





 , Amedeo Amedei 2
, Amedeo Amedei 21 Department of Pharmacological and Biomolecular Sciences, Rodolfo Paoletti, Università degli Studi di Milano, 20133 Milano, Italy
2 Department of Clinical and Experimental Medicine, University of Florence, 50134 Florence, Italy
Abstract
Increasing evidence suggests that the gut microbiome (GM) may exert a seminal role in the maintenance of host health as well as in the pathogenesis of various illnesses, including cardiovascular disease (CVD) and neurodegenerative disorders (NDDs). GM influences host physiology by metabolizing dietary factors and host-derived substrates, thereby producing active molecules that trigger responses at local and systemic levels. The inflammatory process is characterized by a rapid “onset phase” followed by a “resolution phase”, which is essential to curtail inflammation and restore tissue homeostasis. Unique individual and environmental conditions may alter the GM equilibrium as well as impair the “resolution phase” leading to GM dysbiosis and chronic low-grade inflammatory conditions, which eventually promote the development of common/widespread human pathologies, such as inflammatory bowel and autoimmune diseases, cancer, CVD, and NDDs. This review describes the different components of the gastrointestinal tract, namely the enteroendocrine and enteric nervous systems, and the GM. Secondly, it discusses the connections among unresolved and sterile inflammation, GM and human illnesses, namely CVD and NDDs. Finally, we emphasizes the limitations of current evidence and the need for further research to fill the gap in establishing the causal link between the GM and the pathogenesis of both CVD and NDDs.
Keywords
- humans
- gastrointestinal microbiome
- dysbiosis
- cardiovascular diseases
- enteric nervous system
- gastrointestinal tract
- inflammation
- neurodegenerative diseases
It is well documented and established that the human gut microbiome (GM) lives in a mutualistic relationship with its host and the microbial colonization runs in parallel with immune system development [1, 2]. Of note, the limit between the immune system and the microbiome is not an impermeable wall but a porous structure with mutual exchanges and/or influences [3]. Recently, to better define the strong interplay between microbiome and immunity, the term “symmunobiont” has been coined to indicate a new immune system structure, formed by three intertwined pillars: adaptive immunity, innate immunity, and symmunobiome [4].
Inflammation is a conserved process that involves the activation of immune and non-immune cells aimed at protecting the host from microorganisms (e.g., bacteria, viruses, toxins) and injuries, and, consequently, to remove the pathological cause, supporting tissue repair and recovery [5, 6]. The acute inflammatory response starts within minutes after recognition of a harmful signal with an “onset phase” that involves the production of chemokines, cytokines, eicosanoids, proteases, vasoactive amines, neuropeptides and neurotransmitters by resident immune and structural cells [7]. Moreover, this process is characterized by the recruitment of different cell types, such as granulocytes, from blood to the tissue inflammatory site [8]. In this physiological scenario, the inflammatory response is programmed to evolve to an active “resolution phase” characterized by highly coordinated cellular and molecular events, including the release of anti-inflammatory cytokines and specialized pro-resolving mediators (SPMs), the loss of receptors for pro-inflammatory stimuli and the activation of regulatory cells that weaken the activity of pro-inflammatory cells [9]. The effectiveness of the “resolution phase” is conditional on specific cellular mechanisms that are orchestrated by mediators engaged in halting the inflammatory response and starting tissue repair and healing [10, 11].
Nevertheless, the presence of unique individual (age, diet, cigarette smoking, medications, genetic factors) and environmental conditions (such as, geographical location and social status) may negatively impact both the symmunonobiont and the “resolution phase”, eventually leading to a state of chronic low-grade inflammation (Fig. 1) [12, 13, 14].
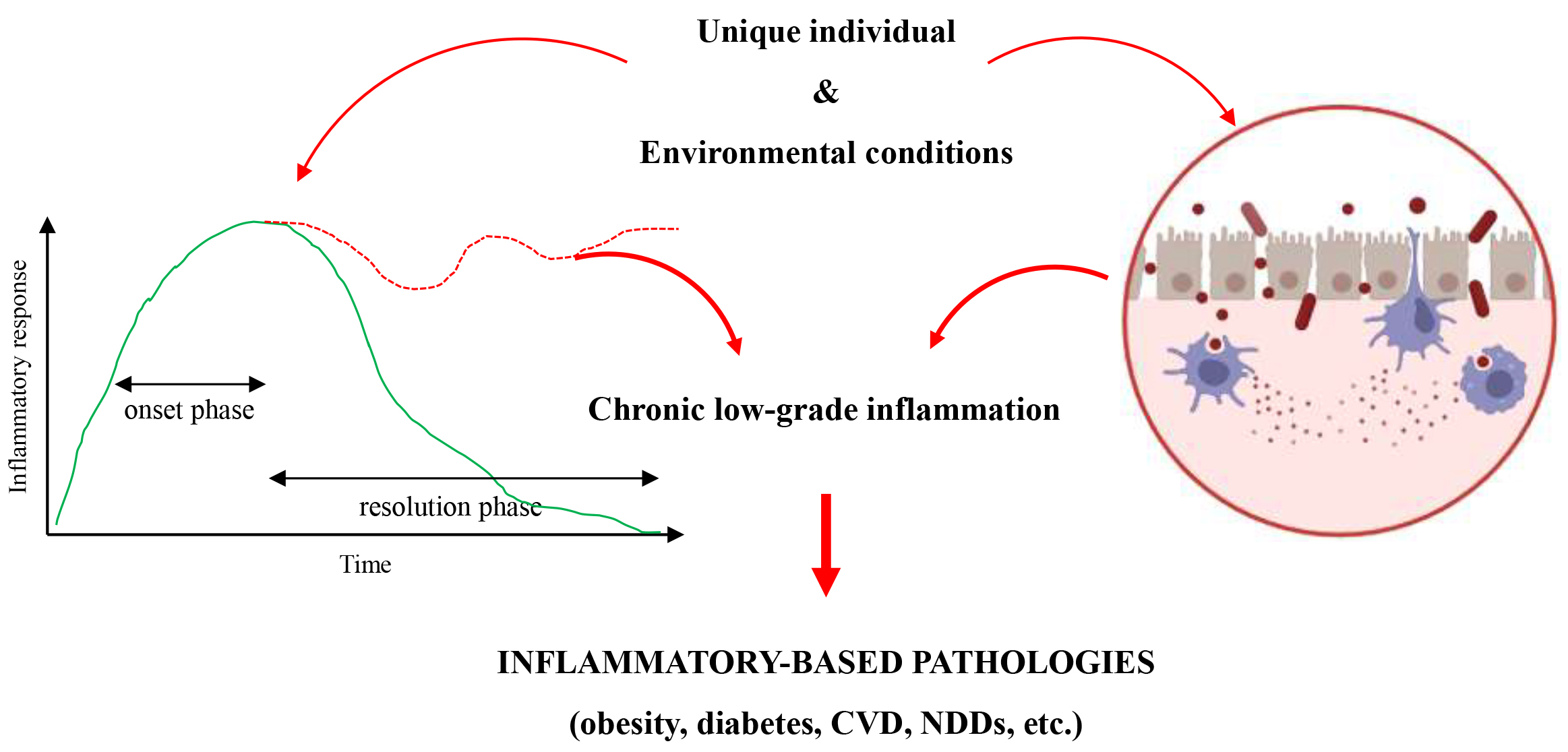 Fig. 1.
Fig. 1.
The process leading to chronicity of inflammation. Unique individual and environmental conditions may cause the failure of the resolution phase and/or the disruption of the symmunobiont equilibrium, leading to an increased susceptibily of diverse inflammatory-based pathologies. CVD, cardiovascular disease; NDDs, neurodegenerative disorders. Created with BioRender.com.
This sterile inflammatory condition is characterized by tissue and organ damage, metabolic disorders (i.e., atherosclerosis, type-2 diabetes, and obesity), and an increased predisposition to diverse inflammatory-based pathologies, such as cardiovascular disease (CVD), inflammatory bowel diseases, cancer, autoimmune and neurodegenerative disorders (NDDs) [11, 15, 16]. Aging seems to be a major risk factor for CVD, NDDs, and leaky gut. Indeed, besides the long persistence of traditional risk factors, intrinsic biological changes, accouring during aging, trigger the activation of the NLRP3 inflammasome, eventually leading to endothelial dysfunction, modification in the composition and function of the intestinal microorganisms (namely GM dysbiosis), and blood-brain barrier (BBB) breakdown. However, the complexity of heart-brain-GM interactions still limits establishing causality between GM dysbiosis and the pathogenesis of both CVD and NDDs [17, 18].
