

 , Anatoly V. Skalny 2,3,4, Svetlana V. Notova 4, Margarita N. Tinkova 5, Rongzhu Lu 6, Andrey A. Skalny 3, Abel Santamaria 7, Ji-Chang Zhou 8, Alexey A. Tinkov 2,4,9,*
, Anatoly V. Skalny 2,3,4, Svetlana V. Notova 4, Margarita N. Tinkova 5, Rongzhu Lu 6, Andrey A. Skalny 3, Abel Santamaria 7, Ji-Chang Zhou 8, Alexey A. Tinkov 2,4,9,*
1 Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY 10461, USA
2 Center of Bioelementology and Human Ecology, Sechenov First Moscow State Medical University, 119435 Moscow, Russia
3 Department of Medical Elementology, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
4 Laboratory of Metallomics, Institute of Bioelementology, Orenburg State University, 460018 Orenburg, Russia
5 Outpatient Department, Orenburg Central District Hospital, 460008 Orenburg, Russia
6 Department of Preventive Medicine and Public Health Laboratory Sciences, School of Medicine, Jiangsu University, 212013 Zhenjiang, Jiangsu, China
7 Laboratorio de Nanotecnología y Nanomedicina, Departamento de Atención a la Salud, Universidad Autónoma Metropolitana-Xochimilco, and Facultad de Ciencias, Universidad Nacional Autónoma de México, 04960 Mexico City, Mexico
8 School of Public Health (Shenzhen), Shenzhen Campus of Sun Yat-sen University, 518107 Shenzhen, Guangdong, China
9 Laboratory of Ecobiomonitoring and Quality Control, Yaroslavl State University, 150003 Yaroslavl, Russia
Abstract
The use of manganese oxide nanoparticles (MnOxNPs) in biomedicine increases the risk of their accumulation in the body, potentially leading to toxicity in various organs and tissues. In addition, occupational exposure to MnOxNPs-containing aerosols may also occur. MnOxNPs have been shown to accumulate in the brain and induce neurobehavioral alterations. However, the specific mechanisms of MnOxNPs toxicity in the brain and other tissues remain incompletely understood. Therefore, the objective of this review is to summarize existing data on the toxicity of MnOxNPs in the brain and other tissues, and to discuss the molecular mechanisms underlying their neurotoxic effects. It has been shown that MnOxNPs induce neuronal death through induction of mitochondrial dysfunction and subsequent apoptosis, and overaccumulation of tau protein and amyloid-β. Neurotoxic effects of MnOxNPs may also be mediated by blood–brain barrier disruption, and dysregulation of dopaminergic and glutaminergic signaling. Exposure to MnOxNPs induces neuroinflammation through activation of nuclear factor kappa B (NF-κB) and p38 mitogen-activated protein kinase (p38 MAPK) pathways in a reactive oxygen species-dependent manner. In vitro studies further demonstrate that MnOxNPs exhibit a dose-dependent cytotoxic effects in alveolar macrophages, as well as in respiratory, colonic, and other epithelial cells, through the promotion of oxidative stress and an inflammatory response. Overexposure to MnOxNPs has significant nephrotoxic, hepatotoxic, and immunotoxic effects, as well as affecting the reproductive system. Smaller particles exhibit more pronounced toxic effects in the brain and other tissues than larger nanoparticles or microparticles. However, the mechanisms underlying the different toxicities of MnOxNPs of different sizes, shapes, and surface modifications remain unclear. These observations highlight the potential of MnOxNP exposure to contribute to neurological disorders and dysfunction of other systems, underscoring the need for further mechanistic studies to ensure their safe application in biomedicine.
Keywords
- manganese
- nanoparticles
- brain
- toxicity
- nanomedicine
Manganese (Mn) is the third most abundant transition metal in the Earth’s crust [1]. It is also considered an essential metal involved in brain development and functioning, immunity, and metabolic regulation [2]. Mn occurs in the environment in various oxidative states (+2, +3, and +4), giving rise to more than 30 minerals [3], including multiple oxides [4]. Manganese oxides (MnOx) have been used as pigments in glass and agents in steel production since ancient Egypt and Greece [5]. In the last decades, the investigation and applications of MnOx nanoparticles (MnOxNPs) have been significantly increased [6]. MnOxNPs are characterized by unique properties including magnetism, semiconductivity, catalytic activity, energy storage, optical properties, biocompatibility, polymorphisms, ion exchange, and phase transformation, which mediate a wide spectrum of applications [7]. Briefly, MnOxNPs are utilized in water [8] and soil [9] remediation, the optoelectronic and catalytic industries [10], the production of Li-ion batteries [11], and semiconductors [12].
Along with other Mn-containing particles like Mn-ferrite NPs [13, 14, 15], MnOxNPs are widely used in biomedicine [16]. Briefly, due to the significant magnetization of MnOxNPs [17], they are used as strong T1 MRI contrast agents [18]. MnOxNPs also have potential applications in treatment of cancer [19, 20] and brain diseases including Parkinson’s disease [21, 22], Alzheimer’s disease [23, 24], and hypoxic brain damage [25]. In addition, MnOxNPs are used for drug delivery [26]. Specifically, MnO2NPs are loaded with small molecules, peptides, nucleic acids, proteins, as well as phytochemicals [27]. Redox activity of Mn mediates its potential in developing MnOx-containing nanoconstructs with enzyme-like activity [26]. The use of MnOxNPs in biomedicine may result their increased body burden, thus raising the issue of their potential toxicity.
Although Mn is an essential metal, it is toxic upon overload, affecting mainly the brain and resulting in neurodegeneration [28]. Congruently, while it is assumed that MnO and MnOxNPs are characterized by low toxicity due to the essentiality of Mn, several findings showed that these particles possess cytotoxic effects in a number of cell lines [29]. In addition, in vivo studies also showed that MnOxNPs adversely affected the liver and kidneys [30], reproductive system [31], and other systems and organs. Given the neurotoxic effects of Mn, MnOxNPs may also exert neurotoxicity [32]. Several studies showed that MnOxNPs impair the viability of brain cells [33, 34] as well as affects behavior in laboratory rodents [35, 36]. However, the mechanisms of MnOxNPs toxicity in various organs, and especially the brain, are still disputable.
Therefore, the objective of the present review was to summarize the existing data on the toxicity of MnOxNPs in the brain and other tissues, as well as discuss the molecular mechanisms underlying their neurotoxic effects.
In view of a wide spectrum of the potential biomedical applications of MnOxNPs, understanding its adverse health effects is crucial for further clinical applications [7]. Several studies showed that MnOxNPs possess cytotoxic effects in various types of cells which may be mediated by ROS overproduction. Specifically, in vitro data showed that Mn3O4NPs induce an increase in intracellular ROS production [37]. Mn2O3NPs also possess a high capacity for cytochrome c oxidation, exceeding that observed for other nanoparticles, including CeO2, TiO2, BaSO4, carbon black, Cu, Ag, and ZnO NPs. This oxidative potential was mediated by reduced glutathione (GSH) and ascorbic acid depletion [38]. Prooxidant effect of Mn2O3NPs characterized by hydroxyl radical generation was also revealed in an assay of L-3,4-dihydroxyphenylalanine (L-dopa) autoxidation [39]. It has also been shown that ability of MnO2NPs to generate ROS through Fenton-like mechanism and oxidize cytochrome c is not associated with Mn2+ dissolution, which was insignificant [40]. Finally, prooxidant activity of Mn-containing welding fume nanoparticles from stainless steel directly correlated with Mn2+ release from the particles [41]. Congruently, commercially available MnOxNPs of various shapes (spherical, rods, flakes) mainly sized around 30 nm induced a significant decrease in viability of A549, HepG2, and J774A.1 cells due to ROS overproduction and caspase 3 activation [29].
