




1 Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Abstract
Each chromosome contains a centromere, the site at which the kinetochore assembles to ensure accurate chromosome segregation during cell division. Centromeric chromatin, which anchors the kinetochore, includes three core proteins: Centromere Protein A (CENP-A), CENP-B, and CENP-C. Among these, CENP-B is unique for its sequence-specific DNA binding to a 17–base pair element known as the CENP-B box within the alpha-satellite DNA. CENP-B contains an N-terminal DNA-binding domain and a C-terminal dimerization domain that together enable juxtaposition of distant CENP-B boxes and promote higher-order centromeric structure. CENP-B also interacts directly with CENP-A and CENP-C, thereby facilitating kinetochore assembly. The CENP-B box includes two CpG dinucleotides that, when methylated, reduce CENP-B binding and limit recruitment of CENP-A and CENP-C. The recently completed human genome assembly (T2T-CHM13) revealed centromeric regions with low CpG methylation, termed centromere dip regions, that coincide with active, unmethylated CENP-B boxes. The uniform density of these unmethylated sites across chromosomes contributes to balanced kinetochore–spindle attachment. The CENP-B gene shows no pathogenic alterations in the American Association for Cancer Research (AACR) GENIE cancer cohort (211,526 patients), underscoring its conserved role in chromosome stability.
Keywords
- human genome
- chromosome
- centromere
- kinetochore
Centromeres are chromosomal regions that define where kinetochores assemble to mediate chromosome movement during cell division. They are characterized by arrays of 171-bp alpha-satellite DNA repeats and a specialized chromatin structure enriched in the histone H3 variant Centromere Protein A (CENP-A) [1, 2, 3, 4, 5, 6, 7]. The centromeric chromatin forms the foundation of the kinetochore and includes three essential proteins—CENP-A, CENP-B, and CENP-C—that together recruit additional centromere-associated complexes (CENP-HIKM, CENP-LN, CENP-OPQUR, and CENP-TWSX) to form the constitutive centromere-associated network (CCAN) [8, 9, 10, 11]. The CCAN then anchors the outer kinetochore KMN network (KNL1C, MIS12C, and NDC80C), which connects to spindle microtubules to drive chromosome segregation.
Despite decades of study, the repetitive nature of alpha-satellite DNA hindered precise mapping of centromeres until recently. This limitation obscured understanding of how CENP-A, CENP-B, and CENP-C coordinate to ensure faithful segregation. With advances such as the T2T-CHM13 genome assembly, these questions can now be addressed in detail. This review summarizes current insights into the roles of CENP-A, CENP-B, and CENP-C in centromere organization and kinetochore function, with particular emphasis on CENP-B.
The CENP-B gene, located on chromosome 20p13, encodes a 65.2-kDa protein of 599 amino acids [12] (Table 1). CENP-B is distinctive among centromeric proteins as it binds DNA directly in a sequence-specific manner. Its N-terminal helix-loop-helix motif (amino acids 1–125) mediates DNA binding, while its C-terminal dimerization domain (amino acids 540–599) enables protein dimerization [13, 14] (Fig. 1). The protein recognizes a 17-bp sequence, the CENP-B box (YTTCGTTGGAARCGGGA) (Y = Cytosine / Thymine; R = Guanine / Adenine) within centromeric alpha-satellite DNA [15, 16]. The presence of the underlined nine-nucleotide core recognition motif is critical for binding. Through dimerization, CENP-B can juxtapose two distant CENP-B boxes, facilitating the formation of higher-order chromatin structures and compact centromeric organization via DNA looping and protein-protein interactions [13, 17] (Fig. 2, Ref. [18]). This intrinsic ability to bridge distant DNA sites positions CENP-B as a key architectural factor that prepares centromeric chromatin for subsequent epigenetic and structural regulation.
| Gene | Chromosome locus | Size (Amino Acids) | Molecular mass (kDalton) |
| CENP-A | 2p23.3 | 140 | 16.0 |
| CENP-B | 20p13 | 599 | 65.2 |
| CENP-C | 4q13.2 | 943 | 106.8 |
CENP, Centromere Protein.
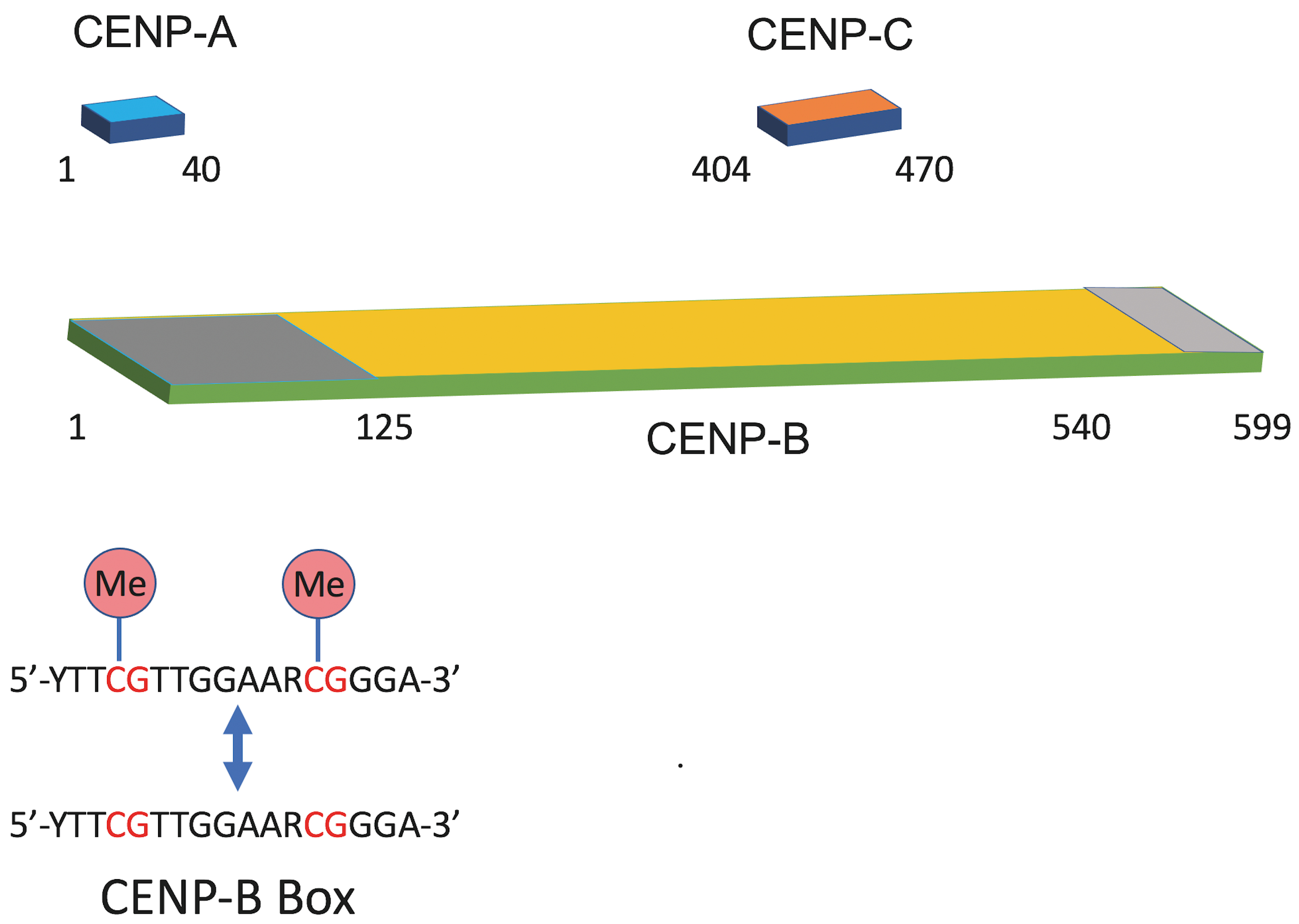 Fig. 1.