Firstly, this review describes the different components of the gastrointestinal tract, namely enteroendocrine and enteric nervous systems, and GM. Secondly, it discusses the connections among unresolved inflammation, GM and human illnesses, namely CVD and NDDs, highlighting the limitations of the available studies and the need for further research to correctly and fully understand the underlying mechanisms. Finally, we present a critical overview of the gap in the causal link between GM and the pathogenesis of both CVD and NDDs.
The gut wall is organized in diverse layers: (1) epithelial with its mucosal barrier; (2) submucosal; (3) muscular; (4) subserosal; (5) serosal [19]. In detail, the intestinal epithelia is constituted of different cell types, including enterocytes (the most prominent cell type), secretory cells (such as Goblet cells that produce mucins), enteroendocrine cells (EECs), Paneth cells that release antimicrobial factors, chemosensory tuft cells and M cells, that shuttle the antigens from the lumen to specialized antigen-presenting cells in Peyer’s patches and lymphoid follicles [20]. The mucosal barrier includes the mucin layer, a polarized epithelial cell layer and the apical junction complex (tight junctions, adherens junctions and desmosomes) [19]. The submucosa harbours blood vessels, lymphatic and nerve branches and the submucosal plexus [19]. The muscular layer is organized into two types of muscle, the circular and the longitudinal muscle. Between these two muscle layers is located the myenteric plexus (Fig. 2, Ref. [19]) [21].
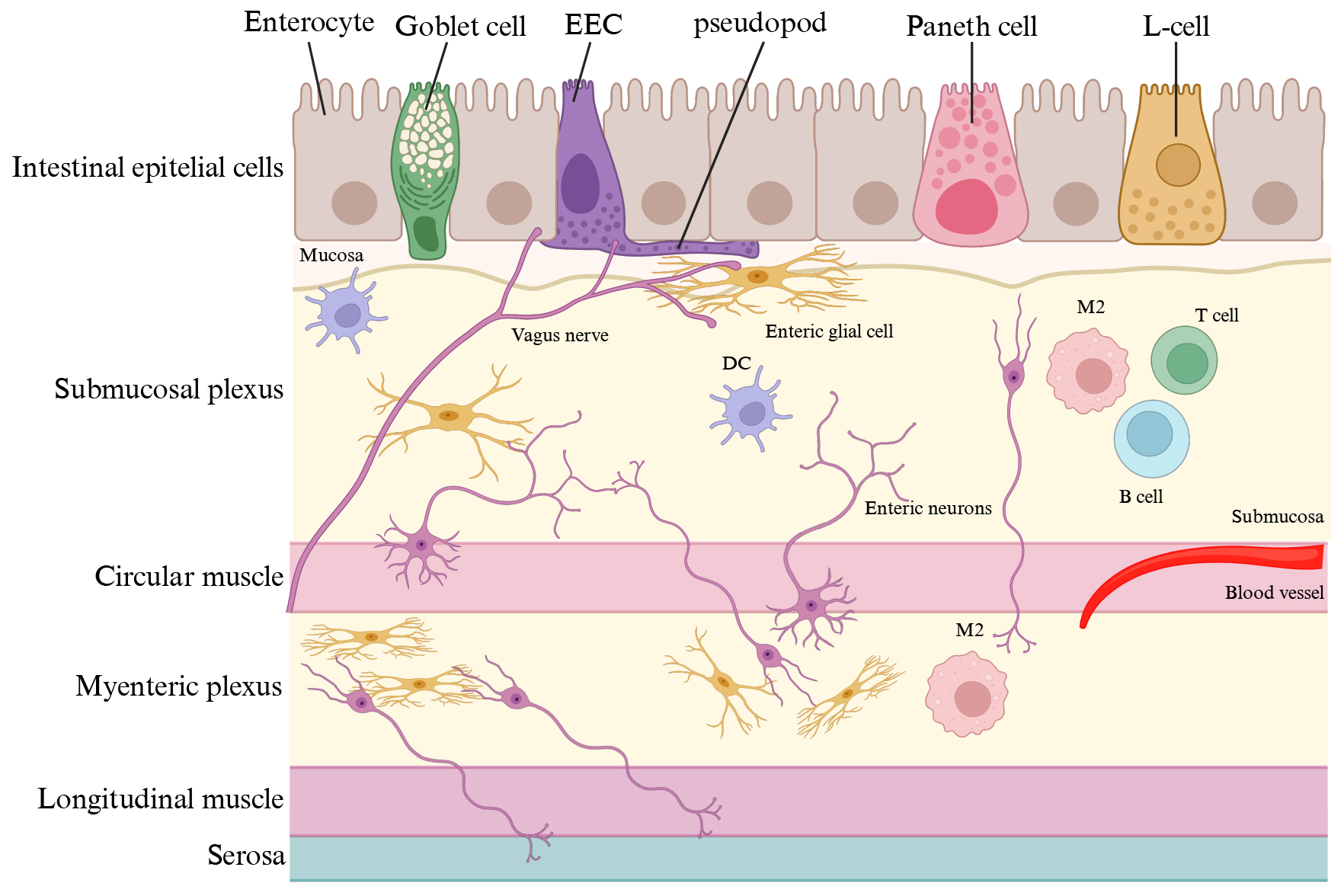 Fig. 2.
Fig. 2.
The gut wall. The gut wall is organized in diverse layers. The intestinal epithelia is constituted of different cell types, including enterocytes (the most prominent cell type), secretory cells (such as Goblet cells that produce mucins), enteroendocrine cells (EECs, L-cells, etc.), and Paneth cells. The submucosa harbours blood vessels, lymphatic and nerve branches and the submucosal plexus [19]. The muscular layer is organized into two types of muscle, the circular and the longitudinal muscle. Between these two muscle layers is located the myenteric plexus. M2, macrophage; DC, dendritic cell. Created with BioRender.com.
Of note, the stronger extrinsic modulator of the mucosal barrier is the gut content, formed by chyme (digested food), drugs and GM [22]. The gut epithelium might be compromised, becoming a “leaky gut”, by aging or varying microbial compositions [23, 24]. A “leaky gut” allows the intrusion of bacteria, their metabolites or food components into the gut wall, eventually leading to the development of chronic inflammatory gastrointestinal disease, metabolic disorders, such as, insulin resistance [25], and neurodegeneration [26, 27]. Of note, the integrity and functionality of the mucosal barrier is guaranteed by the interplay among enterocytes, immune cells and enteric glial cells. Enteric glial cells, like astrocytes, are antigen-presenting cells and have an impact on the mucosal immune system through the expression of cytokines and cytokines receptors [28].
EECs, which account for just 1% of intestinal epithelial cells, comprise at least eight cellular subtypes and collectively form the largest endocrine organ in the body, namely the enteroendocrine system (EES) [29]. EES is the primary sensor of ingested food and produce different gut hormones [cholecystokinin (CCK), GLP1, glucose-dependent insulinotropic peptide (GIP), peptide YY (PYY), somatostatin, ghrelin, and serotonin (5-HT)], implicated in the regulation of different physiological processes, such as gastrointestinal motility and secretion, glucose homeostasis, and appetite [30]. EECs possess a peculiar morphology, characterized by a narrow apical surface facing the gut lumen and a prominent basolateral process, referred to as a “pseudopod”, presenting axon-like features including neurofilaments, secretory vesicles, and physical relationship with enteric glial cells [31] and vagal afferent nerves (Fig. 2) [32]. Of note, the pseudopod forms a synapse with the vagal afferent nerves, allowing sensory stimuli from the gut lumen to reach the brain within milliseconds, using glutamate as a neurotransmitter [32]. EECs are directly activated by nutrients present in the gut lumen due to the expression of sensory transporters and receptor [33]. For instance, glucose stimulates GLP1 and GIP secretion by entering in the EECs through the sodium/glucose cotransporter member 1 (SGLT1) [34, 35]. Dietary proteins are potent stimuli of CCK release by the activity of brush border H+-coupled transporter of dipeptides and tripeptides, peptide transporter 1 (PEPT1) [36] and the G-coupled calcium-sensing receptor (CaSR) [37]. Different GPCRs, GPR120 (FFAR4), GPR119 and FFAR1 (GPR40), have been recognized as receptors of long- and medium-chain fatty acids [38, 39, 40]. Of note, GM diversity strongly influence the EECs activity by affecting the metabolism of the different nutrients [32]. Finally, EECs can be activated by bacterial antigens (i.e., LPS) because they also express Toll-like receptors (TLRs) [41, 42]. Basolateral-released gut hormones sense enteric, vagal, and spinal sensory neurons by interacting with their specific receptors or enter the circulation exerting their endocrine function. In addition, once in the subepithelial space, gut hormones may also act in a paracrine manner to neighbouring epithelial cells [43].
