





1 Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, 67100 L’Aquila, Italy
2 Department of Molecular Medicine, Sapienza University of Rome, 00161 Rome, Italy
†These authors contributed equally.
‡These authors contributed equally.
Abstract
Glaucoma is a complex neurodegenerative disease characterized by the progressive loss of retinal ganglion cells (RGCs) and optic nerve damage. Both mechanical and vascular factors are believed to contribute to the etiology of glaucoma. However, the underlying pathogenic mechanisms are not yet fully understood. In this article, although it is a single component of a multifactorial condition, we argue that neuroinflammation is a significant factor in glaucoma pathogenesis. Glaucoma, at present, is recognized as a neurodegenerative disorder sharing common neuroinflammatory mechanisms with classical neurodegenerative diseases. The involvement of classical immune signaling pathways, such as TLRs and NF-κB, as well as proinflammatory cytokines like TNF-α, aligns glaucoma with other neurodegenerative diseases where inflammation is pivotal (e.g., Parkinson’s and Alzheimer’s diseases). As such, glaucoma should be considered not only an ocular pressure disorder but also a neurodegenerative condition with a strong immune component. This perspective opens new avenues for novel therapeutic intervention, including the targeting of glial cells or modulators of inflammatory signaling. However, the complexity of microglial phenotypes and the timing of their activation relative to astrocytes remain areas that require further clarification. The current M1/M2 paradigm is acknowledged as overly simplistic, highlighting the need for more refined and nuanced models. Although oxidative stress and other interconnected signaling, such as STAT3, are involved in the pathogenesis of glaucoma, here, we focus on the role of the NF-κB signaling pathway within the glaucomatous condition with a special focus on the main characters fostering the neuroinflammation.
Keywords
- glaucoma
- neuroinflammation
- microglia
- NF-κB
- Müller cells
- astroglia
Glaucoma is a chronic neurodegenerative disease characterized by the progressive degeneration of retinal ganglion cells (RGCs), loss of synapses, optic nerve damage and corresponding visual field defects [1]. Elevated intraocular pressure (IOP) is a well-known risk factor; however, glaucomatous neurodegeneration also occurs in patients with normal IOP, a condition commonly referred to as normal tension glaucoma (NTG). The evidence that NTG patients can still develop glaucoma points to mechanisms beyond mechanical pressure, with neuroinflammatory pathways emerging as central contributors (Fig. 1).
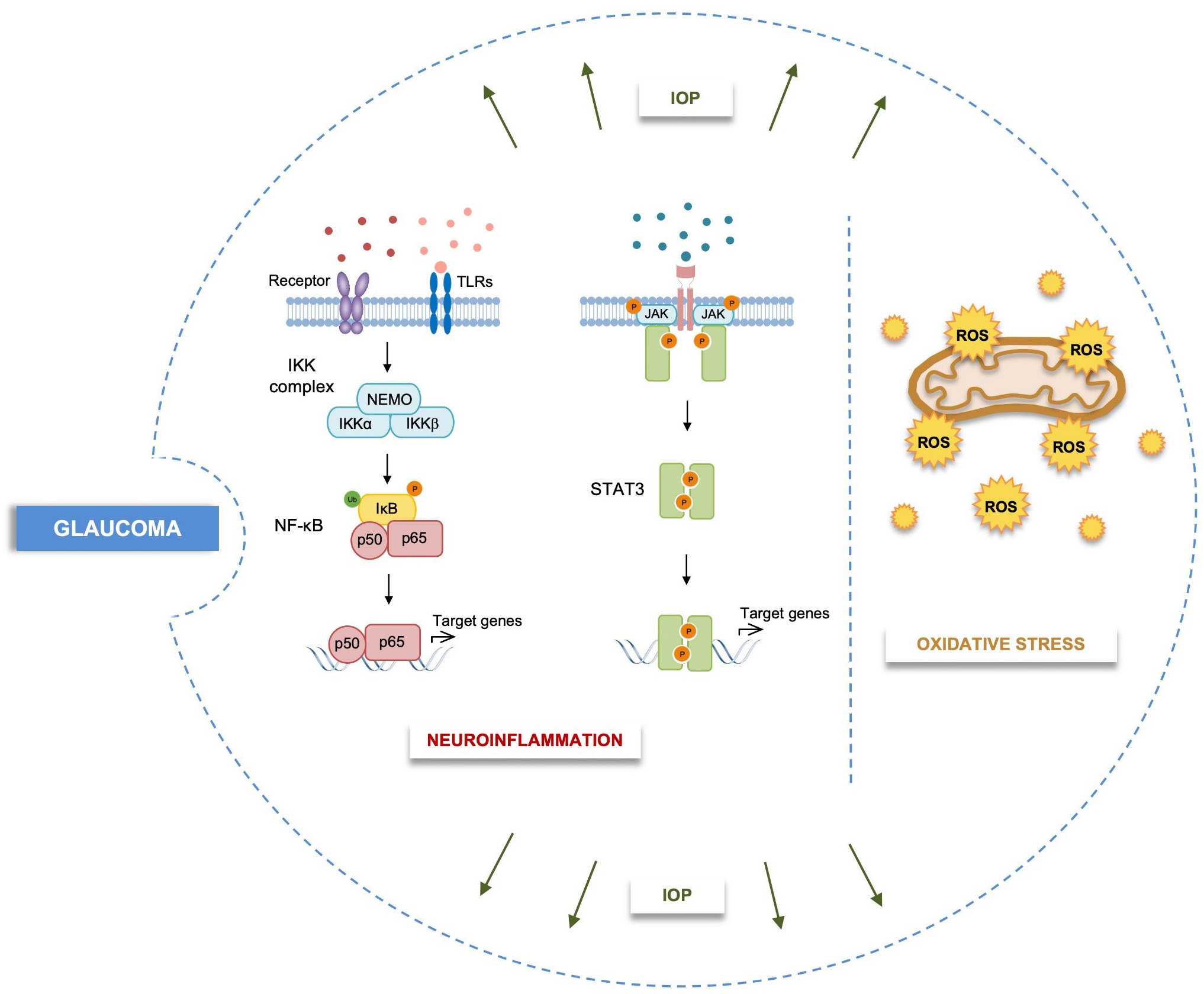 Fig. 1.
Fig. 1.
Schematic representation of multifactorial pathogenic processes
involved in glaucoma. Neuroinflammation, oxidative stress and biomechanical
dysfunction are key factors in the disease onset and progression. IOP,
intraocular pressure; IKK, I
