lncRNAs included in the risk
model. ***p
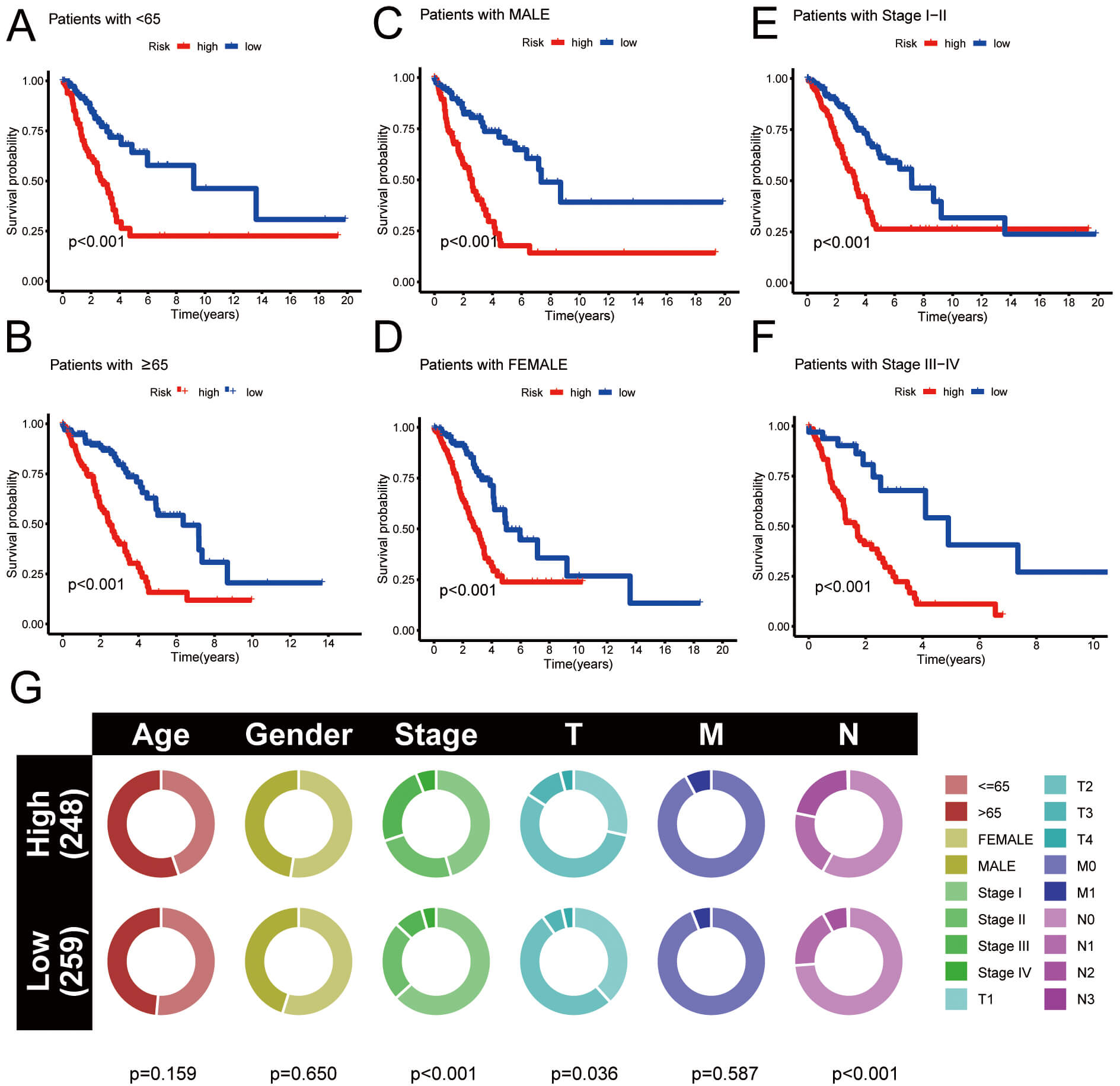
The clinical prognostic analysis for HRG and LRG was conducted. OS rates of HRG individuals with clinical features (age, sex, and stage) were significantly lower than those of LRG individuals (Fig. 6A–F). Fig. 6G illustrates the percentage of diverse clinical data in HRG and LRG of LUAD. Given the limited number of patients, this comparison of clinical data is for reference purposes only. Overall, using risk stratification to predict the prognosis of LUAD patients has potential clinical value, but may be limited by small sample sizes.
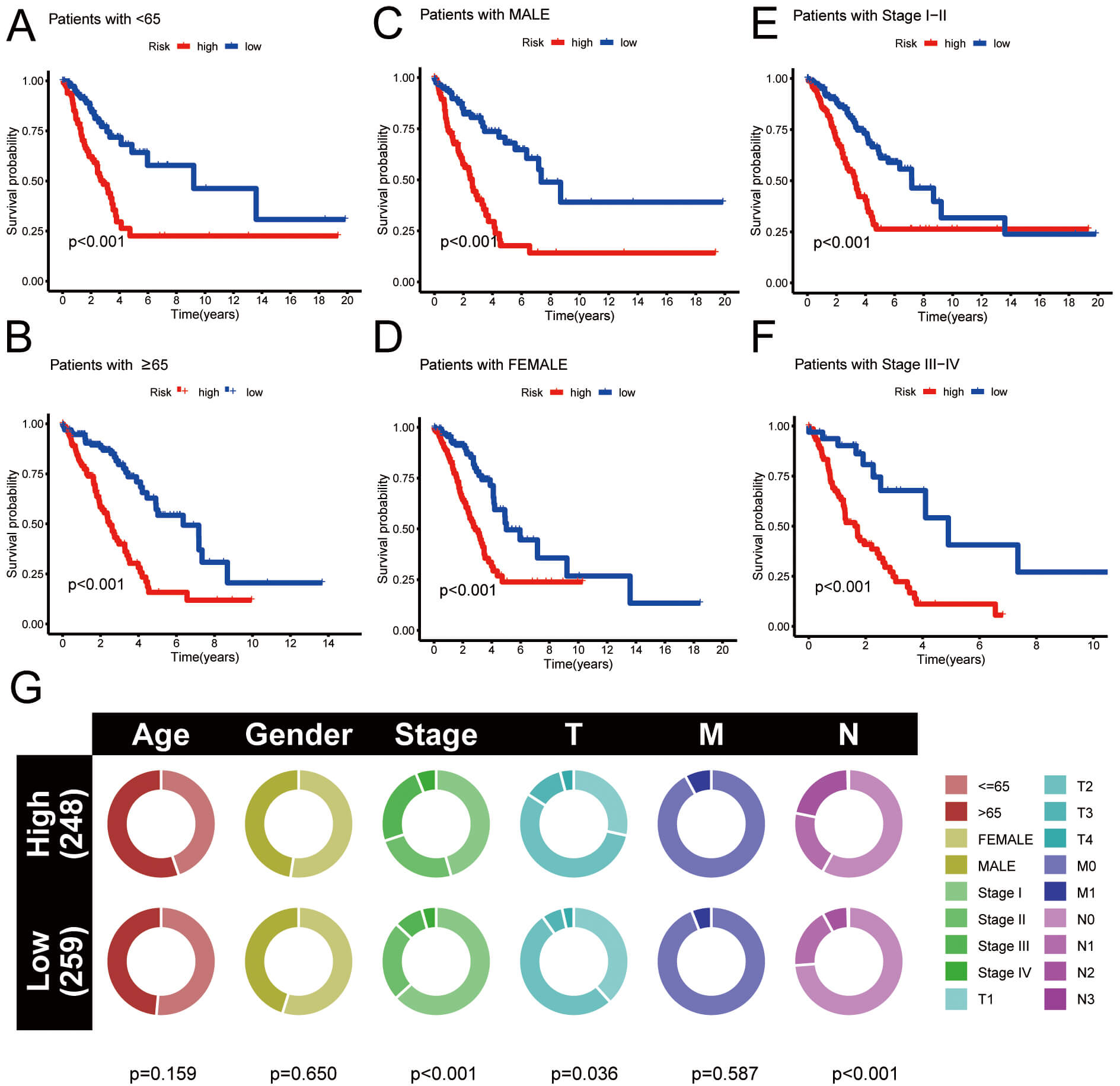 Fig. 6.
Fig. 6.
Clinical prognostic analysis. (A–F) Survival curves for HRG and LRG under various grouping circumstances. (G) The proportion of clinical data in HRG and LRG.