The enteric nervous system (ENS) is the seminal component of the autonomic nervous system, composed of sympathetic, parasympathetic and enteric, and a large division of the peripheral nervous system (PNS) responsible for controlling the gastrointestinal behaviour independently of central nervous system (CNS) input [21]. The ENS is organized into two distinct ganglionated neuronal plexuses, called myenteric and submucosal plexuses, and controls gastrointestinal motility, secretion, nutrient absorption, immune regulation and defence (Fig. 2) [44, 45]. Indeed, the ENS is interrelated with the enteroendocrine and gastrointestinal immune system, the PNS, the CNS, and the GM [45]. Key players in the ENS and gastrointestinal tract are enteric glial cells because they are the first component of ENS in contact with the luminal content and form relay stations with enterocytes or EECs within the gut epithelium [46]. Enteric neuronal phenotypic heterogeneity is robust and almost all the neurotransmitters found in the CNS are also detected in the ENS [44]. Indeed, signalling within the ENS implies the activation of GPCRs by acetylcholine, noradrenaline, many peptide neurotransmitters, such as, calcitonin gene-related peptide (CGRP), vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating protein (PACAP), galanin, bombesin-related neuromedin B (NMB) and gastrin-releasing peptide (GRP), eventually, impacting gut hormone release [31]. Cannabinoid receptors have been also detected throughout the gastrointestinal tract, mainly localized in enteric nerve fibres [47, 48].
ENS is unique having both sensory and motor properties. Therefore, the ENS can function without input from the CNS [49]. Nevertheless, a two-way communication normally occurs, where CNS influences enteric behaviour and the gut also sends information to the brain, namely the gut-brain axis [50, 51]. On note, 90% of vagal fibres between gut and brain are afferent, indicating that the brain is more a receiver than a transmitter [32, 49]. In addition, GM has been recognized as a component critically involved in the gut-brain axis, giving rise to the concept of microbiota-gut-brain communication [52]. Both alterations of enteric neurons and glial cells (namely, enteric neuropathies [53]), and imbalances in the GM composition are involved in the pathogenesis of different gastrointestinal (such as, inflammatory bowel and celiac disease) [54] and non-gastrointestinal disease (such as, obesity, diabetes, and NDDs) [55].
The intestinal microbial community that inhabits the human gut counts
SPMs, such as, resolvins, protectins, maresins, and cysteinyl-SPMs, are endogenously synthesized from EPA (eicosapentaenoic acid), DPA (docosapentaenoic acid) and DHA (docosahexaenoic acid) by the action of specific enzymes. SPMs, upon activation of specific receptors, play a relevant role in the “resolution phase” of the inflammatory response by (1) increasing efferocytosis, phagocytosis and leukocytes egress; (2) accelerating wound healing; (3) inhibiting the release of pro-inflammatory cytokines (mainly TNF, IL-1beta, IL-6) and lipid mediators (prostaglandins and leukotriens); (4) promoting the production of IL-10; and (5) halting platelet aggregation and inflammasome formation [66]. Of note, specific beneficial taxa (Bifidobacterium, Lactobacillus, and Akkermansia) may strongly impact the bioavailability of SPMs, by influencing the activity of different enzymes. Consequently, GM dysbiosis may support the failure of the “resolution phase” of the inflammation due to the low amount of the above mentioned beneficial taxa [67]. In addition, experimental studies have demonstrated that the richness of Akkermansia muciniphila, the most abundant species in the human intestinal microbiota, has been inversely correlated with body weight, mucus thickness, immune/inflammatory index, insulin resistance, and the development of atherosclerosis in several experimental studies [68, 69]. Moreover, in mouse models a decreased relative abundance of Akkermansia muciniphila with age [70, 71]. Conversely, the daily administration of live cells of Akkermansia muciniphila prevented not only weight gain, but also restored epithelial barrier integrity (mucus thickness), counteracted endotoxemia (serum LPS), and improved the metabolic profile in mice fed a high fat diet [72, 73]. However, these data performed in mice were not confirmed in rats later on [71]. The different outcomes are likely the effects of environmental and genetic factors. Similarly, a debate about whether Prevotella copri is beneficial or detrimental in human health is still present [74, 75]. Moreover, further investigations and human trials are required to establish the real causality between these two types of bacteria and the health condition.
Based on both experimental and clinical data, gut metabolites play a seminal role in the interplay between the host and its microbiome [76, 77].
In detail, short-chain fatty acids (SCFAs), mainly acetate, propionate and butyrate, are produced by microbial fermentation of undigested carbohydrates (namely, dietary fibres). They exert both direct and indirect effects. Locally, SCFAs (i) are used as fuel for colonic mucosal epithelial cells [76]; (ii) up-regulate the expression of tight junction proteins (i.e., ZO-1 and occludin) and antimicrobial peptides [78]; (iii) promote mucus production [79]; (iv) have a beneficial impact on gastrointestinal motility [80]. After entering the portal bloodstream they act as hormone-like signalling molecules, through the interaction with host receptors to regulate innate immunity and host metabolism [e.g., promoting the differentiation of regulatory T cells (Tregs) and IL-17-expressing T cells (T helper-Th17), and ameliorating glucose handling] [81, 82, 83]. They can bind to the following receptors: (1) GPR41 [also known as free fatty acid receptor 3 (FFAR3)] expressed on the gut epithelial L-cells, stimulating the production of the endocrine hormone PYY [84]; (2) GPR43 (also known as FFAR2), triggering the secretion of the hormone GLP-1 by intestinal L-cells [84]; (3) GPR109A [also known as hydroxycarboxylic acid receptor 2 (HCAR2)] [85]; (4) olfactory receptor 78 (Olfr78) [86]. Moreover, it has been demonstrated that SCFAs are histone deacetylase (HDAC) inhibitors as well as acetylase activators, resulting in epigenetic changes that promote the growth and function of Tregs. Indeed, butyrate can stimulate acetylation of the Foxp3 gene, which is a key transcription factor for Tregs [77, 87]. Based on these data, it is not surprising that SCFAs are considered as potential disease-preventing or disease-mitigating molecules. However, in vitro and in vivo studies have shown that SCFAs overproduction or accumulation in the bowel may cause obesity, due to increased energy accumulation [88, 89]. In addition, a dysregulated production of SCFAs is associated with more human diseases, including colorectal cancer, celiac disease, amyotrophic lateral sclerosis (ALS) and myocardial infarction [90, 91, 92, 93, 94]. Resolution of these conflicting results needs a comprehensive review of the causal relationships among GM composition, SCFAs abundance and host metabolism.
The consumption of diets rich in meat (such as, the Western-type diet), which are a source of L-carnitine and choline, are directly correlated with the levels of trimethylamine-N-oxide (TMAO), the product of the TMA oxidation by the enzymatic activity of the liver flavin monooxygenase 3 (FMO3) [95]. It has been demonstrated that diverse taxa of faecal microorganisms are associated with serum concentrations of TMAO, such as, Peptostreptococcaceae, Prevotella, Sporobacter, Fusibacter, Clostridium, Lachnospira, and Clostridiales Family XI Incertae Sedis [86]. Different studies have demonstrated that elevated TMAO levels are associated with the development of CVD, kidney dysfunction, and NDDs [96, 97, 98, 99].
