







 , Igor Duquesne 2, Michael A O’Donnell 1
, Igor Duquesne 2, Michael A O’Donnell 11 Department of Urology, University of Iowa Health Care, Iowa City, IA 52242, USA
2 Department of Urology, Henri Mondor University Hospitals, Assistance Publique-Hôpitaux de Paris, 94000 Créteil, France
Abstract
Non-muscle-invasive bladder cancer (NMIBC) accounts for roughly 75% of all bladder cancer cases. For patients with intermediate- and high-risk disease, intravesical Bacillus Calmette-Guérin (BCG) remains the standard treatment, yet it fails in up to 40% of cases. While radical cystectomy is the most effective salvage option, it carries significant morbidity and long-term quality-of-life consequences, highlighting the urgent need for bladder-sparing alternatives. Advancing such therapies requires a deep understanding of the immunologic mechanisms within the tumor microenvironment (TME). This review offers a concise overview of the immunologic mechanisms underlying BCG therapy, along with a detailed examination of the multifactorial immune evasion mechanisms that contribute to its failure in NMIBC. Within the TME, ten principal mechanisms of immune suppression have been identified. These include the activity of myeloid-derived suppressor cells, tumor-associated macrophages, regulatory T cells, and tolerogenic dendritic cells, as well as signaling pathways such as programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1), the natural killer group 2A/human leukocyte antigen-E (NKG2A/HLA-E) checkpoint, and the release of immunomodulatory molecules within the TME. Further contributors to immune evasion include cluster of differentiation 6/activated leukocyte cell adhesion molecule (CD6-ALCAM) signaling, effector T cell exhaustion, and cancer-associated fibroblasts. Collectively, these mechanisms disrupt antigen presentation, suppress cytotoxic immune responses, and facilitate tumor progression, ultimately undermining the efficacy of BCG therapy. In parallel, we highlight emerging intravesical immunotherapies for BCG-unresponsive NMIBC with carcinoma in situ, including nadofaragene firadenovec (Adstiladrin), nogapendekin alfa inbakicept-pmln (Anktiva), cretostimogene grenadenorepvec (CG0070), and detalimogene voraplasmid (EG-70). These agents employ diverse platforms, including gene therapy, cytokine stimulation, oncolytic virotherapy, and plasmid-based immune activation, to enhance antitumor responses. While early and late-phase clinical trials have shown promising response rates and favorable safety profiles for these novel agents, direct comparisons remain limited due to the reliance on single-arm study designs. The lack of comparative data, coupled with the absence of predictive biomarkers of response, complicates treatment selection. Our review underscores that developing effective therapies for BCG-unresponsive disease will require combination strategies targeting multiple immune escape mechanisms that shape immune dynamics within the TME.
Keywords
- non muscle invasive bladder cancer
- urinary bladder neoplasms
- Bacillus Calmette-Guérin
- BCG unresponsive disease
- tumor microenvironment
- immune evasion
- immunotherapy
Bladder cancer is the ninth most common malignancy worldwide, with approximately 75% of cases classified as non-muscle-invasive bladder cancer (NMIBC), for which intravesical therapy remains a cornerstone of treatment. According to major urological guidelines, patients with intermediate- and high-risk NMIBC should receive intravesical Bacillus Calmette-Guérin (BCG) as induction therapy for six weeks, followed by maintenance for up to one to three years [1, 2]. However, BCG fails in up to 40% of cases [3], and the only guideline-endorsed option in this setting remains radical cystectomy (RC) [1, 2], a major surgical procedure associated with a complication rate exceeding 60% within 90 days, including major complications in over 15% of cases [4]. For patients who are unfit for or decline surgery, bladder-sparing therapy (BST) offers a viable alternative without compromising survival. Recent data show no significant difference in overall survival (HR: 1.40, p = 0.4) or cancer-specific survival (HR: 0.88, p = 0.9) between BST and early RC in highly selected BCG-unresponsive NMIBC [5]. BST options include intravesical chemotherapy, gene therapy, such as nadofaragene firadenovec, and cytokine-based immunotherapy, including nogapendekin alfa inbakicept-pmln, an interleukin-15 (IL-15) superagonist. Each of these modalities has demonstrated clinical efficacy in the management of BCG-unresponsive disease [6].
For over four decades, BCG has remained the cornerstone of intravesical therapy for NMIBC, primarily attributed to its ability to elicit robust local innate and adaptive immune responses within the tumor microenvironment (TME). However, recent insights have significantly broadened our understanding of its mechanism of action. BCG can be internalized by urothelial carcinoma cells, enabling them to acquire antigen-presenting capabilities, modulate Programmed Death-Ligand 1 (PD-L1) expression on both tumor and immune cells, and orchestrate a reshaping of the local immune landscape [7]. Additional evidence suggests that prolonged BCG exposure may lead to sustained stimulation of cytotoxic CD8+ T cells, ultimately driving them toward an exhausted or anergic state and facilitating tumor immune escape [8].
Despite its long-standing clinical use, the immunological interplay between BCG and the TME remains incompletely understood. These insights have inspired the development of novel therapeutic strategies for BCG-unresponsive disease. For example, the rationale for using immune checkpoint inhibitors stems from the observation that PD-L1 is frequently overexpressed in tumors resistant to BCG [7]. Similarly, nogapendekin alfa inbakicept-pmln, a recombinant IL-15 superagonist designed to activate and expand NK and CD8+ T cells within the TME, has shown promise in this setting, particularly when combined with BCG to enhance antitumor immunity [9]. Interestingly, recent data suggest that BCG rechallenge in patients with BCG-unresponsive NMIBC may yield acceptable outcomes, which is unexpected given the prior failure of BCG therapy [10]. This paradox may reflect the intricate and dynamic immunological mechanisms at play within TME, or alternatively, highlight the heterogeneity of BCG-unresponsive disease, characterized by distinct immune signatures.
A deeper understanding of immune mechanisms in NMIBC not only facilitates the development of novel therapeutics but also improves the ability to predict treatment response. Systemic inflammatory markers, such as the neutrophil-to-lymphocyte ratio have been investigated as potential predictors of BCG efficacy, reflecting underlying immune activity [11]. Additionally, urinary cytokine profiles may serve as non-invasive biomarkers of immune activation and therapeutic response in NMIBC [12].
The immune landscape of NMIBC is multifaceted, shaped by factors such as field cancerization and heterogeneous tumor lineage characteristics, which may precede intravesical therapy and influence treatment response [7]. A thorough understanding of the TME is essential for advancing the clinical management of NMIBC. In this review, we synthesize key immune mechanisms implicated in NMIBC, revisit BCG-induced immunologic responses previously characterized by our team [7], and examine pathways of immune evasion that contribute to BCG-unresponsive disease. We also highlight emerging immunotherapeutic strategies and their mechanisms of action that are either approved or currently under regulatory evaluation for patients with BCG-unresponsive NMIBC.
Historically, failure of intravesical BCG therapy in NMIBC has been described using heterogeneous terms, including BCG‑refractory, BCG‑relapsing, and BCG‑exposed. These terms broadly refer to persistent high‑grade disease despite BCG, recurrence after an initial response, or prior BCG exposure without meeting failure criteria, respectively, but their inconsistent application has limited comparability across studies [13].
To harmonize patient classification and better identify tumors unlikely to respond to additional BCG, contemporary guidelines from the U.S. Food and Drug Administration (FDA), American Urological Association (AUA), and European Association of Urology (EAU) have adopted the unified category of BCG‑unresponsive NMIBC. Under the FDA definition, BCG‑unresponsive disease encompasses persistent high‑grade T1 disease at the first post‑treatment assessment (typically performed approximately three months after BCG initiation), recurrence of carcinoma in situ (CIS) within 12 months, or recurrence of high‑grade Ta or T1 disease within six months, despite adequate BCG exposure. Adequate therapy is defined as completion of at least five of six induction instillations, in combination with either a minimum of two of three maintenance doses or at least two of six re‑induction instillations [14].
