









 , Xiaoqiang Li 3, Weijie Wang 2, Song-Bai Liu 1,2,*
, Xiaoqiang Li 3, Weijie Wang 2, Song-Bai Liu 1,2,* , Xiaohua Li 1,4,*
, Xiaohua Li 1,4,*1 Jiangsu Province Engineering Research Center of Molecular Target Therapy and Companion Diagnostics in Oncology, Suzhou Vocational Health College, 215009 Suzhou, Jiangsu, China
2 College of Life Science, North China University of Science and Technology, 063210 Tangshan, Hebei, China
3 Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, 215163 Suzhou, Jiangsu, China
4 Thyroid and Breast Surgery, Wuzhong People’s Hospital of Suzhou City, 215128 Suzhou, Jiangsu, China
Abstract
Breast cancer, characterized by distinctive epidemiological patterns and substantial heterogeneity, continues to be among the leading causes of cancer-related mortality in women. As breast tumors progressively acquire resistance to doxorubicin (DOX), DNA damage repair (DDR) pathways are recognized as key determinants of both DOX efficacy and the onset of resistance. Targeting DDR mechanisms in breast cancer patients with specific repair deficiencies offers the potential for personalized therapeutic approaches. This review first discusses the pivotal roles of five major DNA repair pathways (homologous recombination, nonhomologous end-joining, base excision repair, nucleotide excision repair, and mismatch repair) in the development of DOX resistance. This review aims to establish a theoretical framework and reference for future studies on DDR mechanisms in DOX-resistant breast cancer to advance intervention strategies for resistant breast tumors and to promote further research in this area.
Keywords
- breast neoplasms
- drug resistance
- neoplasm
- doxorubicin
- DNA repair
- molecular targeted therapy
Breast cancer is characterized by distinctive epidemiological patterns and marked heterogeneity and remains one of the leading causes of cancer-related mortality in women [1]. Epidemiological data indicate that the annual rate of increase in the incidence of breast cancer among women younger than 50 years (1.4%) is significantly greater than that among women aged 50 years and older (0.7%) and that the disparity in growth rates is even more pronounced in white women [2]. The International Agency for Research on Cancer (IARC) projects that by 2040, the global annual number of newly diagnosed breast cancer cases will exceed 3 million [3]. In recent years, research has increasingly focused on the combination of conventional doxorubicin (DOX) therapy with molecularly targeted interventions against DNA repair or the optimization of treatment sequences. Advances in the “targeted and precise” treatment of breast cancer have significantly reshaped clinical practice. For patients who harbour BRCA1/2 (BRCA1 DNA repair associated/ BRCA2 DNA repair associated) mutations and those who exhibit homologous recombination deficiency (HRD), Poly (ADP‒ribose) polymerase (PARP) inhibitors have emerged as a key class of targeted therapy, as they have demonstrated competitive efficacy in both advanced/metastatic settings, and in some cases, early-stage disease [4]. In p53-deficient breast cancer cell lines, PARP inhibition enhances DOX-induced cytotoxicity, which suggests that impairment in DNA repair processes may underlie increased chemosensitivity [5, 6]. Antibody–drug conjugates (ADCs), such as Enhertu, Trodelvy, and the recently highlighted Datroway, selectively deliver cytotoxic payloads to cancer cells, expanding the population of “targetable” patients and offering novel alternative or combinatorial strategies to overcome DOX resistance [7]. However, evidence remains limited regarding whether DNA damage repair (DDR) pathway inhibitors can reverse DOX resistance driven by enhanced DNA repair mechanisms. Endocrine therapy combined with CDK4/6 inhibitors, as well as immune checkpoint blockade in specific subtypes, has further reshaped therapeutic options and renders the timing, combination, or substitution of DOX increasingly dependent on tumor molecular profiling and individualized clinical decision-making [8].
DOX is an anthracycline antibiotic originally isolated from a mutant strain of Streptomyces peucetius subsp. caesius (ATCC 27952) [9]. The drug and its derivatives were patented by Farmitalia Carlo Erba SpA in 1971 [10, 11]. According to the ClinicalTrials.gov registry (https://clinicaltrials.gov), as of February 2025, approximately 1112 clinical trials involving DOX have been registered worldwide. As a prototypical anthracycline, DOX remains a cornerstone of adjuvant, neoadjuvant, and metastatic breast cancer therapy, as this drug markedly improves both overall survival and disease-free survival in long-term clinical practice. Although rapid advances in molecular stratification and targeted therapies have rendered DOX no longer the default first-line option in certain breast cancer subtypes, it continues to play a central role in curable early-stage disease, locally advanced cases, and patients who lack effective targeted treatments [12, 13, 14].
The antitumor effects of DOX in breast cancer primarily involve its
intercalation into double-stranded DNA, inhibition of topoisomerase II (Topo II, a type II
DNA topoisomerase that modulates DNA topology by generating transient
double-strand breaks (DSBs)), and the consequent induction of DNA damage that
triggers tumor cell apoptosis [15]. Despite its considerable clinical efficacy,
the use of DOX is limited by intrinsic and acquired resistance. Although
triple-negative breast cancer (TNBC) is relatively more sensitive to DOX than are
other subtypes, treatment failure due to resistance still affects approximately
30–50% of patients [16]. Aberrant upregulation of DDR pathways is a central
mechanism of DOX resistance, as tumor cells repair DOX-induced DNA lesions
through increased DNA repair activity, which promotes the development of drug
resistance [17, 18]. The upregulation of drug efflux pumps, particularly
P-glycoprotein (P-gp/ABCB1), ABCG2, and ABCC1, reduces the intracellular
accumulation of DOX and represents one of the central mechanisms underlying
breast cancer chemoresistance [19]. Tumor cells further evade immune clearance
and apoptosis by enhancing the DDR to counteract Topo II inhibition and
ROS–induced DNA lesions. Additional factors that attenuate DOX cytotoxicity
include the following: decreased expression or functional alterations of Topo II,
activation of antiapoptotic pathways such as those mediated by BCL2 apoptosis
regulator (BCL-2), Nuclear Factor kappa-B (NF-
This review describes how DNA repair pathways, homologous recombination (HR), nonhomologous end joining (NHEJ), base excision repair (BER), nucleotide excision repair (NER), and mismatch repair (MMR) contribute to resistance to DOX in breast cancer and critically examines key genes, signalling networks, and their clinical implications [21, 22, 23]. This study also provides a detailed evaluation of strategies to overcome resistance to DNA damage induced by DOX in breast cancer cells, combined targeted therapies, noncoding RNA (ncRNA)-mediated sensitization approaches, and interventions with natural bioactive compounds (NPs), with an aim to provide a theoretical foundation and practical reference for the development of antiresistance interventions and to advance research in this field [24, 25].
The use of VOSviewer (bibliometric mapping software, version 1.6.20, Centre for Science and Technology Studies, Leiden University, Leiden, the Netherlands) [26] to analyse relevant literature from the past five years revealed that research on DOX resistance has evolved from early studies focused on single-cell resistance mechanisms to a broader spectrum that includes nanocarrier-based delivery and ncRNA–mediated regulation, clinical cohort and radiomic (quantitative imaging feature extraction) studies, and integrative multiomics artificial intelligence-based approaches. In 2018–2019, keywords such as “cell viability” and “knockdown” were predominant, reflecting mechanistic investigations into how DOX, through the inhibition of Topo II, induces tumor cell apoptosis, cell cycle arrest, and activation of the DDR and repair processes. From 2020 to 2021, terms such as “nanoparticle”, “liposome”, and microRNA (miRNA) and long noncoding RNA (lncRNA) became markedly more prominent, which indicates that nanoparticle-based delivery strategies and RNA-mediated regulation of the DDR, encompassing lesion detection, signalling, and repair, were needed. Multiple miRNAs and lncRNAs have been reported to modulate resistance by targeting DNA repair genes. Concurrently, the frequent occurrence of “retrospective study” and “disease-free survival” signals the growing application of retrospective cohort analyses for prognostic evaluation. In 2022–2023, an increasing trend was observed towards the integration of radiomics and deep learning with genomics, transcriptomics, metabolomics, and epigenomics as well as combinations with artificial intelligence methods, to enable earlier detection of resistance, risk stratification, and extraction of DDR-related imaging features. In conclusion, literature within the past five years shows a shift from predominantly in vitro or in vivo mechanistic studies towards a more balanced emphasis on nanomedicine, ncRNA regulation, combined imaging/AI and multiomics approaches, and clinical cohort research (Fig. 1A,B).
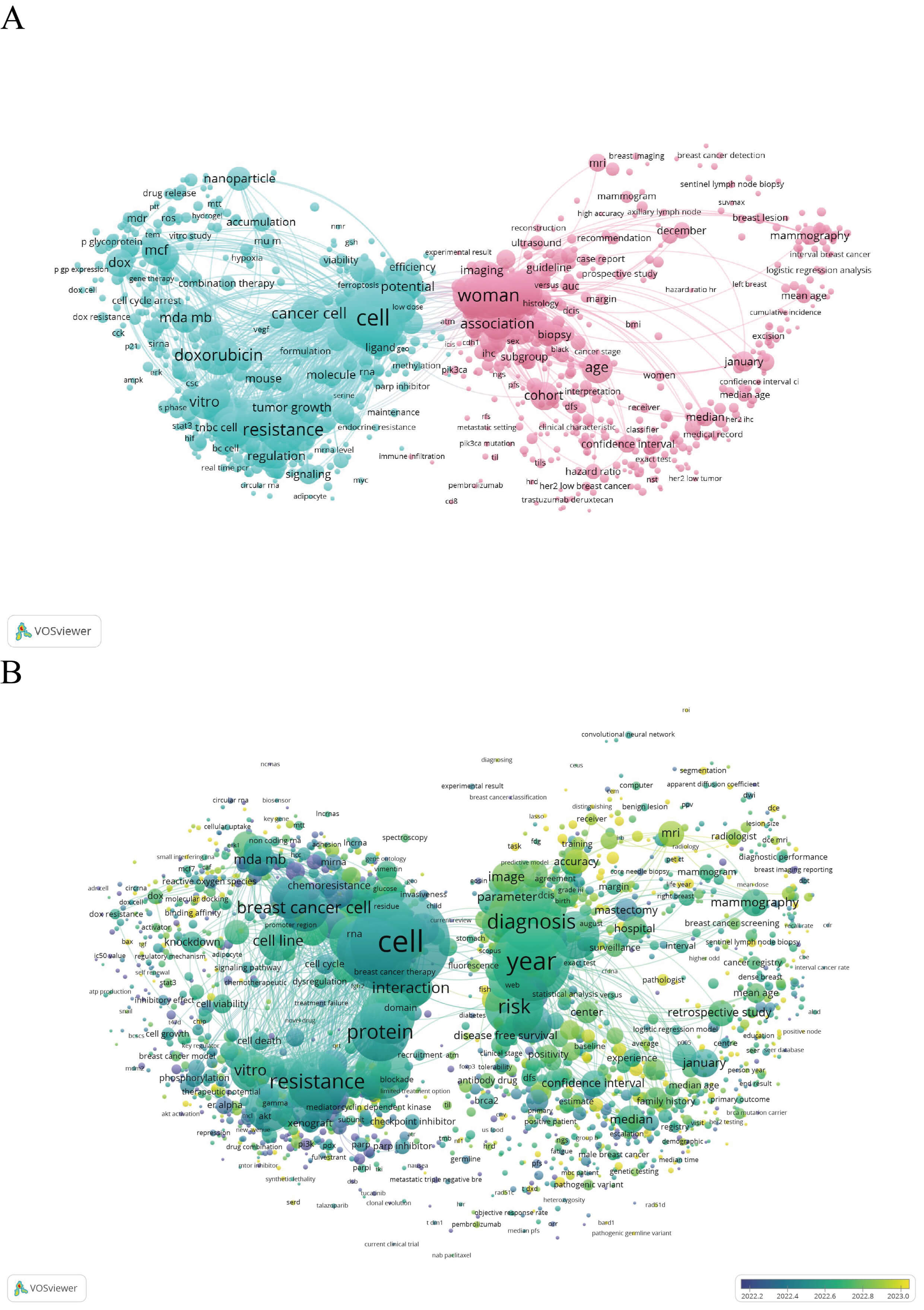 Fig. 1.