Primary bile acids, released into the duodenum from the gall bladder, facilitate the absorption of dietary lipids and lipophilic vitamins [100]. In the colon, through bile salt hydrolysis and bile acid 7alpha-dehydroxylation, bacteria are able to generate secondary bile acids. In addition, bile acids can act as direct antimicrobial agents, due to their hydrophobic and detergent proprieties on bacterial membranes [101]. Therefore, a dynamic balance exists between diet-intestinal, microbiome-bile acid pool size and composition [102]. Of note, in absence of microbial activity, the host bile acid signature is markedly altered potentially leading to diverse gastrointestinal, metabolic and inflammatory pathologies [103, 104]. Once, bacterial-modified bile acids enter the portal blood they can act as signalling molecules by interacting with host bile acid receptors. In detail, Farnesoid X-receptor (FXR) regulates the transcription of genes involved in the metabolic regulation of bile acids, lipids and glucose [105]. Diverse bile acids activate the vitamin D3 receptor (VDR) [106]. Experimental and clinical studies uncovered a role for VDR in CVD disease [107, 108]. Takeda GPCR5 (TGR5) is involved in energy expenditure and improvement of the glycaemic control [109]. Secondary bile acids, mainly lithocholic acid, are endogenous ligand of the pregnane X receptor (PXR), a nuclear hormone receptor that acts as a xenobiotic sensor to regulate xenobiotic and lipid metabolism [110].
Besides these well-studied bile acid-regulated receptors, bile acids are ligands for sphingosine-1-phosphate receptor 2 (S1PR2) [111], muscarinic receptors M2 and M3, N-methyl-D-aspartate receptor (NMDAR) and gamma-aminobutyric acid A receptor (GABAAR) [112]. Of note, different primary and secondary bile acids and their receptors have been detected in the brain. Circulating bile acids produced in the liver and intestine can reach the brain by crossing the BBB, either by simple diffusion or through bile acid transporters, such as OSTalpha/beta [113], OATP [114], ASBT [115], NTCP and BSEP [116]. However, all the genes encoding enzymes required for the synthesis of primary bile acids are expressed in the brain, whereas enzymes responsible for the synthesis of secondary bile acids are exclusively expressed by the intestinal bacteria [117].
Tryptophan is an essential aromatic amino acid, known to be a precursor of many microbial and host metabolites [118]. Specifically, it has been discovered that GM bacteria (such as, Lactobacilli) are able to catabolize the tryptophan into indole and its derivates, which are ligands of the aryl hydrocarbon receptor (AhR) [119]. AhR signalling is considered a critical component of the immunity because it is expressed by innate lymphoid cells group 3 (ILC3s) [120], and when activated upregulates the expression of IL-22, a key cytokine that impacts on intestinal mucosal homeostasis and provides resistance to the fungus Candida albicans [121]. In addition, GM influences the kynurenine-producing IDO (indoleamine 2,3-dioxygenase) pathway, which plays a critical role in inflammatory mechanisms, immune responses and neurobiological functions [122]. Finally, enterochromaffin cells produce a significant amount of the total body 5-HT, including the plasma 5-HT [123]. It has been demonstrated that the 5-HT synthesis is under GM control, specifically mediated by SCFAs [124, 125].
Recent data have showed that dietary patterns can shape the GM diversity, which in turn impacts on CVD outcomes [126]. Indeed, GM dysbiosis, supporting a state of chonic low-grade inflammatory, has been implicated in the CVD pathogenesis (Fig. 3).
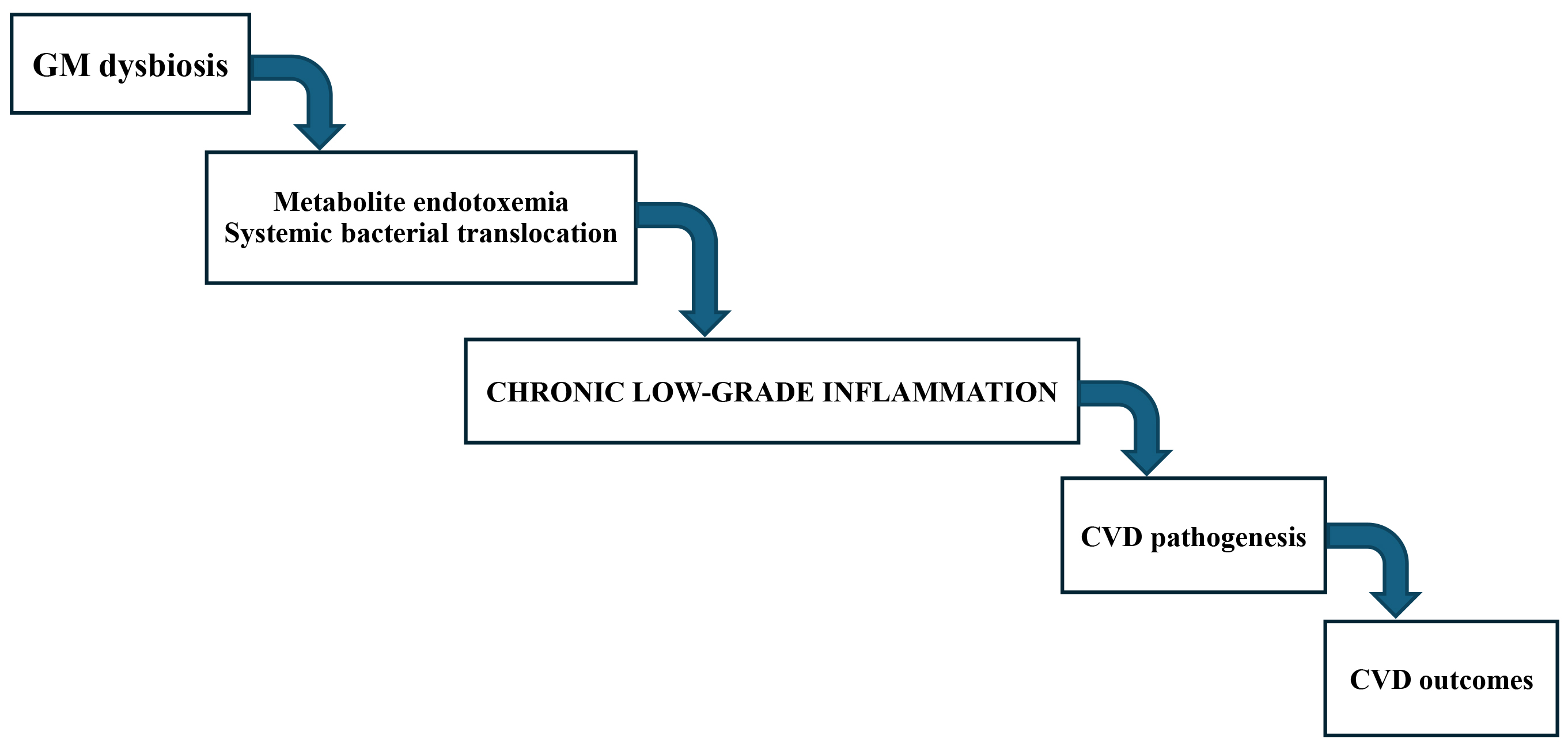 Fig. 3.
Fig. 3.
The GM-inflammation-CVD axis. Alteration in the composition of gut microbiome (GM) and accompanying functional changes in metabolism, such as GM dysbiosis, have been implicated in metabolic endotoxemia and systemic bacterial translocation, chronic low-grade inflammation leading to cardiovascular disease (CVD) pathogenesis and eventually CVD outcomes.
Enteric neuron loss has been observed in both streptozocin-induced animal diabetes models [127] and in the gut wall of patients with type 2 diabetes mellitus [128, 129], explaining, at least in part, the high percentage of people with diabetes who report having gastrointestinal symptoms [55]. Furthermore, experimental and epidemiological studies strongly supported the notion of a correlation between microbial metabolites and CVD, identifying SCFAs and TMAO as crucial ones [81]. However, bile acids, tryptophan metabolites, branched-chain amino acids, vitamins, and polyamines also play critical roles in host pathophysiology [76]. Following we describe the relevant studies performed to investigate the interplay between the different microbial metabolites and the CVD.