Significant cytotoxic effects of MnOxNPs were revealed in cultures of airway epithelial cells. It has been demonstrated that Mn2O3NPs exposure decreases viability of both BEAS-2B and A549 cells through induction of apoptosis [42]. In BEAS-2B cells and RAW 264.7 macrophages, exposure to Mn2O3 resulted in a dose-dependent decline in cell viability, energy production, and membrane leakage. Further analysis also showed that along with membrane damage Mn2O3 treatment induced mitochondrial dysfunction with mitoROS production [43]. Although Mn3O4NPs did not possess cytotoxicity upon short-term exposure (24 h), long-term exposure (9 days) was associated with significant cytotoxicity in A549 and especially Caco-2 cells [44]. It has also been demonstrated that exposure to Mn3O4NPs induces ROS overproduction in airway epithelial A549 cells and colorectal adenocarcinoma HT29 cells, although this prooxidant effect was associated with reduced cell viability only in A549 cells. Furthermore, exposure to a novel nanomaterial, GNA35, based on Mn3O4NPs with enhanced electrochemical properties, induced a more severe prooxidant effect and decrease in A549 cells viability [45]. It has been also demonstrated that Mn2O3NPs exposure significantly reduces A549 viability through induction of apoptosis and slightly inhibits cell proliferation [46]. Another study in a culture of A549 cells demonstrated that exposure to Mn oxide nanoparticles increases ROS production, up-regulates transferrin, ferritin light and heavy chains, and DMT1 mRNA expression, as well as increases IL-6 and IL-8 mRNA and protein expression, although some of these effects were more profound upon exposure to Fe oxide NPs or mixed type NPs [47]. Finally, it has been demosntrated that IC50 of Mn3O4NPs accounts for 98 µg/mL [48]. In another lung cell line (FE-1) MnO2NPs dose-dependently induced DNA damage, exceeding that observed in MnO2MPs-exposed cells [49]. Comparative analysis of Mn3O4NPs and MnSO4 showed that, despite both forms of Mn induce caspase-3-mediated apoptosis in CCL-149 alveolar epithelial cells, only Mn3O4NPs promoted ROS production and GSH oxidation. Noteworthy, exposure to MnNPs resulted in higher intracellular Mn content compared to MnSO4 [50]. Another study also showed that Mn3O4NPs increase ROS production in CCL-149 airway epithelial cells, although not reducing the metabolic activity of cells [51]. Congruently, Mn2O3NPs induced a significant decline in human bronchial epithelial NCI-292 cell viability with up-regulation of IL-8 mRNA and protein expression. At the same time, development of oxidative stress upon exposure to Mn2O3NPs induced up-regulation of SOD2 and HO1 mRNA expression in NCI-292 cells [38]. In another line of human bronchial epithelial cells (16HBE14o-), exposure to Mn2O3 resulted in a dose dependent decrease in cell metabolic activity assayed by MTT test with IC50 of 33 mg/L [52].
Adverse effects of MnOxNPs exposure were also revealed in alveolar
macrophages. Specifically, exposure to Mn3O4NPs induced a significant
increase in ROS production in CRL-2192 alveolar macrophages, resulting in a
significant reduction in cellular metabolic activity and down-regulation of MCP-1
production. Increased caspase-3 activity in Mn3O4NPs-exposed cells is
indicative of apoptosis. Attenuation of these effects by Trolox treatment showed
that cytotoxicity of Mn3O4NPs in alveolar macrophages is ROS-dependent
[51]. Correspondingly, MnO2 NPs reduced the viability of RAW264.7
macrophages, also decreasing their phagocytic activity due to the enhancement of
TNF
Certain studies showed that MnOxNPs are cytotoxic to intestinal epithelial
cells in vitro. It has been also demosntrated that Caco-2 cells (IC50 =
66.7 µg/mL) were more sensitive than lung epithelial A549 cells (IC50 =
136.2 µg/mL) and mouse Balb/c 3T3 fibroblasts (IC50 = 195.0 µg/mL) to
Mn3O4NPs-induced cytotoxicity. The latter was shown to be dependent on
ROS generation, but not metal ion release [54]. In a culture of Caco-2 cells
exposure to various MnOxNPs resulted in a dose-dependent reduction in cell
viability with cytotoxicity decreasing in the following order: Mn3O4
Cytotoxic effects of MnOxNPs were also evident in other epithelial cell lines. Exposure of epithelial MCF-7 and HT1080 cells to MnO2NPs resulted in increased total ROS and mitoROS production, inhibition of SOD, CAT, and glutathione reductase (GR) activity, and depletion of GSH pool. Along with induction of oxidative stress, activation of proapoptotic signaling by up-regulation of p53 and Bax mRNA expression, with repression of Bcl-2 mRNA, and reduction of mitochondrial membrane potential result in a decrease in cell viability. However, the results show that the effects of MnO2NPs were more profound in HT1080 cells compared to MCF-7 cells [57]. In human cervical carcinoma cells (HeLa) and mouse fibroblast cells (L929), exposure to silica-coated MnONPs induced a significant increase in ROS production, mitochondrial dysfunction, and G2/M phase arrest. Along with the up-regulation of p53 protein expression and the reduction of Bcl-2/Bax ratio, these effects were associated with caspase-3 activation, leading to cell apoptosis [58].
Finally, Mn3O4NPs exposure significantly reduced viability of mouse embryonic stem (mES) cells with up-regulation of gene products associated with oxidative stress (Srxn1), DNA damage (Rtkn), endoplasmic reticulum stress (Ddit3) and p53-related stress (Btg2), although this effect was less pronounced than in the case of metallic MnNPs [59].
However, it is demosntrated that synthesis of NPs also significantly affects particle toxicity. In hMSC cells chemically synthesized MnO2NPs induced significant alterations in cell volume and shape, while green-synthesized NPs did not possess adverse effects on cell morphology, thus showing its lower cytotoxicity [60].
Generally, these findings show that MnOxNPs possess cytotoxicity in airway epithelial cells, alveolar macrophages, enterocytes, and other epithelial cells (Table 1, Ref. [38, 42, 43, 44, 45, 46, 48, 49, 50, 51, 52, 53, 54, 55, 57, 58, 59]). Evidence shows that the cytotoxicity of MnOxNPs is mainly mediated by their prooxidant activity. Specifically, overproduction of ROS upon MnOxNPs exposure, along with induction of mitochondrial dysfunction, results induces cell cycle arrest and apoptosis, resulting in reduced cell viability. In addition, MnOxNPs-induced oxidative stress is associated with up-regulation of proinflammatory cytokine expression, which also contributes to cytotoxicity.