Fig. 1.
Domain organization of CENP-A, CENP-B, and CENP-C and sequence features of the CENP-B box. Schematic representations illustrate the relative positions of functional domains within centromere proteins. CENP-A contains an N-terminal domain (residues 1–40). CENP-C is shown with its annotated domain spanning residues 404–470. CENP-B includes an N-terminal DNA-binding domain (dark grey, residues 1–125) and a C-terminal dimerization domain (light grey, residues 540–599). The consensus CENP-B box sequence (5′-YTTCGTTGGAARCGGGA-3′) is shown below, highlighting the two Cytosine Guanine (CG) dinucleotides (red) that can undergo cytosine methylation (Me), which can influence CENP-B binding.
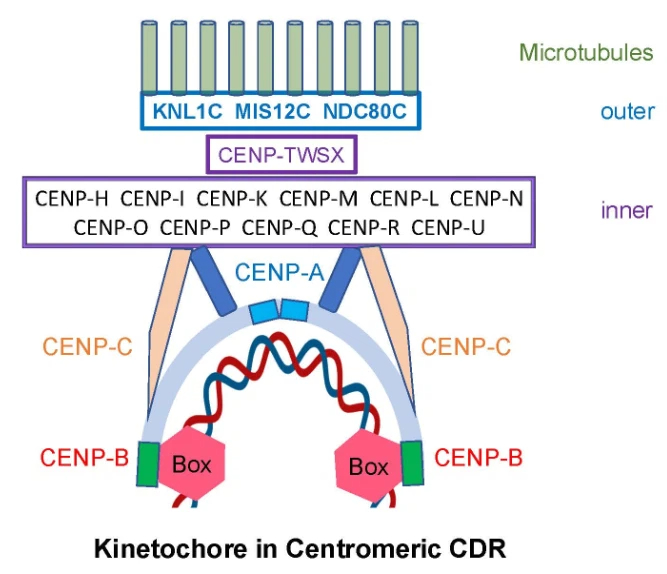 Fig. 2.
Fig. 2.
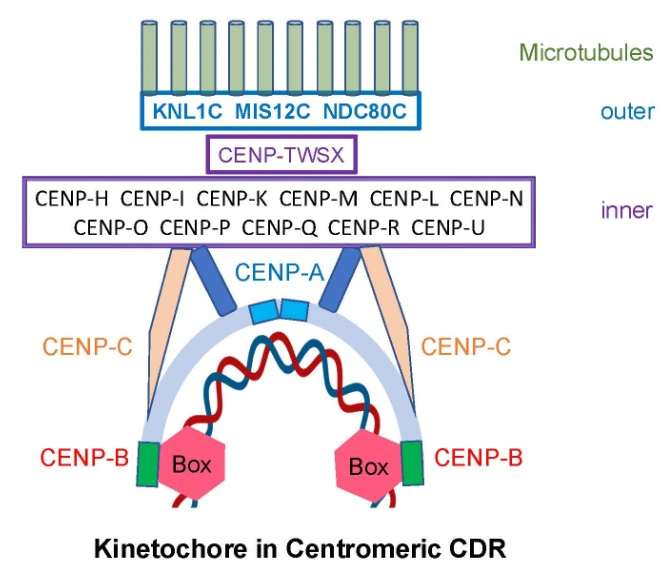
Schematic model of human kinetochore with cornerstone of CENP-B protein
and anchor of unmethylated CENP-B boxes in centromeric dip region (CDR). CENP-B
dimer binds to two CENP-B boxes (red hexagons) within
Kinetic analyses determined a dissociation constant (Kd) of 6.3
| Protein | DNA | Kd (M) | Reference |
| CENP-B | CENP-B box | 6.3 × 10–8 | [19] |
| MSH2/MSH3 | 50-mer Oligonucleotide Duplex | 7.7 × 10–8 | [20] |
| c-Myb | Myb Binding Site | 3.8 × 10–9 | [21] |
| ER alpha | Estrogen Response Element (ERE) | 2.4 × 10–10 | [22] |
| p53 | p53 Consensus Binding Site | 3.1 × 10–11 | [23] |
CENP-B forms homodimers that bind two CENP-B boxes, producing a tetrameric complex [17]. The crystal structure of the N-terminal DNA-binding region (residues 1–129) in complex with the CENP-B box DNA was resolved at 2.5 Å [24]. The domain comprises two helix-turn-helix motifs bound to adjacent major DNA grooves. The DNA adopts a ‘kink-straight-kink’ conformation, with a total bend of 59°, largely due to phosphate bridging by an arginine-rich helix. This unique geometry likely contributes to centromere-specific chromatin architecture.
CpG methylation within the CENP-B box can interfere with binding. Modeling of the CENP-B (1–129)–methylated DNA complex showed that methyl groups on cytosines cause steric clashes with CENP-B residues Thr44 and Arg125 [25]. The short alpha-helix (residues 120–129) includes four arginines (Arg125, 127, 128, 129) that penetrate the DNA major groove. Arg125 specifically forms a hydrogen bond with guanine 16, recognizing the second CpG dinucleotide, while the other arginines bind phosphate backbones. CpG methylation disrupts this precise interaction, reducing binding specificity.
The dimerization domain (residues 540–599), solved at 1.65 Å, consists of two antiparallel alpha-helices forming a symmetric four-helix bundle with extensive hydrophobic contacts [13]. The monomeric domain is unstable in solution, as its hydrophobic surface (~34% of total area) would be solvent-exposed. The N-terminal loops of the dimer project outward, ideally positioned to engage two separate CENP-B boxes through the DNA-binding domains. This structural configuration supports its role in linking distant centromeric DNA sites and promoting chromatin compaction. Together, these structural insights provide a mechanistic basis for how CENP-B integrates DNA sequence recognition with higher-order centromeric architecture.