Fig. 1.
Bibliometric keyword co-occurrence analyses of recent literature on DOX resistance in breast cancer. The literature published during the most recent five-year period was retrieved from the Web of Science database and exported in “Full Record” plain-text (.txt) format. Visual maps were generated in VOSviewer (version 1.6.20) using a minimum keyword occurrence threshold of 10 and were clustered according to node parameters. (A) Network map produced for the search terms “DOX resistance” AND “breast cancer” (n = 7679 records). (B) Network map produced for the search terms “DOX resistance” AND “breast cancer” AND “DDR” (n = 6556 records). In both panels, node size corresponds to keyword frequency, connecting lines indicate keyword co-occurrence relationships (the edge weight reflects co-occurrence strength), and different colours represent thematic clusters identified algorithmically by VOSviewer. No restrictions were applied to the document type, and no additional screening, data processing, or visualization software was used. DOX, doxorubicin; DDR, DNA damage repair. The figure was created using VOSviewer (version 1.6.20, Nees Jan van Eck and Ludo Waltman (Centre for Science and Technology Studies (CWTS) at Leiden University), Leiden, South Holland, The Netherlands). Other figures were created using Adobe Illustrator 2022 (Adobe Inc., San Jose, CA, USA).
The core mechanisms underlying DOX resistance in breast cancer have been elucidated primarily at the cellular level in models using MDA-MB-231 and MCF-7 cells. These mechanisms primarily involve antiapoptotic signalling and the transcriptional and posttranscriptional regulation of genes and proteins, which enhance the proliferative capacity of resistant cells. To overcome resistance, current research has focused on nanodelivery platforms and combination therapeutics. Nanoparticle-based drug delivery systems can improve the pharmacokinetic profile of DOX, increase tumor selectivity, and reduce systemic toxicity and are therefore considered among the most promising approaches for clinical translation. lncRNAs have emerged as key regulators of resistance-associated gene expression, whereas extracellular vesicles serve as carriers of lncRNAs or miRNAs that enable intercellular transfer of resistance phenotypes. With the rapid accumulation of multiomics datasets, systems biology and machine learning approaches are accelerating the identification of resistance-related biomarkers and regulatory networks. This offers new strategies for clinical resistance stratification, prediction of the therapeutic response to DOX, and the development of precision treatment approaches.
DSBs are among the most deleterious forms of DNA damage and are principally repaired via two pathways: HR and NHEJ. The expression levels of key HR factors are significantly associated with DOX resistance in breast cancer cells [27]. Dysregulation of DSB repair commonly manifests as an imbalance in these pathways: aberrant expression or functional impairment of critical genes not only compromises the high-fidelity repair mediated by HR but also drives hyperactivation of NHEJ, which promotes DOX resistance and supports breast cancer cell proliferation.
The HR pathway comprises three principal subpathways—DSB repair,
synthesis-dependent strand annealing (SDSA), and break-induced replication (BIR)
[28]. HR is a high-fidelity, template-dependent mechanism that uses sister
chromatids to accurately repair DSBs [29]. Key HR proteins include RAD51
recombinase (RAD51), RAD52 DNA repair protein (RAD52), DNA-dependent ATPase RAD54
(RAD54), replication protein A (RPA), RAD51 paralog B (RAD51B), RAD51 paralog C
(RAD51C), RAD51 paralog D (RAD51D), X-ray repair cross complementing 2 (XRCC2),
and X-ray repair cross complementing 3 (XRCC3), and the breast cancer
susceptibility proteins BRCA1 and BRCA2. The overexpression of RAD51 and BRCA1
enhances HR-mediated repair, promotes breast cancer cell survival, and alters
cell cycle control, which reduces DOX-induced cell death and contributes to
chemoresistance [30, 31]. For example, Shan Wang et al. [32] reported
that E2F transcription factor 8 (E2F8) upregulates RAD51 in breast cancer cells,
which in turn promotes HR activity and contributes to DOX resistance. epithelial
membrane protein 3 (EMP3) functions as a tumor inhibitor in breast cancer by
inhibiting DNA replication, impairing HR, reducing tumor cell stem-like
properties, and inhibiting oncogenic signalling [33]. For example, Kailing Zhou
et al. [34] demonstrated that EMP3 downregulation is associated with
decreased expression of the cell cycle regulator p21, upregulation of
S–phase–associated factors, reduced phosphorylated histone H2A.X
(
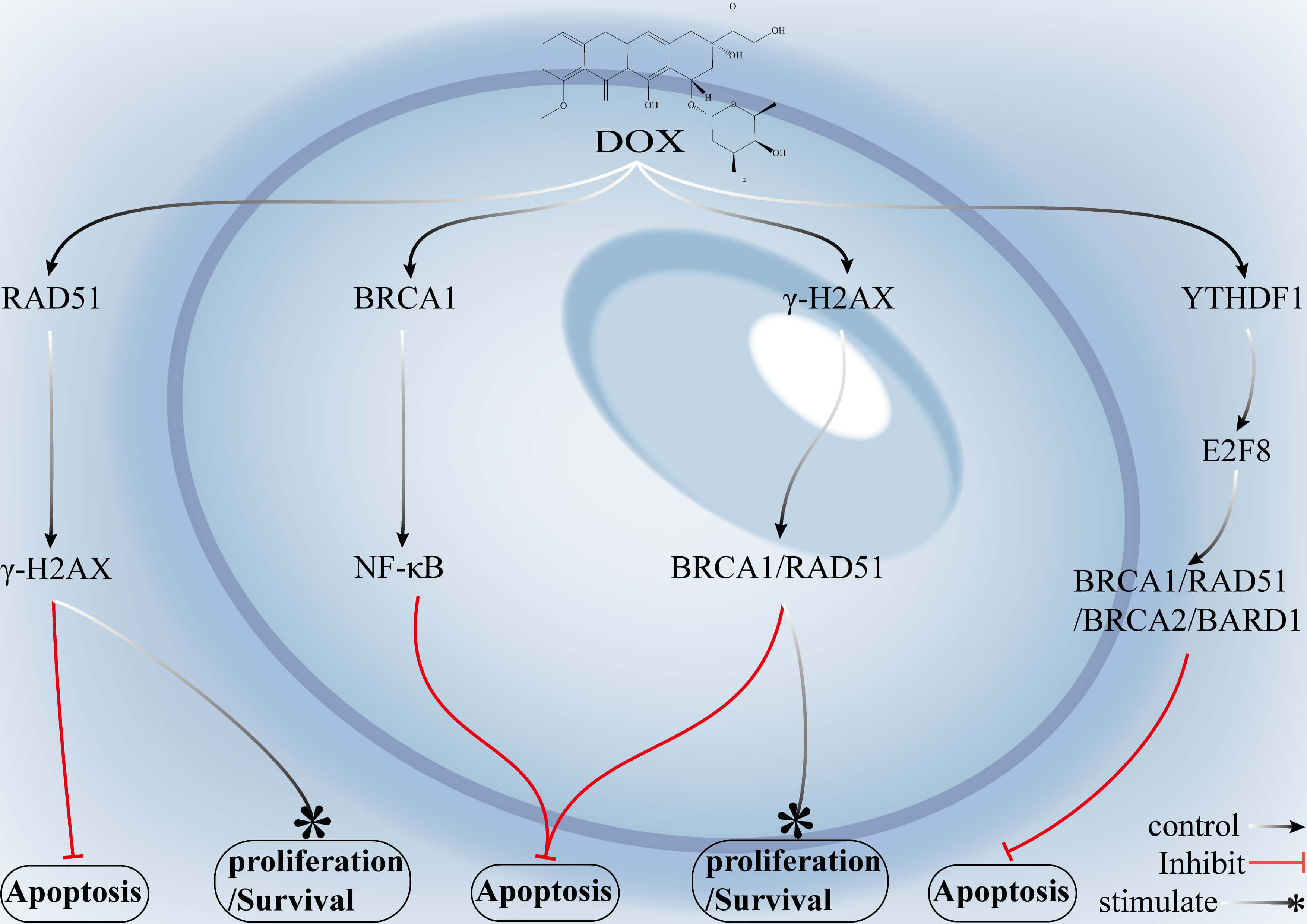 Fig. 2.
Fig. 2.
Mechanisms by which key genes in the HR pathway and associated
signalling networks mediate DOX resistance in breast cancer. DOX induces DSBs
primarily by inhibition of topoisomerase II (Topo II), which subsequently triggers the
phosphorylation of
Overexpression of RAD51 and BRCA1/2 enhances the accuracy of DSB repair
and reduces DOX-induced cell death. Elevated levels of E2F8 and CENPL similarly
promote resistance by upregulating RAD51 expression or increasing DNA repair
capacity. Downregulation of EMP3 leads to decreased p21 expression, increased
S-phase–associated factor expression, reduced
The inhibition of RAD51 prevents its binding to single-stranded DNA (ssDNA) and blocks nucleofilament formation, disrupting a central step of HR and preventing the repair of DSBs. Suppression of the assembly of HR complexes involving BRCA1, BRCA2, or PALB2 diminishes template-dependent repair capacity and drives tumor cells into a state resembling “functional HR deficiency”. Likewise, inhibition of YTHDF1 reduces HR repair capacity by preventing its regulatory effect on E2F8, which in turn governs the expression of BRCA1, BRCA2, and RAD51. Activation of the AkT–mTOR signalling axis enhances DNA repair and cell survival, and blockade of this pathway using AkT or mTOR inhibitors indirectly downregulates HR-related gene expression, which restores the cytotoxic effects of doxorubicin.