| Cell model | Nanoparticles | Effects | Reference |
| NCI-292 | Mn2O3, 20–100 nm size, 2.5–40 µg/cm2 for 24 h | Dose-dependent | [38] |
| BEAS-2B | Mn2O3, spherical, 82 nm size, 5–100 µg/mL for 24 h | Dose-dependent | [42] |
| A549 | |||
| BEAS-2B | Mn2O3, 51.5 nm size, 50–200 µg/mL for 24 h | Dose-dependent | [43] |
| RAW264.7 | |||
| Caco-2 | Mn3O4, |
Dose-dependent | [44] |
| A549 | |||
| Balb/c 3T3 | EC50 (9-days): Caco-2—28.9 µg/mL; A549—47.1 µg/mL; Balb/c 3T3—49.9 µg/mL | ||
| A549 | Mn3O4, spheric, 100 nm size, 1–10 mg/L for 24 h | Dose-dependent | [45] |
| HT29 | |||
| A549 | Mn2O3, spheric, 82 nm size, 20–100 µg/mL for 24–48 h | Dose-dependent | [46] |
| A549 | Mn3O4, tetragonal, 36 nm size, 10–100 µg/mL for 24 h | Dose-dependent | [48] |
| MCF-7 | |||
| IC50 (A549) = 98 µg/mL; IC50 (MCF-7) = 25 µg/mL | |||
| FE1 | MnO2, 40–60 nm size, vs MnO2MPs 5–10 µm size, 5–100 µg/mL for 2–4 h | Dose-dependent | [49] |
| CCL-149 | Mn3O4, 30 nm size, 5–20 µg/mL for 24 h | [50] | |
| CRL-2192 | Mn3O4, 20 nm size, 6 µg/cm2 cell surface for 4–24 h | [51] | |
| CCL-149 | |||
| 16HBE14o- | Mn2O3, 40–60 nm size, 13–500 mg/L for 48 h | IC50 = 33 mg/L | [52] |
| Raw 264.7 | MnO2, spheric, 30–50 nm size, 6.25–100 µg/mL for 24 h | Dose-dependent | [53] |
| 12.5 µg/mL: | |||
| Caco-2 | Mn3O4 14.9 nm size, 3–100 µg/mL for 24 h | Dose-dependent | [54] |
| A549 | |||
| Balb/c 3T3 | |||
| IC50 (Caco-2) = 66.7; IC50 (A549) = 136.2; IC50 (Balb/c 3T3) = 195.0 | |||
| Caco-2 | MnO2, Mn2O3 and Mn3O4, spherical, 50 nm size, 10–500 µM for 24 h | Dose-dependent | [55] |
| Upon H2O2 exposure | |||
| MCF-7 | MnO2, nanoflakes, diameter—10–20 nm, length—80–120 nm, 25–100 µg/mL for 24 h | Dose-dependent | [57] |
| HT1080 | |||
| 100 µg/mL: | |||
| Effects in HT1080 |
|||
| HeLa | SiO2-coated MnO, 39.3 nm size, 10–200 µg/mL for 24 h | Dose-dependent | [58] |
| L929 | |||
| Effects in L929 |
|||
| mES | Mn3O4, cubic, 20–180 nm and rods 8000 × 400 nm, 1–100 µg/mL for 24 h | Dose-dependent | [59] |
Agreeing with cellular models, in vitro studies also show potential
toxicity of MnOxNPs exposure for various organs. Specifically, exposure to
MnNPs via intraperitoneal injection in rats induced histopathological damage,
including hepatocyte degeneration, central vein congestion, sinusoid dilatation,
and hemorrhages in the liver, capsule rupture, inflammatory infiltration, and
glomerulosclerosis in the kidneys, and epithelial desquamation, impaired tubular
morphology, and vacuoles in the testis [31]. Correspondingly, intraperitoneally
injected Mn3O4NPs induced severe liver injury characterized by hepatic
inflammation and cell exfoliation along with induction of hepatocyte apoptosis
through up-regulation of caspase-3 protein expression and Bax/Bcl2 ratio. In
addition, Mn3O4NPs exposure up-regulated expression of genes involved
in endoplasmic reticulum stress (GRP78, Climp63) xenobiotic metabolism
by cytochrome P450 (CYP1A2, Sult2a1). The latter may contribute
to ROS overproduction, playing a key role in hepatocyte apoptosis, as evident
from inhibition of apoptosis upon treatment with antioxidant NAC [61]. In yellow
catfish, Pelteobagrus fulvidraco, exposure to MnO2NPs induced a more
profound toxic effect in liver characterized by up-regulation of pro-inflammatory
(il1
In addition, intravenous injection with Mn3O4NPs (via tail vein)
weekly for 120 days resulted in accumulation of Mn in testes and alteration of
testicular morphology characterized by reduced thickness of the germinative layer
and degeneration of seminiferous tubules. These effects were associated with
reduced levels of testosterone and FSH, as well as decreased sperm count in
ejaculate, reduced sperm motility, and a higher rate of abnormalities. Adverse
reproductive effects of Mn3O4NPs exposure may be mediated by
overproduction of ROS, induction of mitochondrial dysfunction, and subsequent
apoptosis, as observed in a culture of TM4 cells. Furthermore, transcriptional
analysis of testes revealed up-regulation of genes involved in PPAR-signaling
(Fabp1, Apoa2, Apoa3, and Pck1), steroid hormone synthesis (LOC100361547,
Cyp2c12, Cyp2c6v1, and Ugt2b37), and cytochrome P450-mediated metabolism (Gsta4,
LOC102550391, Ugt2b37, Sult2a2) [67]. Another study showed that MnO2NPs also
induce testicular damage characterized by a decline in the number of
spermatogonial cells, primary spermatocytes, spermatids, and Leydig cells,
resulting in a reduction sperm number, motility, and viability, along with
increased sperm abnormalities, and altered reproductive hormone levels. The
latter may be mediated by MnO2NPs-induced down-regulated mRNA expression of
StAR, HSD-3
Intratracheal instillation of Mn3O4NPs in a dose of 0.25 mg per animal resulted in a significant increase in the number of neutrophils and increased neutrophil-to-alveolar macrophage ratio in BALF, as well as increased amylase and LDH activity, altogether indicating pulmonary inflammation and toxicity [71]. Correspondingly, exposure of CB57BL/6 mice to Mn2O3NPs significantly increased BALF neutrophil count, as well as MCP-1 and IL-6 levels [43].
Certain studies show that MnOxNPs exposure may adversely affect other
organs and tissues. Specifically, oral exposure to 125 mg/kg and 250 mg/kg BW
Mn3O4NPs for 20 days did not induce histopathological effects in the
liver, spleen, kidneys, or colon, while inducing an inflammatory response in the
colon by up-regulating IL-1
Taken together, the increasing body of evidence shows that exposure to MnOxNPs possesses reproductive toxicity, induces hepatic and kidney damage, promotes intestinal inflammation, as well as induces immunotoxicity (Table 2, Ref. [30, 43, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 74]). In contrast, smartly engineered casein MnOxNPs generated with MnCl2 and casein under alkaline conditions [77] did not induce toxic effects upon both acute and chronic exposures [78]. These findings show that modified MnOxNPs are characterized by lower toxicity, while unmodified particles possess cytotoxic effects, and thus dissolution of MnOxNPs from modified particles must be limited [26]. These findings are in agreement with the role of metal nanoparticle surface coating in reducing its toxicity and facilitating their therapeutic effect in treatment of brain diseases [79].