A short inverted repeat (TTGGAA) within the CENP-B box contains CpG dinucleotides at both ends, which can be methylated (Fig. 1). This methylation significantly reduces CENP-B’s binding affinity to its box—almost to the level of nonspecific DNA interaction—since CENP-B preferentially binds to unmethylated DNA [25]. Thus, CpG methylation establishes an epigenetic switch that modulates whether CENP-B can contribute to chromatin organization, linking sequence-specific binding to broader regulatory programs.
Human centromeres consist of 1500–30,000 copies of imperfectly repeated alpha-satellite DNA sequences [26]. Until 2022, their repetitive nature hindered complete sequencing. The Telomere-to-Telomere (T2T) Consortium resolved this by mapping all human centromeres, including their epigenetic features, in the T2T-CHM13 reference genome [27, 28].
Using these data, the locations of CENP-B boxes were identified as kinetochore anchor points [18]. Notably, each centromere contains a single hypomethylated region called the centromeric dip region (CDR), which colocalizes with CENP-A and CENP-B enrichment [27, 28]. Most CENP-B boxes are methylated except those within the CDR, where CENP-B dimers bind to adjacent unmethylated boxes. These unmethylated regions likely play a crucial role in kinetochore assembly. This epigenomic organization provides a functional transition from CENP-B’s molecular properties to its chromosome-level role in defining the kinetochore-competent domain.
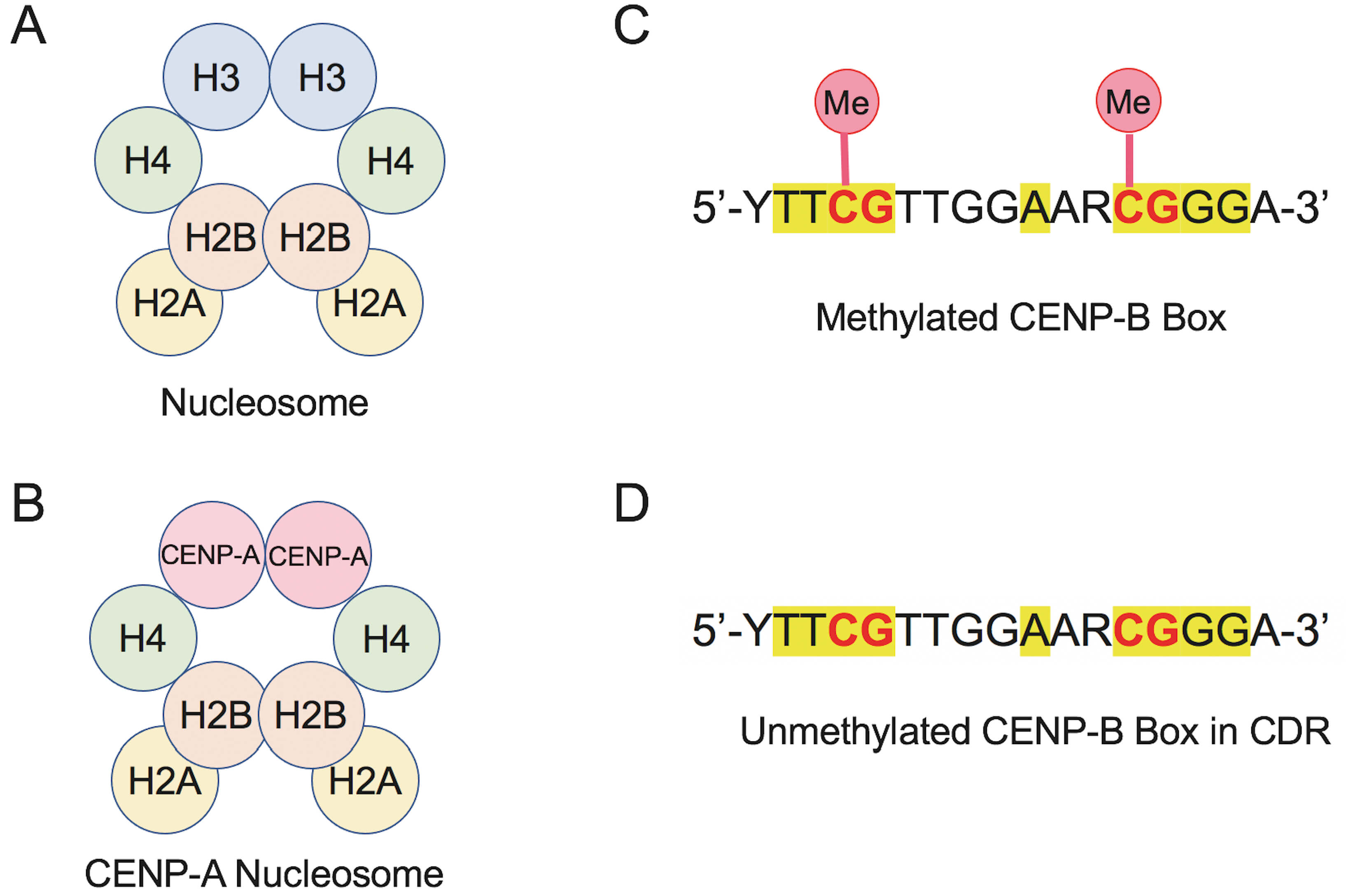
Two key epigenetic processes define human centromeres: histone modification and DNA methylation [1, 18, 25, 29]. The former involves replacement of canonical histone H3 with CENP-A, which establishes the foundation for centromere-specific chromatin, while the latter regulates CENP-B binding through methylation status of the CENP-B box. Hypomethylation within the CDR enhances CENP-B binding and supports kinetochore formation, illustrating how DNA sequence and epigenetic state cooperate to define centromere identity (Fig. 3A–D). Together, these mechanisms create a chromatin environment that enables subsequent recruitment of key kinetochore proteins.