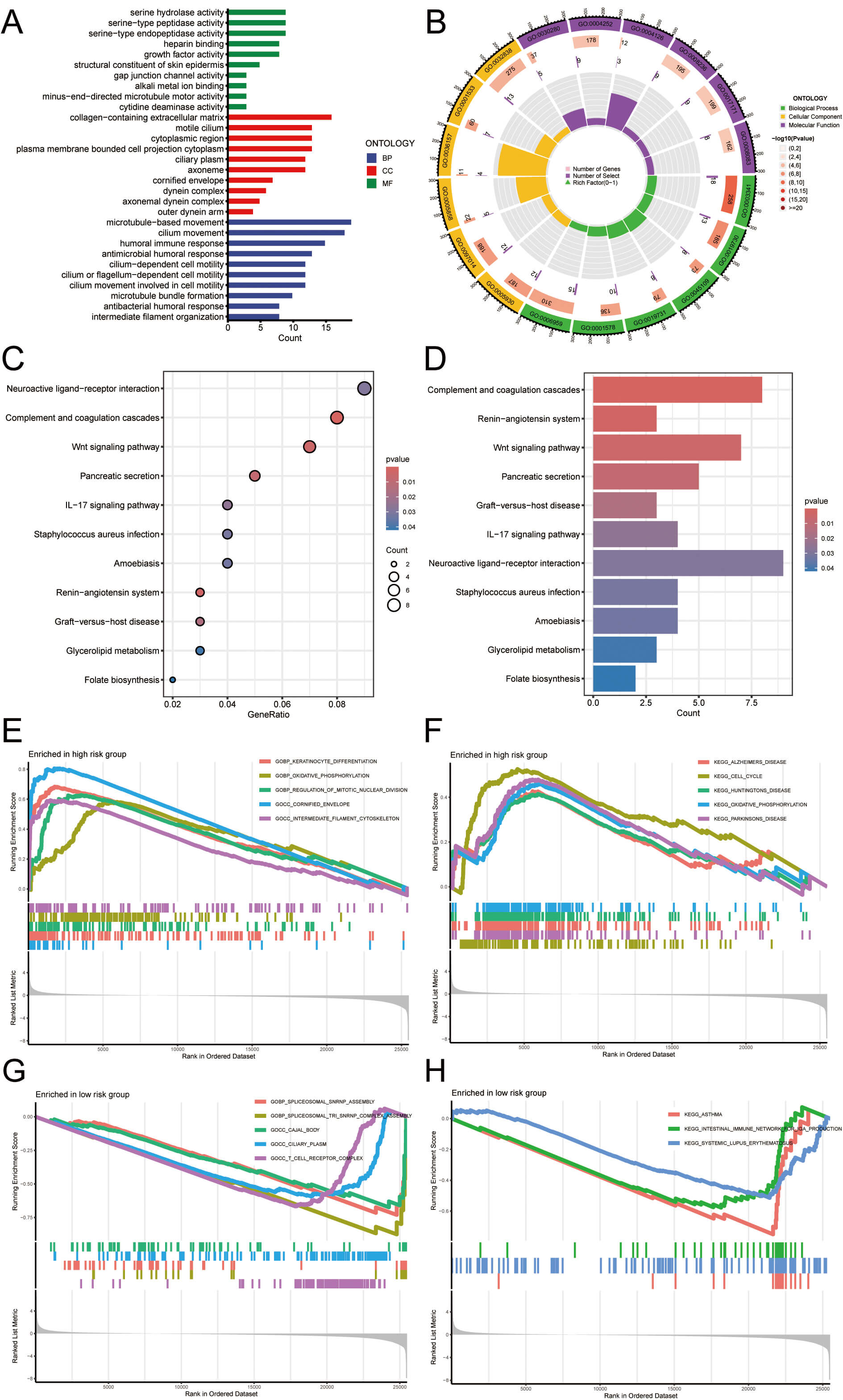
Functional enrichment analysis was performed to identify the significantly enriched GO terms in the HRG. For biological processes (BP), the enriched terms included microtubule-based movement, cilium movement, humoral immune response, antimicrobial humoral response, cilium-dependent cell motility, antibacterial humoral response, cilium or flagellum-dependent cell motility, cilium movement participating in cell motility, microtubule bundle formation, and intermediate filament organization. For cellular components (CC), the enriched terms included collagen-containing extracellular matrix, motile cilium, cytoplasmic region, plasma membrane-bounded cell projection cytoplasm, ciliary plasm, axoneme, cornified envelope, dynein complex, axonemal dynein complex, and outer dynein arm. For molecular functions (MF), the enriched terms included serine hydrolase activity, cytidine deaminase activity, serine-type peptidase activity, serine-type endopeptidase activity, heparin binding, growth factor activity, structural constituent of skin epidermis, gap junction channel activity, alkali metal ion binding, and minus-end-directed microtubule motor activity. The results are shown in Fig. 7A,B. KEGG enrichment analysis was performed to identify significantly enriched pathways in the HRG. The enriched pathways included neuroactive ligand-receptor interaction, complement and coagulation cascades, Wnt signaling pathway, pancreatic secretion, IL-17 signaling pathway, Staphylococcus aureus infection, folate biosynthesis, amoebiasis, renin-angiotensin system, graft-versus-host disease, and glycerolipid metabolism. Fig. 7C,D depict the results. Furthermore, to further examine the most significantly enriched functional terms between HRG and LRG patients, GSEA was conducted. Keratinocyte differentiation, regulation of mitotic nuclear division, intermediate filament cytoskeleton, Alzheimer’s disease, cell cycle, Huntington’s disease, cornified envelope, Parkinson’s disease, and oxidative phosphorylation were enriched in HRG. However, spliceosomal snRNP assembly, ciliary plasm, spliceosomal tri snRNP complex assembly, Cajal body, T cell receptor complex, asthma, intestinal immune network for IgA production, and systemic lupus erythematosus were enriched in LRG patients (Fig. 7E–H). These results suggest that the functional characteristics of HRG patients are primarily associated with cellular metabolism, the cell cycle, and neurodegenerative diseases, whereas LRG patients are more closely linked to immune responses and gene splicing. These mechanisms suggest that LUAD patients, especially those in HRG, may face a broader range of health issues, including an increased risk of metabolic disorders and neurodegenerative diseases. In contrast, LRG may be more stable in immune defense and in the regulation of cellular function. However, the specific risks need to be further analyzed in conjunction with the patient’s clinical characteristics and treatment process.
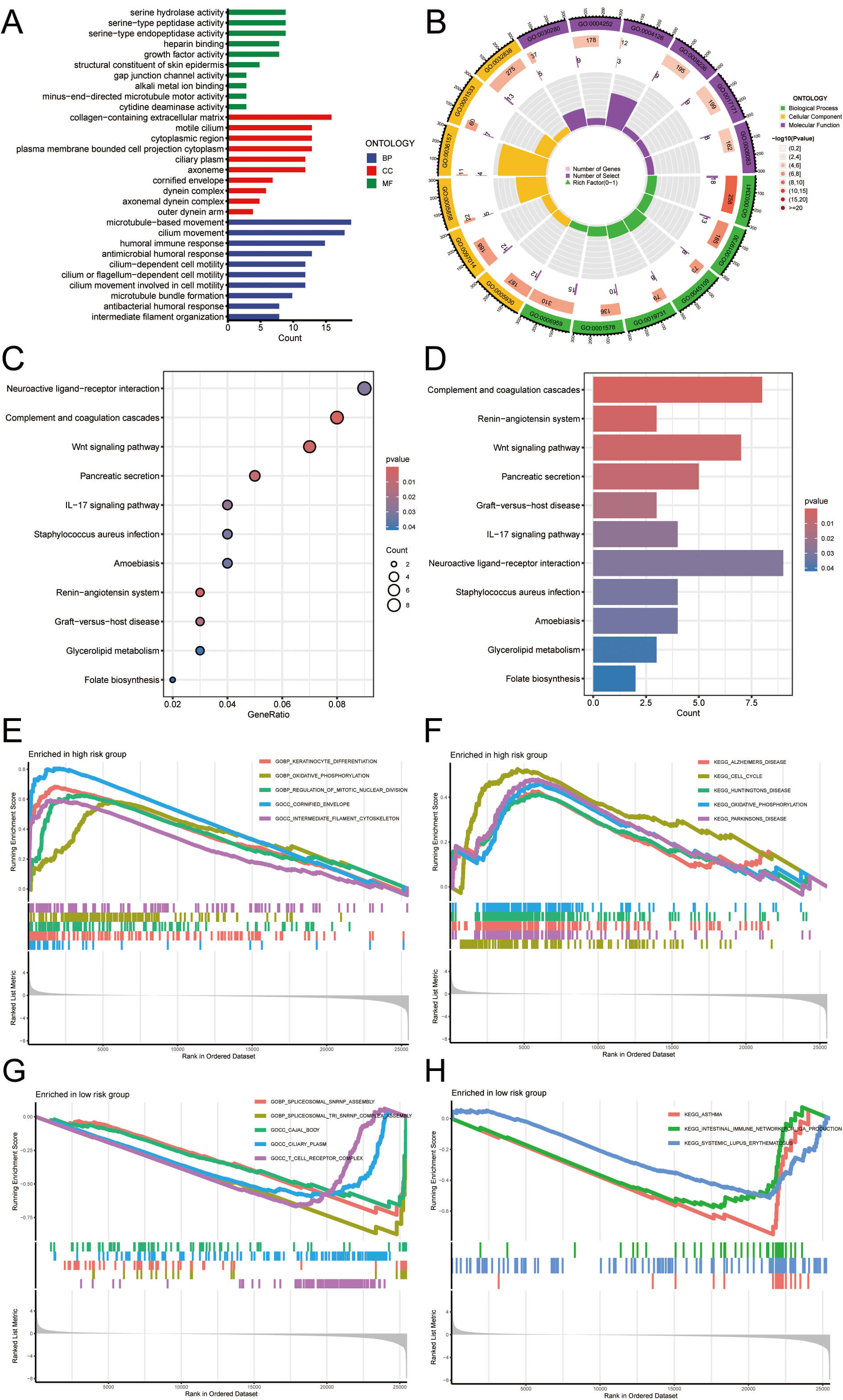 Fig. 7.