| Model | Exposure | Effects | Reference |
| Sprague-Dawley rats | Mn3O4, 15 nm size, 20 mg/kg i.p. for 120 days | Dose-dependent: | [61] |
| P. fulvidraco | MnO2, 50 nm size, or MnSO4, 6 mg Mn/kg feed for 10 weeks | Vs MnSO4 | [62] |
| P. fulvidraco | MnO2, 50 nm size, or MnSO4, 6 mg Mn/kg feed for 10 weeks | vs MnSO4: | [63] |
| P. fulvidraco | MnO2, 50 nm size, 20–80 mg/kg diet for 8 weeks | [64] | |
| White feather chickens | Mn2O3, polygonal, 200 nm size, 900 mg Mn/kg diet for 3 weeks | [65] | |
| BALB c mice | MnO2, spherical, |
Serum: | [30] |
| Wistar rats | MnO2, 25–85 nm size, and MnO2MPs subcutaneous injection 100 µg/kg every 2 weeks for 14 weeks | [66] | |
| Sprague-Dawley rats | Mn3O4, spherical, 20 nm size, injected via tail vein every week 10 mg/kg/week for 60–120 days | [67] | |
| Sprague-Dawley rats | MnO2, lattice fringes shape, 50–100 nm size, subcutaneous injection with 100 mg/kg b.w. for 56 days | [68] | |
| testicular degeneration, absence of germinal cells lining, exfoliation of immature germ cells, vascular congestion, edema | |||
| Wistar rats | Mn2O3, 70 nm size, 100–400 ppm orally for 14 days | Dose-dependent: | [69] |
| Interstitial edema of seminiferous tubules and vacuolar degeneration of epithelium | |||
| Wistar rats | MnO2, 25–85 nm size, subcutaneous injection with 100 mg/kg once a week for 4 weeks | [70] | |
| Albino rats | Mn3O4, 18.4 nm size, intratracheal instillation with 0.25 mg per animal | [71] | |
| C57 BL/6 mice | Mn2O3, 51.5 nm size, 20 µg by oropharyngeal installation for 40 h | [43] | |
| Kunming mice | Mn3O4, 6.0 to 10.5 nm size, via gavage 125–250 mg/kg BW for 20 days | [72] | |
| Wistar rats | Mn2O3, 40–60 nm size, replacing MnCO3 in the diet at a dose of 65 mg Mn/kg diet for 16 weeks | Bone: | [74] |
| fibrosis, steatosis |
Given the role of Mn2+ release from MnOxNPs as one of the mechanisms of its biological activity [80], as well as neurotoxic effects of Mn2+ [28], it has been assumed that exposure and brain accumulation of MnOxNPs results in brain damage [32].
Mn3O4NPs induce cell death in dopaminergic PC12 cells by induction of oxidative stress and apoptosis through up-regulation of mitochondrial calcium (Ca2+) uniporter (MCU) and subsequent increase in mitochondrial Ca2+ concentration. The role of Ca overload in cytotoxic effects of Mn3O4NPs in PC12 cells was also confirmed by the observed inhibition of apoptosis and oxidative stress by treatment with the MCU inhibitor. Furthermore, a decrease in dopamine content in Mn3O4NPs-exposed cells and rats was mediated by downregulation of DOPA decarboxylase (DDC) expression [33]. Correspondingly, MnNPs effectively internalized by PC12 cells induce a significant decrease in cellular dopamine, DOPAC, and HVA contents, and this effect was similar to that observed upon exposure to soluble Mn2+ (Mn acetate). Although exposure to MnNPs induced only a slight mitochondrial dysfunction, it resulted in a more than 10-fold increase in ROS production, exceeding the respective values observed for Mn2+ [34]. In addition, a comparative study showed that Mn2O3NPs resulted in more profound decrease in PC12 cell viability compared to Mn3O4NPs and especially Mn5O8NPs. This effect was associated with a species-specific decrease in cellular GSH levels by Mn2O3NPs followed by Mn3O4NPs and Mn5O8NPs [81].
In another model of dopaminergic neurons, N27 cells, exposure to MnNPs also
induced cell death associated with ROS overproduction and caspase-3-mediated
proteolytic cleavage and activation of protein kinase C