In canonical NHEJ, the Ku heterodimer (Ku70/Ku80) serves as the core DNA end
recognition factor that rapidly binds to DNA double-strand break ends and
recruits downstream repair proteins to initiate end joining [37, 38]. The
catalytic subunit DNA-dependent protein kinase catalytic subunit (DNA-PKcs)
associates with the Ku complex and phosphorylates downstream effectors to promote
DSB repair. Reduced Ku80 expression impairs NHEJ activity and attenuates the DNA
repair capacity of otherwise resistant tumor cells, sensitizing them to DOX.
Conversely, high Ku70/Ku80 expression facilitates rapid end stabilization, the
recruitment of downstream factors, and efficient NHEJ, which enables tumor cells
to evade apoptosis or necrosis [39, 40]. For example, Min-Gu Lee et al.
[41] reported that the upregulation of MDR1 enhances DOX efflux and decreases
intracellular drug accumulation, contributing to chemoresistance. Cotreatment
with DOX and the natural product arctigenin (ATG) substantially increases
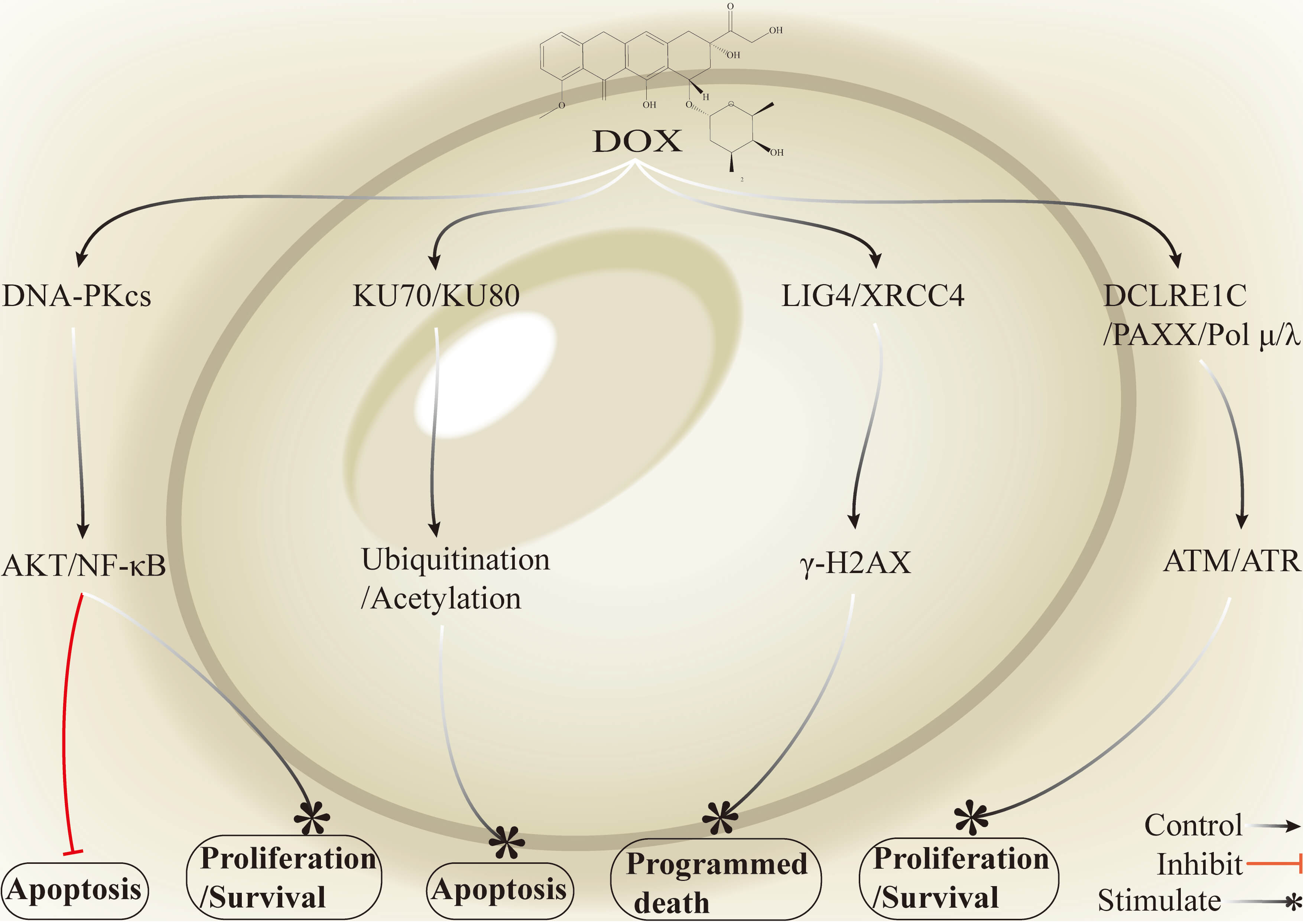 Fig. 3.
Fig. 3.
Mechanisms by which key genes in the NHEJ pathway and associated
signalling networks mediate DOX resistance in breast cancer. Activation of
DNA-PKcs promotes the phosphorylation and activation of AKT and interactions with
the NF-
High expression of Ku70/Ku80 and DNA-PK enhances end-break stabilization and repair efficiency, and thus they constitute key molecular nodes that promote DOX resistance. Suppression or functional impairment of these factors markedly decreases NHEJ efficiency and increases cellular sensitivity to DOX. MDR1 further reinforces the resistant phenotype by increasing DOX efflux and reducing drug accumulation in the nucleus. Treatment with ATG or the DNA-PK inhibitor peposertib downregulates the expression of repair proteins such as Ku80 and RAD51, which blocks NHEJ repair, promotes the accumulation of DOX-induced DNA damage, and subsequently activates the ATM–p53–p21 axis to trigger cell cycle arrest and apoptosis. The inhibition of the Ku70/Ku80–DNA-PK–mediated NHEJ pathway, together with overcoming MDR1-driven drug efflux, has been consistently shown in extensive preclinical and early clinical studies to potentiate DOX cytotoxicity and reverse resistance. Peposertib is already under evaluation in phase I and combination trials. However, challenges remain, including toxicity to normal tissues due to a narrow therapeutic window and compensatory and heterogeneous repair pathway activation [45, 46]. Improvements in safety will require biomarker-guided patient stratification, short-pulse or precisely timed combination dosing, optimized drug administration and delivery strategies, and the development of tumor-targeted delivery systems or low-toxicity efflux inhibitors.
The BER pathway is a major DNA repair mechanism responsible for the removal of oxidative, alkylation, and base loss (depurination/depyrimidination) lesions that do not typically grossly distort the DNA helix. BER is generally initiated by a family of DNA glycosylases that excise damaged bases and generate apurinic/apyrimidinic (AP) sites [47, 48, 49]. Apurinic/apyrimidinic endonuclease 1 (APE1) contains two functional domains: a C-terminal domain that mediates canonical BER activity and an N-terminal domain that performs redox (reduction–oxidation) regulatory functions [50]. APE1 possesses intrinsic redox enzyme activity. This activity increases under hypoxic conditions and contributes to tumor cell resistance to DOX. The inhibition of the redox function of APE1 can markedly increase the cytotoxic efficacy of DOX [51, 52]. For example, Ísis Salviano Soares de Amorim et al. [52] reported that under hypoxia, treatment with the APE1 inhibitor APX2009 in combination with DOX significantly increases apoptosis and caspase-3/7 activity in MDA-MB-231 cells. This combination also promotes intracellular DOX accumulation. These findings further demonstrate that caspase-3 is the key mediator of the cytotoxic effects induced by this combination treatment.
The overexpression of flap endonuclease 1 (FEN1) has been reported to accelerate BER-mediated resolution of DOX-induced abasic sites and single-strand breaks (SSBs); this reduces DNA damage accumulation and allows breast tumor cells to clear lethal lesions rapidly and evade DOX cytotoxicity, which ultimately promotes chemoresistance [53, 54, 55]. For example, Xiao Lu et al. [56] reported that the transcription factor Yin Yang 1 (YY1) normally activates miR-140 expression; in DOX-resistant breast cancer cells, the regulation of miR-140 by YY1 is weakened, leading to the downregulation of miR-140 expression, the consequent upregulation of FEN1 expression, increased DNA repair efficiency, and the development of resistance.
Nei-like 2 (NEIL2), a DNA glycosylase central to the BER pathway, is upregulated in breast cancer stem cells and confers protection through two synergistic mechanisms: (1) direct excision of DOX-induced base lesions and SSBs and (2) mitigation of secondary DNA damage that arises from reactive oxygen species (ROS) and contributes to DOX resistance in breast cancer [57, 58, 59]. For example, Banerjee et al. [58] demonstrated that by inhibiting p300-mediated acetylation (p300 is a histone acetyltransferase), DOX induces NEIL2 expression at both the transcriptional and translational levels, enabling cells to withstand DNA damage and ROS-induced stress and acquire chemoresistance. Furthermore, studies have demonstrated that the survival and DOX resistance of breast cancer stem cells are enhanced through the regulation of redox homeostasis [58] (Fig. 4).