Fig. 7.
Immune-related and functional enrichment analysis. (A,B) GO
enrichment results: outer to inner circles represent GO terms, gene counts,
significance (deeper red = higher significance), co-expressed genes, and gene
heat ratios. (C,D) KEGG enrichment results: bar color indicates p-value
(darker = more significant), and point size reflects the number of enriched
genes. (E–H) GSEA identified key pathways with p
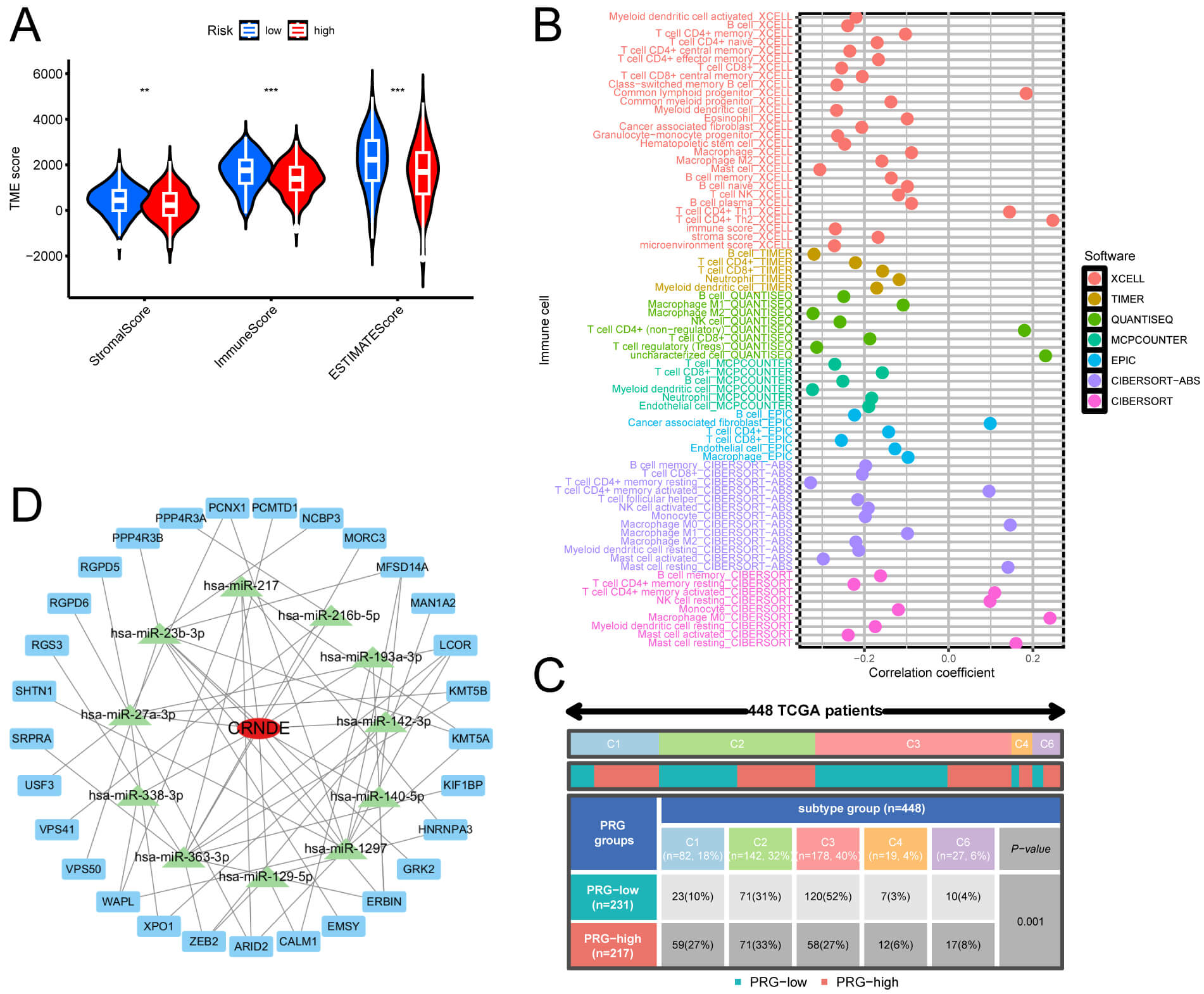
LUAD samples in HRG showed a significant reduction in StromalScore, ImmuneScore,
and ESTIMATEScore compared to samples in LRG (Fig. 8A). To evaluate the
percentage of ICs within LUAD patient samples, we utilized methodologies (XCELL,
EPIC, TIMER, QUANTISEQ, MCPCOUNTER, CIBERSORT-ABS, and CIBERSORT), utilizing
marker genes and deconvolution algorithms. A Pearson correlation test was
performed to examine the relationship between the risk coefficient model and
tumor-infiltrating ICs, applying a screening criterion of p
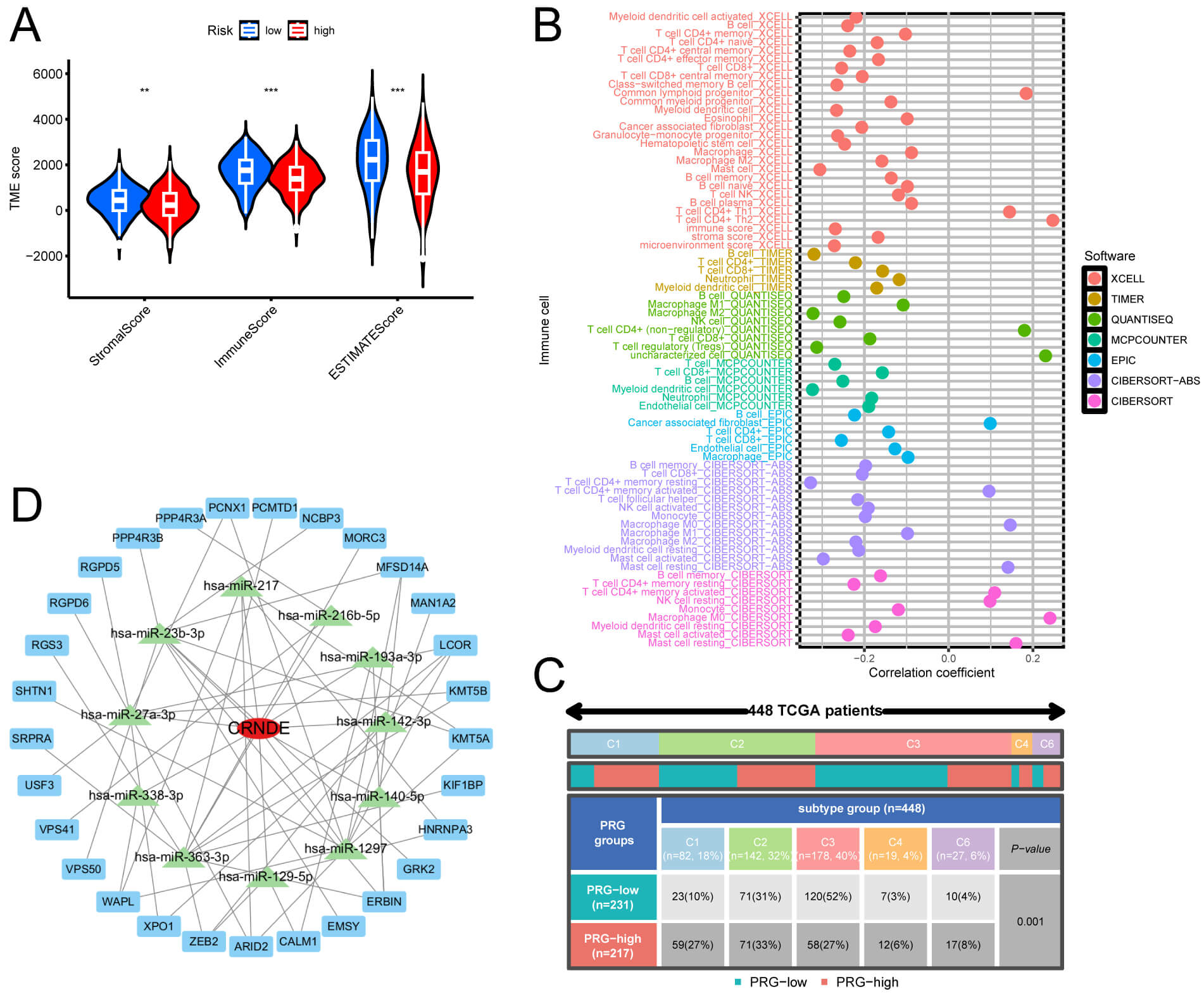 Fig. 8.