Exposure of neuronal SH-SY5Y cells to MnNPs led to GSH depletion and a
significant increase in cellular ROS production due to mitochondrial dysfunction,
altogether resulting in oxidative DNA damage, chromosome fragmentation,
phosphatidylserine translocation, and caspase-3 activation, contributing to
cellular apoptosis [84]. Correspondingly, Mn2O3NPs exposure induced
both necrosis and apoptosis in a dose-dependent manner in SH-SY5Y neuroblastoma
cells. The latter was mediated by caspase-3 and caspase-9 activation and
increased Bax/Bcl-2 ratio. In addition, molecular docking and molecular dynamic
studies showed that Mn2O3NPs can bind tau protein and promote its
folding, potentially contributing to neurotoxic effects [85]. Mn3O4NPs,
especially small-sized, were shown to have more profound effects in dividing
ST-14 striatal neuroblasts due to induction of mitochondrial dysfunction and ROS
production with subsequent increase in NF-
Cellular studies also show that MnOxNPs contribute to neuroinflammation.
Specifically, in BV2 microglial cells MnO2NPs exposure resulted in a
significant increase in ROS production and p38 MAPK pathway activation,
ultimately leading to overexpression of IL-1
Taken together, the results of in vitro studies highlight cytotoxic
effects of pristine MnOxNPs in both neuronal and non-neuronal brain cells
(Table 3, Ref. [33, 34, 82, 83, 84, 85, 86, 87, 88]). The existing data show that MnOxNPs induce
neuronal death through a variety of mechanisms including oxidative stress,
mitochondrial dysfunction, altered Ca2+ homeostasis, induction of tau and
A
| Cell model | Nanoparticles | Effects | Reference |
| PC12 | Mn3O4, 25 nm size, 5–20 µg/mL for 24 h | Dose-dependent: | [33] |
| PC12 | Mn, 40 nm size, irregularly shaped, 5–50 µg/mL for 24 h | Dose-dependent: | [34] |
| N27 | Mn, 20 nm size, 25–400 µg/mL for 24 h | [82] | |
| PC12 | Mn, 52 nm size, spherical shape, 10 µg/mL for 24 h | [83] | |
| SH-SY5Y | MnO2, 40.6 nm size, 10–60 µg/mL for 24 and 48 h | Dose-dependent: | [84] |
| SH-SY5Y | Mn2O3, spherical, 30 nm size, 1–200 µg/mL for 24 h | Dose-dependent: | [85] |
| IC50 = 88.45 µg/mL | |||
| ST-14 | Mn3O4, 70–120 (A), 150–220 (B), and 200–350 (C) nm size, 1–10 µg/mL for 24 h | Dose-dependent: | [86] |
| BV2 | MnO2, 2.5–10.0 µg/mL for 12 h | [87] | |
| CATH.a and C8-B4 coculture | Mn, 65.8 nm size, irregular morphology, 10 or 50 µg/mL for 24 h | Dose-dependent: | [88] |
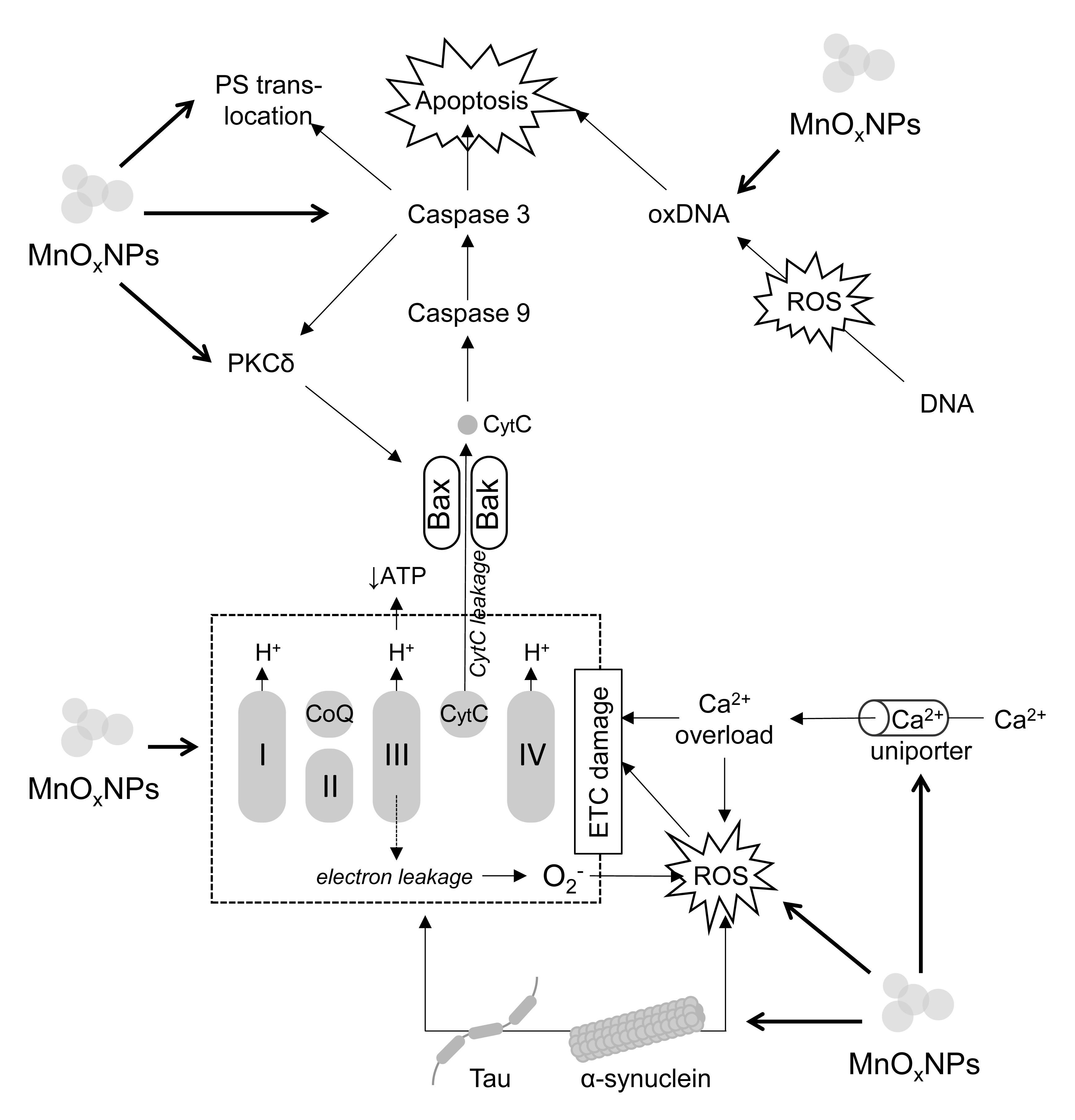
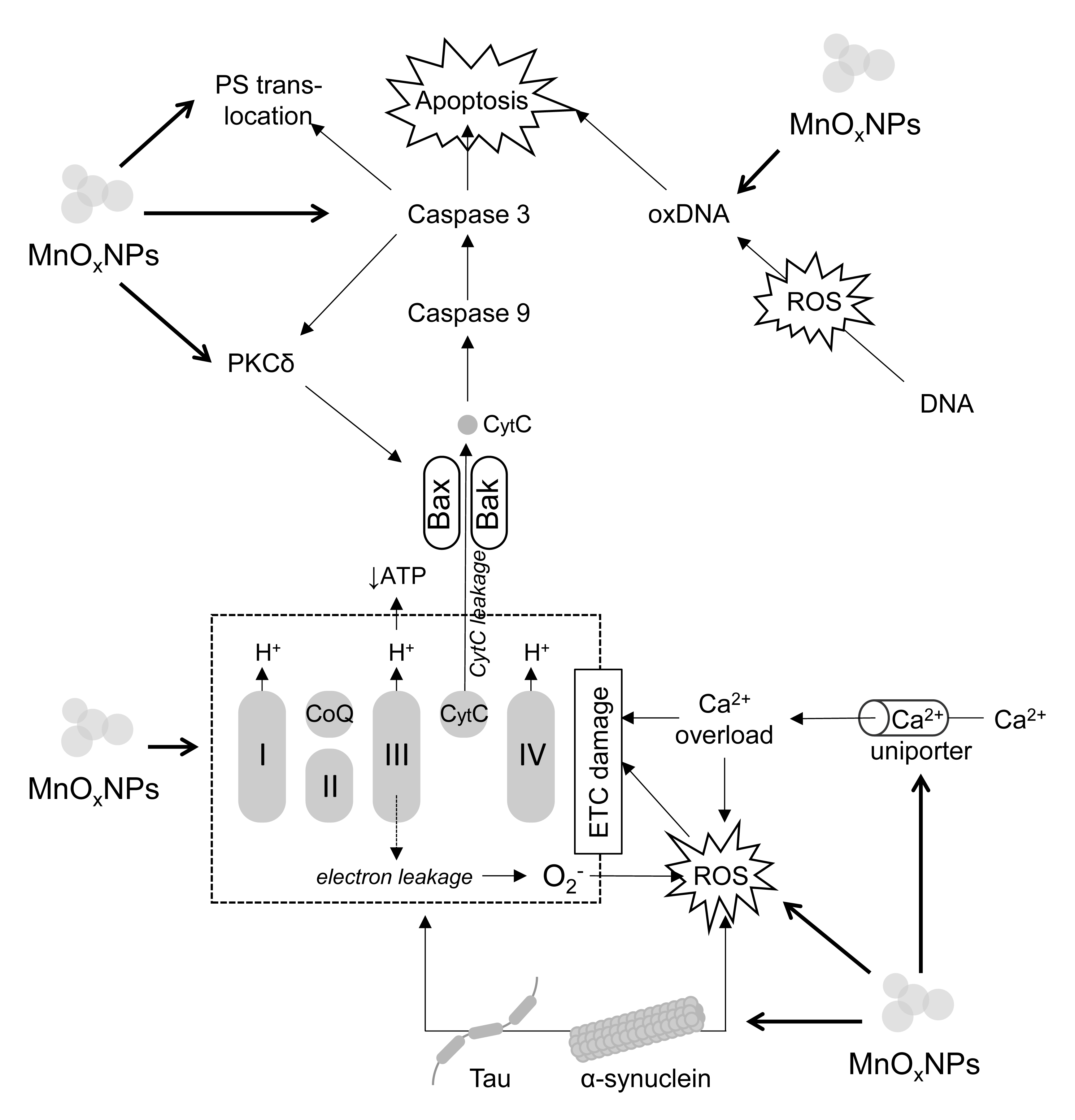 Fig. 1.
Fig. 1.
The molecular mechanisms of MnOxNPs-induced neuronal death.
Briefly, MnOxNPs exposure inhibits electron transport chain (ETC)
functioning resulting in reduction of ATP production and increased electron
leakage at complexes I and III with subsequent formation of ROS. In addition to
ROS formation, MnOxNPs increase activity of Ca2+ uniporter resulting in
mitochondrial Ca2+ overload. Both ROS overproduction and Ca2+ overload
further affect ETC functioning. MnOxNPs induce tau and
Findings from animal models generally corroborate the results of in vitro studies. Intratracheal exposure to MnO2NPs results in a more than 5-fold increase in Mn content in brain, whereas the level of Mn in the liver is elevated by less than 2-fold, indicative of the role of the brain as a target for Mn accumulation following MnNPs exposure [89]. An in vivo study demonstrated that intratracheal instillation with MnO2NPs resulted in a significant increase in both blood and brain Mn accumulation after 9 weeks of exposure [90, 91].