Genetic factors have been identified as prodromal in the development of glaucomatous dysfunction (e.g., MYOC, OPTN, CYP1B1). Furthermore, it has been demonstrated that key genes contribute to the onset of the neuroinflammatory state associated with the disease. Gain-of-function mutations in the pro-inflammatory gene TBK1 play a crucial role in driving neuroinflammation in glaucoma pathogenesis, independent of IOP elevation [2, 3, 4]. Moreover, genome-wide association studies (GWAS) have implicated the ATP-binding cassette (ABC), subfamily A member 1 (ABCA1) gene in primary open-angle glaucoma (POAG), the most commonly diagnosed form of glaucomatous dysfunction [5, 6]. ABCA1 is expressed in glaucoma-relevant ocular tissues such as the iris, ciliary body, retina, optic nerve head, optic nerve and trabecular meshwork. It has been demonstrated that ABCA1 may regulate neuroinflammation and neurodegeneration in several murine models, and some studies have shown its involvement in retinal pathogenesis and POAG [7, 8]. ABCA1 has been implicated in retinal inflammation and RGC apoptosis, and has also been shown to play a role in IOP regulation through modulation of aqueous humor dynamics [9, 10].
The inflammatory process involves the activation of resident glial cells, the production of proinflammatory cytokines and chemokines, and the infiltration of peripheral cells into the central nervous system due to the disruption of the blood–brain barrier. Initially, this response serves as a protective mechanism against various pathogens [11]. Glial cells continuously monitor the retina microenvironment and immediately respond to even minor changes to maintain tissue homeostasis. Macroglia (astrocytes and Müller cells) and microglia are resident immune cells that perform innate immune functions within the retina and optic nerve [12]. In the healthy, undamaged retina, these cells provide nutritional and structural support, participate in metabolism and regulate homeostasis. They also coordinate with each other to regulate neuronal activity through phagocytosis and the secretion of inflammatory cytokines and neurotrophic factors [13]. Functional dysfunction and neuronal damage occur when this homeostasis is perturbed and glial cells are unable to return to their resting state, as demonstrated by morphological, functional and molecular deregulations. This process is commonly referred to as “reactive gliosis” and is marked by specific morphological features such as hypertrophic cell bodies and thickened processes [14, 15, 16]. The dynamic interplay between microglia and macroglia further complicates the inflammatory milieu, potentially creating feedback loops that can either exacerbate or resolve damage [17]. Overall, these findings strongly suggest that glial cells are a driving force in the retina’s inflammatory process, and a deeper understanding of their role within the glaucomatous dysfunction could potentially support the identification of new therapeutic approaches.