Accordingly, this review focuses primarily on patients with BCG‑unresponsive NMIBC, with legacy terminology referenced only when discussing historical studies conducted prior to standardization.
BCG instillation initiates a potent innate immune response marked by the
recruitment and activation of macrophages, neutrophils, natural killer (NK)
cells, and dendritic cells (DCs). Macrophage polarization is central to the
immunologic outcome: M1 macrophages promote antitumor activity through
pro-inflammatory cytokine release and direct tumoricidal effects, whereas M2
macrophages facilitate immune evasion and tumor progression via immunosuppressive
signaling. The dynamic interchange between M1 and M2 phenotypes is regulated by
cytokines and interactions with regulatory T cells (Tregs) within the TME.
Neutrophils contribute to tumor cell death through the release of tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) and tumor necrosis factor-alpha
(TNF-
Fig. 1 summarizes the key components and interactions involved in the innate immune response following BCG instillation.
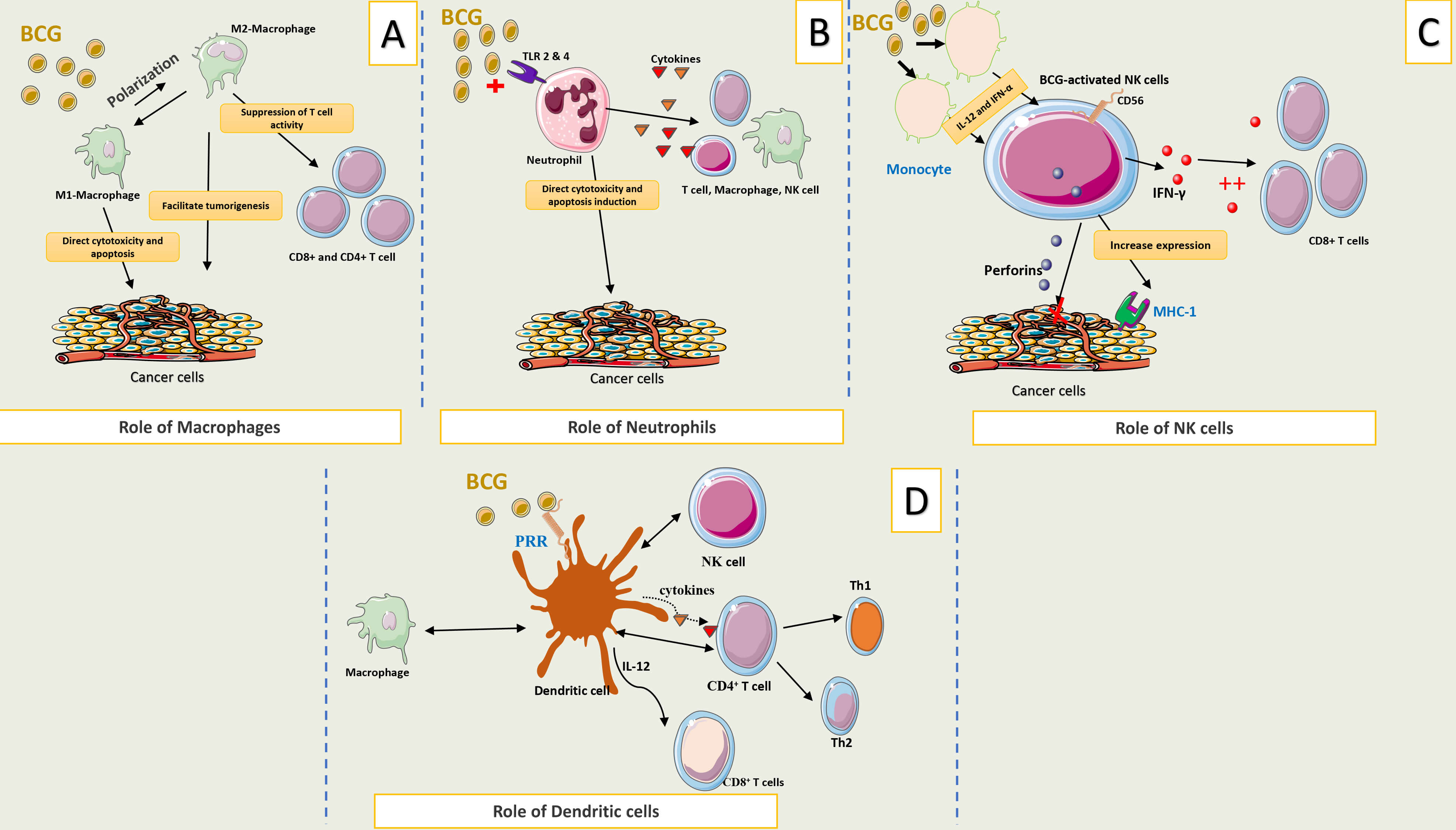 Fig. 1.
Fig. 1.
Brief description of innate immune response following BCG
instillation. (A) Macrophages exhibit a dual role in the bladder tumor
microenvironment, with their antitumor or pro-tumorigenic effects largely
dependent on their polarization state. M2 macrophages promote immune evasion and
tumor progression, while M1 macrophages support antitumor immunity. (B)
Neutrophils contribute to tumor cell death through the release of TRAIL and
TNF-
The adaptive immune response to BCG therapy involves the coordinated activation
of multiple immune cell subsets, including CD4+ and CD8+ T cells,
Tregs, gamma delta T cells (
Fig. 2 summarizes the key cellular components and interactions involved in the adaptive immune response following BCG instillation.
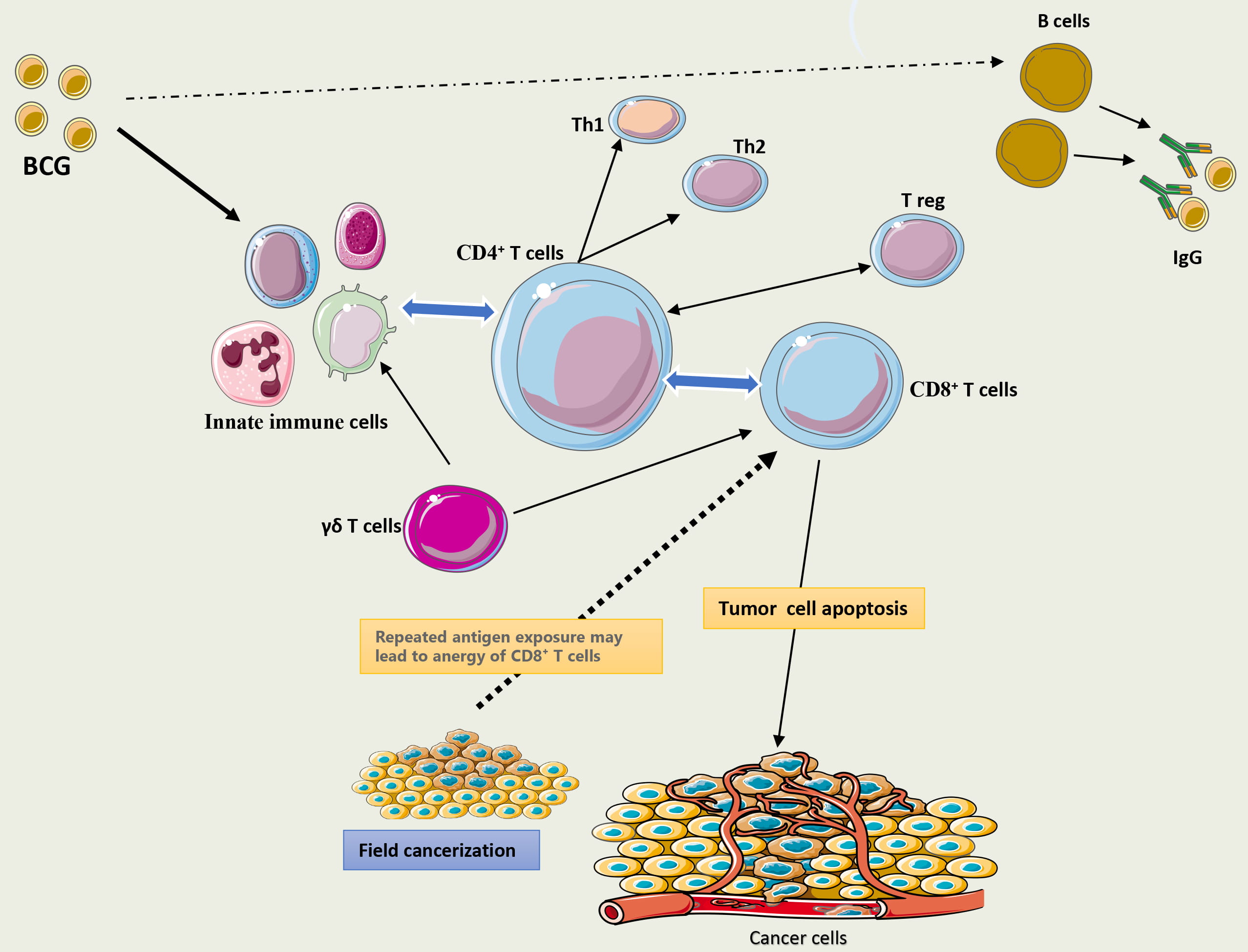 Fig. 2.