Elder et al. (2006) [92] demonstrated that the olfactory neuronal
pathway is the predominant mechanism of transport of MnNPs into the brain upon
inhalation. Specifically, the authors observed a more profound increase in the
olfactory bulb Mn content (3.5-fold) compared to lungs (2-fold) upon exposure to
MnNPs. Furthermore, in the exposed animals, up-regulation of TNF
Additional studies showed that exposure to MnOxNPs results in significant alteration in brain functioning. Specifically, intratracheal instillation with MnO2NPs significantly reduced ambulation and rearing with increased local activity and immobility due to increased frequencies of spontaneous cortical activity, elongation of cortical evoked potential latency, as well as decreased nerve conduction velocity [90, 91]. This effect was dependent on the size of the exposing particles. It has been shown that alterations in spontaneous cortical activity were more profound upon exposure to smaller MnO2NPs (9 and 42 nm) compared to larger (118 nm) particles [94]. Furthermore, the changes in spontaneous and evoked cortical activity and peripheral nerve action potential positively correlated not only with cortical Mn2+ content, but also cortical TBARS level, being indicative of the role of lipid peroxidation in MnO2NPs-induced neuro-functional alterations [95]. When compared to the effects of soluble MnCl2, MnNPs exposure exerted less profound effects on the latency of cortical evoked potentials and tail nerve conduction velocity [96]. However, combined exposure of MnCl2 (orally) and MnNPs (intratracheally) for 3 weeks possessed more profound effects on cortical sensory evoked potential and nerve action potential compared to 6-week exposure to MnCl2 at the same cumulative dose [97].
MnNPs-induced alterations in evoked potential latency and nerve conduction velocity were abolished by antioxidant-rich green tea, whereas other adverse effects of MnNPs exposure, like reduced body weight gain, were not reversed by green tea administration [95]. Respectively, administration of antioxidants attenuated adverse effects of MnNPs, with rutin being the most effective, whereas ascorbic acid only abolished alterations in cortical evoked potentials, and curcumin did not exert any protective effects [98]. Intraperitoneal injection of Mn3O4NPs resulted in a significant accumulation of Mn in brain, and dose-dependent reduction in the number of head-dips into holes, brain weight as well as an increase in the number of cells without a nucleolus in caudate nucleus and hippocampus (CA1) [99], which were completely abrogated by administration of a complex of antioxidants (N-acetylcysteine, vitamins A, C, E, selenium) and other bioactive compounds, including pectin, glutamate, glycine, iodide, and omega-3 polyunsaturated fatty acids [100]. These findings show that the neurotoxic effects of MnNPs depend on the prooxidant effect of the particles, although the different efficiencies of various antioxidants may be indicative of the contribution of various sources of ROS in MnNPs neurotoxicity.
Mitochondrial dysfunction is a primary source of ROS upon MnOxNPs exposure. Specifically, Mn particles induced a significant increase in mitochondrial ROS production at respiratory complexes I and III, being more profound in MnO2NP-exposed mitochondria compared to MnO2MPs. Furthermore, exposure to MnO2NPs, but not MnO2MPs, possessed inhibitory effects on complex II and IV activity in the brain [101].
Acute exposure to MnNPs induced significant neurotoxic effects characterized by depression and loss of reaction to auditory stimuli [102], which were associated with severe cerebellar damage characterized by neuronal and glial cell shrinkage and pyknosis, ischemia, and edema [103]. In addition, the route of MnOxNP exposure induces distinct changes in brain pathomorphology. Specifically, acute inhalation exposure to MnO2NPs (15–29 nm) in rats resulted in brain and especially cerebellum damage, characterized by ischemic damage to neurons and microglia, endothelial swelling in cerebral blood vessels, and neurite defects. In turn, acute oral exposure resulted in brain edema, vascular hyperemia in the brain cortex and cerebellum, neurite demyelination, as well as alterations in the granular cortex layers [104].
Exposure of adult male Wistar rats to 50–100 µg/kg MnO2NPs through intraperitoneal injection resulted in a significant increase in immobility time and reduced sucrose preference. Further analysis showed that MnO2NPs induced a significant increase in hippocampal ROS production and lipid peroxidation. These effects were also associated with neuronal apoptosis and necrosis in the hippocampus, as well as a dose-dependent decrease in hippocampal catecholamine content [105]. Surprisingly, adverse neurophysiological effects of MnNPs were attenuated when co-exposed to Fe2O3NPs, while this effect was independent of brain Mn accumulation [106].
Further, it has been shown that alterations in sensory motor functions induced by intraperitoneal MnO2NPs exposure in rats were associated with BBB damage with the resulting brain edema and reductions in blood flow in the sensory-motor cortex, hippocampus, caudate putamen, cerebellum, and thalamus, and to a lesser extent in hypothalamus, pons, medulla, and spinal cord. In addition, a dose-dependent increase in neuronal damage characterized by vacuolation, chromatolysis, and Nissl substance loss was observed predominantly in parietal and temporal cortex, hypothalamus, and thalamus, followed by other brain areas [35]. In addition, MnO2NPs-induced alterations in learning and memory are associated with hippocampal lesions characterized by edematous cells with karyopyknosis and irregular arrangement, cell connections loss, increased number of irregular cells, as well as damage to the choroid plexus, a blood–cerebrospinal fluid barrier structure. Transcriptomic analysis of the choroid plexus showed that MnNPs exposure affects expression of transporter, ion channel proteins, and ribosomal genes, including BMP/Retinoic Acid Inducible Neural Specific 1 (Brinp), synaptoporin (Synpr) and Collapsin response mediator protein 1 (Crmp1) [36].
It has also been demonstrated that MnO2NPs exposure induces
neuroinflammation due to astrocyte activation, evidenced by increased glial
fibrillary acidic protein (GFAP) immunoreactivity, which was more profound in
cerebellum, hippocampus, and thalamus [35]. Induction of astrocyte activation and
neuroinflammation with the increased number of GFAP and iNOS-positive cells upon
injection of MnO2NPs to the substantia nigra was also observed [107].
Furthermore, replacement of Mn2+ (as MnCO3) in the diet with
Mn2O3NPs induced neuroinflammation, evidenced by a significant increase
in brain TNF
Several studies show that MnOxNPs impair neurotransmitter metabolism in vivo. Specifically, injection of MnO2NPs to the substantia nigra induced a significant decrease in the number of TH-positive neurons, which was similar to that observed upon exposure to dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA) [107], indicating altered dopaminergic neurotransmission. Metabolomic analysis of brain tissues of rats intravenously injected with MnNPs showed that, irrespective of the dose and period of exposure, MnNPs exposure altered brain guanosine, phenylalanine, GABA, myo-inositol, uracil, and propionate levels. Upon short-term exposure, MnNPs at both doses also increased brain glutamate content. These findings demonstrate that exposure to MnNPs altered the glutamine–glutamate/GABA cycle in the brain [110]. Replacement of dietary MnCO3 with the respective dose of Mn2O3NPs resulted in a significant reduction in brain 5-HT levels along with an increase in norepinephrine and dopamine levels, which may be associated with gut microbiota alterations. The latter was characterized by reduced intestinal levels of SCFA, including acetate, propionate, butyrate, decreased enzymatic activity of caecal bacteria, as well as elevated ammonia concentrations and increased pH in caecum [111]. MnO2NPs-induced Mn accumulation in the brain was also associated with down-regulation of brain AChE, Na/K-ATPase, Mg2+-ATPase, and Ca2+-ATPase activities, as well as neuronal vacuolation and inflammatory cell infiltration [112].