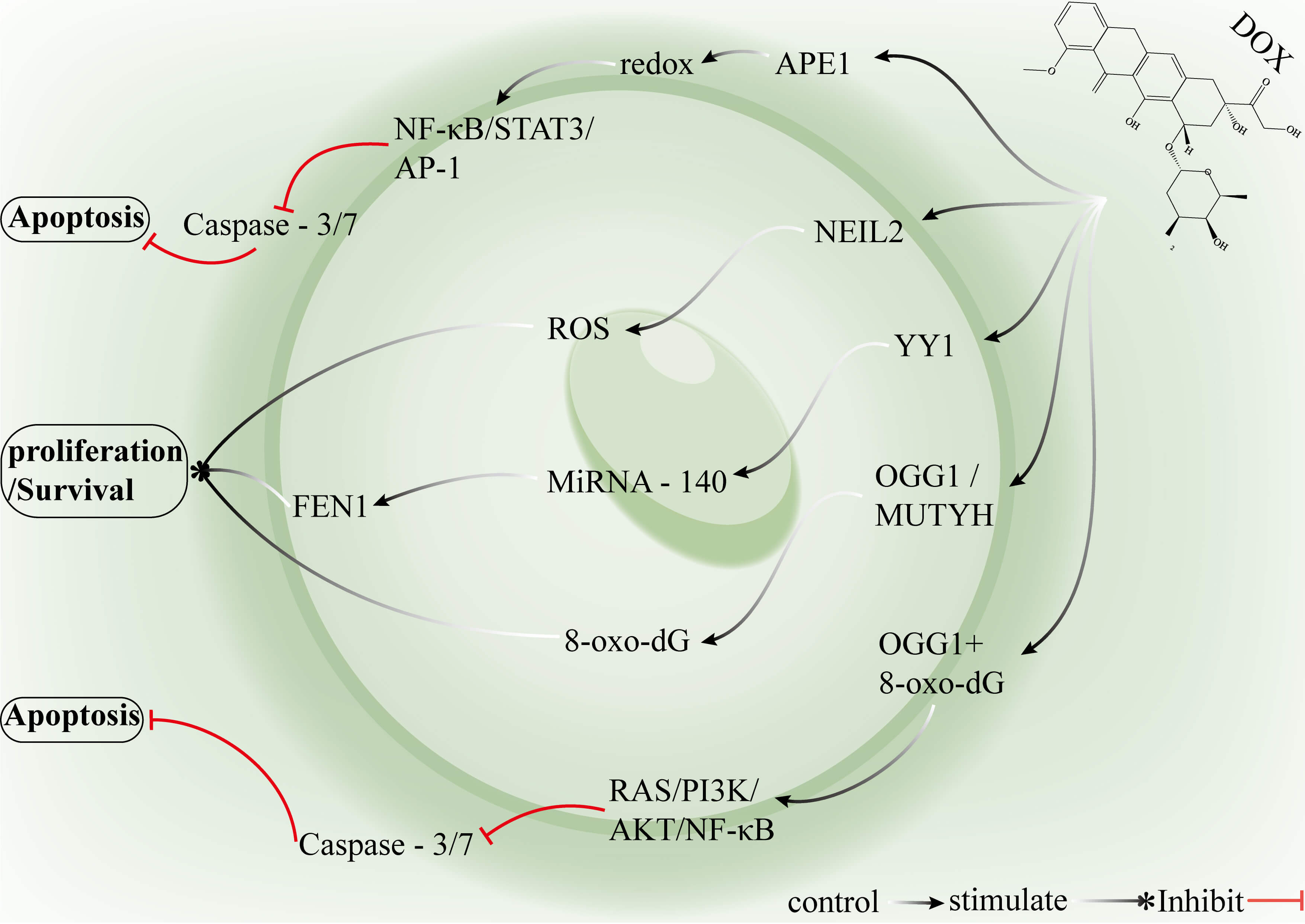 Fig. 4.
Fig. 4.
Mechanisms by which key genes in the BER pathway and associated
signalling networks mediate DOX resistance in breast cancer. The redox activity
of APE1 enhances the DNA-binding capacity of NF-
APE1 not only participates in the DNA BER pathway but also markedly upregulates redox activity in the hypoxic tumor microenvironment, where it can inhibit caspase-3/7–mediated apoptosis and reduce intracellular DOX accumulation, contributing to the development of a drug-resistant phenotype in breast cancer cells. FEN1 overexpression enhances BER-mediated repair of DOX-induced base lesions and strand breaks, decreases DNA damage accumulation and enables tumor cells to rapidly eliminate lethal lesions. NEIL2 is upregulated in breast cancer stem cells and promotes cell survival and drug resistance by removing DOX-induced DNA damage and suppressing ROS-mediated secondary damage cascades. Suppression of YY1 diminishes its inhibitory effect on miR-140, leading to FEN1 upregulation, whereas DOX-induced inhibition of p300-mediated acetylation increases NEIL2 expression, which further promotes resistance. Inhibition of the redox function of APE1 may restore caspase-3/7 activity and increase the effective intracellular concentration of DOX, resensitizing cells to the drug. The inhibition of FEN1 expression or the modulation of miR-140 expression may reduce BER efficiency and enhance drug cytotoxicity. The combination of BER-targeted inhibitors with DOX can selectively increase tumor sensitivity while minimizing the toxicity to single targets. Nanoparticle-based delivery systems or ADCs can specifically deliver these inhibitors to breast tumors or cancer stem cells. The measurement of APE1, FEN1, or NEIL2 expression or activity enables the identification of patients at high risk of resistance, which allows for personalized clinical treatment. Tumor heterogeneity results in variable target expression, the protective role of BER in normal cells may induce systemic toxicity, and resistance mechanisms often exhibit redundancy [46, 60]. Future strategies may require combined targeting of core BER enzymes alongside conventional chemotherapy to achieve higher selectivity and efficacy in DOX-based therapy.
Compared with BER, NER is a more complex, multistep pathway that removes bulky, helix-distorting adducts and certain crosslinks. NER comprises two subpathways, transcription-coupled NER (TCR-NER; repairs lesions on actively transcribed strands) and global-genome NER (GGR-NER; surveys and repairs lesions across the entire genome) [61, 62]. Excision repair cross-complementation 1 (ERCC1), a core NER excision factor, heterodimerizes with endonuclease XPF (3′-flap repair endonuclease Xpf) (ERCC4) to form an endonuclease complex that incises damaged oligonucleotide fragments. ERCC1 overexpression increases NER capacity in breast cancer cells and has been implicated in DOX resistance [63]. For example, Fu et al. [64] reported that the upregulation of ERCC1 promotes the acquisition of DOX resistance in breast cancer cells by accelerating the repair of drug-induced DNA lesions. Current in vitro models of DOX resistance, typically generated by stepwise dose escalation or selection with high drug concentrations, do not fully recapitulate clinical resistance because the durability of resistance and the proliferative capacity of cells during the posttreatment recovery phase are often not comprehensively evaluated [65, 66, 67]. Compared with control cells, breast cancer cells may achieve more efficient repair of DOX-induced lesions through the overexpression or activation of key NER genes (e.g., XPA, XPC, CSB/ERCC6, XPF/ERCC4, and ERCC1), which reduces DOX cytotoxicity and drives drug resistance [68]. For example, Busatto et al. [69] reported that the upregulation of NER factors (XPA, XPC, ERCC6/CSB, XPF) during DOX exposure enhances NER activity, enables the clearance of extensive DOX-induced damage, and attenuates drug toxicity. After drug withdrawal and prolonged culture, NER gene expression rebounds in parallel with proliferative recovery, culminating in a resistant phenotype.
BMP/OP-responsive gene (BORG) expression promotes the reactivation of
proliferation-associated genes in dormant breast lesions, is strongly induced by
DOX treatment, and enhances survival and resistance in TNBC cells [70, 71]. For
example, Gooding et al. [72] demonstrated that under DOX exposure or
metabolic stress, BORG forms a complex with the ssDNA–binding protein
replication protein A1 (RPA1). The BORG–RPA1 complex activates the
NF-
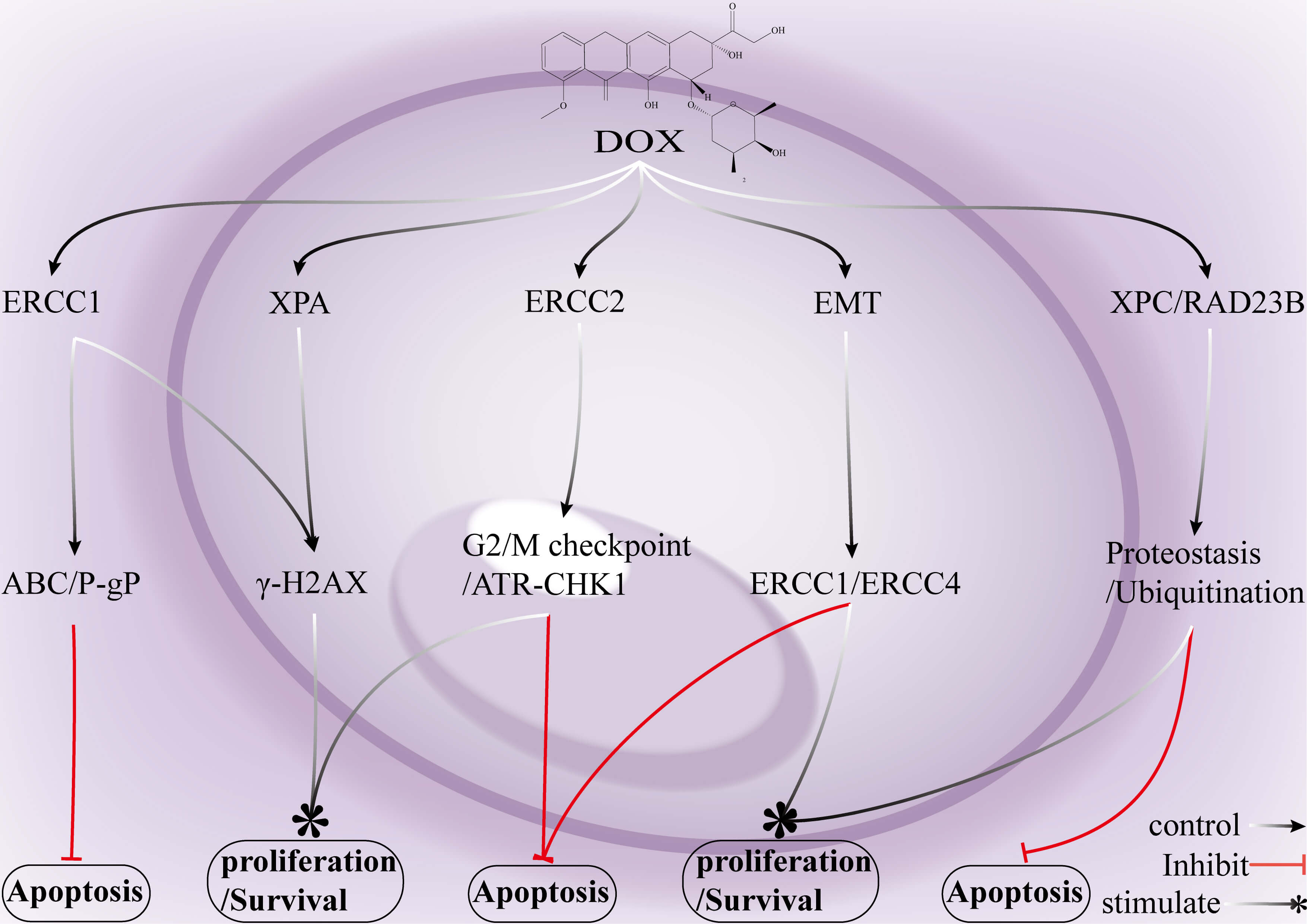 Fig. 5.
Fig. 5.