Fig. 2.
Brief description of adaptive immune response following BCG
instillation. BCG therapy activates a range of innate immune cells, including
natural killer cells, macrophages, neutrophils, and dendritic cells. Dendritic
cells function as key antigen-presenting cells, driving CD4+ T-cell
differentiation toward a Th1 phenotype and initiating IFN-
Myeloid-derived suppressor cells (MDSCs), which are immature precursors of
neutrophils and basophils, undergo abnormal expansion in response to
tumor-derived signals that inhibit their differentiation. Unlike transient
expansion during acute inflammation, cancer-associated MDSCs remain persistently
activated and inhibit T-cell responses [15]. Beyond their immunosuppressive
functions, MDSCs contribute to tumor progression by enhancing angiogenesis,
remodeling the extracellular matrix, and supporting tumor growth [16].
Accordingly, bladder cancer patients demonstrate increased circulating MDSCs and
robust infiltration of these cells within tumor tissue [17]. In a clinical study
of NMIBC patients undergoing BCG therapy, urine samples collected before and four
hours after BCG instillation revealed that a low T-cell to MDSC ratio (
BCG has been shown to induce the production of chemokines such as C‑X‑C motif chemokine ligand 8 (CXCL8) and C‑C motif chemokine ligand 22 (CCL22) by urothelial cells, macrophages, and dendritic cells, thereby promoting the recruitment of MDSCs to the tumor microenvironment [19]. Furthermore, combining BCG with anti‑PD‑L1 therapy has been shown to reduce MDSC infiltration within the tumor, highlighting the complex dynamics of immune modulation in the TME [20]. Together, these observations suggest that BCG’s capacity to reshape the TME through modulation of MDSC populations may play an important role in determining its therapeutic efficacy.
Tumor‑associated macrophages (TAMs) play a central role in tumor progression,
particularly when polarized toward the M2 phenotype. M2‑TAMs suppress CD8+
T‑cell activity through PD‑L1 expression and the secretion of immunosuppressive
cytokines such as IL‑10 and TGF‑
There are four distinct subtypes of M2-TAMs, each with unique phenotypic characteristics. Some subtypes secrete epidermal growth factor (EGF), which activates epidermal growth factor receptor (EGFR) signaling in tumor cells, enhancing pseudopod formation and promoting metastasis. Importantly, TAMs can facilitate DNA repair in tumor cells following chemotherapy-induced damage, thereby contributing to the development of drug resistance [23]. TAMs originate from hematopoietic stem cells in the bone marrow or from erythromyeloid progenitors in the yolk sac and fetal liver, and their functional roles may vary depending on their origin [24]. Clinically, a high density of TAMs infiltrating the TME has been associated with poor outcomes in patients with NMIBC undergoing BCG therapy. In a study involving 71 patients with NMIBC, higher TAM counts within tumors were associated with decreased recurrence-free survival [25]. Another study involving 99 NMIBC patients showed that high TAM infiltration in the tumor stroma was linked to a twofold increased risk of BCG failure [26]. Additional data suggest that the presence of M2-polarized TAMs within the TME may compromise the therapeutic effectiveness of BCG in NMIBC, with their abundance being linked to tumor recurrence as demonstrated by immunofluorescence-based profiling of M1 and M2 macrophage subsets [27].
Regulatory T cells (Tregs) are immunosuppressive CD4+CD25+ lymphocytes that arise either as thymus-derived natural Tregs or from naïve CD4+ T cells in the periphery. Their differentiation is driven by T-cell receptor recognition of self-antigens presented by MHC molecules, and they are characterized by high expression of the transcription factor FOXP3 and dependence on IL-2 for survival [28].
Tregs suppress antitumor immunity through multiple complementary mechanisms,
including secretion of immunosuppressive cytokines such as TGF-
In patients with NMIBC treated with intravesical BCG, increased intratumoral Treg infiltration has been associated with shorter recurrence‑free survival, suggesting a potential predictive role for Treg density in tumor recurrence [25]. Consistently, elevated levels of PD‑L1+ Tregs were detected in urine during BCG therapy, indicating that BCG may promote Treg recruitment and generate an alternative source of PD‑L1-expressing cells that could facilitate recurrence [32]. Histologic analyses of BCG‑unresponsive tumors further demonstrate dense Treg infiltration in non‑responding lesions [33].
The PD‑1/PD‑L1 axis represents a central immune‑regulatory pathway within the
bladder cancer TME. Tumor‑mediated engagement of PD‑1 on effector T cells by
PD‑L1 suppresses T‑cell activation and facilitates immune evasion [34]. BCG
therapy induces PD‑L1 expression on tumor cells and APCs, including macrophages
and DCs, through IL‑6- and IL‑10-mediated Signal Transducer and Activator of
Transcription 3 (STAT3), activation, as well as toll-like receptor 4
(TLR4)‑dependent extracellular signal-regulated kinases (ERK) signaling [35]. In
parallel, BCG stimulates immune cells to release IFN-
In bladder cancer models, BCG increases PD‑L1 expression on tumor cells, and its
combination with anti‑PD‑L1 therapy enhances both the number and function of
CD8+ T cells [20, 37]. Clinically, PD‑L1 expression correlates with
high‑grade tumors, disease progression, and BCG unresponsiveness [38, 39].
Importantly, in a cohort of NMIBC patients identified as BCG-unresponsive, tumor
specimens collected before and after BCG instillations revealed PD-L1
overexpression. Interestingly, these tumors also showed dense infiltration by
CD8+ T cells, suggesting that BCG may impair T cell function without
affecting their recruitment to the TME [40]. A recent study analyzing tissue
microarrays from 432 patients with BCG-naïve HR-NMIBC found that only 7% of
patients had tumors that expressed PD-L1. PD-L1 positivity was defined as
membranous staining in
In high-grade bladder cancer tissues, the malignant phenotype-associated glycan sialyl-Tn (STn), a tumor-associated antigen, is markedly overexpressed. This overexpression is associated with increased infiltration of DCs within the TME, which exhibit an immature phenotype characterized by low expression of MHC class II, CD80, and CD86. These immature DCs demonstrate impaired antigen-presenting capacity and fail to effectively activate immune effector cells, thereby contributing to a diminished antitumor immune response [42].