Noteworthy, a study in iridescent shark (Pangasianodon hypophthalmus) demonstrated that MnNPs exposure resulted in a significant down-regulation of acetyl choline esterase (AChE) activity in the brain, accompanied by significant up-regulation of brain catalase, glutathione-S-transferase, and glutathione peroxidase activity with a concomitant decrease in SOD activity. Furthermore, a dose-dependent increase in hepatic cortisol level may also be indicative of altered hypothalamus-pituitary-inter-renal axis activity. Noteworthy, these effects of MnNPs were nearly similar to those of Mn2+, although the latter possessed higher toxicity (LC50= 111.75 mg/L) compared to MnNPs (LC50 = 93.81 mg/L) [113]. These findings assert the role of Mn2+ release in mediating the toxic effects of MnNPs. This is corroborated by the observed formation of stress-granules through eIF2a phosphorylation and inhibition of oxidative phosphorylation in human glioblastoma U87 MG cells, which were also induced by Mn2+ exposure [114].
Taken together, the existing data show that MnOxNPs exposure induces neurotoxicity through a variety of mechanisms (Table 4, Ref. [35, 36, 89, 94, 95, 96, 99, 101, 102, 105, 107, 108, 110, 111, 112]). Along with induction of ROS production with subsequent development of oxidative stress or overproduction of proinflammatory cytokines clearly demonstrated in cellular models, in vivo studies showed that MnOxNPs induce brain damage through blood-brain barrier damage, alterations in neurotransmitter metabolism, and dysregulation of gut-brain axis.
| Model | Exposure | Effects | Reference |
| Wistar rats | MnO2, 23.2 nm size, intratracheal instillation with 2.63–5.26 mg Mn/kg b.w. 9 weeks | [89] | |
| Wistar rats | MnO2, 9.14 (A), 42.36 (B), and 118.31 (C) nm size, intratracheal instillation with 3–6 mg/kg b.w. for 6 weeks | [94] | |
| Wistar rats | MnO2, 27.4 nm size, intratracheally instilled with 4 mg/kg b.w. for 6 weeks | [95] | |
| Wistar rats | MnO2, 23 nm size, or MnCl2 in a dose of 2.53 mg Mn per rat intranasal instillation | [96] | |
| Outbred white rats | Mn3O4, spherical, 18.4 nm size, intraperitoneally injected with 0.25–0.50 mg 3 times a week up to 18 injections | [99] | |
| C57 mice | MnO2, |
Vs MPs: | [101] |
| Wistar rats | MnOx, 15–29 nm size, inhalation with 0.029 mg/dm3 for 4 h | [102] | |
| neuronal and glial cell shrinkage and pyknosis, ischemia, and edema | |||
| Wistar rats | MnO2, 30 to 60 nm size, intraperitoneal injection with 50–100 µg/kg for 15 days | [105] | |
| Sprague-Dawley rats | MnO2, 10 nm size, intracranial injection with 1 µL of 87 µg/µL | [107] | |
| Wistar rats | MnO2, 30–40 nm size, intraperitoneal injection with 10–20 mg/kg for 7 days | [35] | |
| Sprague-Dawley rats | MnO2, 50 nm size, intratracheal injection with 200–400 mg/kg b.w. once a week for 3 months | [36] | |
| Choroid plexus: epithelium nucleus disappearance, vacuolation, impaired cell junctions | |||
| Hippocampus: edematous cells with karyopyknosis, irregular cell form and arrangement, cell connections loss | |||
| Wistar rats | Mn2O3, 40–60 nm size, 65 mg/kg replacing MnCO3for 12 weeks | Plasma: |
[108] |
| Jejunum: |
|||
| Brain: |
|||
| SD rats | MnO, 10 nm size, single intravenous injection with 10–40 mg Mn/kg b.w. | [110] | |
| Brain metabolomics (both doses): | |||
| Wistar rats | Mn2O3, 40–60 nm size, 65 mg/kg replacing MnCO3 For 12 weeks | Caecum: | [111] |
| Plasma: |
|||
| Intestine: |
|||
| Brain: |
|||
| Wistar rats | MnO2-NPs 42.63 nm size, at 30, 300, 1000 mg/kg b.w. or MnO2MPs at 1000 mg/kg single dose | [112] | |
| Brain: |
|||
| RBC: |
|||
| Liver, serum: |
|||
| Kidney: |
Collectively, current evidence indicates that MnOxNPs, despite their