Of note, the links between SCFAs and CVD risk factors have been derived from animal studies [130, 131, 132] and partly from Mendelian Randomization (MR) analyses [18] but the clinical validation is still lacking [81]. However, a recent study demonstrated that propionate treatment lowers serum total and LDL-cholesterol concentrations, in both mice and humans [133]. In addition, the authors reported that in apoE-null mice fed high-fat diet, propionate decreased cholesterol absorption and atherosclerosis development [133]. Mechanistically, propionate by raising Treg cell numbers and IL-10 intestinal levels, downregulated the expression of Niemann-Pick C1-like 1 (Npc1l1), a key intestinal cholesterol transporter [133]. Therefore, additional well-designed and tailored clinical investigations are needed to establish if SCFAs modulation would improve the risk in CVD patients, aiming at drawing a definitive statement (Table 1, Ref. [109, 130, 131, 133, 134, 135, 136, 137, 138]).
| Class | Type of metabolite | Model | Effects | Reference |
| SCFAs | Mix of SCFAs | Rats | [130] | |
| Acetate + | Mice | [131] | ||
| Propionate + | ||||
| Butyrate | ||||
| Propionic acid | ApoE null mice | [133] | ||
| Humans | ||||
| TMAO | Choline | Mice | [134] | |
| TMAO | ||||
| Microbial transplantation | ||||
| DMB | ApoE null mice | Shift of bacterial taxa | [135] | |
| Bile acids | 6-EMCA | Macrophages | [109] | |
| LDL null mice | ||||
| Lithocholic acid | PXR null mice | [136] | ||
| Amino acids | Imidazole propionate | Primary hepatocyte | Glucose control impairment | [137] |
| HEK293 cell line | Lack of response to metformine | |||
| Mice | ||||
| ECs | [138] | |||
| ApoE null mice |
Note: SCFAs, short-chain fatty acids; TMAO, trimethylamine-N-oxide;
Zhu et al. [134] showed that microbial transplantation of a high TMAO producing bacteria could transmit TMAO production and promotion of thrombosis into recipient germ-free mice. In addition, the TMAO pathway has also been linked with cardiac hypertrophy and fibrosis, chronic kidney disease, type2 diabetes, and obesity [139, 140, 141, 142, 143, 144, 145]. In vitro and in vivo studies have demonstrated that TMAO may: (1) increase thrombin-induced Ca2+ release, enhancing platelet hyperreactivity [146]; (2) promote vascular inflammation by inducing the activation of MAPKs and NF-κB [135] as well as the NLRP3 pathways [147, 148], in endothelial and smooth muscle cells; (3) activate the TGF-beta/SMAD3 (Small mother against decapentaplegic 3/transforming growth factor) signalling pathway, starting profibrotic processes in the heart and kidney [141]; (4) inhibit the reverse cholesterol transport (RCT) at least in part by downregulating the hepatic expression of cholesterol 7alpha-hydroxylase (CYP7A1), the rate-limiting enzyme in bile acid synthesis, and some bile acids transporters [95]; (5) stimulate macrophage cholesterol accumulation by enhancing cell surface expression of CD36 and scavenger receptor class A (SRA) [95]. Altogether, these data suggest that TMAO may also negatively impact seminal HDL functions, eventually leading to a raise of the susceptibility to the atherosclerosis development. Chronic dosing of 3,3-dimethyl-1-butanolo (DMB), a non-lethal inhibitor of microbial TMA lyase, to apoE-null mice drove a shift in the amounts of some bacterial taxa and a marked decrease of TMAO concentrations, macrophage cholesterol storage, foam cell formation, and atherosclerosis progression, in the absence of toxicity or adverse effects (Table 1) [149, 150]. Conversely, some recent studies did not confirm the previous ones [136, 151, 152], suggesting that well-designed experiments are needed to completely understand the interplay between TMAO levels and CVD risk factors and validate the novel therapeutic approach of TMAO-lowering strategies.
It has been established that, microbial metabolism of bile acids alters their bioavailability and impacts on the metabolic responses they are involved [153]. Sato et al. [154] found a correlation between the presence of unique secondary bile acids, generated from novel bile acid miocrobiome-mediated metabolic pathways, and the decreased susceptibility to chronic inflammatory- and age-associated diseases in centenarians. In agreement with these data, Pols et al. [109] have demonstrated that TGR5 activation in macrophages inhibits proinflammatory cytokines expression and atherosclerotic lesion formation. These effects are mediated by TGR5-induced cAMP signalling, eventually leading to NF-kB inhibition [76, 109]. Mice lacking PXR are significantly less prone to atherosclerosis development compared to wild-type animals, and peritoneal macrophages isolated from these mice are characterized by diminished expression of CD36, lipid accumulation, and CD-36-mediated oxidized LDL uptake (Table 1) [155]. Finally, taurine-conjugated bile acids may sense through M2 and M3 muscarinic receptors, the latter being involved in the modulation of the heart rate recovery after myocardial infarction [156]. Interestingly, all conjugated bile acids can bind and activate S1PR2 [157], which has been recognized as a negative modulator of atherosclerosis in apoE-null mice [158].
Microbiome-derived tryptophan metabotites by sensing throught the AhR modulate the mucosal immune homeostasis in the gut [121]. In addition, some studies have shown that microbiome-derived branched-chain amino acids are associated with metabolic inflammation and insuline resistance [159, 160]. Coherent with these results, two independent studies discovered GM pathways able to metabolize histidine and phenylalanine, two essential amino acids, into imidazole propionate and phenylacetylglutamine, respectively, that are upregulated in patients with insulin resistance and type 2 diabetes [161, 162]. In detail, Molinaro et al. [161] found a direct correlation between the increased production of imidazole propionate and altered GM [137, 163] and higher amount of specific bacteria, such as Clostridium bolteae, Clostridium symbiosum, and Ruminococcus gnavus. Moreover, in vitro (primary hepatocytes and human embryonic kidney cell line HEK293) and in vivo (wild-type mice) experiments demonstrated that treatment with imidazole propionate promote the phosphorylation of p38gamma, a subset of the p38 MAPKs [164], and the subsequent activation of two different downstream signalling pathways, p62-mTORC1-S6K1 and AKT-AMPK, causing glucose control impairment and lack of response to metformin, respectively [165, 166]. These data suggest a causal role for circulatory imidazole propionate in the development of type 2 diabetes. In addition, in two large and independent clinical cohort serum concentrations of imidazole propionate are independently correlated with decreased ejection fraction and heart failure [138]. In line with this data, a recent study demonstrated that imidazole propionate exerts deleterious effects on endothelial cells, such as, inhibition of their proliferative and migratory capacities, by impairing the PI3K/AKT/FOXO1 signalling axis [167]. Eventually, this effect leads to a low regenerative activity for endothelial cells following arterial injury and accelerate progression of atherosclerosis in apoE-null mice (Table 1) [167]. These findings were confirmed in humans, where these authors showed a link between imidazole propionate levels and increased risk for prevalent atherosclerotic CVD [167].
Similarly to what was observed by Molinaro et al. [161], using untargeted metabolomics Nemet et al. [162] discovered that phenylacetylglutamine, a GM-derived metabolite, was associated with CVD and incidence of major adverse CV events (myocardial infarction, stroke, or death). Functional and genetic engineering studies with human commensals demonstrated that the microbial porA gene induces dietary phenylalanine conversion into phenylacetic acid, with subsequent host synthesis of phenylacetylglutamine in the liver facilitating platelet responsiveness and thrombosis potential. Mechanistic experiments reveal phenylacetylglutamine exerts these cellular events through GPCRs, including alpha2A, alpha2B, and beta2-adrenergic receptors [162].
Therefore, it can be speculated that both the imidazole propionate and phenylacetylglutamine-producing gut microbial pathways may represent a new therapeutic target in type 2 diabetes and CVD prevention.
The most common NDDs in humans, such as, amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD), are still incurable and lack detailed etiologies. However, inflammatory processes and gastrointestinal dysregulation, mainly, ENS deficits and GM dysbiosis, are now starting to be reported as triggering and/or causal factors [55, 168, 169]. Indeed, abnormal neuroinflammation can lead to cognitive impairment and neurodegeneration [170]. Microglial cells, the resident immune cells of the CNS, exert a seminal role in the neuroinflammation, being able to release both anti and pro-inflammatory molecules, stimulate phagocytosis and immunosuppression, and tissue repair. In addition to microglia, the most numerous cell type in the CNS, namely astrocytes, are key players in neuroinflammation, CNS trauma and ischemic stroke, because they regulate the BBB permeability and blood flow. Specifically, these cells react to a CNS insult by reactive astrogliosis, that is rising the numbers of astrocytes and modifying their activity [169]. Different studies have shown that these disorders are associated with dysbiosis and dysfunctional gastrointestinal manifestation may occur even before CNS symptoms occur, suggesting that GM alteration may contribute to disease pathogenesis (Fig. 4) [171].