In a cohort of 94 bladder cancer patients treated with intravesical BCG, primary tumors from patients who experienced disease recurrence exhibited reduced STn expression. Although STn may impair DC-mediated antitumor immunity, it could potentially enhance BCG internalization into tumor cells, supporting the hypothesis that BCG may exert a direct cytotoxic effect on cancer cells. In certain cases, this direct cellular effect may compensate for, or even surpass, the antitumor immune response elicited by BCG therapy. However, these findings should be interpreted with caution due to the limited sample size and the possibility that the observed outcomes may reflect a direct tumoricidal effect of BCG, rather than an immune-mediated mechanism, in a subset of cases [43].
One key mechanism in cancer progression is the upregulation of cyclooxygenase-2 (COX-2) within tumor tissues, which enhances the inflammatory response through increased production of prostaglandin E2 (PGE2), a pro-tumorigenic molecule. PGE2 impairs the function of APCs, promotes a Th2-skewed immune response, and contributes to immunosuppression. Additionally, PGE2 exerts paracrine effects within the TME by inducing angiogenesis, primarily through the upregulation of VEGF secretion [44].
In a bladder cancer model, PGE2 has been shown to be overproduced within the TME, leading to increased infiltration of MDSCs and inhibition of APC function, collectively contributing to a weakened antitumor immune response [45]. In vitro studies have shown that COX-2 inhibitors suppress bladder cancer cell proliferation in a COX-2-dependent fashion. Additional data suggest that COX-2 inhibitors may also exert antitumor effects through mechanisms beyond PGE2 inhibition, including the direct induction of cancer cell apoptosis [46]. Celecoxib was evaluated as an adjuvant therapy for bladder cancer in a randomized clinical trial involving patients with intermediate- or high-risk NMIBC, known as the BOXIT trial. Participants (n = 472) received standard-of-care treatment, BCG induction and maintenance for high-risk disease, and mitomycin C induction for intermediate-risk disease, and were randomized 1:1 to receive either celecoxib 200 mg twice daily or placebo for two years. While recurrence-free rates were similar between the two groups, time to recurrence in patients with T1 tumors was longer in the celecoxib arm. Despite comparable induction and maintenance protocols, not all patients completed the full treatment course as indicated. Furthermore, COX-2 expression levels in tumor tissues were not assessed, which may have influenced the observed outcomes [47]. As PGE2 primarily exerts its effects PGE2 receptor (EP), selective inhibition of these receptors may enhance antitumor strategies and offer a safer alternative to celecoxib, particularly given the cardiovascular safety concerns associated with its use in elderly cancer patients [46].
NANOG, a transcription factor essential for maintaining stem cell pluripotency, may contribute to tumor progression when aberrantly expressed in the cytoplasm of cancer cells. This atypical localization has been implicated in the upregulation of HDAC1 (Histone Deacetylase 1), an epigenetic modifier known to repress immune-related gene transcription. One consequence of this regulatory axis is the suppression of CXCL10, a chemokine that plays a key role in recruiting CD8+ cytotoxic T lymphocytes to the TME. In patients with NMIBC receiving intravesical BCG therapy, elevated expression of NANOG and/or HDAC1 is associated with unfavorable clinical outcomes, including reduced recurrence-free and progression-free survival. From an immunological perspective, tumors co-expressing these markers exhibit diminished infiltration of CD8+ T cells and a lower abundance of granzyme B+ cytotoxic cells, indicative of a more immunosuppressive TME and compromised anti-tumor immunity [48].
Natural Killer Group 2A (NKG2A) is an inhibitory receptor expressed on NK cells,
CD8+ T cells, and other subsets of T cells. It forms a heterodimeric complex
with CD94, another membrane protein also found on immune effector cells. This
NKG2A/CD94 complex specifically interacts with HLA-E, a non-classical MHC class I
molecule that is frequently overexpressed on cancer cells. The nature of the
peptide presented by HLA-E critically influences the outcome of this interaction,
potentially delivering either inhibitory or stimulatory signals to immune cells.
Notably, cancer cells may present peptides via HLA-E that engage the NKG2A/CD94
receptor complex on cytotoxic immune cells, such as NK cells and CD8+ T
cells, leading to their functional inhibition and contributing to immune evasion
[49, 50]. Upon engagement, the intracellular immunoreceptor tyrosine-based
inhibitory motifs (ITIMs) of NKG2A become phosphorylated, leading to suppression
of activating signals from receptors such as NKG2D and the T‑cell receptor (TCR),
thereby inhibiting the cytotoxic activity of effector immune cells [51]. However,
the interaction between NKG2A and HLA-E within the TME is complex and modulated
by various factors. These include other HLA molecules and cytokines such as
IFN-
Recent studies have investigated the role of the NKG2A/HLA-E axis in patients with NMIBC treated with intravesical BCG. In BCG-unresponsive cases, tumors were heavily infiltrated by NKG2A+ NK and T cells, and the addition of autologous tumor organoids restored antitumor activity, suggesting a potential therapeutic role for NKG2A blockade in overcoming BCG failure [52].
Monalizumab is a humanized monoclonal antibody that blocks NKG2A, thereby enhancing NK cell cytotoxic function. It can be combined with cetuximab, an anti- EGFR antibody, to promote antibody-dependent cellular cytotoxicity (ADCC), or with durvalumab, an anti-PD-L1 antibody, to further boost NK and CD8+ T cell-mediated cytotoxicity. This therapeutic strategy has shown promising results in metastatic colorectal cancer and non-small cell lung cancer [49, 53], with potential for extension to NMIBC and advanced bladder carcinoma.
CD6 is a surface glycoprotein expressed on T lymphocytes, some NK cells, and subsets of B cells. It binds to CD166, also known as activated leukocyte cell adhesion molecule (ALCAM), which is expressed on tumor cells, APCs, and endothelial cells. CD6 plays a key role in T cell activation, proliferation, and adhesion to APCs and endothelial cells. Additionally, CD6 can modulate TCR-mediated signaling and, under certain conditions, exert a negative regulatory effect on T cell responses [54].
A study using single cell RNA sequencing of NMIBC tumor in patients, both BCG-naïve and BCG-unresponsive disease, has revealed elevated expression of the CD6-ALCAM immune-regulatory pathway prior to BCG therapy. These findings suggest that pre-existing inflammatory signaling within the TME may contribute to resistance to BCG [55]. These findings suggest that inhibition of the CD6-ALCAM axis may restore antitumor immunity and represents a promising therapeutic strategy to overcome BCG unresponsiveness.
In a separate study on T-cell lymphoma, intratumoral injection of a CD6-targeted antibody-drug conjugate (CD6-ADC) led to regression of subcutaneous nodules, demonstrating potent antitumor activity through CD6-ALCAM pathway inhibition [56]. These insights may inform future in vitro and clinical investigations targeting CD6-ALCAM signaling in BCG-unresponsive NMIBC.
Chronic stimulation of CD8+ T cells leads to functional exhaustion,
characterized by reduced cytotoxicity and diminished secretion of TNF‑
Strandgaard et al. [60] performed a multiomics analysis of urinary and
tumor samples collected before and after BCG therapy in a cohort of 156 patients
with NMIBC. They identified an immune activation signature following BCG therapy,
characterized predominantly by elevated levels of IFN
Collectively, these findings suggest that BCG‑unresponsive disease is characterized by a paradoxical immune landscape in which intense pro‑inflammatory signaling coexists with profound T‑cell exhaustion following BCG stimulation. In contrast, durable responders appear to harbor a more favorable baseline TME, defined by lower CD4:CD8 and Th2:Th1 ratios [61].
Cancer‑associated fibroblasts (CAFs) in the bladder cancer TME arise from
resident fibroblasts activated by tumor‑derived factors, particularly
TGF‑
In addition, CAFs release extracellular vesicles (EVs) that suppress CD8+ T
cell proliferation and reduce the secretion of key pro-inflammatory cytokines,
including IFN-
Following BCG therapy, fibroblasts contribute to granuloma formation and inflammatory signaling within the TME, yet their direct modulation by BCG remains unstudied, representing an important knowledge gap given their role in immune evasion and therapeutic resistance [65].