Recent findings indicate that glaucomatous optic neuropathy is a neurodegenerative disorder that shares neuroinflammatory mechanisms with classical neurodegenerative diseases, highlighting the role of glial cell-mediated neuroinflammation in the pathophysiology of glaucoma [18, 19].
Glial activation is crucial for restoring tissue homeostasis, facilitating repair and providing neuroprotection in the central nervous system (CNS), where astrocytes and microglia mediate the innate immune response [20]. The strong interplay between neurons and glial cells suggests that a dysfunction in either can result in harmful processes that may disrupt neuron–glia communication or jeopardize neuronal health [21, 22]. The retina is an immune-privileged site where the impact of the systemic immune system is strictly regulated [23]. This immune privilege helps maintain retinal homeostasis and protects it from persistent and detrimental inflammation.
At least three types of glial cells exist in the mammalian retina: Müller cells and astrocytes, collectively referred to as macroglia, and microglia. Each of these cell types plays distinct roles in supporting retinal function and responding to injury or disease by carefully modulating immune responses. Astrocytes and microglia are now recognized to display various activation phenotypes, with their shift from protective to potentially harmful roles closely associated with the severity and progression of neurodegeneration [24].
Microglia are the resident immune cells of the CNS, including the retina, where they are found in several layers such as the nerve fiber layer (NFL), ganglion cell layer (GCL), inner plexiform layer (IPL) and outer plexiform layer (OPL), acting as neuropathological sensors and as a first line of defense against injury. Microglia originate from primitive erythromyeloid progenitors that enter the CNS via the primitive bloodstream during early embryonic development.
Microglial cells can defend neural tissue from harmful stimuli like
pathogen-associated molecular patterns (PAMPs) and damage-associated molecular
patterns (DAMPs) by detecting them through their specialized receptors, including
TLRs [25, 26]. In response to neural damage, microglia undergo morphological
changes, proliferate, migrate and release inflammatory cytokines such as
IL-1
In the context of glaucoma, reactive microglial cells initially engage in the phagocytosis of debris from damaged RGCs, thereby contributing to maintaining a toxin-free microenvironment. Additionally, these cells release neurotrophic factors, such as brain-derived neurotrophic factor (BDNF) and ciliary neurotrophic factor (CNTF), which provide neuroprotection and promote tissue regeneration [29].
A rapid increase in IOP is able to induce changes in microglial morphology, such
as retraction of processes, enlargement of the soma and increased expression of
activation markers like CD68, in just 60 minutes [30]. Activated microglia
produce pro-inflammatory and cytotoxic molecules, including complement factors,
nitric oxide (NO), TNF-
Indeed, microglial activation is one of the earliest events in glaucoma,
preceding RGC loss, as demonstrated in the DBA/2J animal model, where early
microglial alterations correlate with the extent of neurodegeneration. Treatments
that inhibit microglial activation, like minocycline, have been shown to reduce
RGC death in this model, underscoring the critical role of activated microglia in
the pathogenesis of glaucoma [16, 34, 35]. Overall, these findings highlight the
role of microglial activation in glaucomatous neurodegeneration. Within this
context, it is now widely accepted that this microglial-activation status in the
CNS can shift among a plethora of phenotypes (M0-M1-M2a-M2b-M2c-M2d) that may
exacerbate the neurodegeneration or rescue the stress-induced damage with a
strong neuroprotective effect. These microglia subsets are conventionally divided
into M0 (resting), M1-like (classical activated or pro-inflammatory) induced by
lipopolysaccharide (LPS), interferon-gamma (IFN
Retinal astrocytes mainly reside in the NFL and GCL layers and are classically identified by the expression of Glial fibrillary acidic protein (GFAP), vimentin and nestin. Paired box 2 (Pax2) and SRY-Box Transcription Factor 2 (Sox2) instead are used as nuclear markers of astrocytes, which are present as well in the optic nerve [13, 42, 43, 44, 45, 46, 47, 48].