Mechanisms by which key genes in the NER pathway and associated signalling networks mediate DOX resistance in breast cancer. ERCC1 upregulation cooperatively enhances drug efflux via P-gp/ABCB1, which reduces DOX-induced cytotoxicity and promotes the survival of breast cancer cells. Elevated ERCC1 typically facilitates the DDR, enabling tumor cells to withstand DOX treatment and acquire chemoresistance. Although DOX induces extensive DNA lesions in breast cancer cells, activation of the XPA-dependent NER machinery augments the repair capacity of these cells and increases the likelihood of cell survival. ERCC2 (XPD) strengthens the ATR–CHK1 signalling axis, maintaining tumor cells in a “pause-and-repair” state that further contributes to drug resistance. EMT enhances the expression of ERCC1/ERCC4 (XPF), which enables breast cancer cells to survive even under conditions of severe DNA damage. The XPC –RAD23B complex stabilizes NER protein homeostasis, which allows cells to “maintain equilibrium and continue survival” under DOX-induced stress. DNA damage recognition and repair factor (XPA): A core NER protein that recognizes bulky DNA lesions and recruits downstream repair factors. ERCC2 (XPD): A DNA helicase within the TFIIH complex that unwinds damaged DNA to initiate excision repair. XPC complex subunit (XPC): A DNA damage sensor that detects helix-distorting lesions and triggers global-genome NER. RAD23 nucleotide excision repair protein B (RAD23B): An accessory NER that forms a complex with XPC and stabilizes its lesion-recognition function. ERCC4 (XPF): A structure-specific endonuclease that cleaves the 5′ side of DNA lesions during NER. ATP-binding cassette transporters (ABC transporters): A family of membrane proteins that are responsible for mediating ATP-dependent substrate transport. P-gp (ABCB1): A pivotal ABC transporter that functions as a drug efflux pump to reduce the intracellular accumulation of chemotherapeutic agents. NER, nucleotide excision repair; ERCC1, ERCC excision repair 1, endonuclease non-catalytic subunit; ATR, ATR checkpoint kinase; CHK1, protein kinase Chk1; EMT, epithelial–mesenchymal transition.
ERCC1 forms a nuclease complex with XPF, and its overexpression significantly
enhances NER efficiency, which reduces drug-induced cytotoxicity and promotes a
resistant phenotype. XPA, XPC, CSB, and XPF are upregulated during both DOX
treatment and the withdrawal phase, which enables breast cancer cells to rapidly
repair NER lesions and maintain their proliferative capacity. BORG interacts with
the ssDNA-binding protein RPA1 to activate the NF-
MMR corrects replication-associated errors, including single-base mismatches and small insertion/deletion loops (IDLs), and is executed by two principal protein complexes: the MutS complex, which recognizes mismatches, and the MutL complex, which coordinates downstream repair events and the recruitment of accessory factors [73, 74, 75]. The perturbation of MMR function or MMR-dependent cellular responses to DOX, such as defective G2-phase cell cycle arrest, reduces tumor cell sensitivity to DOX [76, 77]. In breast cancer, mutL homolog 1 (MLH1) and mutS homolog 2 (MSH2) can be inactivated by genomic deletion or epigenetic mechanisms (promoter hypermethylation), and the methylation status of the MLH1/MSH2 promoter has been proposed as a potential biomarker for DOX responsiveness [78]. Epigenetic silencing of MMR genes, most notably promoter hypermethylation of MSH2, can also drive DOX resistance [79]. For example, Logeswari Ponnusamy et al. [80] demonstrated by pyrosequencing that multiple CpG sites within the MSH2 promoter are hypermethylated in DOX-resistant MCF-7 and MDA-MB-231 sublines, which substantially reduce MSH2 mRNA and protein levels, disrupt MMR-dependent apoptotic signalling, and promote a resistant phenotype. Importantly, treatment with the DNA-demethylating agent 5-Aza-2′-deoxycytidine (5-Aza-2dC) or the histone deacetylase inhibitor trichostatin A (TSA) largely restores MSH2 expression, re-establishes MMR-dependent apoptosis, and significantly increases the sensitivity of resistant cells to DOX (Fig. 6).
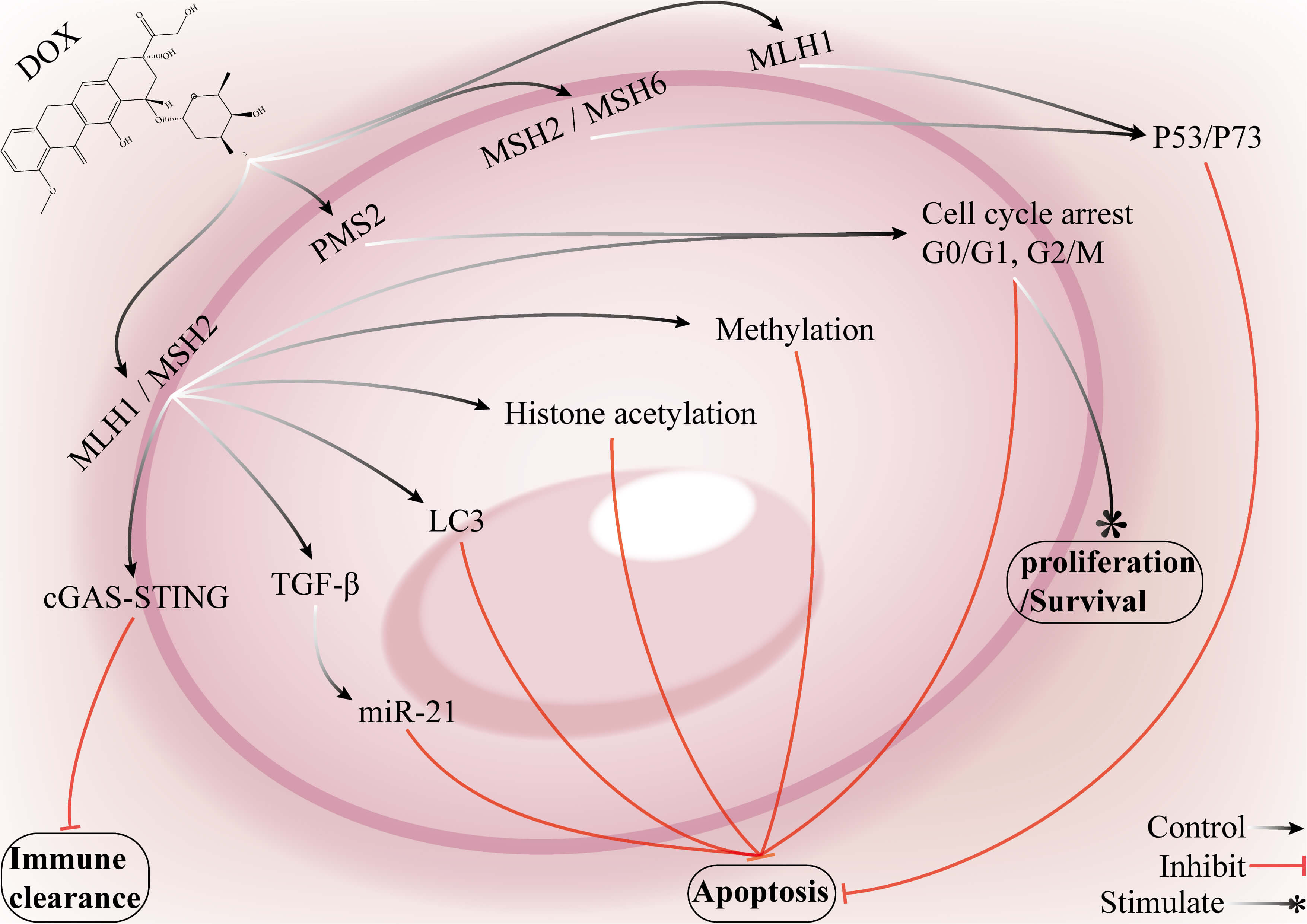 Fig. 6.
Fig. 6.
Mechanisms by which key genes in the MMR pathway and associated
signalling networks mediate DOX resistance in breast cancer. The promoter
methylation of MSH2 and MLH1 can lead to reduced mRNA and protein expression,
impairing the ability of the cell to sense DNA damage and attenuate apoptosis,
which ultimately promotes DOX resistance in breast cancer cells. MSH2 activity is
also modulated by epigenetic mechanisms such as histone acetylation, which
affects its capacity for DNA damage recognition and repair and further reinforces
the drug-resistant phenotype. Loss of MSH2 or MLH1 function suppresses the
cGAS–STING signalling pathway, diminishing antitumor immune clearance; this loss
may also activate TGF-
In MCF-7 and MDA-MB-231 cells exposed to DOX, hypomethylation of the MLH1 and MSH2 promoter regions leads to upregulation of mRNA and protein expression, which is thought to contribute to DOX tolerance. Conversely, hypermethylation of the MSH2 promoter results in reduced expression, accompanied by impaired MMR-dependent cell death. Treatment with demethylating agents or histone deacetylase inhibitors can restore MSH2 expression and significantly reverse resistance. These findings suggest that the methylation status of the MLH1 and MSH2 promoters may serve as a biomarker and that these genes may be therapeutic targets. The downstream mechanisms through which MMR activation influences resistance include the regulation of cell cycle checkpoints, such as the G2/M checkpoint, and the induction of cell death. Reliance on a single biomarker to predict resistance remains challenging because of tumor heterogeneity, which may compromise therapeutic evaluation. Future strategies may involve combination therapies that target epigenetic regulation and the establishment of precise molecular diagnostic systems for the dynamic monitoring of MLH1/MSH2 methylation and expression levels. This approach could guide personalized therapy and improve both the clinical feasibility and the reversal of drug resistance.