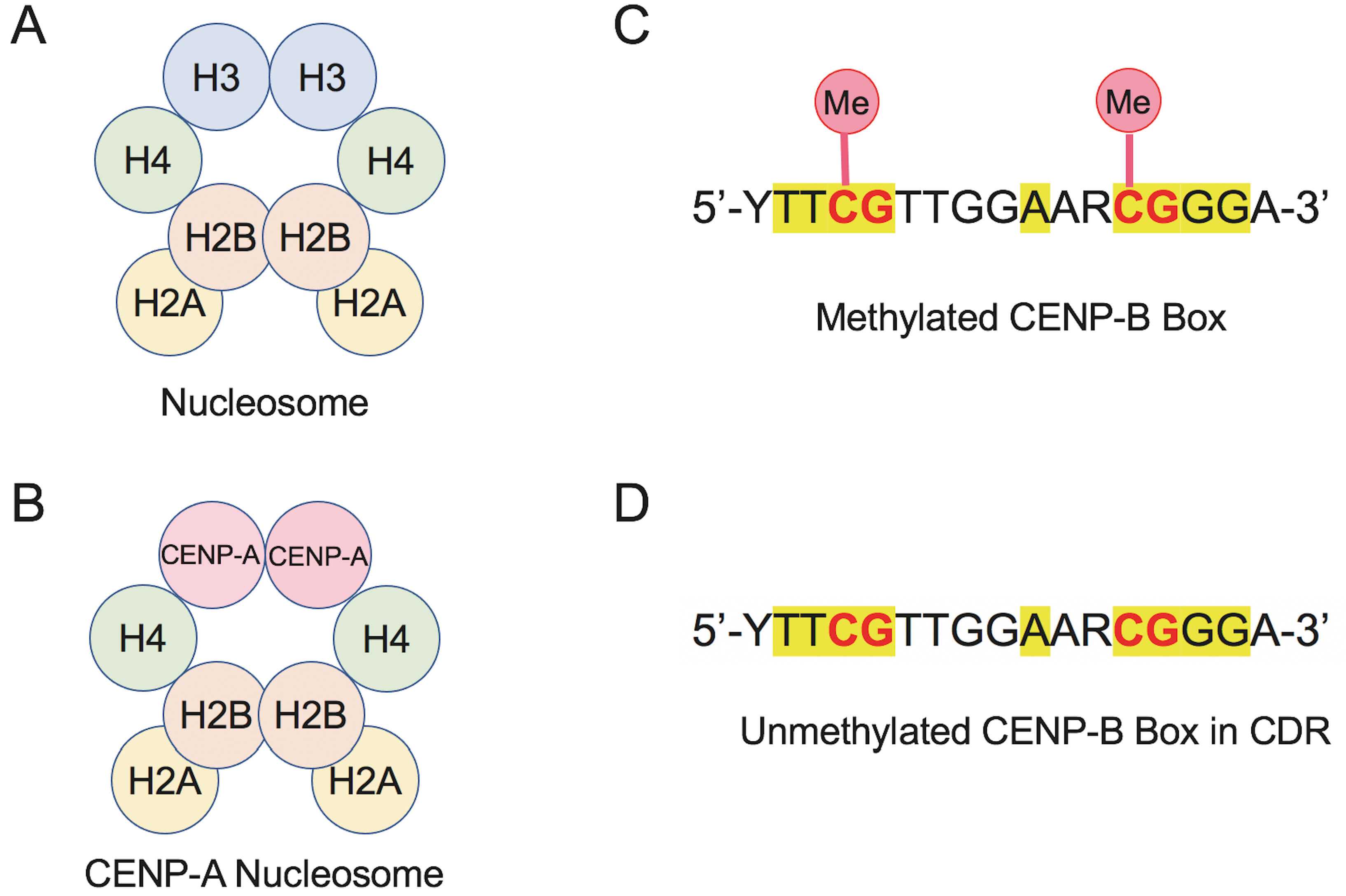 Fig. 3.
Fig. 3.
Comparison of canonical and CENP-A nucleosomes and methylation states of the CENP-B box to illustrate epigenetic mechanisms in human centromeres. (A) Schematic of the canonical nucleosome containing histones H3, H4, H2A and H2B. (B) CENP-A nucleosome in which canonical H3 is replaced by the centromere-specific histone variant CENP-A. (C) Sequence of the methylated CENP-B box, highlighting CpG dinucleotides (yellow) with cytosine methylation (Me), which reduces CENP-B binding. (D) Unmethylated CENP-B box variant found in centromeric dip region (CDR), retaining CpG motifs but lacking cytosine methylation, which increases the binding affinity for CENP-B protein.
CENP-A, CENP-B, and CENP-C play central roles in chromosome segregation. Binding of CENP-B dimers to adjacent unmethylated CENP-B boxes in centromeric DNA initiates kinetochore assembly. These boxes act as anchors, positioning CENP-B as a cornerstone of the kinetochore by directly linking CENP-A and CENP-C [26] (Figs. 1,2). This anchoring function provides a mechanistic bridge between the epigenetic landscape described above and the structural organization of the kinetochore. The interaction between CENP-B and CENP-A occurs through their N-terminal tails, while CENP-B binds to CENP-C via regions homologous to the human CENP-C central domain and CENP-C motif [26, 30]. These coordinated interactions ensure that epigenetically defined centromeric chromatin is translated into a stable and functional kinetochore complex.
The kinetochore is a large protein complex connecting centromeric DNA to spindle microtubules during mitosis and meiosis, ensuring proper genome segregation [29]. Its primary function is to form stable, load-bearing attachments between sister chromatids and spindle fibers, transmitting the pulling forces generated by depolymerizing microtubules to separate chromatids toward opposite centrosomes during anaphase [29, 31, 32].
Classical micromanipulation experiments using calibrated glass needles measured
the maximum force exerted by the spindle on a single chromosome at 7
Across all human chromosomes, centromeres span about 407 Mb, with kinetochores
comprising 5.0 Mb (1.2%) [27, 28, 37]. Although there is no correlation between
centromere and kinetochore size (p = 0.77), the number and density of
unmethylated CENP-B boxes—anchoring kinetochores to centromeric DNA—are
crucial for uniform spindle mechanics. While box numbers vary four-fold among
chromosomes, their density varies less than two-fold, averaging 2.61
Because CENP-B engages directly with centromeric DNA and contributes to kinetochore positioning, this uniformity in CENP-B cassette density produces a regularized and predictable array of anchor points for kinetochore formation. In turn, it supports the generation of consistent spindle traction forces across chromosomes of very different sizes and repeat architectures. By minimizing heterogeneity in the number of microtubule attachment sites per unit DNA, centromeres avoid imbalances in spindle pulling strength that could otherwise distort kinetochore geometry, increase merotelic attachments, or elevate segregation error rates.
Thus, the uniform density of unmethylated CENP-B boxes acts as a force-balancing mechanism: it standardizes kinetochore-DNA coupling strength across the karyotype, ensuring that each chromosome experiences comparable spindle forces despite inherent structural differences. This design principle likely represents an evolutionary solution to mitigate mechanical asymmetry and maintain robust genome transmission during mitosis.