In order to counteract glaucomatous dysfunction, the morphological structure of astrocytes, characterized by long processes that envelop RGCs, plays a structural role in counteracting IOP-related biomechanical stress and maintaining retina homeostasis. This is achieved through their ability to remove cellular debris and increased phagocytic activity in glaucoma onset and progression [49].
Although astrocytes continue to remain enigmatic in terms of retinal functions, they share common features with microglia, such as plasticity and the ability to change their phenotype toward either neurodegenerative or neuroprotective states under pathological conditions, as well as reactivity and ability to migrate to injury sites, as demonstrated by several studies [50, 51, 52, 53]. Morphological changes in astroglia populations are classically recognized as key markers of reactive astrogliosis, which is also characterized by a strong upregulation of GFAP [54, 55]. Among the first to link glaucomatous conditions to the inflammatory insults, Stevens et al. [54] demonstrated that retinal astrocytes become reactive and increase the complement component 1q (C1q) expression in RGCs, contributing to the glaucoma inflammatory-related degeneration and vision loss [54].
Astrocytes and microglia do not function independently in healthy or pathological tissue; rather, they team up to drive and intensify neuroinflammation [20, 56]. A mutual communication between microglia and astrocytes has been well demonstrated during the onset, progression and amplification of the glaucomatous neuroinflammation [20, 56, 57, 58]. This teamwork arises from both direct molecular interactions and their shared responses to environmental cues. In glaucoma, stress from increased IOP and damage to RGCs seem to be the key triggers that synchronize the astrocyte–microglia partnership. In addition to their close relationship, the molecules produced by these glial cells can also recruit immune cells from the bloodstream, further fueling neuroinflammation [15].
Astrocytes and microglia communicate with each other through different mechanisms. First, both cell types can sense and respond to the same signals at the same time. They have similar immune receptors that enable them to communicate and work together during inflammation. These cells can detect stress in other cells by recognizing damage signals. Both cells respond to ATP, which is released from stressed or dying RGCs. This ATP triggers inflammation through purinergic signaling. Besides purinergic receptors (e.g., ionotropic P2X7R-P2X purinoceptor 7), astrocytes and microglia also express pattern recognition receptor (PRR)-like toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs). When these receptors are activated by damage signals, they can quickly trigger an inflammatory response [59].
Astrocytes and microglia crosstalk can either increase or decrease their responses. They do this by releasing molecules like cytokines, chemokines, or complement molecules. Astrocytes mainly produce chemokines, which bind to receptors on microglia and help microglia migration. Cytokines from astrocytes can also assist microglia in removing synapses. Complement components, like C3 from astrocytes, can support microglia with their eating function. Astrocytes and microglia also keep their numbers balanced. Microglia can control astrocyte numbers by phagocytosing them in the retina.
Communication between astrocytes and microglia can also affect their behavior.
Congruently, microglia can induce astrocytes to become harmful, thus promoting
inflammation [27]. In glaucoma, microglia can shift astrocytes from a protective
anti-inflammatory A2 state to a harmful pro-inflammatory A1 state [27, 60].
Interleukin-1alpha (IL-1
Müller cells are a type of glial cell found only in the retina, stretching throughout its full thickness. Müller cells are strategically positioned transversely across all nuclear and plexiform layers, with their endfeet making contact with the vitreous cavity and their microvilli extending into the subretinal space. Within the IPL and OPL, these processes envelop synapses and form connections with blood vessels [61]. These cells play a crucial role in directing light to photoreceptors and in mitigating mechanical stress within the retinal tissue. They are classically identified by the expression of glutamine synthetase (GS), cellular retinaldehyde acid-binding protein (CRALBP), glutamate–aspartate transporter (GLAST), S100b and vimentin. Müller cells’ soma is located in the inner nuclear layer (INL), with its processes extending in both directions towards the inner and outer retinal layers [62]. Although classically used as a marker of astrocytes, GFAP has been shown to be highly expressed by reactive Müller cells in glaucoma patients [63].