Fig. 8.
Comparison of TME and ICs between risk groups. (A) Violin plots
of three TME scores in HRG and LRG. (B) Correlation of IC infiltration in LUAD.
(C) Distribution of PRG high and low expression groups across different subtypes.
(D) CRNDE-centered lncRNA-miRNA-mRNA network diagram. **p
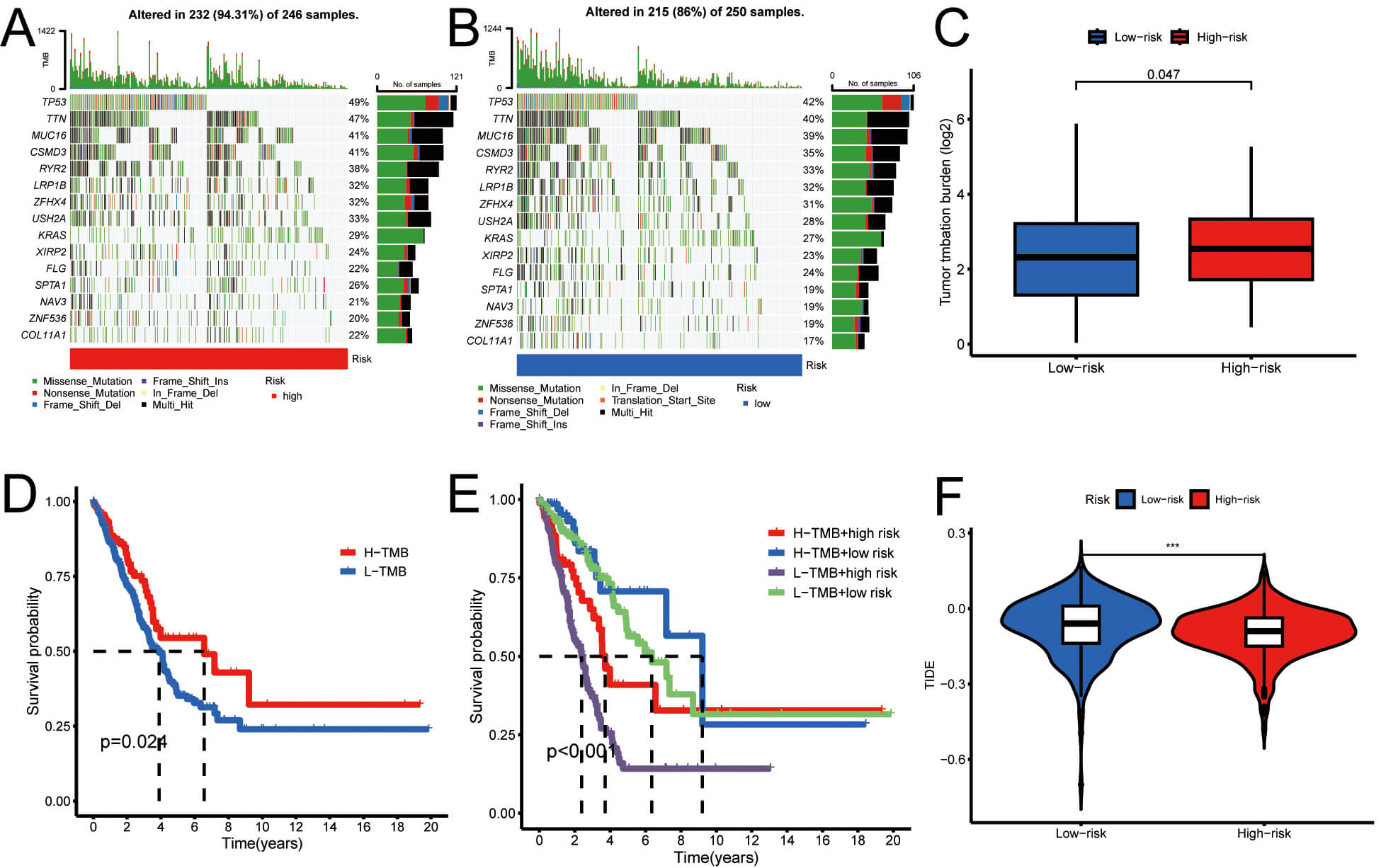
Using tumor mutation load data from the TCGA database, in combination with the risk values for the samples, we employed a waterfall plot to visualize tumor mutation load. In HRG, the genes with the highest mutation frequencies were TP53 (49%), TTN (47%), and CSMD3 (41%), while in LRG, the most regularly mutated genes were TP53 (42%), TTN (40%), and MUC16 (39%). Fig. 9A,B depict these findings. This analysis indicates that tumors within HRG may harbor a greater number of genetic mutations, potentially correlating with increased tumor aggressiveness and poorer prognosis. A comparative analysis of mutation loads was conducted between HRG and LRG, and the mutation discrepancies in modeled genes across these groups were examined. The results in Fig. 9C demonstrate a higher mutation rate in the HRG than in the LRG.
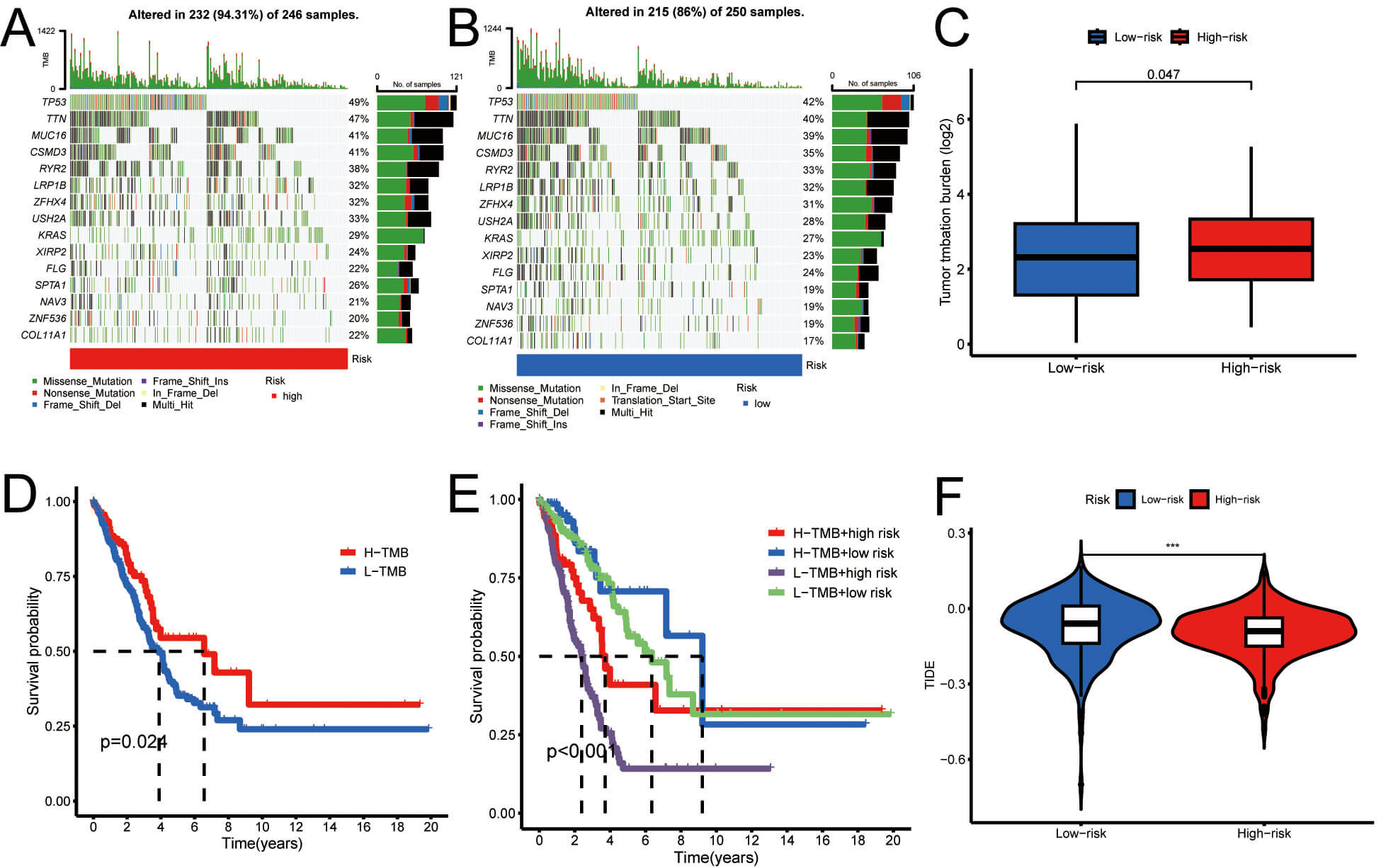 Fig. 9.