Combination therapies that pair DOX with inhibitors of DDR pathways can overcome resistance driven by enhanced repair capacity. Poly (ADP‒ribose) polymerase (PARP) inhibitors block the repair of SSBs, allowing unrepaired SSBs to collapse into lethal DSBs. Inhibitors of ataxia telangiectasia and Rad3-related (ATR) or checkpoint kinases 1 and 2 (CHK1/CHK2) disrupt the replication-stress response and S/G2 checkpoint, which promotes replication-fork collapse. Ataxia telangiectasia mutated (ATM) inhibitors combined with DNA-PKcs inhibitors cooperatively impair HR and NHEJ. WEE1 (a tyrosine kinase that restrains CDK activity) inhibitors and cyclin-dependent kinase 1/2 (CDK1/2) inhibitors abrogate the G2M checkpoint, forcing DNA-damaged tumor cells into mitosis and precipitating mitotic catastrophe and apoptosis [81, 82]. Inhibitors of ATR, CHK1/2, and DNA-PK sensitize TNBC cells by disabling key repair mechanisms and increasing DOX cytotoxicity, which can help overcome drug resistance [83]. For example, Kevin J. Lee et al. [68] reported that ATM inhibition impairs the DSB-repair capacity of breast cancer cells, increasing their DOX sensitivity and apoptosis rates. ATR inhibitors block ATR kinase activity and the cellular response to single-strand lesions and stack replication forks, which leads to the accumulation of DNA damage and cell death. CHK1/CHK2 inhibitors attenuate checkpoint control during DNA damage and replication stress, which prevents timely repair and promotes apoptosis. DNA-PK inhibitors inhibit DNA-PK kinase activity and the cellular DSB response, which results in unrepaired lesions and subsequent cell death. Targeting HR directly enhances DOX efficacy: RAD51 inhibitors markedly increase DOX cytotoxicity by blocking HR-mediated repair and inducing cell cycle arrest, mitochondrial dysfunction, and apoptotic signalling [84]. For example, Schürmann et al. [30] demonstrated that the RAD51 inhibitor B02 induces G2/M arrest, causes mitochondrial damage, and activates apoptosis, which increases DOX-induced cytotoxicity in breast cancer cells.
Deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTPase) inhibitors promote the incorporation of aberrant uracil into DNA and induce DNA damage. When DNA damage is induced by fluoropyrimidines or DOX, inhibition of dUTPase impairs the ability of the cell to prevent uracil incorporation and to process the resulting lesions, which results in the accumulation of lethal DNA damage and triggers apoptosis in breast cancer cells [85]. Compared with DOX monotherapy, the combination of DOX with a dUTPase inhibitor increases the number of DNA DSBs and suppresses their repair, which helps to overcome DOX resistance [86, 87]. For example, Davison et al. [88] reported that DOX induces DSBs in breast cancer cells and that TNBC cells adapt to persistent DNA damage through metabolic reprogramming, which leads to the upregulation of new pyrimidine biosynthesis, a change that supports resistance. dUTPase inhibitors drive uracil incorporation, generate base mismatches, and cause the accumulation of fatal DNA lesions that ultimately induce apoptosis in TNBC cells (Fig. 7).
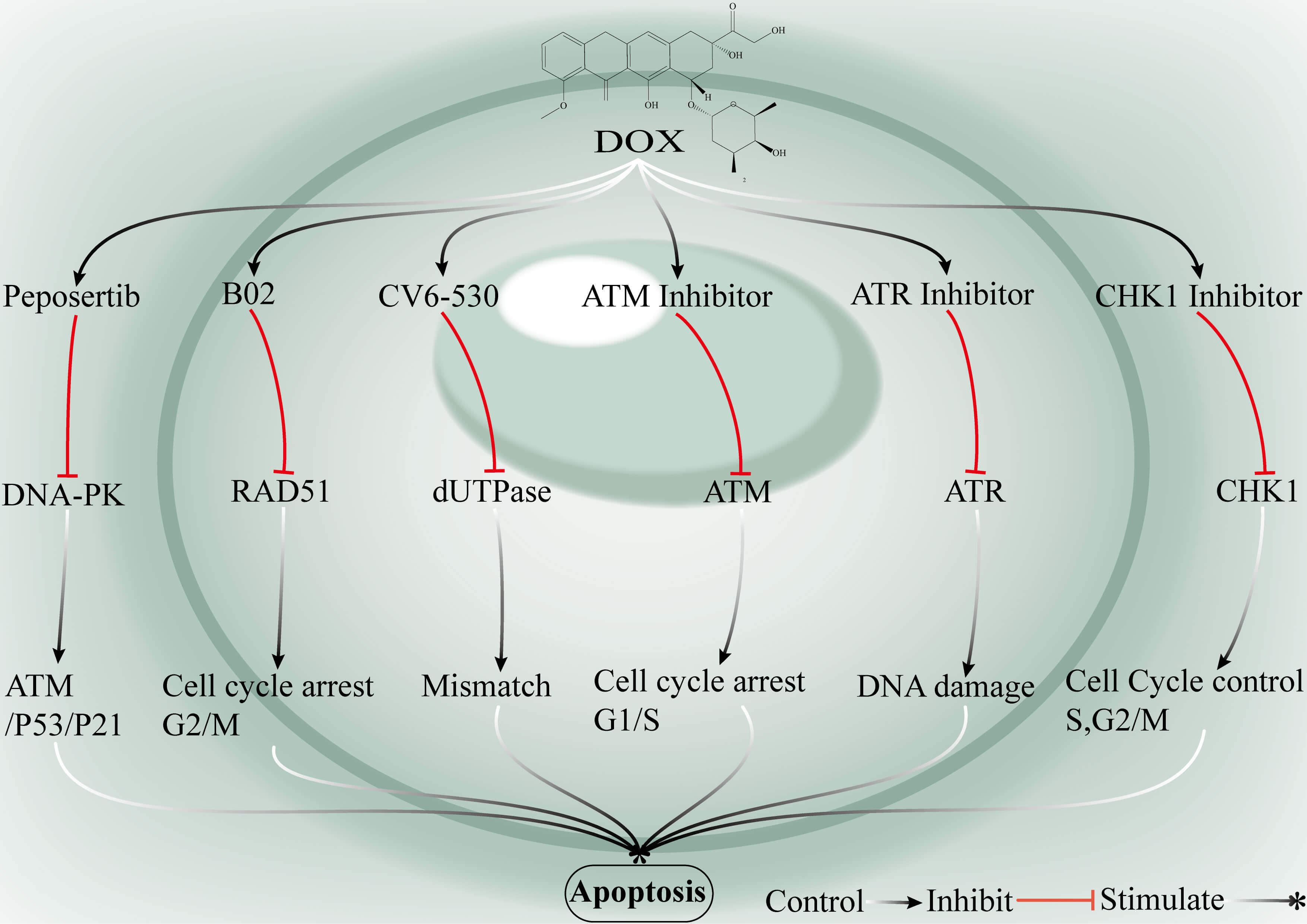 Fig. 7.
Fig. 7.
Synergistic strategies to overcome breast cancer chemoresistance via targeted inhibition of DDR pathways. DNA-PK inhibitors suppress DNA-PK activity and consequently activate the ATM–P53–P21 signalling axis, which induces cell cycle arrest and apoptosis. B02, a RAD51 inhibitor, disrupts HR repair and perturbs G2/M phase regulation. CV6-530 inhibits deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTPase) activity, which leads to the accumulation of uracil misincorporation and triggers the DDR. ATM inhibitors block ATM activity, causing cell cycle arrest at the G1/M checkpoint, suppress ATR signalling and impair the progression of DDR. CHK1 inhibitors inactivate CHK1, resulting in S-phase and G1/M arrest. When combined with DOX, these inhibitors synergistically enhance apoptotic responses in breast cancer cells, ultimately promoting tumor cell death. Ataxia-telangiectasia mutated (ATM): A master kinase that senses DSBs and activates the DDR, including the P53/P21 pathway. P21: A P53-inducible cyclin-dependent kinase inhibitor that blocks cell cycle progression into S phase or G2/M. ATM and Rad3-related (ATR): A key kinase that detects replication stress and initiates DNA damage checkpoint signalling. Checkpoint kinase 1 (CHK1): A checkpoint regulator essential for the initiation of the DDR and cell cycle arrest. CV6-530: A dUTPase inhibitor that induces uracil misincorporation and DNA base mispairing.
The combination of DOX with inhibitors of DDR pathways, such as PARP, ATR, CHK1/2, ATM, DNA-PKcs, RAD51, and dUTPase inhibitors, can increase DOX-induced DNA damage accumulation through multiple mechanisms, including blockade of single-strand break repair (SSBR), disruption of the replication stress response, inhibition of HR and NHEJ, and abrogation of the G2/M checkpoint. This leads to a significant increase in apoptosis in breast cancer cells. The rationale for such targeted combination strategies lies in their strong synergistic effects, which allow for enhanced antitumor activity at reduced doses of DOX while overcoming resistance associated with elevated DNA repair capacity [88, 89, 90]. Clinical translation remains challenging because of factors such as limited drug bioavailability, off-target effects, toxicity to normal cells, and the potential for tumor cells to develop new resistance via alternative repair pathways. Currently, this strategy is primarily at the preclinical stage, but some ATR inhibitors have entered phase I trials where the goal is to demonstrate tolerability and preliminary efficacy, although large-scale registration trials have not yet been conducted [91]. The combination of DOX with DDR inhibitors has substantial therapeutic potential in breast cancer, particularly TNBC, but further optimization of drug properties and combination regimens is needed to facilitate clinical application.
NcRNAs are functional RNA species transcribed from the genome that are not translated into proteins. Major classes include lncRNAs, miRNAs, and circular RNAs (circRNAs) [92, 93]. MiRNAs are short (~18–25 nt), single-stranded RNAs whose expression is initiated by RNA polymerase II-mediated transcription. miRNAs regulate gene expression in a posttranscriptional manner by base pairing with target sites (most commonly within 3′ untranslated regions, 3′UTRs) to repress translation or promote mRNA decay [94]. In p53-mutant breast cancer cells, dysregulation of miR-30c enables more effective activation of DDR mechanisms in response to DOX-induced lesions, which reduces DOX cytotoxicity. Conversely, miR-30c can act as a sensitizer of DDR proteins and increase DOX sensitivity in resistant cells [95]. For example, Shu Lin et al. [96, 97] demonstrated that miR-30c directly targets the 3′ UTRs of FA complementation group F (FANCF) and the translesion-synthesis polymerase deoxycytidyl transferase (REV1), which suppresses their expression and increases breast cancer cell sensitivity to DOX. p53 mutation is associated with reduced miR-30c expression and the concomitant upregulation of FANCF and REV1 expression, which contributes to drug resistance. The expression of miR-21 has been reported to inversely correlate with the levels of DDR proteins under certain conditions: high miR-21 expression reduces DNA repair activity and increases DOX sensitivity. Circular RNA circ-21 (a covalently closed circRNA) modulates this axis by sponging miR-21; through the regulation of miR-21, circ-21 may alter PARP-1a expression and affect tumor cell sensitivity to DOX [98, 99]. For example, Ana R. Rama et al. [100] reported that miR-21 overexpression in DOX-resistant cells decreases PARP-1 expression, impairs the DDR and influences cell survival. Conversely, circ-21 relieves the miR-21-mediated repression of PARP-1, which restores PARP-1 levels and enhances DDR capacity; this is associated with the suppression of proliferation, migration, and angiogenesis, increased apoptosis, and increased DOX sensitivity [85, 86, 87]. These reports underscore the context-dependent role of the miR-21/circ-21/PARP-1 axis in the modulation of DNA repair and cell fate and highlight ncRNA-based approaches as promising strategies to sensitize breast tumors to DOX.
lncRNAs regulate DOX resistance through complex molecular mechanisms, primarily by promotion of tumor cell proliferation, inhibition of apoptosis, upregulation of drug efflux transporters, modulation of DDR pathways (either activation or repression of repair genes), induction of autophagy, transfer of resistance factors through exosomes, and an increase in invasion and metastasis [101, 102]. Some lncRNAs act as negative regulators of resistance and increase tumor cell sensitivity to DOX. The lncRNA H19 negatively regulates PARP1 expression, which is positively correlated with breast cancer cell sensitivity to DOX, and can function as a sensitizing target to increase DOX efficacy [90]. For example, Yu Wang et al. [103] reported that H19 downregulates PARP1 in DOX-treated breast cancer cells and that H19 knockdown increases PARP1 expression and enhances cellular sensitivity to DOX, whereas H19 overexpression reduces PARP1 expression and restores resistance [103, 104] (Fig. 8).