biomedical applications as contrast or therapeutic agents, can exert toxic
effects across multiple organs and tissues. Laboratory findings demonstrate
hepatotoxicity, reproductive toxicity, as well as nephrotoxic and immunotoxic
effects, while the brain emerges as a primary target of MnOxNPs accumulation
(Fig. 2). Within the central nervous system, MnOxNPs trigger neuronal death
through excessive ROS generation, oxidative stress, apoptosis, glial activation,
and neuroinflammation. Specifically, MnOxNPs-induced mitochondrial
dysfunction or direct prooxidant activity of the particles result in ROS
overproduction which promotes cytochrome c release from the mitochondria and
promote DNA oxidation, thus activating apoptosis. On the other hand,
MnOxNPs-induced ROS generation, both directly and through MAPK signaling,
promotes activation of redox-sensitive transcription factor NF-
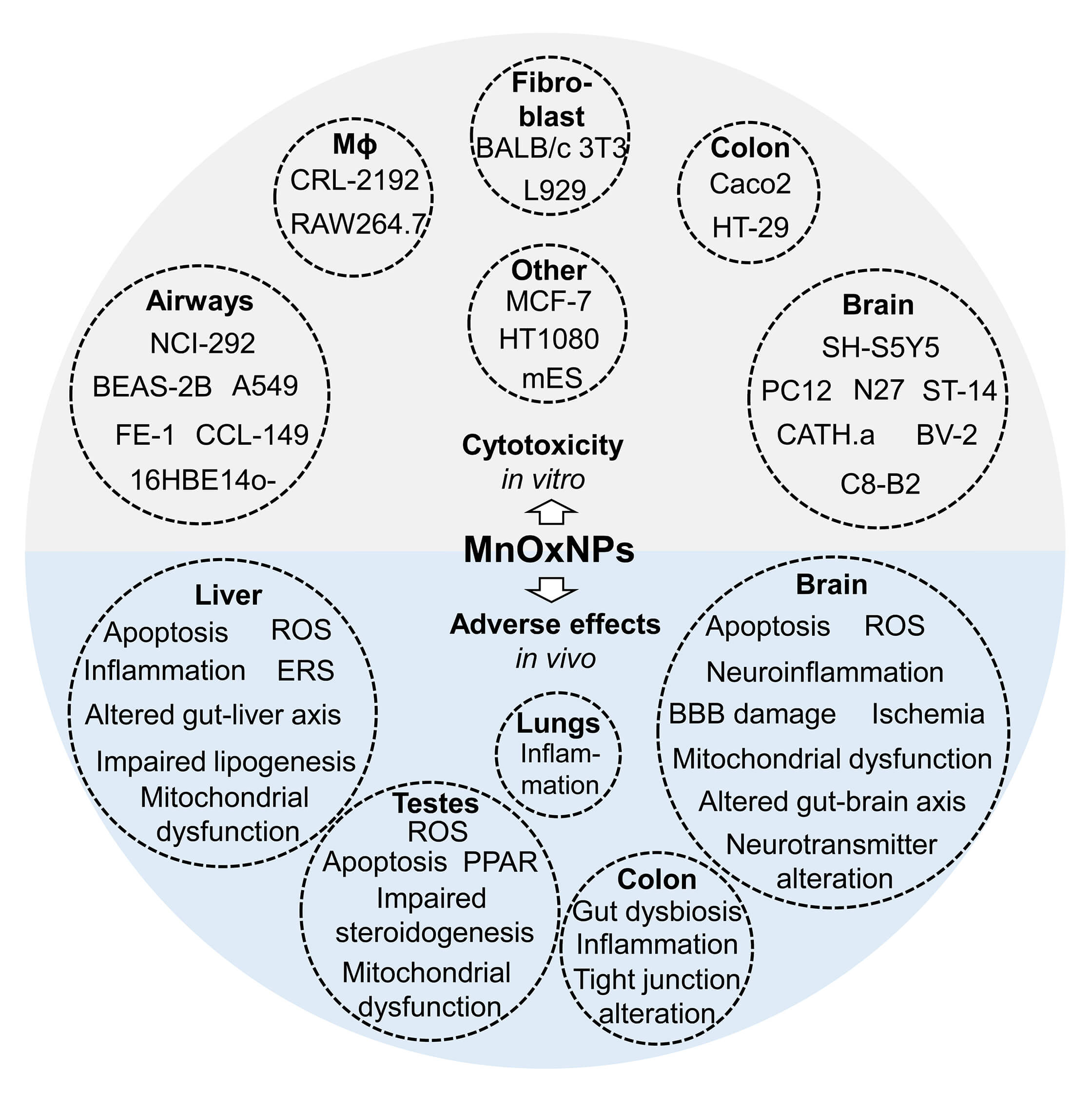
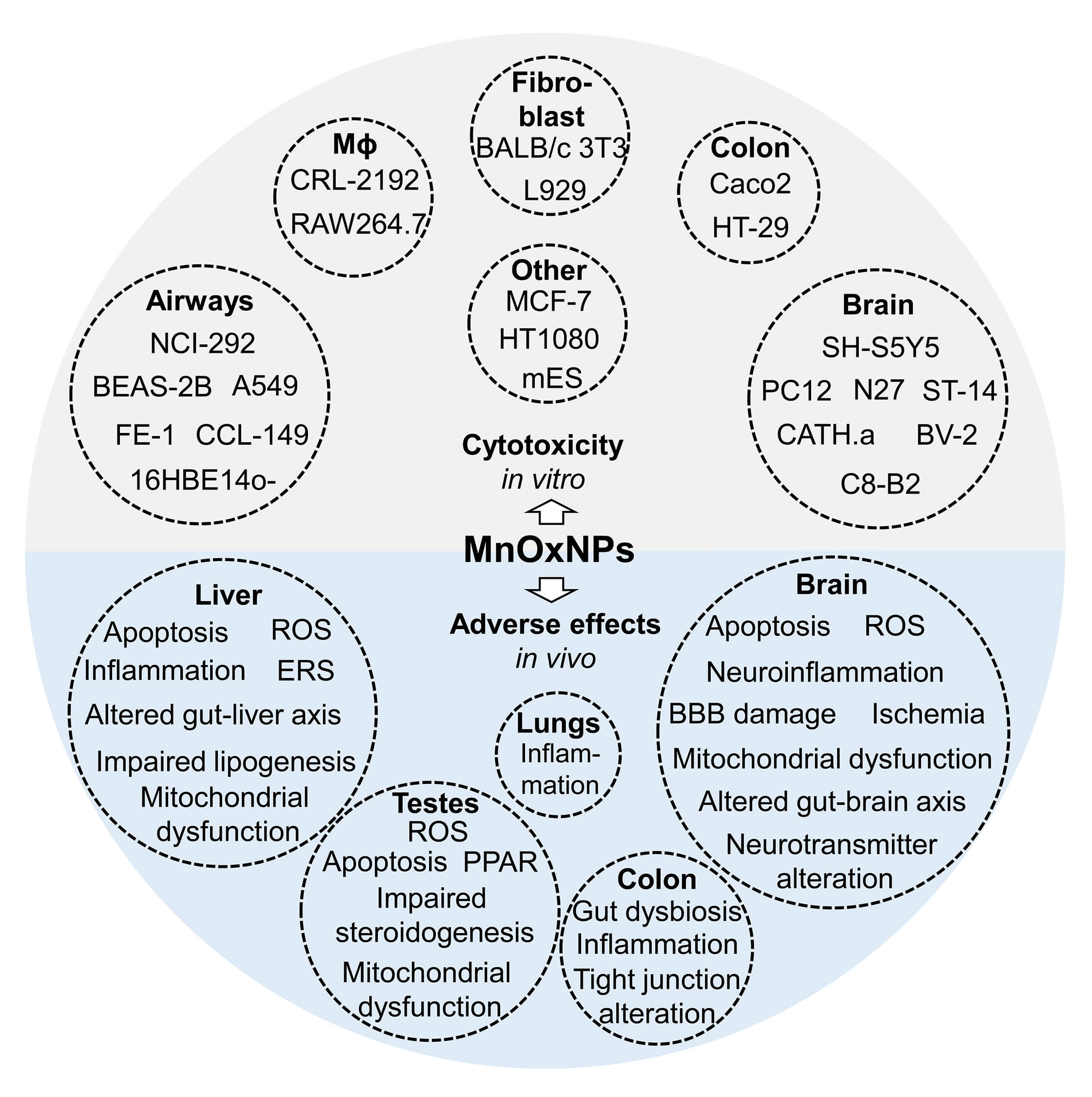 Fig. 2.
Fig. 2.
A summary of adverse effects of MnOxNPs in in vitro and in vivo models. The existing in vitro studies show that exposure to pristine MnOxNPs possesses cytotoxicity in brain, airway and lung, and colon cells, as well as macrophages, fibroblasts and other cell lines. The results from in vivo studies generally corroborate in vitro findings, demonstrating adverse effects of MnOxNPs exposure on brain, liver, testes, colon, and lungs.
Despite being limited, the certain studies show that MnOxNPs possess comparable or even higher toxicity than soluble Mn2+ compounds, being indicative of the role of not only metal release, but also particle-specific effects in MnOxNPs-induced neurotoxicity. Furthermore, small-sized MnOxNPs possess more profound toxic effects compared to larger size NPs or microparticles. The existing data show that surface modifications like silica-coating or formation of nanocomposites of MnOxNPs reduces toxicity of the particles [17]. However, comparative analysis of toxic properties of pristine and modified MnOxNPs has not been performed. Although previous findings show that the shape of metal-containing NPs significantly affects their biological activity, data from comparative analysis of toxicity of MnOxNPs of various forms are lacking, while the majority of existing studies investigated the effects of spheric particles. Along with physical characteristics of the particles, chemical composition of MnOxNPs also affects its toxic properties. Although the data are scarce and controversial, it appears that Mn2O3NPs or Mn3O4NPs are more toxic than Mn5O8 and MnO2in vitro. Finally, the way of particle synthesis also affects their toxicity, with green synthesis resulting in less toxic MnOxNPs compared to chemically synthesized particles.
Therefore, further studies are required to investigate the particular characteristics of MnOxNPs affecting its toxicity including size, shape, surface characteristics, and chemical composition. Specifically, comparative studies investigating the toxic effects of various MnOxNPs in a similar in vivo and in vitro models are highly required. Furthermore, more detailed studies investigating the molecular mechanisms of toxicity of MnOxNPs with distinct physicochemical characteristics are warranted to reveal the potential adverse effects of MnOxNPs exposure and ensure their safe application in biomedicine.
MA and AAT designed the study. AVS, SVN, MNT, RL, AAS, AS, JCZ, and AAT performed the literature search. MA, AVS, SVN, MNT, RL, AAS, AS, JCZ, and AAT wrote the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The study was supported by the Russian Ministry of Science and Higher Education, Russia (075-15-2024-550).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.