Figs. 3,4 summarize the key mechanisms of immune evasion that contribute to BCG failure in patients with NMIBC.
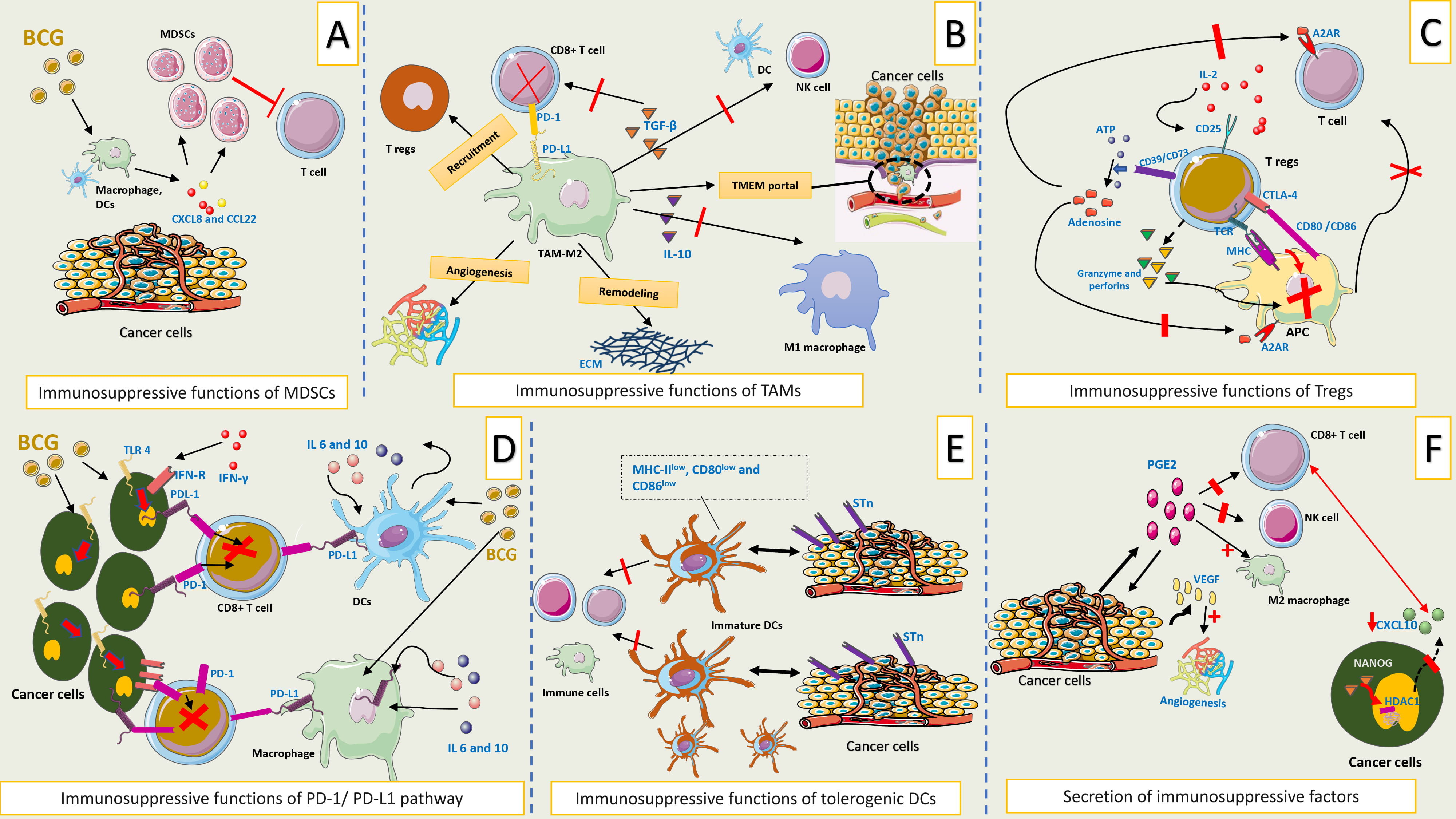 Fig. 3.
Fig. 3.
Mechanisms of immune evasion contributing to BCG failure in
NMIBC. (A) Immunosuppressive functions of MDSCs. BCG stimulates the release of
chemokines such as CXCL8 and CCL22 from urothelial cells, macrophages, and
dendritic cells. These chemokines recruit MDSCs to the tumor microenvironment,
where they remain persistently activated and suppress T-cell responses,
contributing to immune evasion. (B) Immunosuppressive functions of TAMs.
M2-polarized TAMs contribute to tumor progression by suppressing immune responses
and remodeling the extracellular matrix within the tumor microenvironment. These
cells express PD-L1 and secrete immunosuppressive cytokines such as IL-10 and
TGF-
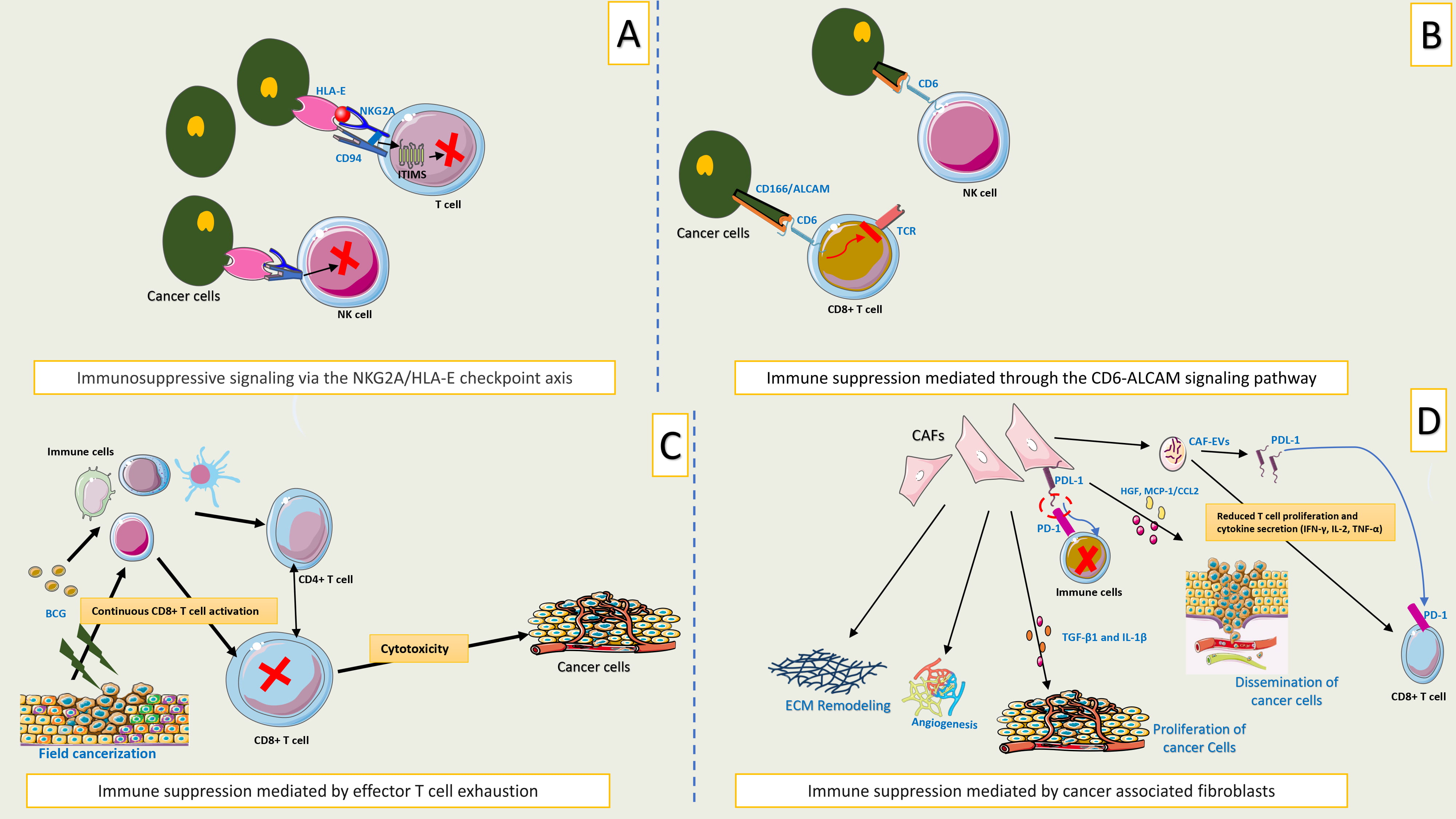 Fig. 4.