In detail, Müller cells play a crucial role in the rapid clearance of excess glutamate from the extracellular environment via amino acid transporters, particularly excitatory amino acid transporters. This process is essential for maintaining low glutamate levels, thereby preventing excitotoxicity. The conversion of glutamate to glutamine is facilitated by the enzyme glutamine synthetase, which is specifically expressed by Müller cells. Glutamine then acts as a precursor for the synthesis of glutamate in neurons. In glaucoma, both in human patients and in animal models, an increase in the expression of glutamine within Müller glia has been observed, suggesting a heightened activation of the glutamate–glutamine cycle. It is proposed that the rise in glutamine concentrations within Müller glia could be attributed to a diminished need for glutamine in injured RGCs. Beyond their function in clearing excess glutamate from the synaptic cleft, Müller glia are also capable of metabolizing glutamate as a substrate [64].
This support is vital for the survival of photoreceptors and neurons, as it enables the efficient uptake and recycling of neurotransmitters and their precursors, thereby ensuring precise neuronal communication [65].
Under stress conditions, Müller cells show cellular hypertrophy, which
involves the thickening of both their somas and processes. This condition is also
associated with increased cellular proliferation and an upregulation of
cytoskeletal proteins, such as vimentin and a reduction in the expression of GS
[66]. Similar to microglia and astrocytes, Müller cells initially display
neuroprotective phenotypes through the release of factors such as pigment
epithelium-derived factor (PEDF) or vascular endothelial growth factor (VEGF).
However, they can also fuel neuroinflammation by releasing IL-1
Although Müller glia are more numerous than astrocytes in the retinal environment, both macroglial subtypes exhibit a range of shared functions alongside distinct attributes, many of which remain to be elucidated. Moreover, timing and site of macroglial activation are crucial to induce different activation states and to determine the beneficial or detrimental effect, thus making it more difficult to choose an appropriate therapeutic approach [69, 70]. It is imperative for forthcoming research to explore both the unique and common roles of astrocytes and Müller glia. Furthermore, understanding their potential collaborative interactions is vital for gaining a comprehensive insight into their functions.