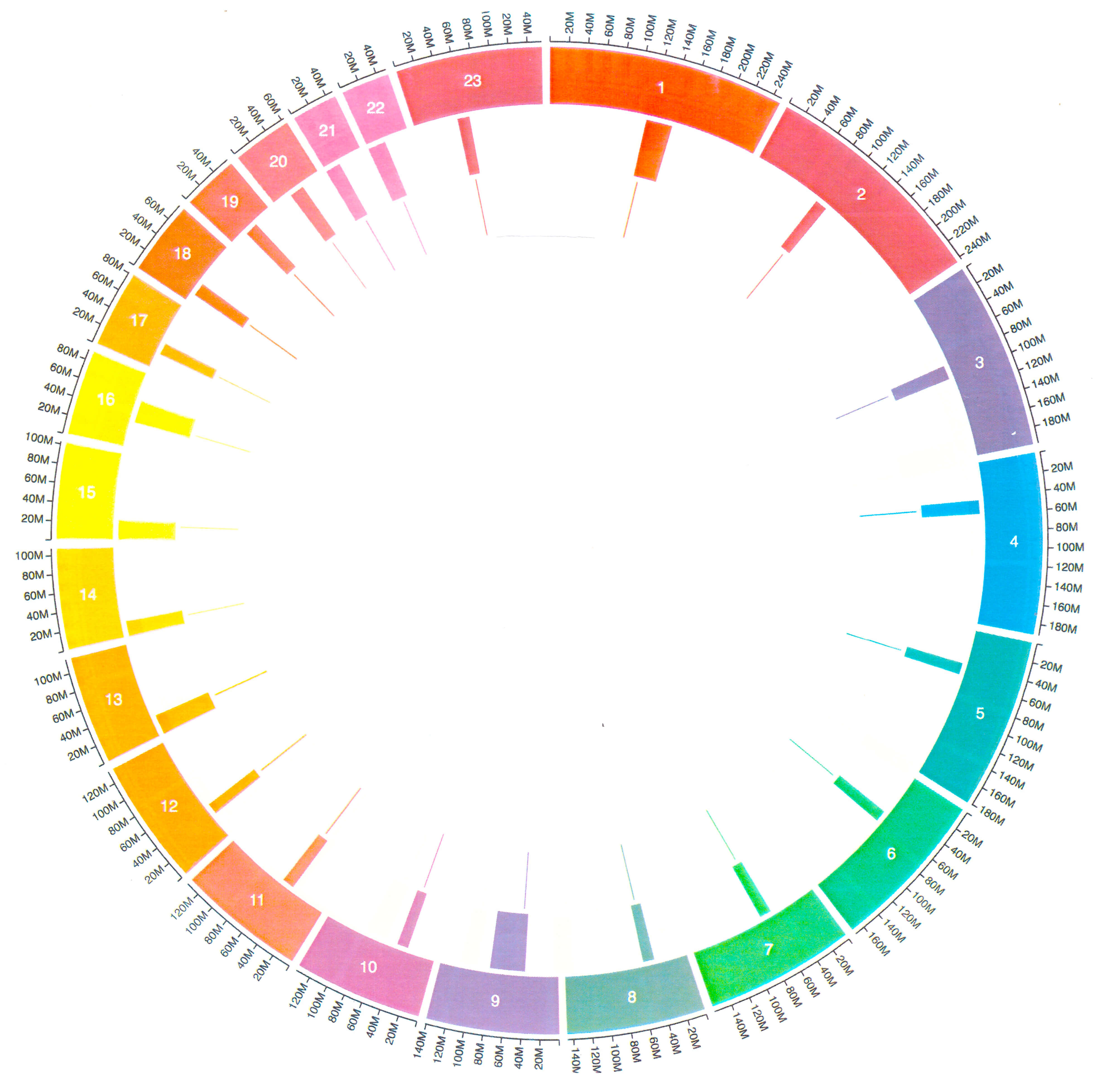
Circos plots illustrate kinetochore positions within centromeres of human chromosomes (Fig. 4, Ref. [18]). Although overall patterns are inconsistent, acrocentric chromosomes (13–15, 21, 22) show kinetochores positioned asymmetrically within centromeres, displaced away from telomeres.
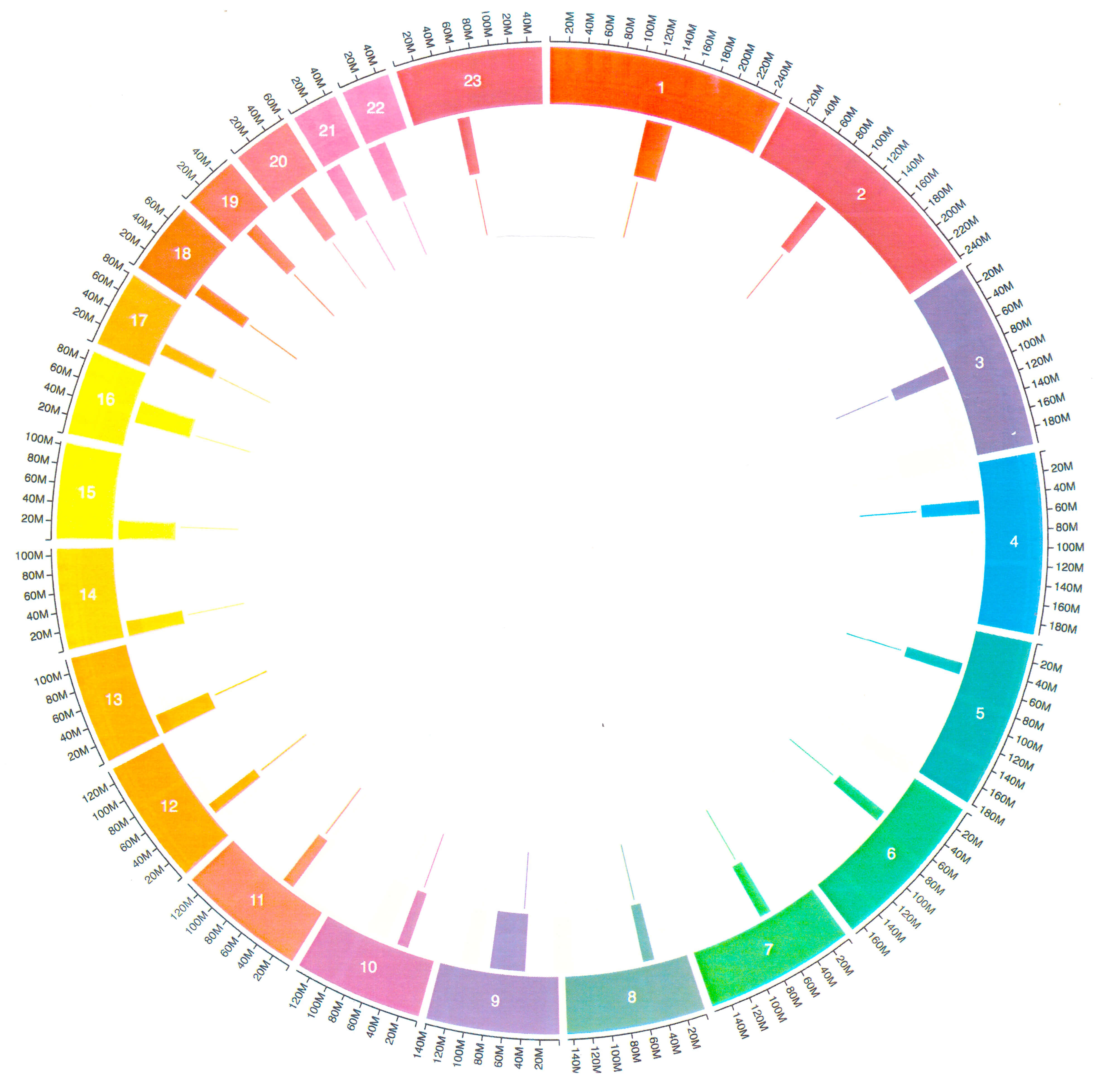 Fig. 4.