Fig. 4.
Mechanisms of immune evasion contributing to BCG failure in
NMIBC (continued). (A) Immunosuppressive signaling via the NKG2A/HLA-E checkpoint axis. This
involves the inhibitory receptor NKG2A, which is expressed on NK cells and
CD8+ T cells. NKG2A forms a heterodimer with CD94 that binds to HLA-E, a
non-classical MHC class I molecule frequently overexpressed on cancer cells.
Peptides presented by HLA-E modulate this interaction, triggering phosphorylation
of the ITIMs within NKG2A, thereby suppressing cytotoxic immune responses in the
tumor microenvironment. (B) Immune suppression mediated through the CD6-ALCAM
signaling pathway. Immune suppression in bladder cancer may be mediated through
the CD6-ALCAM signaling pathway, wherein CD6+ T cells and NK cells interact
with ALCAM-expressing tumor cells. This interaction modulates TCR signaling and
may negatively regulate T cell activation and proliferation within the tumor
microenvironment. (C) Immune suppression mediated by effector T cell
exhaustion. Prolonged stimulation of CD8+ T cells by tumor antigens leads to
functional exhaustion, characterized by reduced cytokine secretion and increased
expression of inhibitory receptors on T cells, such as PD-1 and CTLA-4. (D)
Immune suppression mediated by cancer associated fibroblasts. CAFs within the
tumor microenvironment promote tumor progression by remodeling the ECM and
secreting cytokines and growth factors that enhance cancer cell proliferation and
metastasis. They also suppress CD8+ T cell function through the release of
EVs carrying PD-L1 and by directly expressing PD-L1, leading to impaired
cytotoxic activity and diminished pro-inflammatory cytokine production. TCR, T
Cell Receptor; NKG2A, Natural killer group 2A; NK cells, Natural killer cells;
CD8+ T cells, Cluster of differentiation 8 positive T cells; CD, Cluster of
differentiation; HLA-E, Human leukocyte antigen-E; MHC, Major histocompatibility
complex; ITIMs, Immunoreceptor tyrosine-based inhibitory motifs; ALCAM, Activated
leukocyte cell adhesion molecule; PD-1, Programmed cell death protein 1; CTLA-4,
Cytotoxic T-lymphocyte-associated protein 4; CAF, Cancer-associated fibroblast;
TGF-
Rather than acting independently, the ten suggested immune escape mechanisms identified in BCG‑unresponsive disease operate as an integrated and self‑reinforcing immune evasion network within the TME. BCG‑induced chemokine release promotes the recruitment of suppressive myeloid cells, including myeloid‑derived suppressor cells and M2‑polarized tumor‑associated macrophages, which in turn secrete immunosuppressive cytokines and angiogenic factors that impair antigen presentation and favor regulatory T‑cell accumulation. These suppressive immune circuits are further reinforced by checkpoint signaling through the PD‑1/PD‑L1 axis and alternative inhibitory pathways such as NKG2A/HLA‑E and CD6-ALCAM, which collectively attenuate cytotoxic lymphocyte function despite continued immune infiltration. In parallel, tumor‑ and stromal‑derived immunomodulatory mediators, including PGE2 and NANOG‑HDAC1-dependent epigenetic repression of CXCL10, restrict effective immune cell recruitment and polarization. Chronic antigen exposure within this suppressive environment ultimately drives effector T‑cell exhaustion, while cancer‑associated fibroblasts augment immune exclusion through extracellular matrix remodeling and PD‑L1-mediated suppression. Together, these mechanisms form a hierarchical and interconnected network (Fig. 5) that simultaneously impairs immune activation, effector function, and immune persistence, providing a biological explanation for BCG-unresponsive disease and highlighting why targeting isolated pathways is unlikely to yield durable responses.
 Fig. 5.
Fig. 5.
Integrated immune evasion in BCG‑unresponsive disease. This schematic illustrates how multiple immune escape mechanisms operate as an interconnected and self‑reinforcing network within the TME of BCG‑unresponsive NMIBC. Suppressive myeloid cells, immune checkpoint and inhibitory signaling, soluble and epigenetic immunosuppression, and stromal‑mediated immune exclusion interact dynamically to undermine effective antitumor immunity. These pathways converge on shared functional outcomes, impaired antigen presentation, reduced cytotoxic immune activity, and loss of immune persistence, ultimately contributing to resistance to BCG therapy. BCG, Bacillus Calmette-Guérin; NMIBC, non-muscle-invasive bladder cancer; TME, tumor microenvironment; MDSCs, Myeloid‑derived suppressor cells; M2‑TAMs, M2‑polarized tumor‑associated macrophages; DCs, Dendritic cells; ECM, extracellular matrix; PD-L1, Programmed death-ligand 1; NKG2A, Natural killer group 2A; HLA‑E, Human leukocyte antigen E; CD6, Cluster of differentiation 6; ALCAM (CD166), Activated leukocyte cell adhesion molecule; PGE2, Prostaglandin E2; VEGF, Vascular endothelial growth factor; Tregs, regulatory T cells; PD-1, Programmed Death-1; EVs, Extracellular vesicles; HDAC1, Histone Deacetylase 1; NANOG, Nanog Homeobox (a transcription factor involved in stem cell pluripotency); CXCL10, C-X-C Motif Chemokine Ligand 10.
Our aim in this section is not to compare the efficacy of emerging therapies for BCG‑unresponsive disease (Table 1). Rather, given that the available evidence is derived from heterogeneous single‑arm clinical trials, response rates are presented descriptively to illustrate overall efficacy within the context of individual studies. These studies vary substantially in trial design, eligibility criteria, endpoints, and duration of follow‑up.
| Parameter | CG0070 (Cretostimogene) | N-803 + BCG | Nadofaragene firadenovec | Intravesical EG-70 (Detalimogene voraplasmid) |
| BOND-003 | QUILT-3.032 | NCT02773849 | LEGEND | |
| Trial phase | III | II | III | II |
| Therapy type | Oncolytic adenovirus | IL-15 superagonist + BCG | Adenoviral gene therapy | Non-viral plasmid-based immunotherapy |
| Route | Intravesical | Intravesical | Intravesical | Intravesical |
| Sample size (CIS cohort) | 112 | 84 | 103 | 26 |
| CR (any time), (CIS cohort) | 75% | 71% | 55% | 71% |
| CR at 3 mo, (CIS cohort) | 68% | 55% | 53% | 67% |
| CR at 12 mo, (CIS cohort) | 46% | 45% | 24% | N/A |
| CR at 24 mo, (CIS cohort) | 42% | 37% | 19% | N/A |
| Grade |
0% | 2% | 4% | 0% |
NMIBC, non-muscle‑invasive bladder cancer; BCG, Bacillus Calmette‑Guérin; CIS, carcinoma in situ; CR, complete response; TRAE, treatment‑related adverse event.