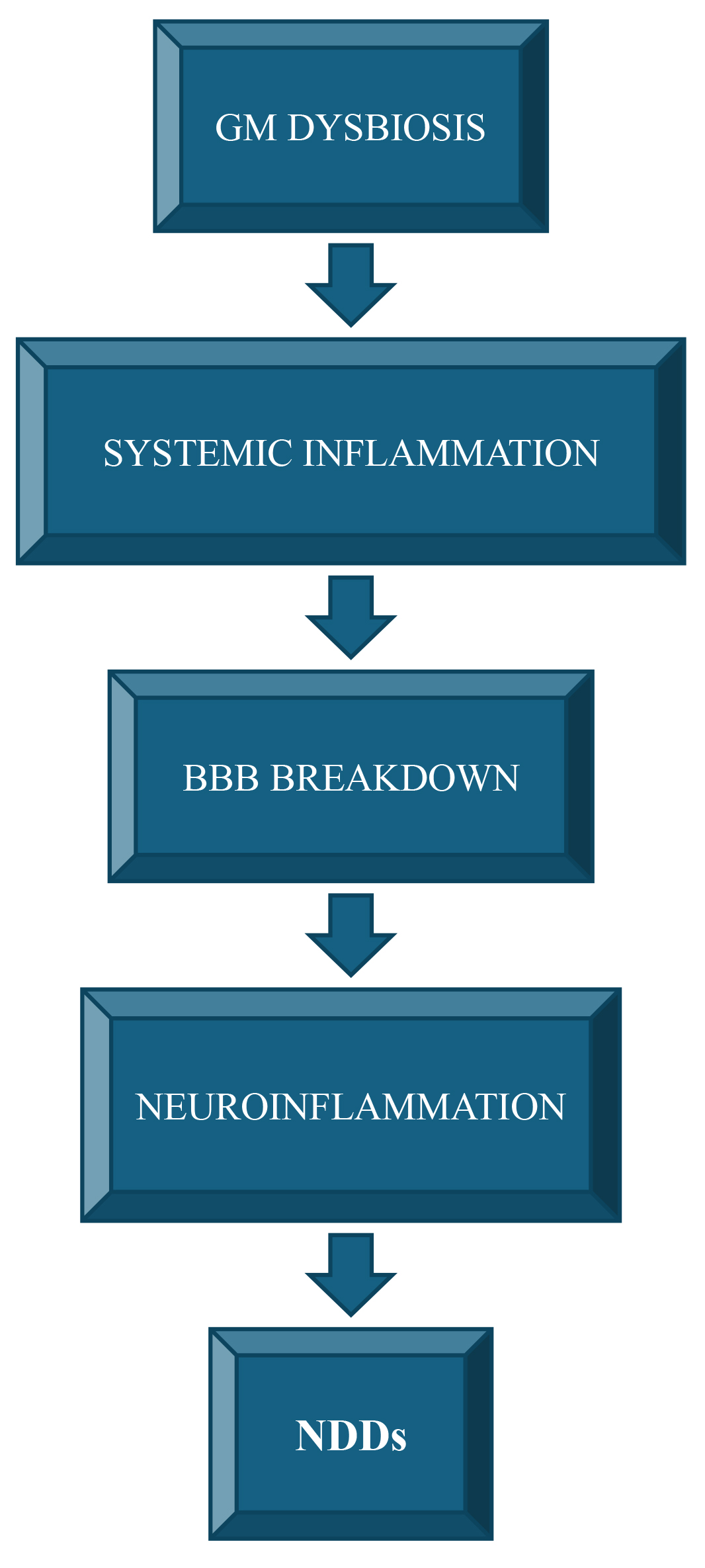 Fig. 4.
Fig. 4.
The GM-inflammation-NDDs axis. Gut microbiome (GM) dysbiosis, have been implicated in systemic inflammatory conditions leading to alteration of blood-brain barrier (BBB) and neuroinflammation, eventually contributing to neurodegenerative disorders (NDDs) development/progression.
Moreover, patients suffering from neurodegenerative disease are also prey to nutritional deficiency caused by dysphagia and other gastrointestinal problems [56]. Conversely, in the elderly, poor diet has been correlated with altered microbiota composition, inflammation and disability [172]. In animal models and human studies, the administration of prebiotics and probiotics proved efficacient in modulating stress reactivity, cognitive processes and behaviour [173].
Studies have demonstrated that SCFAs can influence gut-brain communication and brain function directly or indirectly through immune, endocrine, and vagal pathways [174, 175]. Specifically, SCFAs act locally, by activating FFARs, expressed in epithelial and immune cells, or inhibiting histone deacetylases or increasing the activity of histone acetyltransferases, enhancing intestinal mucosal immunity, barrier function and integrity. Peripherally. SCFAs modulate both systemic and neuroinflammation, eventually affecting emotion, cognition, and pathophysiology of mental disorders. In addition, through the endocrine pathway they promote the release of GLP1 and PYY, which might influence learning, memory and mood. SCFAs can directly activate vagal afferent nerves via FFARs, thereby signalling to the brain, cross the BBB, influencing the BBB integrity by halting the pro-inflammatory responses [176]. However, there is a dearth of human research and low coherence with experimental results. Well-designed and tailored clinical studies should therefore be performed before making claims on a mechanistic role of SCFAs in the microbiota-gut-brain axis [176].
Another GM-derived metabolite involved in neuroinflammation is TMAO. An in vivo study found that TMAO levels in plasma and brain are directly related to aging [177]. In addition, an age-dependent influence on inflammatory responses and BBB integrity was detected. Of note, young mice fed TMAO-rich diet had a significant drop of the performance in the novel objective recognition test compared to animals fed chow diet [177]. In addition, experimental exposure of human astrocytes to TMAO elicited a dramatic modification of cellular morphology and protein expression, mirroring astrocyte activation [177]. Studies reported that these signs of neuroinflammation induced by TMAO were correlated with the activation of the NF-kB signalling pathway, eventually increasing the expression of IL-6, and TNF-alpha [178, 179], and the acceleration of cellular senescence in endothelial cells and astrocytes cause BBB dysfunction [180]. These correlations were confirmed by a clinical study performed in middle-aged and older humans [177].
Finally, a growing number of studies have been performed to uncover the actions of the neuroactive activites of bile acids and their influence on brain function. However, additional experimental and clinical experiments are necessary since there are data supporting a detrimental role of bile acids in cerebral pathological conditions and cognitive decline [181].