Fig. 4.
Circos plots illustrating centromeric and kinetochore-associated features across human chromosomes. Circular genomic maps display chromosomes 1–5 (top), 6–12 (middle), and 13–23 (bottom), with each chromosome represented as a color-coded arc in the outer track. Centromeres (middle track) and kinetochores (inner track) are indicated by inward-facing blocks, while outward tick marks denote positional labels along each chromosome. The plots highlight the organization and relative positioning of centromeres and kinetochores across chromosome groups, enabling visual comparison of variation in centromeric structure throughout the human genome. Figure reprinted with permission from [18].
In addition to the CENP-B box, human alpha satellite DNA contains a second
conserved sequence, the pJ
The pJ
Cancer cells often display an abnormal number of chromosomes—aneuploidy—a condition linked to centromere instability and chromosome segregation errors [40]. Given that CENP-B is crucial for accurate chromosome segregation, researchers have investigated whether alterations in the CENP-B gene contribute to aneuploidy and cancer development [42, 43].
To explore this, I examined the CENP-B gene in the American Association for Cancer Research (AACR) Genie Cohort, the largest publicly available cancer database, accessed through cBioPortal [44, 45, 46]. Version 18.0 includes 211,526 patients and 250,018 tumor samples analyzed for genetic alterations such as point mutations, copy number variations, and structural variants [47]. Remarkably, the dataset revealed a complete absence of genetic alterations in the CENP-B gene across all samples (Table 3).
| Gene | GENIE AACR Cohort | |
| 211,526 patients | 250,018 samples | |
| CENP-A | 230 ( |
239 ( |
| CENP-B | 0 (0%) | 0 (0%) |
| CENP-C | 1 ( |
1 ( |
AACR, American Association for Cancer Research.
The complete absence of CENP-B mutations, copy-number changes, or structural variants in the AACR GENIE cohort stands out as exceptionally rare for a protein involved in chromosome segregation, especially given the widespread aneuploidy characteristic of cancer. This finding strongly suggests that CENP-B is highly intolerant to genetic disruption.
Several biological principles may explain this intolerance.
CENP-B binds directly to the CENP-B box within centromeric alpha-satellite DNA, contributing to nucleating centromere structure, reinforcing kinetochore positioning, and stabilizing centromere–microtubule interfaces. Because centromere integrity must be preserved on every chromosome in every cell division, even partial loss of CENP-B function may be incompatible with long-term cell survival. Many cancers tolerate extensive chromosomal chaos—but only to a threshold. Loss of a core centromeric architectural protein may push cells beyond that tolerance, resulting in catastrophic mitotic failure rather than tumor evolution.
Most cancer-driving mutations are positively selected, while neutral or deleterious ones can accumulate. The complete absence of CENP-B mutations suggests: CENP-B loss may cause early cell death, preventing a clone from expanding enough to appear in tumor datasets. Mutant cells may undergo mitotic catastrophe, apoptosis, or be competitively outcompeted by cells with intact centromeres. Thus, CENP-B may be under purifying selection even within cancer, a context where genomic instability is often tolerated.
CENP-B contains a DNA-binding helix–turn–helix domain, a dimerization domain,
and a centromere-targeting domain. Mutations in any of these regions could
disrupt DNA recognition, protein–protein interactions, or kinetochore linkage.
Such multidomain proteins often exhibit ultra-low mutational tolerance because
small amino acid changes can propagate structural breakdown across domains. This
helps explain why CENP-A and CENP-C show rare mutations (
CENP-B boxes—when unmethylated—provide consistent anchoring points that normalize spindle traction across chromosomes. A mutation that disrupts either binding affinity or dimer stability could alter spacing or stability of kinetochore attachment sites, produce asymmetric spindle forces, and increase merotelic attachments or lagging chromosomes. Cancer cells already operate near the edge of mitotic tolerance; losing CENP-B may push them into unsustainable levels of instability.
Some centromere proteins (e.g., CENP-A) have partial redundancy through chaperones and deposition machinery. In contrast CENP-B is the only protein that binds a defined DNA motif within alpha-satellite and it plays unique roles in organizing higher-order centromeric arrays. This lack of functional redundancy means that no other protein can compensate for partial or complete CENP-B dysfunction, making mutations more deleterious.
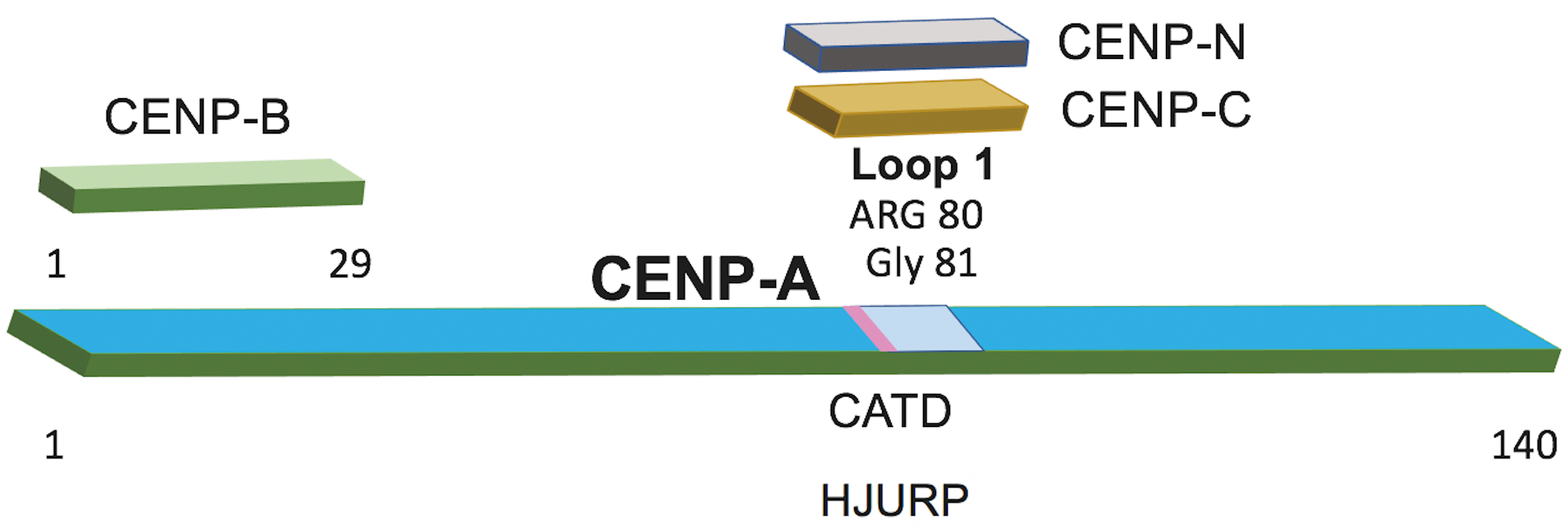
The CENP-A gene, located at 2p23.3, encodes a 16-kDa protein of 140 amino acids (Table 1) [48, 49]. Biochemical studies have shown that CENP-A is a variant of histone H3, one of the four core histones (H2A, H2B, H3, H4) present in nucleosomes. The C-terminal two-thirds of CENP-A comprise the histone-fold domain, which shares over 60% sequence identity with canonical histone H3, while the N-terminal one-third is unique [50, 51] (Fig. 5). This histone-fold domain is essential for targeting CENP-A to centromeric regions [51, 52]. CENP-A is the only centromere-specific histone variant, marking centromeric chromatin in most eukaryotes [53]. Thus, centromeres are specialized chromatin domains defined by the enrichment of CENP-A, which replaces histone H3 in centromeric nucleosomes [1, 29]. CENP-A nucleosomes are interspersed with canonical H3 nucleosomes containing histone modifications (e.g., H3K4me2), together forming the centromeric structure [5, 54]. This mosaic organization provides the molecular backdrop for the specialized structural and regulatory roles that CENP-A nucleosomes play in shaping functional centromeres.