Fig. 9.
The outcome of the tumor mutation load. (A,B) Waterfall plots
of tumor mutations in LRG (blue) and HRG (red). (C) Box plot showing differential
mutation analysis between groups. (D) Kaplan-Meier curves comparing high- vs.
low-TMB groups. (E) Kaplan-Meier survival curves across four combined subgroups.
(F) Violin plot of tumor immune dysfunction and exclusion in LRG (blue) and HRG
(red). ***p
Additionally, LUAD patients were stratified into high- and low-TMB groups based on the median TMB, followed by Kaplan-Meier survival analysis. The findings indicated that individuals with LUAD who had a high TMB demonstrated improved prognostic outcomes, suggesting that TMB is a potential prognostic biomarker for this patient cohort (Fig. 9D). To further explain the synergistic effects of risk scores and TMB on OS, LUAD patients were stratified into four distinct subgroups for survival analysis. The outcomes revealed that those classified as low TMB and HRG had the most unfavorable prognosis, thereby validating our predictive model and delineating the most favorable prognostic subgroup for clinical application (Fig. 9E). In addition, Fig. 9F shows that the TIDE score is lower in the high-risk group compared to the low-risk group.
Each sample’s drug sensitivity was evaluated using database-derived data alongside our own findings. We compared HRG and LRG sensitivities to different treatment drugs by combining the risk scores for each sample. Through rigorous screening and exclusion of outcomes with no significant differences, we identified disparities in drug-sensitivity outcomes across 16 drugs between HRG and LRG (Fig. 10). HRG exhibits increased sensitivity to drugs including 5-fluorouracil, cisplatin, erlotinib, foretinib, savolitinib, trametinib, ERK_6604, AZD6738, BI-2536, and VX-11e, whereas LRG demonstrates heightened sensitivity to axitinib, doramapimod, ribociclib, sinularin, BMS-754807, and ZM447439 [46, 47, 48, 49, 50, 51]. These findings suggest a significant association between risk scores and drug sensitivity in lung adenocarcinoma patients.
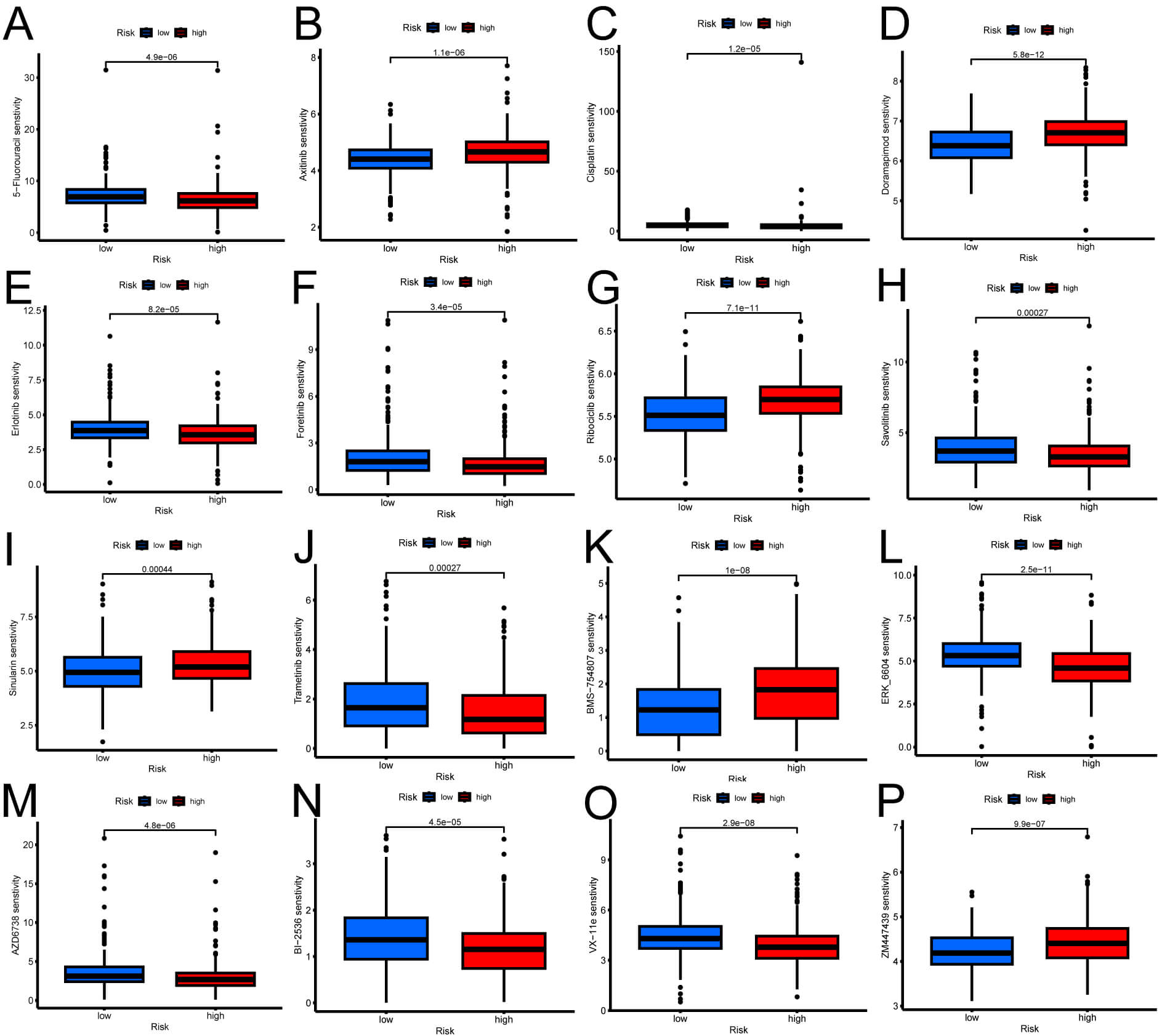 Fig. 10.
Fig. 10.
Drug sensitivity analysis. (A–P) Comparison of the variations in sensitivity to chemotherapy medications (5-fluorouracil, axitinib, cisplatin, doramapimod, erlotinib, foretinib, ribociclib, savolitinib, sinularin, trametinib, BMS-754807, ERK_6604, AZD6738, BI-2536, VX-11e, ZM447439) between LRG and HRG.
Additionally, the relationship between high- and low-expression groups of AL157895.1 in the prognostic model and drug sensitivity was further evaluated. The results show that the high-expression group exhibits greater sensitivity to axitinib, cisplatin, doramapimod, ribociclib, BMS-754807, and ZM447439, while the low-expression group is more sensitive to 5-fluorouracil, erlotinib, foretinib, sorafenib, TAF1-5469, talazoparib, ERK_6604, AZD6738, BI-2536, and VX-11e (Supplementary Fig. 2).