Nadofaragene firadenovec (Adstiladrin) is an intravesical gene therapy that
employs a non-replicating adenoviral vector to deliver the human interferon
alpha-2b (IFN
The Phase III multicenter clinical trial (NCT02773849) evaluating the efficacy
of intravesical nadofaragene firadenovec led to its FDA approval for patients
with high-risk BCG unresponsive disease with CIS [71]. Patients received 75 mL of
nadofaragene firadenovec at a concentration of 3
Nogapendekin alfa inbakicept-pmln (Antkiva) (N-803), also known as IL-15
superagonist or ALT-803, is a cytokine complex composed of IL-15, the sushi
domain of the interleukin-15 receptor alpha (IL-15R
In the phase II, open-label, multicenter QUILT 3.032 study, patients with BCG-unresponsive NMIBC with CIS received intravesical therapy consisting of 400 µg of N-803 combined with 50 mg of BCG. Treatment was administered once weekly for six weeks, followed by additional courses once weekly for three weeks at months 4, 7, 10, 13, and 19. Among the CIS cohort (n = 84), the CR rate at any time was 71%. CR rates at 12 and 24 months were 45% and 37%, respectively. In a safety analysis of patients who received intravesical N-803 monotherapy (n = 10), one patient experienced a Grade III TRAE (cerebrovascular accident). No adverse events exceeding Grade III were reported [76].
Cretostimogene grenadenorepvec also known as CG0070 is a tumor-selective oncolytic adenovirus designed to target bladder cancer cells with dysregulated retinoblastoma (Rb) protein pathways. The virus selectively replicates in these malignant cells, leading to direct tumor cell lysis. This process releases tumor-associated antigens, including neoantigens, into the local TME. Concurrently, the virus delivers a transgene encoding granulocyte-macrophage colony-stimulating factor (GM-CSF), which is synthesized by infected tumor cells. GM-CSF promotes the recruitment and activation of APC, such as DCs and macrophages, facilitating the uptake and presentation of neoantigens to T lymphocytes [77, 78]. This dual mechanism, tumor destruction and immune stimulation, enhances both local and systemic anti-tumor immune responses.
In the phase III BOND-003 trial, patients with BCG-unresponsive NMIBC with CIS
(n = 112) received intravesical cretostimogene grenadenorepvec (CG0070) at a dose
of 1
EG-70 (detalimogene voraplasmid) is a non-viral plasmid-based immunotherapy administered directly into the bladder. Following instillation, the plasmid is primarily internalized by bladder epithelial cells. Once inside, the plasmid is transcribed, leading to the expression of two key immunostimulatory components: IL-12, a cytokine that promotes adaptive immune responses, and double-stranded RNA (dsRNA) mimics, which activate retinoic acid-inducible gene I (RIG-I), initiating innate immune signaling pathways [80]. EG-70 comprises a plasmid, a dually derivatized chitosan polymer that complexes with the plasmid, and a polyethylene glycol-polyglutamic acid coating that surrounds the resulting complex [81].
In the phase II LEGEND trial involving patients with BCG-unresponsive CIS NMIBC (n = 26), EG-70 was administered intravesically at a dose of 40 mg per treatment, delivered in a 50 mL volume at a concentration of 0.8 mg/mL. The treatment schedule included four instillations given during weeks 1, 2, 5, and 6 of a 12-week induction cycle. Patients who remained free of disease progression after this initial cycle were eligible to receive up to three additional 12-week cycles of therapy. Preliminary results demonstrated an overall CR rate of 71%, with CR rates of 67% at 3 months and 47% at 6 months. Notably, no TRAEs of grade III or higher were reported [82].
Fig. 6 illustrates emerging immunomodulatory therapies for BCG-unresponsive NMIBC, along with their proposed mechanisms of action.
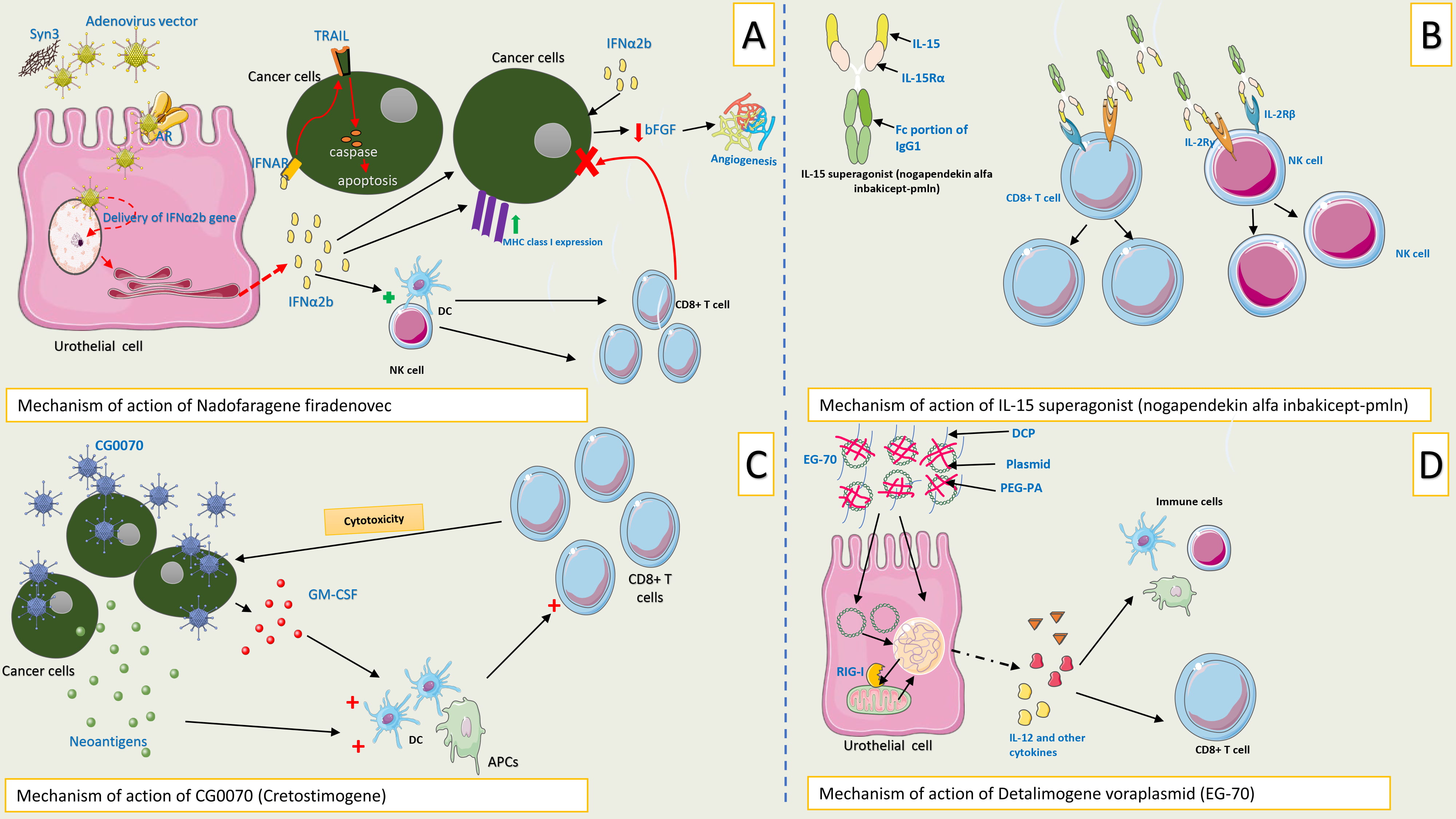 Fig. 6.
Fig. 6.
Mechanisms of action of emerging immunomodulatory therapies for
BCG unresponsive NMIBC. (A) Mechanism of action of intravesical nadofaragene
firadenovec: Following bladder instillation, the adenoviral vector utilizes Syn3,
a surfactant, to facilitate entry into urothelial cells. Once internalized, the
vector delivers the IFN
Multiple immune escape mechanisms have been implicated in resistance to intravesical BCG therapy, although the relative contribution of individual pathways varies among experimental and clinical studies. In this review, we examine ten core immune evasion processes identified through a comprehensive synthesis of the literature, prioritizing pathways supported by reproducible preclinical or translational evidence, associations with reduced responsiveness to BCG or adverse clinical outcomes, and relevance as established or emerging therapeutic targets in NMIBC.