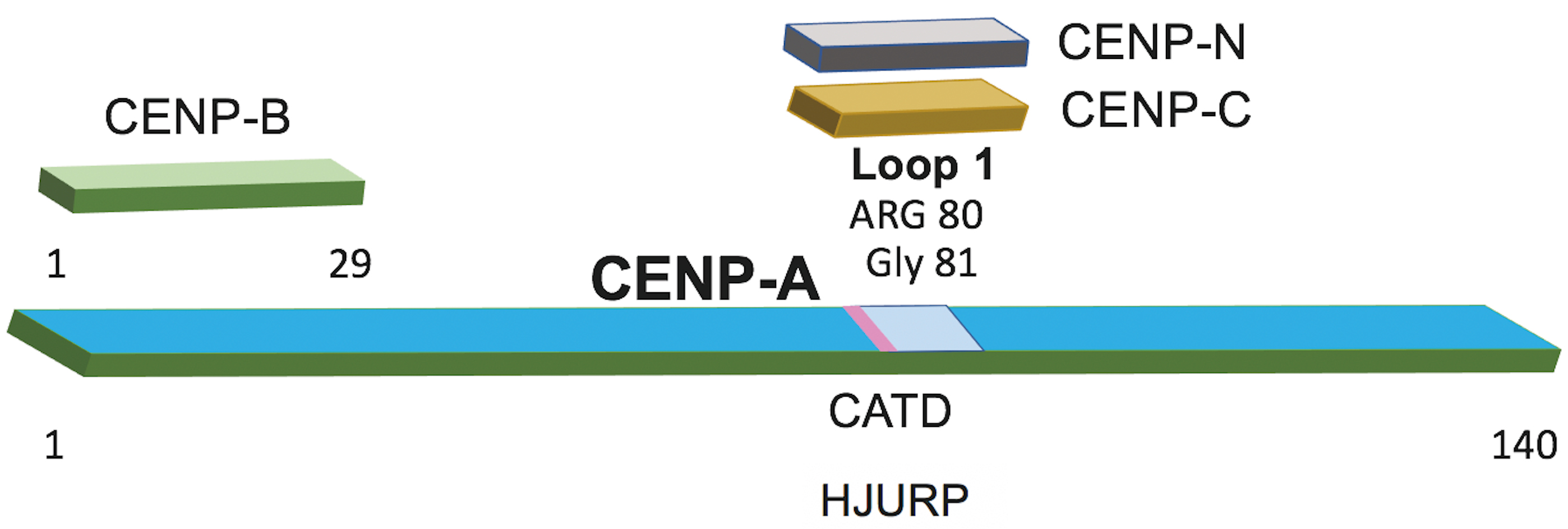 Fig. 5.
Fig. 5.
Domain architecture of CENP-A and its interactions with centromeric proteins. A schematic representation of CENP-A (residues 1–140) indicating regions required for binding to centromere-associated factors. The N-terminal segment (residues 1–29) corresponds to the CENP-B–interacting region. Loop 1 (pale blue), containing Arg80 and Gly81, mediates contacts with CENP-C and CENP-N. The CENP-A targeting domain (CATD; pink) marks the region recognized by the CENP-A chaperone HJURP and is essential for centromere-specific deposition of CENP-A.
The crystal structure of human CENP-A bound to its alpha-satellite DNA (147 bp)
has been resolved [55]. The DNA is wrapped around a histone octamer composed of
H2A, H2B, H4, and CENP-A, in a left-handed orientation. Unlike canonical H3
nucleosomes, only the central 121 base pairs of DNA are visible, while the
terminal 13 base pairs at each end are detached from the histone surface. Two
regions of CENP-A differ structurally from H3 [56]: the N-terminal
CENP-A interacts with CENP-B and CENP-C. The N-terminal tail (residues 1–29) mediates binding to CENP-B [26], while the RG-loop interacts with CENP-C [58]. CENP-A also recruits CENP-N and the Holliday junction recognition protein (HJURP) during centromeric chromatin assembly. Loop 1 of CENP-A binds both CENP-C and CENP-N in a cell cycle-dependent manner [59]. This loop alternates between concealed (compacted chromatin) and exposed (open chromatin) states, enabling CENP-N recruitment during the G1/S transition before cell division. These dynamic interactions position CENP-A as the central coordinator of chromatin architecture and protein recruitment at the centromere.
HJURP acts as a key assembly factor for CENP-A deposition at centromeres [61, 62]. The CENP-A-targeting domain (CATD), encompassing loop 1 and the adjacent
two-helix region, is critical for HJURP binding. An amino-terminal fragment of
HJURP is sufficient to assemble CENP-A nucleosomes in vitro, confirming
its role as a chromatin assembly factor [63]. HJURP directly binds
Mis18
Cyclin-dependent kinase 1 (CDK1), the master regulator of mitosis,
phosphorylates both CENP-A and HJURP to control centromere assembly timing [67].
CENP-A is phosphorylated at Ser68 during early mitosis by CDK1 [68], disrupting
its interaction with HJURP and preventing premature loading. Concurrently,
CDK1-mediated phosphorylation of HJURP weakens its binding to Mis18
To assess whether CENP-A plays a role in cancer, I examined the gene in the AACR
Genie Cohort, Version 18.0 [47]. CENP-A alterations were identified in 230 of
211,526 patients (
The CENP-C gene is located at 4q13.2 [69]. It encodes a 106.8-kDa protein composed of 943 amino acids (Table 1). This protein is larger than other kinetochore components such as CENP-A and CENP-B. Its functional size doubles through dimerization, allowing it to act as a structural scaffold that connects multiple kinetochore elements, including CENP-A, CENP-B, and CCAN components in the inner kinetochore, as well as MIS12C and NDC80 in the outer kinetochore (Fig. 2). Through this extensive connectivity, CENP-C forms the principal architectural bridge between centromeric chromatin and the microtubule-binding machinery.