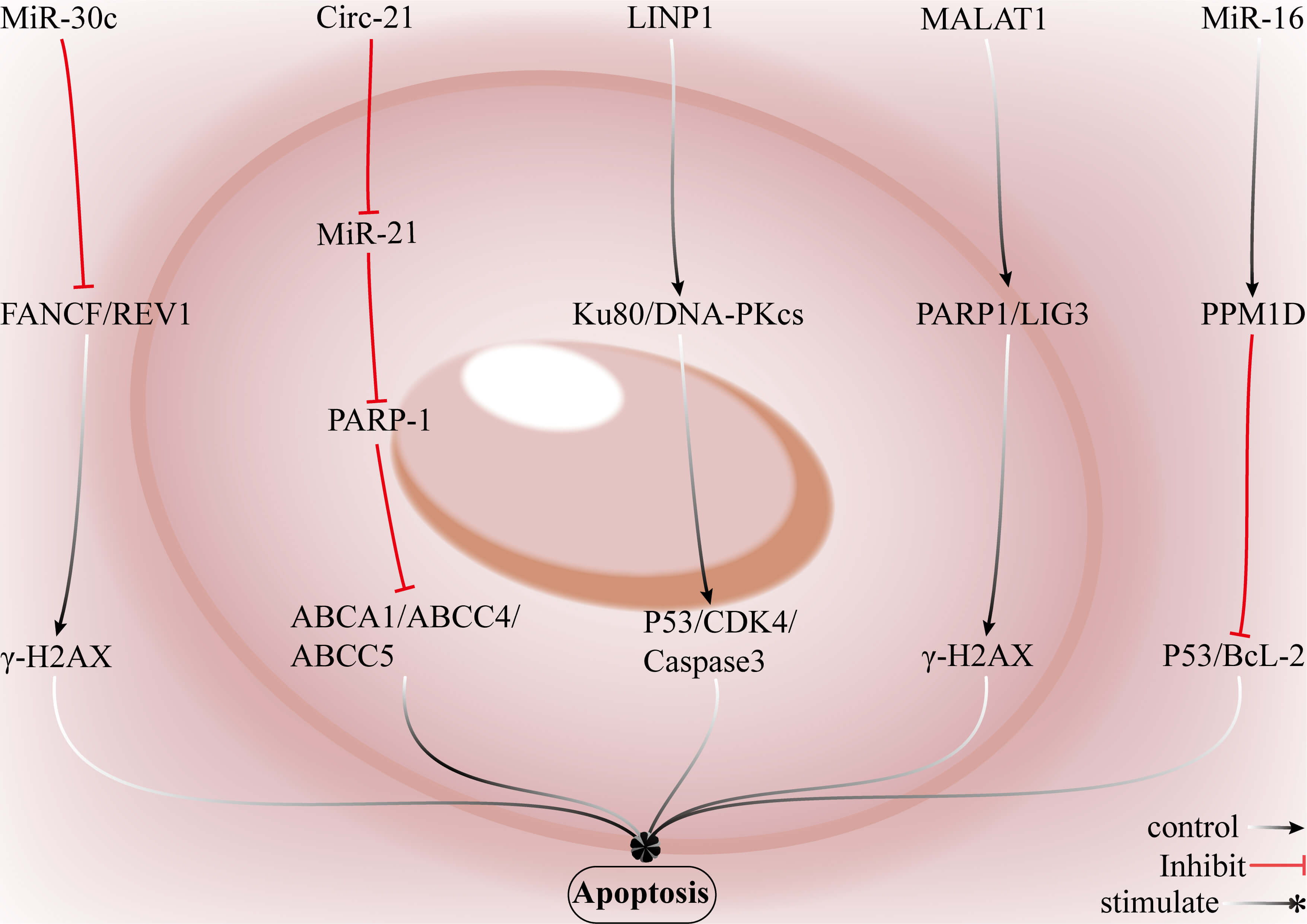 Fig. 8.
Fig. 8.
Strategies for reversing DOX resistance in breast cancer through
ncRNAs. LncRNAs, such as lncRNA in non-homologous end joining pathway 1 (LINP1),
increase cellular resistance to DOX-induced DNA DSBs by upregulating key
components of the NHEJ pathway, including Ku80 and DNA-PKcs, to promote cell
survival. CircRNAs, such as circ-21, modulate miR-21 to suppress PARP-1
expression or activity and may synergistically regulate multidrug
resistance–associated ATP-binding cassette transporters (ABCA1, ABCC4, and
ABCC5), which drives tumor cell proliferation and survival. miR-30c impairs the
DDR by targeting core factors of the Fanconi anaemia pathway and translesion
synthesis machinery, namely, FA complementation group F (FANCF) and deoxycytidyl
transferase (REV1), resulting in
Restoration or overexpression of miR-30c suppresses FANCF and REV1 expression, which increases the sensitivity of breast cancer cells to DOX. Circ-21 enhances the antitumor efficacy of DOX by sequestering miR-21 and subsequently restoring PARP-1 expression. The lncRNA H19 negatively regulates PARP1, and silencing H19 upregulates PARP1 expression and sensitizes breast cancer cells to DOX. Most therapeutic miRNAs, lncRNAs, and circRNAs are prone to nuclease degradation, exhibit restricted tissue distribution, and display low cellular uptake in vivo. Achieving effective intratumoral concentrations often requires dose escalation, which in turn amplifies their inherent pleiotropic activities; this leads to gene expression perturbations in normal tissues and increases the risk of off-target effects. Although delivery technologies [105], such as lipid nanoparticles, polymeric nanocarriers, viral vectors, and chemically modified nucleic acids, can improve molecular stability and tumor uptake, they may also introduce new challenges, including nonspecific accumulation in normal tissues, immune activation, and long-term alterations in gene-regulatory networks [106].
NPs are chemical entities isolated from plants, animals, microorganisms, or marine sources and are characterized by high structural complexity and diverse biological activities [107]. Compared with typical synthetic small molecules, NPs generally have higher molecular weights, a larger fraction of sp3-hybridized carbons, more hydrogen-bond donors and acceptors, and lower predicted cLogP values [108]. As they act on multiple targets in a synergistic manner, NPs can disrupt the DDR equilibrium in drug-resistant breast cancer cells and can markedly potentiate DOX-mediated cytotoxicity [109, 110]. Isorhamnetin (IS) is a naturally occurring flavonoid found in vegetables, fruits, and medicinal herbs. As a sensitizer of DOX-resistant breast cancer cells, IS enhances DOX cytotoxicity and inhibits tumor growth by increasing intracellular DOX accumulation (via downregulation of P-gp/ABCB1), impairing DDR pathways to reduce repair efficiency and modulating multiple signalling cascades that suppress cell proliferation [111, 112]. For example, T. Yang et al. [113] demonstrated that in DOX-resistant breast cancer cells, IS induces ROS production and consequent DNA damage, leading to G₂/M cell cycle arrest. IS also downregulates BCL-2, promotes caspase-3 cleavage, and triggers apoptosis. In addition, IS activates AMPK in a ROS-dependent manner and inhibits the mTOR/p70S6K signalling axis. These coordinated effects synergize to sensitize resistant cells to DOX to effectively overcome DOX resistance.
Morin is a naturally occurring flavonoid found in elm and mulberry species
(family Ulmaceae and Morus spp.) and in traditional medicinal herbs such
as Phellodendron and Glycyrrhiza (liquorice) [114]. Morin
markedly sensitizes DOX-resistant breast cancer cells; combined treatment with
morin and DOX inhibits proliferation and promotes the apoptosis of resistant
cells in vitro, and this cotreatment also significantly reduces tumor
volume and weight in mouse xenograft models [115]. Moreover, morin disrupts both
the accumulation and the repair of DNA damage in breast tumor cells, which
impairs the ability of resistant cells to resolve drug-induced lesions, reverses
resistance and enhances antitumor efficacy [116]. For example, Maharjan
et al. [117] demonstrated that morin increases
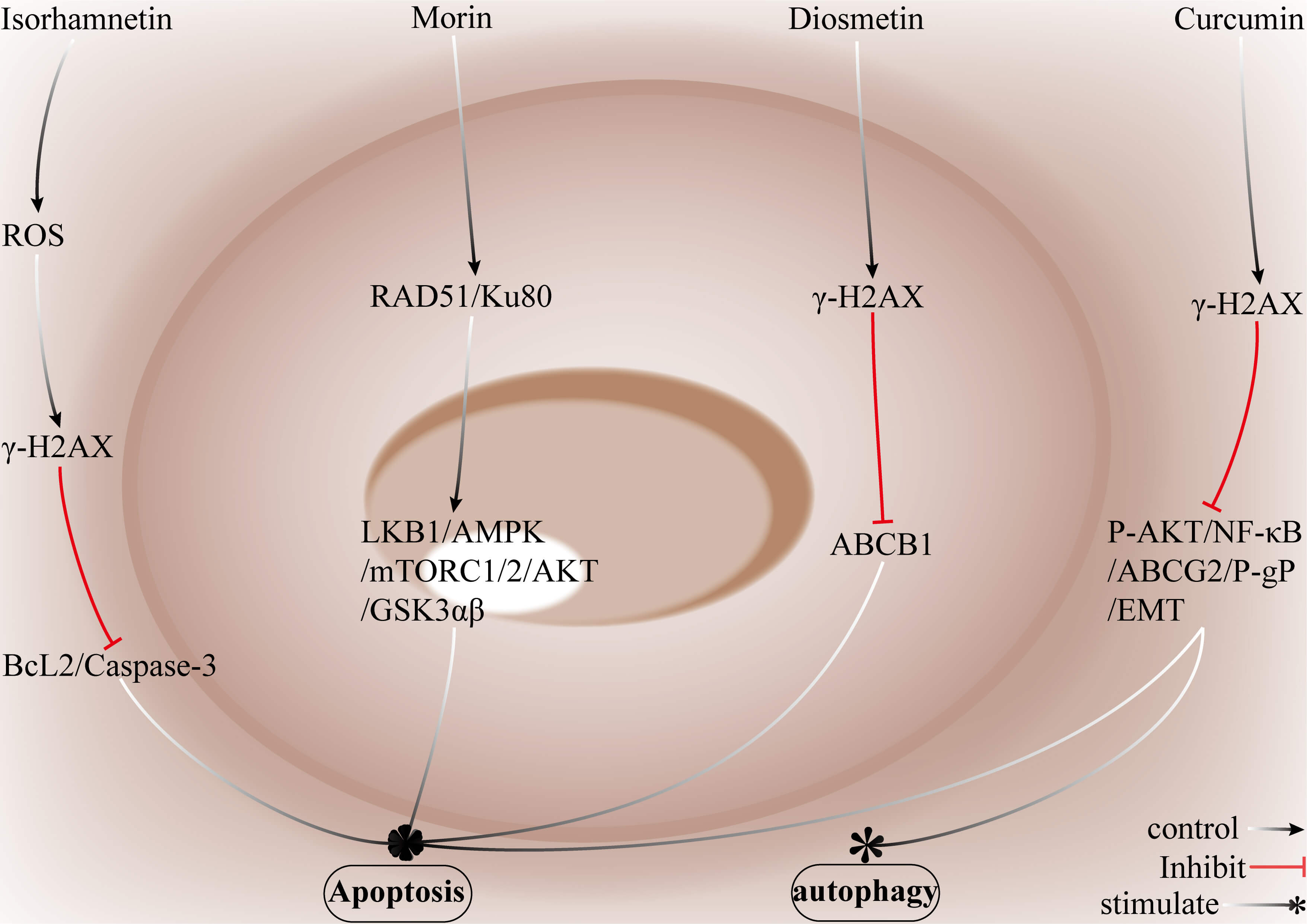 Fig. 9.