These mechanisms should not be considered isolated or mutually exclusive. Rather, resistance to BCG reflects a complex and interconnected immune landscape encompassing the proposed ten evasion processes. The framework presented here is intended to provide a clinically grounded and translationally relevant model for understanding BCG‑unresponsive disease, rather than a definitive or exhaustive mechanistic classification.
Notably, much of the literature linking immune dysregulation to BCG‑unresponsive disease is based on observational clinical data or findings from preclinical experimental models. Consequently, for many proposed pathways, the current evidence indicates correlation rather than definitive causal relationships. While mechanistic studies in vitro and in vivo offer valuable insight and biological plausibility, their direct applicability to human BCG‑unresponsive disease has yet to be fully established. Future prospective, biomarker‑guided studies and interventional trials will be critical to validate these pathways, define their relative contributions, and guide the rational development of targeted therapeutic strategies.
As discussed above, BCG‑unresponsive NMIBC results from the interplay of multiple immune evasion mechanisms that evolve across time and space within the TME. Although emerging intravesical immunotherapies activate antitumor immunity through diverse modalities, none currently addresses the full spectrum of resistance pathways, accounting for both their observed efficacy and inherent limitations.
Defects in antigen presentation and tolerogenic dendritic cell states (Fig. 3E)
are most directly addressed by cretostimogene grenadenorepvec (CG0070) and
detalimogene voraplasmid (EG‑70). CG0070 promotes immunogenic tumor cell lysis
and delivers GM‑CSF, enhancing dendritic cell recruitment and priming, thereby
counteracting deficient antigen presentation. EG‑70 activates innate immune
sensing through RIG‑I signaling and induces IL‑12 expression, favoring Th1
polarization and potentially reversing tolerogenic DC phenotypes. In contrast,
nadofaragene firadenovec indirectly enhances antigen presentation by upregulating
MHC class I expression via interferon‑
Effector lymphocyte dysfunction and exhaustion (Fig. 4C) are most effectively targeted by nogapendekin alfa inbakicept‑pmln, which selectively expands NK cells and CD8+ T cells while limiting regulatory T‑cell proliferation. This mechanism directly counteracts effector cell exhaustion‑associated functional decline, particularly in the setting of BCG‑induced chronic immune stimulation. Nadofaragene firadenovec also enhances cytotoxic lymphocyte activity through interferon‑mediated NK‑cell activation, although it does not directly reverse exhaustion programs driven by persistent antigen exposure.
Regulatory T‑cell- and checkpoint‑mediated suppression (Fig. 3C,D) are only
partially addressed by current intravesical agents. While nogapendekin alfa
inbakicept‑pmln minimizes Treg expansion by avoiding IL‑2 receptor
Myeloid‑driven immunosuppression (Fig. 3A,B), including MDSC recruitment and M2‑polarized TAM activity, remains a major unmet therapeutic need. While GM‑CSF delivery by CG0070 and interferon signaling induced by nadofaragene firadenovec may indirectly modulate myeloid function, none of the available intravesical immunotherapies specifically deplete or reprogram suppressive myeloid populations. Similarly, CAF‑mediated immune suppression (Fig. 4D) is not directly addressed by existing agents.
Soluble immunosuppressive mediators (Fig. 3F), such as PGE2‑mediated signaling that suppresses APCs function, are also largely unaffected by current intravesical therapies, potentially limiting immune cell recruitment and persistence despite immune activation.
An important point to consider is that advanced age has been identified as a
potential factor contributing to reduced responsiveness to BCG therapy in
patients with NMIBC. Clinical data indicate that older individuals (
In addition, BCG efficacy may be diminished in immunosuppressed patients, including transplant recipients, individuals with autoimmune conditions, and those receiving systemic chemotherapy. This is biologically plausible given the reliance of BCG on host immune activation. While BCG appears to be safe in these populations, lower efficacy, particularly in transplant recipients, has been reported, though available data are limited by small sample sizes and short follow‑up [85]. Importantly, the retained activity of BCG in some immunosuppressed patients suggests the presence of immune‑independent mechanisms, including direct cytotoxic effects on tumor cells, as previously discussed by our group [7]. Consequently, BCG‑unresponsive disease may reflect tumor‑intrinsic resistance mechanisms in addition to impaired immune stimulation.
To date, two novel intravesical therapies have received FDA approval for the treatment of BCG-unresponsive NMIBC with CIS: nadofaragene firadenovec (Adstiladrin) in 2022 and nogapendekin alfa inbakicept-pmln (Anktiva) in 2024 [86]. While both agents have demonstrated promising efficacy, long-term outcomes remain under investigation. As additional therapies are expected to gain regulatory approval, clinicians will increasingly face the challenge of selecting the most appropriate treatment in the absence of predictive biomarkers. The reliance on single-arm trials for regulatory approval further complicates direct comparisons between available agents [87]. Caution is warranted when considering these therapies for patients with BCG-unresponsive papillary disease without CIS, as robust data in this subgroup are still lacking. In such cases, clinicians should engage in shared decision-making with patients, discussing the current evidence, potential benefits, tolerability, and the off-label nature of use. A comparative assessment of efficacy outcomes among emerging immunomodulatory therapies for BCG-unresponsive NMIBC suggests that relying on a single immune-enhancing mechanism may be suboptimal. This underscores the importance for clinicians to critically evaluate the underlying mechanisms of action rather than passively interpreting outcome data.
A deeper understanding of the immune mechanisms underlying BCG failure, along with the immunologic basis of emerging therapies in this context, is essential for advancing treatment strategies. This is particularly complex due to the dynamic interactions within the TME, which we have highlighted in this review. Given the heterogeneity of immune signatures across bladder tumors, an optimal therapeutic strategy should address the diverse mechanisms of immune evasion contributing to BCG unresponsive disease, ten of which have been identified as key contributors in our analysis. Ultimately, combination therapies capable of addressing several of these mechanisms simultaneously may offer the most promising path forward. For example, combining an intravesical immune‑priming therapy such as cretostimogene grenadenorepvec or detalimogene voraplasmid with systemic PD‑1/PD‑L1 inhibition may simultaneously enhance antigen presentation while alleviating checkpoint‑mediated T‑cell dysfunction. Alternatively, pairing intravesical interferon‑based therapies with agents targeting myeloid‑driven or NK‑cell inhibitory pathways (e.g., TAM reprogramming or NKG2A blockade) could produce more durable antitumor immunity by addressing both immune activation and suppression within the TME.
BCG-unresponsive NMIBC presents a multifactorial clinical and immunological challenge, driven by diverse mechanisms of immune escape within the TME. Emerging intravesical therapies, including gene-based, cytokine-driven, and oncolytic platforms, offer promising bladder-sparing alternatives. However, the absence of predictive biomarkers for treatment response and the lack of head-to-head comparative data among these novel agents complicate clinical decision-making. Future strategies should prioritize combination approaches that target multiple immune evasion pathways implicated in BCG failure. This multi-targeted strategy may enhance therapeutic efficacy and help overcome resistance to single-agent therapies. Immunologists must play a central role in guiding therapeutic development and translating immunologic insights into clinical practice for the treatment of BCG-unresponsive disease. A multidisciplinary approach is essential to optimize outcomes and advance personalized care in this evolving therapeutic landscape.
MAC: Conceptualization, methodology, literature collection, original draft writing. ID: literature collection, manuscript review and editing. MAO: Conceptualization, methodology, manuscript review and editing. All authors: Reviewed and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Some elements of the figures were created using Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
This research received no external funding.
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.