In the previous section of this study, we identified 13 effective
palmitoylation-related lncRNA genes in the model. Through further differential
expression analysis in LUAD (
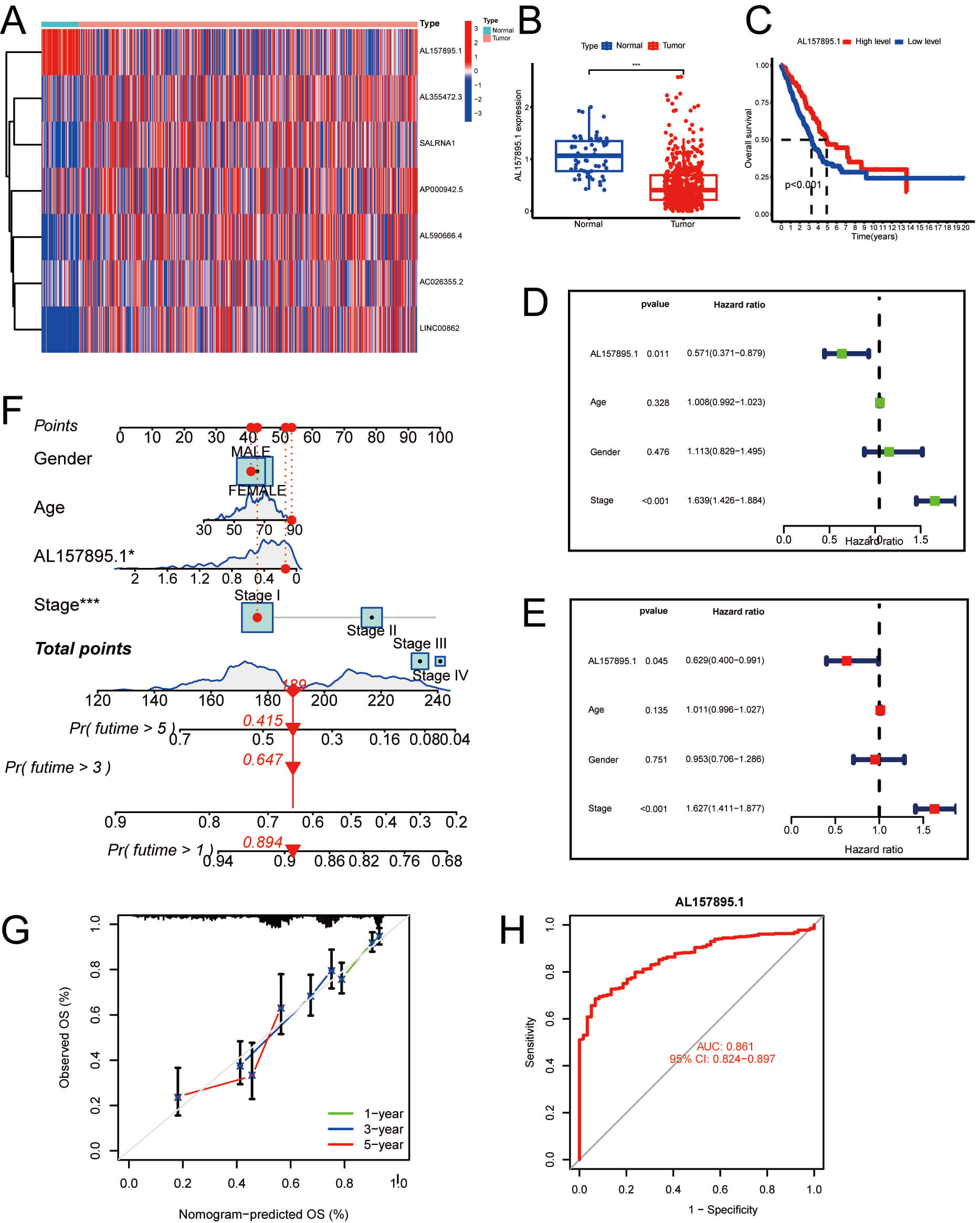 Fig. 11.
Fig. 11.
Evaluation of the predictive ability of the
palmitoylation-associated lncRNA risk model in LUAD patients. (A) A heatmap
showcasing the expression of seven palmitoylation-associated lncRNA genes in LUAD
compared to normal tissues, specifically highlighting AL157895.1, AL355472.3,
SALRNA1, AP000942.5, AL590666.4, AC026355.2, and LINC00862. (B) The expression of
the lncRNA AL157895.1 in LUAD was analyzed using the TCGA-LUAD dataset. (C) The
link between the AL157895.1 levels and OS in the TCGA database was also examined.
Additionally, forest plots were generated for (D) univariate and (E) multivariate
Cox regression analyses. (F) A column line graph was utilized to forecast patient
survival, estimating the 1-, 3-, and 5-year survival rates through a scoring
system, supplemented by the correction curve depicted in (G). (H) ROC analysis of
AL157895.1 expression revealed a robust capacity to differentiate between tumor
and non-tumor samples. *p
Seven DEGs were identified when comparing gene expression between individuals in HRG and LRG groups, allowing detection of variations in biological functions. GO analysis showed that lncRNAs related to palmitoylation were mostly enriched in BP, including pathways for platelet formation, mast cell degranulation regulation, and myeloid cell differentiation. Regarding the CC, the voltage-gated sodium channel complex, the sodium channel complex, and the glial cell projection were the primary foci of interest. The MF was primarily concerned with MAP kinase activity, C2H2 zinc finger domain binding, and the minor groove of adenine-thymine-rich DNA binding (Fig. 12A). DEGs were primarily involved in several signaling pathways, as determined by KEGG enrichment analysis. These pathways include Fc epsilon RI, sphingolipid, fluid shear stress and atherosclerosis, and MAPK (Fig. 12B). Further investigation into the functional terms most significantly enriched in HRG and LRG patients was conducted using GSEA. RNA biology and gene expression regulation were enriched in the HRG (e.g., gene silencing by RNA, Sm-like protein family complex, spliceosomal snRNP complex, RNA-binding involved in post-transcriptional genes, u6 snRNA binding) (Fig. 12C).
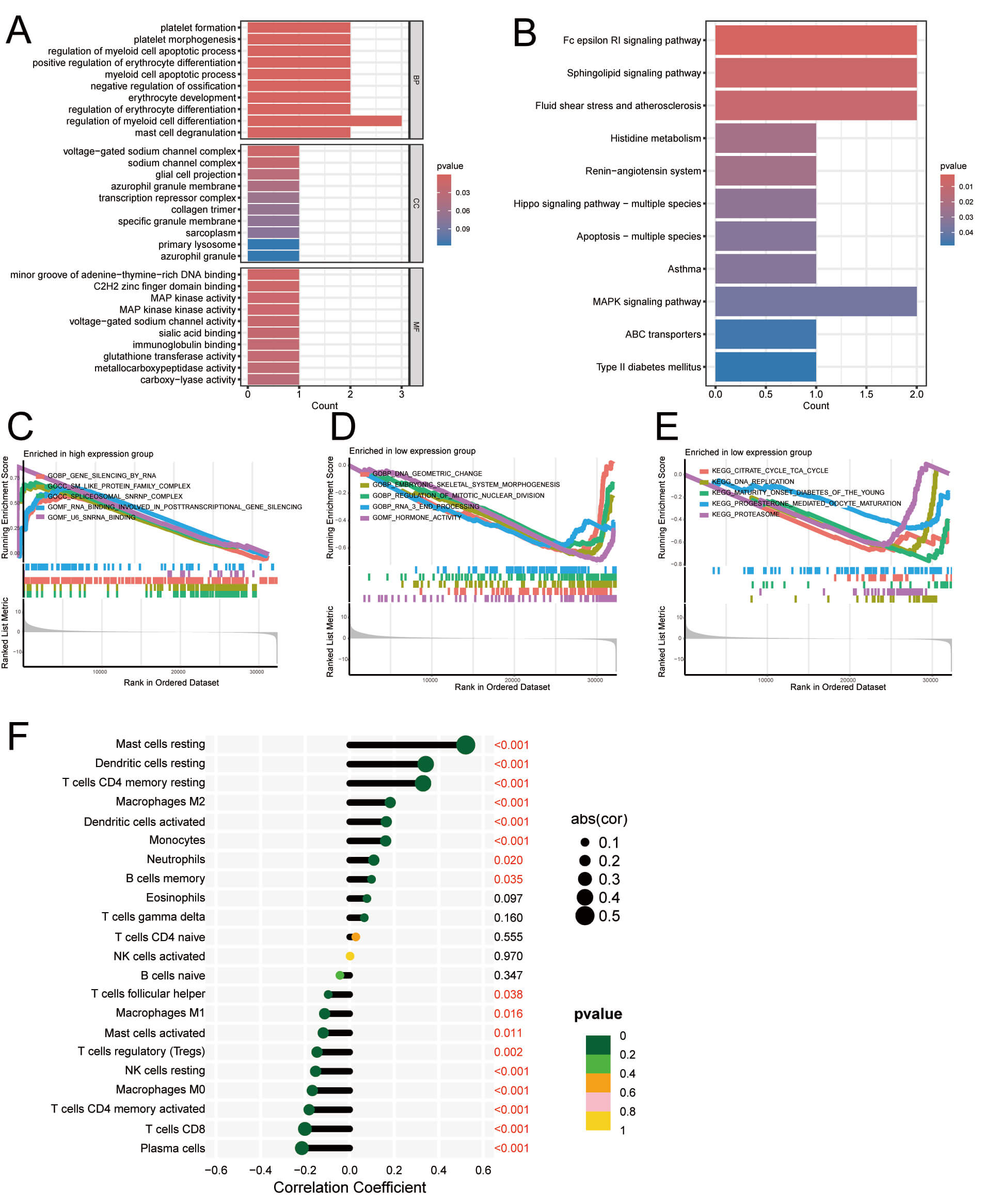 Fig. 12.