Fig. 9.
Strategies by which natural compounds restore DOX sensitivity in
breast cancer. IS increases the generation of ROS, induces DNA damage
characterized by
IS enhances the cytotoxicity of DOX by inhibiting P-gp/ABCB1, increasing
intracellular DOX accumulation, inducing the generation of ROS, and triggering
substantial DNA damage. This is accompanied by G2/M cell cycle arrest,
downregulation of BCL-2, and activation of caspase-3. IS further sensitizes
resistant cells through ROS-mediated activation of AMPK, which suppresses
mTOR/p70S6K signalling. Morin promotes apoptosis and inhibits cell proliferation
through the upregulation of
The poor clinical efficacy of DOX in the treatment of breast cancer is largely attributable to DDR-mediated resistance.
Different DDR pathways do not operate in isolation, but rather, they show competitive, cooperative, and compensatory interactions. These interactions depend on the cell cycle phase, end-processing status, and availability of key repair factors [120]. When HR is impaired or inhibited, tumor cells often compensate by upregulating classical NHEJ and the more error-prone alternative end-joining (alt-EJ/TMEJ), which is mediated mainly by DNA polymerase theta (POLQ). These pathways then “hijack” DSB repair and sustain survival at the cost of increased genomic instability [121]. This plasticity among pathways explains why single-target therapies often result in rapid resistance. This includes reversion mutations in HR genes, such as BRCA, restoration of HR activity, or increased use of the alt-EJ pathway as an escape mechanism. In HR-deficient contexts, combined strategies based on “synthetic lethality plus blockade of backup pathways” can help. For example, pairing PARP inhibitors with POLQ or DNA-PK inhibitors can prevent pathway switching and increase therapeutic lethality [122]. However, simultaneous inhibition of multiple DDR pathways markedly increases toxicity in normal rapidly dividing tissues. Successful clinical translation, requires precise patient stratification, careful optimization of dosing and scheduling, and dynamic monitoring through longitudinal tumor sequencing or circulating tumor DNA to detect early signs of resistance or pathway rewiring [123] (Table 1).
| Strategies | Drugs | Advantages | Disadvantages | Applicable scenarios | Challenges |
| Small molecule inhibitor | PARP inhibitors, ATR inhibitors, CHK1 inhibitors, ATM inhibitors, WEE1 G2 checkpoint kinase (WEE1) inhibitors, CDK1/2 inhibitors, RAD51 inhibitors, dUTPase inhibitors. | (1) Direct targeting of ATM, ATR, CHK1, CHK2, DNA-PK, or RAD51 can induce synthetic lethality or suppress compensatory escape pathways. | (1) Inhibition of multiple DDR pathways may lead to toxicity in normal, rapidly proliferating tissues, such as the bone marrow and intestinal epithelium. | (1) The applicability of these strategies in HR–deficient tumors relies on the compensatory upregulation of NHEJ or POLQ-mediated microhomology-mediated end joining (MMEJ). | (1) Clinical toxicity is difficult to manage, with narrow therapeutic windows and a lack of highly selective small-molecule inhibitors—particularly for targets such as DNA polymerase theta (POLQ). |
| (2) These agents exhibit well-defined pharmacological activity, allowing for controlled dose modulation, and are suitable for combination therapy with DOX in clinical settings. | (2) Single-target interventions are prone to the emergence of DOX resistance. Substantial patient heterogeneity underscores the need for precise molecular stratification to guide therapeutic selection. | ||||
| DNA-PKcs inhibitors | (2) In patients with recurrent or DOX-resistant breast cancer, long-term and dynamic monitoring of pathway rewiring is required to guide treatment. | ||||
| (2) Inadequate patient stratification further complicates clinical trial design, especially when combination strategies, safety assessment, and resistance monitoring must all be simultaneously addressed. | |||||
| ncRNA | miR-30c, circ-21, LINP1, metastasis associated lung adenocarcinoma transcript 1 (MALAT1), MiR-16 | (1) These molecules may regulate DDR pathways and exhibit flexible mechanisms of action. | (1) Delivery remains challenging, including issues of stability, target specificity, and achieving effective intratumoral concentrations. | (1) The context-dependent functionality of ncRNAs, the intricate nature of their regulatory networks, limited efficiency of in vivo delivery, and the risk of non-specific effects remain major obstacles to the successful clinical translation of ncRNA-targeted therapeutic strategies in breast cancer. | (1) Delivery platforms require further optimization, and comprehensive studies on clinical dosing, safety, and off-target effects, as well as validation of biomarkers are needed. |
| (2) They can also serve as biomarkers, as miRNAs are relatively stable in body fluids and may be used to predict DOX resistance in breast cancer. | |||||
| (2) Nonspecific effects are a concern, as a single miRNA may target multiple genes, leading to off-target consequences. | |||||
| (2) Regulatory and manufacturing challenges also remain significant for the development of ncRNA therapeutics. | |||||
| (2) Immune responses and in vivo toxicity are not yet fully understood, contributing to the slow pace of clinical translation. | |||||
| Natural bioactive compounds (NPs) | Isorhamnetin (IS), Morin, Diosmetin, Curcumin | (1) These compounds generally exhibit low toxicity and good biocompatibility. | (1) Poor bioavailability and limited stability, such as low oral absorption and inefficient in vivo delivery, pose significant challenges. | (1) These strategies are applicable to DOX-resistant breast tumors, particularly when transport proteins and ABC pumps are upregulated. | (1) Enhancing bioavailability remains a key goal, while comprehensive studies on systemic toxicity and pharmacokinetics are still needed. |
| (2) They can serve as sensitizers in combination with DOX, enhancing the susceptibility of resistant breast tumors to DOX while mitigating adverse effects. | |||||
| (2) Their complex pharmacological mechanisms may lack sufficient specificity, and combination with DOX could carry potential risks. | (2) Long-term combination therapy may be required to delay the onset of resistance, with the potential to reduce DOX-associated toxicities, such as cardiotoxicity. | (2) Clinical translation is challenging, particularly regarding dosing, combination regimens, and formulation strategies; clinical trial data remain relatively scarce. | |||
| (3) Their multitarget activity not only modulates DDR but also affects transport proteins and key signalling pathways. | |||||
| (3) Clinical evidence remains limited, as most studies are still confined to in vitro or animal models. |
Note: This table summarizes the key points described in the text. The relevant references are cited in the corresponding section.
Careful selection and scheduling of DDR inhibitors are essential for maximizing antitumor benefits while minimizing detrimental effects on normal tissue repair. Promising complementary strategies to overcome DOX resistance include early detection and longitudinal monitoring of chemoresistance, optimization of drug formulation and dosing to increase intratumoral exposure, rational combinations of natural products with targeted small-molecule inhibitors and direct targeting of breast cancer stem cells. Natural phytochemicals, such as curcumin, piperine, and quercetin, have been reported to synergize with DOX to increase antitumor efficacy and delay the onset of resistance; moreover, these compounds also generally exhibit low toxicity and favourable biocompatibility. Ligand-coated polymeric nanoparticles, magnetic nanoparticles, liposomes, micelles, nanocages, and nanorods can enable more precise tumor targeting and improved DOX uptake. Targeting resistance-associated transporters and signalling networks, notably through ncRNAs, offers an additional route to suppress DDR activity, limit the development of resistance, and promote tumor cell apoptosis.
This review systematically summarizes the roles of MMR, BER, NER, HR, and NHEJ as central determinants of DOX resistance and evaluates the translational potential of DDR-targeted agents, ncRNA-based approaches, and NPs in reversing resistance. Future advances in multiomics technologies and biomarker discovery should enable DDR defect-driven molecular subtyping of breast cancer and the design of personalized regimens to inform clinical decision-making. The reclassification of tumors according to DNA repair status is likely to yield novel insights for next-generation targeted therapies and more informative clinical trial designs. Elucidating DDR-mediated DOX resistance will provide both the theoretical foundation and practical guidance needed to accelerate the development of precision interventions for the treatment of DOX-resistant breast cancer.
YJW, XQL and WJW conducted the literature search and wrote the original draft. SBL and XHL designed the research and integrated and refined the key points. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
The Jiangsu Higher Education Institution Innovative Research Team for Science and Technology (2021), the Program of Jiangsu Vocational College Engineering Technology Research Center (2023), the Program of Suzhou People’s Livelihood Technology Projects (Grant No. SYWD2024099), the Natural Science Key Foundation of the Jiangsu Higher Education Institutions of China (Grant No. 24KJA310008), the Research Foundation of Jiangsu Commission of Health (Grant No. Z2023031), the Dongwu Health Talent Program (DWWS2024002), the Programs of the Suzhou Vocational Health College (Grant No. szwzy202406), and the Project of Jiangsu Province Engineering Research Center of Molecular Target Therapy and Companion Diagnostics in Oncology (SGK1202413) provided funding for this study.
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.