ALS is a debilitating and rapidly fatal neurodegenerative disease characterized by the progressive loss of the upper and lower motor neurons, eventually leading to respiratory paralysis and death within 3–5 years [182]. ALS shows sexual dimorphism, with males having higher disease incidence than female individuals [183, 184, 185]. Similarly to other neurodegenerative illnesses, i.e., frontotemporal dementia and AD, ALS is described as a proteinopathy because aberrant protein aggregates have been detected in the cytoplasm of motor neurons of patients affected by this pathology [186, 187, 188]. About 90% of ALS cases are sporadic ALS (sALS) lacking identifiable genetic cause, while the rest of affected patients are characterized by mutations in specific genes, leading to familial inheritance (fALS) [189, 190]. The fALS majority are due to mutations in superoxide dismutase 1 (SOD1), chromosome 9 open reading frame 72 (C9orf72), fused in sarcoma (FUS), and transactive response DNA-binding protein 43 (TARDBP) [189]. ALS pathophysiology arises from a complex interplay between genetic and environment over time, associated with dysregulated immune system and/or inflammatory responses [190]. Indeed, this pathology involves astrocytes, oligodendrocytes, microglia and different peripheral immune cells [189]. In addition, altered energetic metabolism, mainly hyperlipidemia, and a hypermetabolic state have been observed in ALS patients [191, 192]. Lastly, gastrointestinal and autonomic symptoms are frequently observed but often neglected and could contribute to lowered food intake, quality of life and survival [193, 194]. As described, GM dysbiosis could play a crucial role in the development of the gastrointestinal alteration observed, modulating luminal fluid balance, bile acid metabolism, SCFA synthesis and intestinal motility [56]. A 2-sample MR study assessed the causal effects of potential microbiome modulators of human ALS and discovered evidence linking some GM genera with the possibility of impacting on the susceptibility to ALS. In addition, this study confirmed the bidirectional relationship between the GM and ALS and hypothesized that a transsynaptic, glutaminergic, excitotoxic mechanism may play a key role in the ALS pathogenesis [195]. Overall, bacterial metabolites and neuropeptides by affecting ENS and CNS have been implicated in ALS development and progression, also because emerging evidence suggests that gastrointestinal symptoms manifest before the appearance of motor failures [196]. Notably, the TARDBP gene is constitutively expressed by enteric neurons and glial cells [197] and mice carrying the mutation Ala315Thr are characterized by neuron and glial cell loss in the myenteric plexus [198]. However, the clinical confirmation of the ENS pathology is still lacking [55]. Nevertheless, the SOD1G93A transgenic mouse model, an established ALS experimental model, displays early GM shifts, leaky gut, abnormal Paneth cell presence and reduced butyrate-producing bacteria, all of which were linearly related with disease progression (Table 2, Ref. [93, 199, 200, 201, 202, 203, 204]) [199]. Moreover, the supplementation of Akkermansia muciniphila improved ALS symptoms, at least in part by increasing cerebrospinal levels of nicotinamide. While detrimental effects on motor neurons and survival rate were observed treating the mice with Ruminococcus torques and Parabacteroides distasonis (Table 2) [199]. In line with these data, Niccolai et al. [200], using the same mouse model found that elevated medium-chain fatty acids and decreased SCFAs correlated with a more rapid ALS progression. The same lipid shifts were also observed in patients with ALS [93].
| Model | Intervention | Effects | Reference |
| SOD1G93A mice | Akkermansia muciniphila | [199] | |
| Ruminococcus torques and Parabacteroides distasonis | |||
| SOD1G93A mice | – | [200] | |
| ALS patients | – | [93] | |
| C9orf72-/- mice | Broad-spectrum antibiotics | [201] | |
| FMT | |||
| SOD1G93A mice | L-carnitine | [202] | |
| SOD1G93A mice | TMAO | Neuroprotection | [203] |
| ALS patients | Acetyl-L-carnitine | [204] |
Note:
Of note, Burberry et al. [201] found that inflammation and immune responses were reduced by treating the C9orf72-/- mice with broad-spectrum antibiotics or faecal microbiota transplantation (FMT), suggesting that microbiota modulation of peripheral inflammation may impact the disease course. Contrasting data come from investigation of GM in ALS patients, mainly due to the small size, variable methodologies, different dietary regimen and disease stage [205]. However, some research groups detected reductions in butyrate-producing bacteria and a higher Firmicutes-to-Bacteroidetes ratio, both features linked to shorter survival [93, 206, 207, 208]. Of note, differently from what was observed for atherosclerosis and CVD, L-carnitine and TMAO levels seem to possess a protective role. Indeed, Kira et al. [202] demonstrated that oral treatment of L-carnitine delay the ALS onset and progression and extended the life span of SOD1G93A transgenic mice. Coherent with these data, a lipophilic derivate of TMAO proved to be efficacious in the neuroprotection of ALS mice as well as in reducing the formation of misfolded proteins in an in vitro experiment [203]. However, the antibiotics’ use may be associated with a higher risk of developing ALS, suggesting that GM dysbiosis could be a potential ALS cause [209]. A clinical study found that the concentrations of TMAO and its precursors in ALS patients and their spouse were significantly different compared to healthy controls, suggesting that the changes in GM composition occurred before disease onset [210]. In addition, a negative relationship between the TMAO levels and the involvement of upper motor neuron was observed [210]. In line with these data, a randomized placebo-controlled trial demonstrated that the treatment of ALS patients with acetyl-L-carnitine improved the symptoms of the disease in a safe and well tolerated manner (Table 2) [204].
AD is a complex neurodegenerative pathology and the primary cause of dementia in the aging population [211]. Dementia is the second leading cause of “years lived with disability” among people over age 60, heavily incrementing the burden on health care and social system [212, 213]. It is characterized by a progressive decline in memory, language, spatial orientation, cognitive function, and daily self-care. There are two the pathological AD hallmarks: (1) abnormal extracellular deposition of amyloid plaques (manly constituted by amyloid beta-42 peptide); (2) neurofibrillary tangles (NFTs) aggregation within neurons [214, 215]. The accumulation of misfolded proteins in the brain triggers oxidative stress and neuroinflammation, leading to impairment of synaptic transmission, neuronal death, and brain atrophy [216, 217]. The unresolved neuroinflammation activate microglia and astrocytes and enhance T cell infiltration and the secretion of pro-inflammatory mediators, such as nitric oxide, TNF-alpha, IL-1beta, and IL-6, without a compensatory rise in the levels of SPMs or their receptors [218, 219, 220]. Remarkably, low DHA levels have been found in brain and serum of AD patients [221] and, more notably, it has been demonstrated that in the brain DHA has a dual function, being a structural component of the synapses and a signalling molecule [222]. Both modifiable (i.e., diet, lifestyle) and non-modifiable (such as, age, sex, and genetic background) risk factors have been implicated in the AD etiology. However, the Global Burden of Disease Study stated that evidence supports a causal link between high body mass index, high fasting plasma glucose, smoking, high intake of refined sugar and AD development [211]. In addition, studies have shown that diet and specific nutrients may impact the production and/or aggregation of amyloid proteins by altering the GM composition [223]. Therefore, GM has been recognized as a seminal factor for the maintenance of host CNS health. Experimental studies performed in AD-like mouse models, such as, APP/PS1, 5XFAD and SAMP8 mice, demonstrated that GM composition was significantly different from that of wild-type animals [224, 225, 226]. Moreover, Cheng et al. [226] revealed gut microbiome and metabolites interactions in APP/PS1 mice. Human studies have reported alterations in GM composition in AD patients compared to controls, with a lower abundance of SCFA-producing species in AD individuals [227]. However, contradictory results were found among the types of specific microbiota modifications [228, 229, 230]. A recent clinical study evaluated the relationship between GM composition, measured with 16S rRNA sequencing, and features of AD, including cerebrospinal fluid (CSF) biomarkers and magnetic resonance imaging (MRI) measures of vascular burden and neurodegeneration [231]. The authors found that lower abundance of SCFAs-producing bacteria was associated with higher odds of AD pathology. Moreover, the analyses performed allowed the researchers to differentiate between predictors for amyloid and p-tau status: microbial strain from the Eubacterium and Ruminococcus genera were the highest ranked predictors for amyloid status, whereas different Lachnoclostridium spp. were the highest predictors for p-tau status [231]. As already reported, SCFAs may exert different beneficial activity, proving a putative target for treatment. Indeed, the treatment of AD-like mice with sodium butyrate proved efficacious in lowering the signs of AD pathology [232]. Coherent with these data, FMT from wild-type mice to AD-mouse models, such as APP/PS1 and ADLPAPT mice, caused an improvement of cognitive deficits and a reduction of the brain deposition of amyloid beta together with a recovering of GM homeostasis and an increase of SCFAs levels [233, 234]. On the other hand, colonization of Tg2576 animals with Bacteroides increased amyloid depositions, hypothesizing an interplay between GM and AD pathology (Table 3, Ref. [233, 234, 235, 236]) [234].
| Model | Intervention | Effects | References |
| APP/PS1 mice | FMT | [233] | |
| ADLPAPT mice | FMT | [234] | |
| SAMP8 mice | TMAO | [235] | |
| APP/PS1 mice | DMB | [236] | |
Note:
In addition, TMAO has been associated with cognitive decline in AD and neurodegenerative changes in p-tau status [237, 238, 239]. Specifically, TMAO has been shown to impact on the conformational changes in the C-terminal segment of tau protein, leading to aggregation/fibre formation, eventually promoting tau-dependent microtubule assembly [240, 241, 242]. Furthermore, it has been demonstrated that TMAO can cause brain aging, neural degeneration, and cognitive impairment. In detail, in comparison with control animals, TMAO-treated SAMP8 mice were characterized by (i) significant reduction of spatial working memory and cognitive performance; (ii) greater neuronal senescence; (iii) accelerated amyloid plaque formation [235]. An integrated computational approach analysis highlighted a role of different microbial metabolites in AD etiology. The top ranked metabolite was TMAO [237]. Finally, a genome-wide study found that 927 single-nucleotide polymorphisms were correlated with AD as well as type 2 diabetes, suggesting the presence of common pathogenic mechanisms [243]. Of note, by treating APP/PS1 mice with DMB in the drinking water caused a decrease of TMAO plasma concentrations, amyloid beta-42 peptide and beta-secretase, along with an improvement of the cognitive decline, long-term potentiation and pathological deterioration (Table 3) [236].