Fig. 12.
Functional enrichment and IC analysis of palmitoylation-related
lncRNAs in LUAD. (A) GO analysis. (B) KEGG analysis showing 11 enriched
pathways. (C–E) GSEA-identified representative pathways (p
However, regulating BP, metabolism, and hormones was enriched in LRG patients (e.g., DNA geometric change, embryonic skeletal system morphogens, regulation of mitotic nuclear division, RNA 3 end processing, hormone activity, citrate cycle TCA cycle, DNA replication, maturity onset diabetes of the young, progesterone-mediated oocyte maturation, proteasome) (Fig. 12D,E). The above findings suggest that RNA biology and gene expression regulation play a significant role in HRG, possibly indicating a more active or disrupted transcriptional environment in these patients. On the other hand, the enrichment of BP related to metabolism, hormones, and cell division in LRG may reflect a more stable or controlled physiological state. This contrast could offer insights into the underlying molecular mechanisms contributing to the different risk profiles, highlighting the importance of targeted interventions based on these biological pathways for better patient management and prognosis. The content of each sample was detected. We determined the link between ICs and risk score via the risk values from each sample in our developed prognostic model (Fig. 12F). ICs that were significantly positively related include: Mast cells resting, dendritic cells activated, dendritic cells resting, T cells CD4 memory resting, macrophages M2, and monocytes. ICs that were significantly negatively correlated include: T cells CD8, T cells CD4 memory activated, plasma cells, macrophages M0, and NK cells resting. The analysis results indicate that RNA biology and gene expression regulation play a significant role in HRG, suggesting that transcriptional activity in these patients is more complex or abnormal, affecting immune response and disease progression. In contrast, LRG may be in a more stable physiological state, in which metabolism and hormone regulation could help maintain a stable disease state.
Utilizing the CIBERSORT algorithm, the link between lncRNA AL157895.1 and palmitoylation-related genes was evaluated. The scatter diagrams illustrate that there was a positive relationship between lncRNA AL157895.1 and palmitoylation-related genes (ABCA6, ANKRD44, C1QTNF7, CPA3, ERVFRD-1, FAT4, GATA1, GCSAML, HDC, HPGDS, MAPK10, MEF2C, MS4A2, RAB44, RGS13, RHEX, SCN7A, and SIGLEC6) (Supplementary Fig. 3). These analyses may suggest that the levels of these palmitoylation-associated genes show a synergistic relationship with AL157895.1 in certain BP, or that lncRNA AL157895.1 may play a vital role in regulating the expression or function of these genes. We further analyzed the expression of Palmitoylation-related lncRNA AL157895.1 in HRG and LRG of 18 palmitoylation-related genes. The palmitoylation-related lncRNA AL157895.1 is correlated with these palmitoylation-associated genes, and there are significant differences in its expression between HRG and LRG (Supplementary Fig. 4). This analysis may suggest that AL157895.1 plays a crucial role in the risk assessment of tumors or diseases.
The qRT-PCR method was utilized to examine AL157895.1, AL355472.3, SALRNA1, AL590666.4, AC026355.2, and LINC00862 expression levels in LUAD A549, PC9, H1299, and HCC827 cell lines and Beas-2B control cell line (the primer sequence of AP000942.5 is missing). The results revealed that AL355472.3, SALRNA1, AL590666.4, AC026355.2 [52, 53], and LINC00862 [41] were overexpressed in LUAD A549, PC9, H1299, and HCC827 cell lines, while AL157895.1 was suppressed in these cell lines, which was consistent with the TCGA cohort (Fig. 13) [38, 52]. These outcomes suggest that these six genes may play a vital role in tumor progression and could serve as potential biomarkers for the diagnosis, prognosis, or treatment response in LUAD.
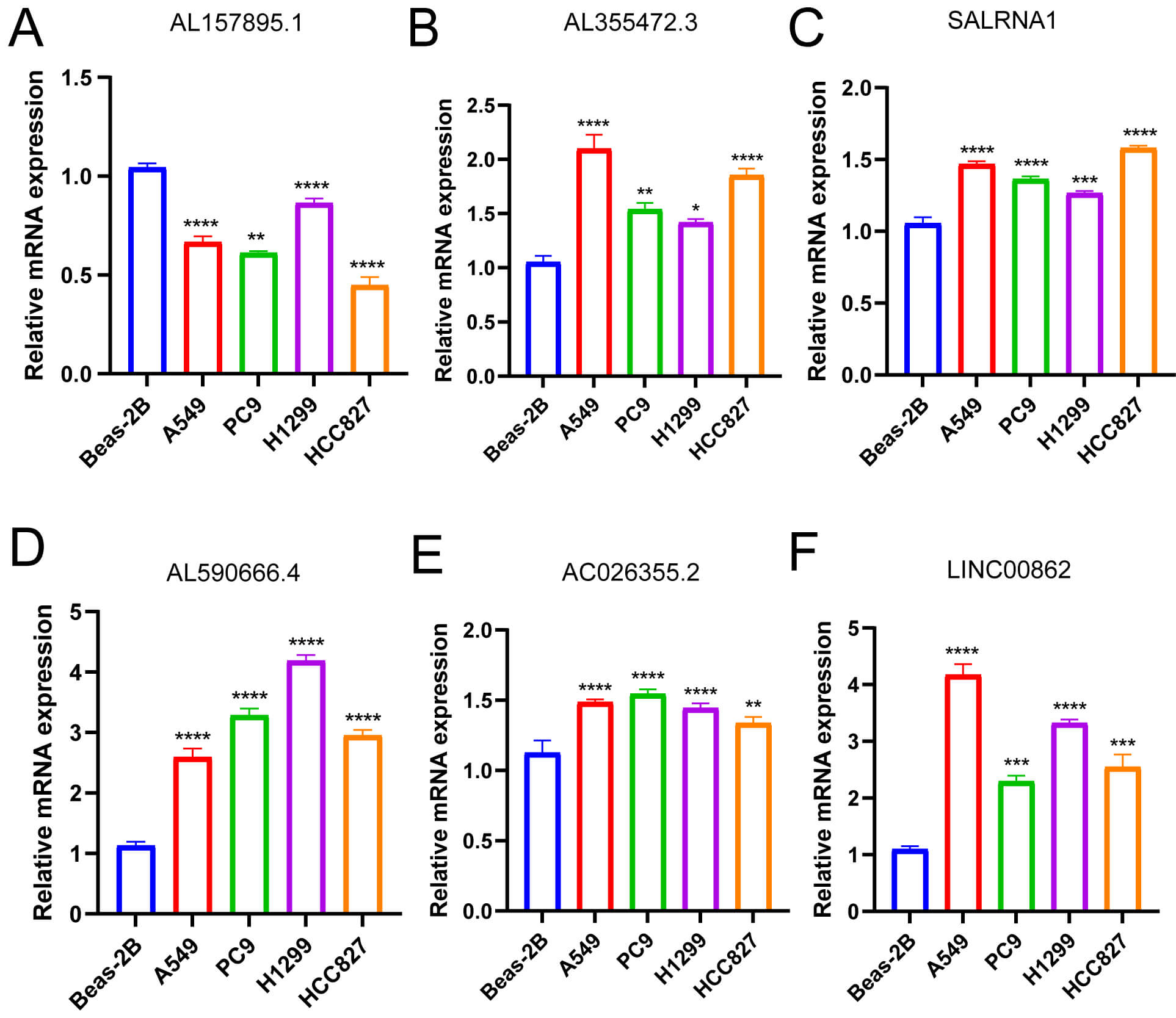 Fig. 13.