CENP-C dimerization is mediated by the cupin domain at its C-terminal end (Fig. 6). This domain has a characteristic
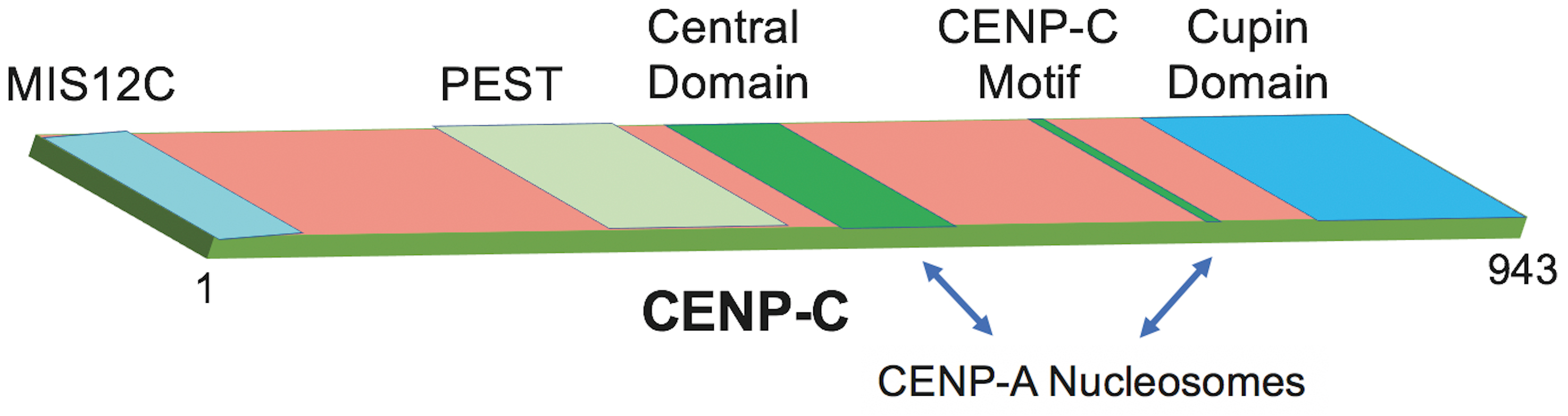 Fig. 6.
Fig. 6.
Domain architecture of CENP-C and regions required for CENP-A nucleosome engagement. Schematic representation of CENP-C (residues 1–943) highlighting major structural and functional domains. The N-terminal MIS12C-interacting region (pale blue) is followed by a PEST sequence (pale green) and a central domain (green). The conserved CENP-C motif (green) lies C-terminal to the central domain and contributes, together with the adjacent cupin domain (blue), to binding CENP-A nucleosomes, as indicated by arrows. The cupin domain additionally mediates CENP-C dimerization, supporting kinetochore assembly.
Human CENP-C contains two CENP-A binding regions: the central domain (amino acids 426–537) and the CENP-C motif (amino acids 735–755), both interacting with the C-terminus of CENP-A [58, 72]. This interaction is strengthened by CDK1-mediated phosphorylation at T734, a key mitotic regulator [73]. CENP-C also interacts with CENP-B via conserved Mif2-related regions homologous to those in budding yeast [26, 30]. These multivalent binding interfaces enable CENP-C to function as an integrator of centromeric signals and structural elements.
CENP-C organizes CENP-A nucleosomes and recruits components of the constitutive
centromere-associated network (CCAN) [72]. Its PEST-rich region (proline,
glutamate, serine, threonine) in the N-terminal half binds directly to the
CENP-HIKM complex [74]. A conserved N-terminal motif also associates with MIS12C,
linking the inner and outer kinetochore [75, 76, 77]. Cryo-EM studies have confirmed
CENP-C’s binding to CENP-N within the CENP-LN complex [9, 78], and together with
CENP-T, it bridges the inner kinetochore to NDC80 [29]. Finally, CENP-C binds
The potential role of CENP-C in cancer has been explored primarily in cell
models [42]. In the AACR Genie Cohort (v18.0), CENP-C alterations are
rare—identified in only one tumor sample (
Centromeres are a focal point of chromosome biology due to their indispensable role in ensuring accurate chromosome segregation and maintaining genomic integrity. Centromere identity is not defined by primary DNA sequence but instead is governed by epigenetic mechanisms. These include the incorporation of the canonical histone variant CENP-A within centromeric chromatin and the presence of a defined centromeric domain (CDR) that facilitates strong CENP-B binding and the assembly of a functional kinetochore [1, 18, 25, 29]. Collectively, these features reflect a multilayered regulatory system that integrates chromatin composition, DNA methylation, and sequence-specific protein interactions to ensure stable centromere function.
Despite CENP-B’s well-documented contributions to centromere structure and chromosome stability, its role in pathological chromosome mis-segregation—including cancer-associated aneuploidy—remains unresolved. The analysis of the AACR GENIE cohort presented here, encompassing more than 211,000 patients and 250,000 tumor samples [47], reveals that CENP-B is remarkably conserved in cancers, with no detectable genetic alterations during tumorigenesis. This striking absence of mutation suggests that CENP-B is not typically compromised at the level of coding sequence variation. Rather, if CENP-B contributes to oncogenic chromosomal instability, the disruption is likely to arise from epigenetic mis-regulation or altered chromatin dynamics instead of direct mutation [18]. A second hypothesis is that the hypomethylated CDR, required for proper kinetochore assembly, becomes progressively destabilized during early tumorigenesis, promoting chromosome segregation errors. Finally, centromere-associated proteins may form compensatory networks, such that only multi-component perturbations—not single-gene mutations—result in catastrophic mitotic failure.
These hypotheses emphasize that future research should expand beyond sequence-level analysis toward dissecting the regulatory mechanisms that maintain CENP-A deposition, CDR methylation, and CENP-B binding in vivo. Addressing these questions will not only promote a deeper understanding of the epigenetic foundations of chromosomal stability but may also identify novel diagnostic and therapeutic targets. Key directions include: High-resolution epigenomic profiling (e.g., DNA methylation, histone acetylation) of centromeres in normal, premalignant, and malignant tissues will be essential for identifying early chromatin alterations associated with aneuploidy. Functional assays that measure CENP-B binding dynamics under controlled methylation states will help determine how CDR epigenetics influence kinetochore stability. Integrating multi-omic data to define how structural and regulatory centromere features are rewired under pathological conditions. Evaluating whether restoring normal centromere chromatin states can reduce aneuploidy or chromosomal instability in model systems. Finally, exploring whether chromatin-modifying enzymes that act on centromeric regions represent therapeutic targets could open new strategies to mitigate chromosome mis-segregation in cancer. Overall, examining the epigenetic and interaction networks that preserve centromere identity offers a powerful path toward understanding—and potentially mitigating—chromosomal instability in human cancers.
FP: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Software, Resources, Supervision, Validation, Visualization; FP: Writing – original draft; Writing – review & editing. FP: Read and approved the final manuscript. FP: Has participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.