It has been demonstrated the presence of the amyloid precursor protein (APP, the molecule from which amyloid beta-42 peptide is derived) in the ENS and its relevant role in gastrointestinal motility, immunity and secretion [244, 245]. Moreover, amyloid-beta staining was detected in gut tissue harvested from AD patients and in the ENS of healthy elderly people [246]. Transgenic mice expressing mutant forms of APP displayed the accumulation of amyloid-beta in enteric neurons that lead to a reduction of neuronal tissue and increased vulnerability to inflammation and functional gastrointestinal deficits, along with a modification in GM composition [247, 248, 249]. Therefore, by these data we can speculate that an altered GM could locally trigger amyloid-beta accumulation. However, the lack of human studies limited the validation of the phenotypes observed in the animal models. Given the widespread AD incidence and its dramatic financial burden, clinicians should start to include gastrointestinal investigations into the new protocols to clarify the link between AD and ENS pathophysiology.
As described earlier, there are plenty of papers demonstrating the interplay among GM dysbiosis and CVD or NDDs. The common denominators are chronic and sterile low-grade inflammation, characterized by elevated levels of cytokines and chemokines in the systemic circulation and/or in the brain, and modifications in the levels of GM-metabolites, such as SCFAs, TMAO, bile acids, etc. Of note, obesity and insulin resistance have been defined as CNS- and ENS-related disorders, respectively [250]. Indeed, hypothalamic inflammation [250], characterized by a high number and reactivity of microglial cells, and ENS neuropathy [251], has been detected after high-fat feeding. More notably, these signs of neuroinflammation are already present when the body weight starts to increase [250]. In addition, it has been demonstrated that high fat diet-induced obesity triggers interruption of the hypothalamic-pituitary-adrenal axis, eventually altering the release of different hormones from the adrenal cortex [252, 253]. For example, increased adiposity has been associated with an overactivity of the hypothalamic-pituitary-adrenal axis, leading to elevated adrenocorticotropic hormone and cortisol release [254, 255, 256]. Conversely, obese subjects have blunted epinephrine release and reduced adrenergic-stimulated lipolysis in visceral fat depots [257, 258, 259]. The mechanisms supporting dysregulated adrenergic signalling with high fat feeding are still matter of investigations. However, the high fat diet-induced loss in catecholamine signalling in the adipose tissue could derive from (1) adrenal medulla fatigue and decreased epinephrine release [260] and/or (2) direct inhibition of epinephrine release [64] and/or (3) enrichment in the adipose tissue of sympathetic neuron-associated macrophages characterized by the expression of the noradrenaline transporter SLC6A2 and the catecholamine catabolizing enzyme monoamine oxidase-A (MAO-A) [261].
As has been well demonstrated, CVD and NDDs share different risk factors [17, 262]. Mid-life obesity (body mass index
The forgotten player appears to be the GM since no studies have been conducted to demonstrate the causal relationship between GM dysbiosis and the pathogenesis of both CVD and NDDs, yet. The reasons for this gap are uncountable, ranging from the lack of the appropriate animal models to use to the patients’ populations that should be selected, and the influence of confounding factors, such as lifestyle, medication use and genetic predisposition. Furthermore, future research should focus on the identification of causal mechanisms through which dysfunction in one organ contributes to impairment in another. Population studies to date on the GM genetics are still underpowered to capture the limited genetic component that has been estimated for GM characteristics. Indeed, these analyses unveil the need for standardized protocols for the harvesting and storing of the sample along with data analyses of large sample sizes. A better understanding of the complex mutual relationship between GM and host metabolism will demand a widening of current methodologies and the ability to include measures of circulating biomarkers. The need for looking for NDD-biomarkers measurable in the blood is another challenge that should be taken up. Current and future microbiome-wide association studies, namely expanded microbiome-genetic studies, will generate many hypotheses that will need to be systematically screened by MR or another standardized tool for causal inference that can guide and complement more direct experimental approaches, namely FMT and animal models. Nowadays, human brain and heart&intestine organoids can be generated in vitro and connected using microfluid systems or co-culture methodologies to analyse inter-organ communications. In addition, advancements in microbiome research have led to the development of diverse technologies for microbial identification and functional analysis [273, 274]. Consequently, the humongous amount of gut metagenomic data available now allows artificial intelligence (AI) to be applied for the identification of many uncharacterized microbial genomes and proteins. Predictive models on how microbes interact within the gastrointestinal tract, and their impact on the metabolism and immunity of the host may guide precision medicine, leading to the formulation of personalized therapeutics [275]. Finally, new data highlighted that aged women display a higher risk of developing both CVD and NDDs compared to age-matched males [276]. Hypothetical gender or sex specific studies are also urgent to fully understand and improve the management of these widespread illnesses.
Growing information from pre-clinical and clinical studies suggest that both chronic and sterile low-grade inflammation and GM modifications are crucial in controlling the health of the cardiovascular and nervous systems. In addition, it has been demonstrated that CVD and NDDs share different common risk factors able to cause endothelial and arterial dysfunction, increased myocardial thickness and BBB permeability and compromised neurovascular coupling, eventually raising the susceptibility to the development of these illnesses. However, contrasting results about the causal relationship between SCFAs and/or TMAO and the CVD incidence are still present, along with contradictory data on the role played by TMAO in AD vs ALS. The lack of a clear picture of the mechanisms regulating these correlations could, in part, explain these inconsistencies, and prompt us to plan further well-designed and tailored studies. Moreover, it is still a matter of debate whether GM dysbiosis is a cause or a consequence of disease development/progression. Understanding the mechanisms that regulate the complexity of the gut-heart-brain interplay is mandatory to develop new integrated strategies for predicting, preventing and managing CVD and NDDs. Furthermore, often the clinical translation of the experimental results represents a challenge. Indeed, the different outcomes are likely the effects of environmental (i.e., diet-microbiome interactions), genetic (i.e., individual microbiome variability) and methodological (i.e., reproducibility across populations) factors. However, no studies have been published up-to-now to define the causal connection between GM dysbiosis and the pathogenesis of these widespread pathologies. Further investigations and widening of current methodologies are required together with new standardized protocols ensuring the correctness of the results obtained. Finally, a multidisciplinary approach, including advanced biological systems and AI tools, may help to define the “ideal longitudinal and multi-omics trial” aiming at establishing the causal link between GM and both CVD and NDDs. In parallel, the application of AI and machine learning to multi-omics data enable deeper insights into exposure-microbiome-inflammation interactions as well as mechanistic GM understanding, eventually leading to personalized and precision therapies. However, data standardization and clinical applicability along with data privacy, patient consent, and algorithmic bias are the current major concerns raised that need to be addressed for the approval and clinical adoption of AI in healthcare [275].
Not applicable.
AA and CP designed, reviewed and edited the paper. CP wrote the paper and prepared figures and tables. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We deeply thank Prof. Robert S. Kiss for improving the English style of our manuscript.
This research was funded by Il bando si inserisce nelle iniziative finanziate dall’Unione Europea – Next Generation EU - CUP B55F21007810001, and by Project Tuscany Health Ecosystem (THE), CUP: B83C22003920001.
The authors declare no conflict of interest. Given his role as the Editorial Board member and Guest Editor, Amedeo Amedei had no involvement in the peer-review of this article and has no access to information regarding its peer review. As a Guest Editor, Cinzia Parolini also had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Wei-Lin Jin.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.