Fig. 13.
The degree of six palmitoylation-related lncRNAs prognostic
signature mRNA expression. The mRNA levels of (A) AL157895.1; (B) AL355472.3;
(C) SALRNA1; (D) AL590666.4; (E) AC026355.2; (F) LINC00862 in Beas-2B, A549, PC9,
H1299, and HCC827 cell lines. *p
LUAD is a significant NSCLC subtype characterized by its heterogeneous nature and poor prognosis. LUAD, as a main cause of cancer-associated death worldwide, poses substantial challenges in terms of early diagnosis and successful treatment strategies. Identifying palmitoylation-related lncRNAs has significant implications for the prognosis of LUAD patients. Our investigation indicates that these lncRNAs serve as prognostic biomarkers and elucidate potential roles associated with tumor behavior.
Herein, we identified palmitoylation-associated lncRNAs with prognostic significance in LUAD and constructed a predictive model based on these lncRNAs. Utilizing co-expression analysis, we identified 1662 palmitoylation-related lncRNAs and visualized their relationships with palmitoylation genes through a Sankey diagram. We subsequently divided 522 LUAD samples from the TCGA database into training and validation cohorts, using one-way Cox regression and LASSO regression to develop a prognostic model comprising 13 lncRNAs. Our findings indicate that this risk-score-based model effectively stratifies individuals into HRG and LRG groups, which are associated with survival outcomes and immune response profiles. Furthermore, we conducted comprehensive analyses, including functional enrichment, IC infiltration, and TMB, to elucidate the biological implications of the identified lncRNAs. The results underscore the potential of palmitoylation-associated lncRNAs as prognostic and therapeutic-response biomarkers in LUAD.
The functional enrichment analysis revealed significant insights into the biological pathways related to palmitoylation-associated lncRNAs in LUAD. Notably, the KEGG enrichment analysis highlighted the Wnt signaling pathway, which plays a vital role in cell aggressiveness [54, 55, 56]. It is believed that LUAD is one of several malignancies in which dysregulation of Wnt signaling contributes to tumor growth and metastasis [57, 58, 59, 60]. Additionally, the analysis identified the neuroactive ligand-receptor interaction pathway as significantly enriched in the HRG. This pathway is essential for cell communication and has been linked to interactions within the tumor microenvironment, influencing immune evasion and tumor growth [61, 62].
Furthermore, the complement and coagulation cascades pathway emerged as another critical enrichment finding. This pathway is known to be involved in inflammatory responses and has been associated with cancer progression and metastasis [63, 64]. In summary, these findings underscore the significance of palmitoylation-associated long non-coding RNAs in LUAD and suggest potential directions for future mechanistic investigations.
Here, the analysis of immune-associated roles revealed significant differences in IC infiltration between HRG and LRG in LUAD patients. Notably, the HRG showed a lower overall immune score, suggesting a reduced presence of ICs in the tumor microenvironment. This finding aligns with earlier investigations demonstrating a link between low IC infiltration and poor prognosis in various cancers, including LUAD [65, 66]. Specifically, a negative relationship was detected between the risk score and several IC types (CD8+ T and NK cells), which are vital for anti-tumor immunity. The diminished presence of these effector ICs in the HRG suggests a potential immune evasion mechanism that may contribute to the aggressive nature of the tumors in this cohort [67]. The reduced infiltration of CD8+ T cells in the HRG may indicate a compromised anti-tumor immune response, which could be a contributing factor to the poorer survival outcomes observed in these patients [68]. Mast cells, another IC type identified in our analysis, displayed a positive relationship with the risk score. While traditionally associated with allergic responses, mast cells play complex roles in tumor biology, including promoting angiogenesis and modulating immune responses [69, 70]. In summary, our findings suggest that the immune landscape in LUAD is markedly altered by risk stratification, with the HRG displaying features of immune evasion and a less favorable prognosis.
Our drug-sensitivity analysis showed that individuals in the LRG displayed heightened sensitivity to multiple chemotherapeutic agents, including 5-fluorouracil and erlotinib. This finding suggests that the risk score derived from palmitoylation-related lncRNAs may serve as a valuable predictor of treatment response, enabling more personalized therapeutic strategies. Furthermore, the identification of AL157895.1 as a significantly downregulated lncRNA in LUAD tissues presents an intriguing avenue for therapeutic exploration. The association between AL157895.1 expression and OS rates suggests its potential as a predictive biomarker. In summary, our comprehensive analysis elucidates the intricate relationships between palmitoylation-related lncRNAs, immune response, and drug sensitivity in LUAD. Lastly, the expression of six palmitoylation-associated lncRNAs in LUAD cell lines was validated by qRT-PCR. The outcomes illustrated that AL157895.1 was significantly suppressed in LUAD tissues, whereas other lncRNAs were upregulated, further supporting the potential biomarker roles of these lncRNAs in LUAD progression.
In summary, our investigation elucidates the significant role of palmitoylation-associated lncRNAs in LUAD patient prognosis, highlighting their potential as biomarkers for clinical outcomes. Identifying 13 key lncRNAs, particularly AL157895.1, underscores their differential expression and prognostic value, with high expression correlating with improved OS. Constructing a robust prognostic model, validated through multiple statistical analyses, demonstrates its independence as a prognostic factor and outperforms traditional clinical features. Furthermore, functional enrichment analyses reveal distinct biological pathways associated with HRG and LRG, suggesting that HRG may be in a more active transcriptional environment, while LRG shows stability in metabolic and hormonal processes. In summary, while this study provides new insights into the personalized treatment of lung adenocarcinoma, the clinical applicability of these findings requires further validation.
This study has several limitations. First, the analysis relies on publicly available datasets, which may introduce potential biases due to sample heterogeneity and data quality [71]. Furthermore, the study is based solely on the TCGA-LUAD cohort, limiting its ability to fully capture the complex genetic and phenotypic characteristics of lung adenocarcinoma. Second, the prognostic model was validated only within this dataset, lacking independent external cohort validation. This may lead to overfitting and compromise the model’s generalizability.
Additionally, palmitoylation-associated long non-coding RNAs were identified solely through co-expression analysis, without functional experiments to verify their direct impact on protein palmitoylation or related enzyme activity. We acknowledge that due to technical challenges in primer design, the key component AP000942.5 in the model could not be experimentally validated via qRT-PCR. Future studies should incorporate large-scale, multicenter data and validate both model efficacy and lncRNA function through in vitro experiments and independent cohort validation.
In conclusion, this investigation created a prognostic model depending on palmitoylation-related lncRNAs, providing innovative insights and approaches for risk stratification and prognosis prediction in LUAD patients, with significant clinical application value. Future studies may further investigate the functions of these lncRNAs in LUAD and explore their potential as therapeutic targets.
OS, Overall survival; ROC, Receiver Operating Characteristic; GO, Gene Ontology; LUAD, Lung adenocarcinoma; KEGG, Kyoto Encyclopedia of Genes and Genomes; GSEA, Gene Set Enrichment Analysis; LRG, Low-risk group; HRG, High-risk group; TMB, Tumor mutational burden; PCA, Principal component analysis; AUC, Area under the curve; ICs, Immune cells; TME, Tumor Microenvironment; lncRNAs, Long non-coding RNAs; NSCLC, Non-small cell lung cancer; qRT-PCR, Quantitative reverse transcription polymerase chain reaction; WGCNA, Weighted gene co-expression network analysis; LASSO, Least absolute shrinkage and selection operator regression analysis; TIDE, Tumor immune dysfunction and exclusion; TCGA, The Cancer Genome Atlas; GDSC2, Drug Sensitivity in Cancer 2; FDR, False discovery rate.
The dataset employed for this study is readily available from online repositories. The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
YHL and LM: review & editing, Conceptualization, Methodology, Project administration; WXD and SXW: Writing — original draft, Conceptualization, Methodology, Validation; LJZ, ZXF, YML, XYY and PW: Writing — original draft, Conceptualization, Methodology. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We extend our gratitude to GEO and TCGA database, and all contributors who generously shared their data on these platforms. We thank Home for Researchers editorial team for language editing service.
This research was funded by the Scientific Research Fund Project of the Hunan Provincial Health Commission (No. 20253699) and supported by the Hunan Provincial Graduate Research and Innovation General Project under grant number LXBZZ2024254.
The authors declare no conflicts of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL48267.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.