Generally, in the glaucomatous retina, proinflammatory cytokines play a crucial
role in both microglia and macroglia. Although microglia act as primary
responders to neural damage, initiating an inflammatory response that leads to
neuronal loss, macroglia provide essential structural and metabolic support to
neurons but can become reactive under chronic stress, contributing to
neuroinflammation and to the RGC degeneration. Within this context, the
involvement of the NF-
The nuclear factor kappa-light-chain-enhancer of activated B cell
(NF-
Various exogenous signals, such as proinflammatory cytokines (e.g.,
IL-1
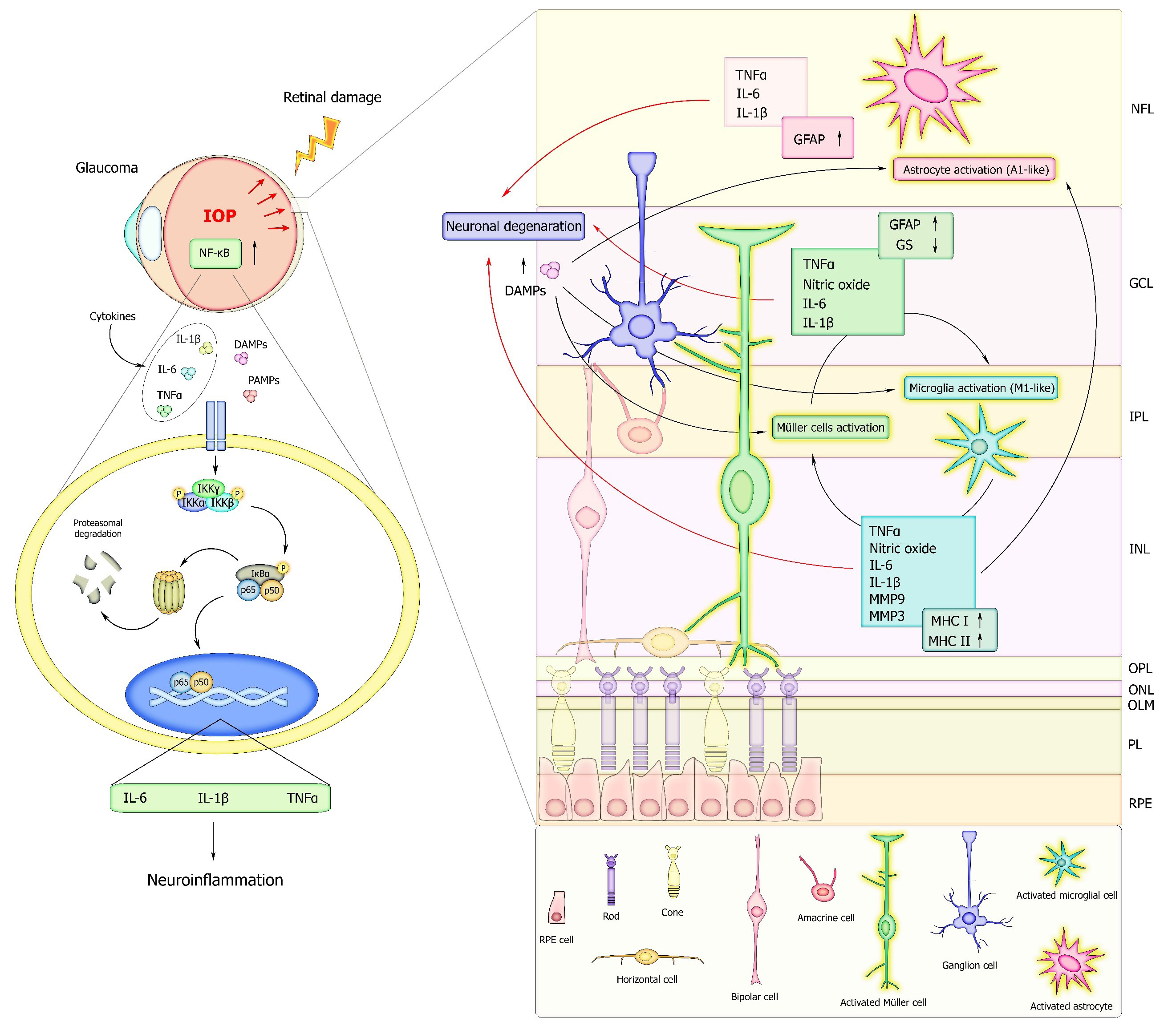 Fig. 2.
Fig. 2.
Schematic representation of the NF-
| NF-κB PATHWAY IN GLIAL CELLS | ||||||
| Functions | Cell type | Cytokines | Receptors | Diseases | Model | Refs |
| Neuroinflammation | Müller glia, Microglia, Astrocytes | TNF- |
CD44, TLR, TNFR1, P2X7R, TLR4, IL-1R | Glaucoma, Retinitis pigmentosa | In vitro, In vivo, Ex vivo | [18, 61, 97, 103] |
| Neuroinflammation, neurodegeneration | Müller glia, Microglia, Astrocytes | TNF- |
TLR, CNTFR, CD44, TRPV4, TNFR1 | Glaucoma, Hypertensive Retinopathy | In vitro, In vivo, Ex vivo | [102, 104, 106] |
| Glial reactivity, neurodegeneration | Müller glia, Microglia | TNF- |
TNFR, IL-1R1, TLR, CD44, RAGE | Glaucoma, Neurodegenerative disease | In vitro, In vivo, Ex vivo | [100, 105] |
| Oxidative and neurotoxic response | Astrocytes | TNF- |
RAGE | Glaucoma, Neurodegenerative disease | In vitro | [99, 102] |
| Cell survival | Microglia, Astrocytes | IL-1 |
p75 | Glaucoma | In vitro | [102] |
| NF-κB PATHWAY IN OTHER RETINAL CELLS | ||||||
| Functions | Cell type | Cytokines | Receptors | Diseases | Model | Refs |
| Neuroinflammation, neurodegeneration | RGC | TNF- |
TRPV4, TNFR1 | Glaucoma | In vitro, In vivo | [104] |
| Neurodegeneration | RGC, Optic nerve | IL-1 |
TLR4 | Glaucoma | In vitro, Ex vivo | [101] |
CNTF, ciliary neurotrophic factor; TRPV4, transient receptor potential vanilloid 4; HMGB1, High Mobility Group Box 1; OPN, Optineurin; CD44, Cluster of Differentiation 44; TNFR1, Tumor Necrosis Factor Receptor 1; P2X7R, P2X purinoceptor 7; CNTFR, Ciliary Neurotrophic Factor Receptor; RAGE, Receptor for Advanced Glycation Endproducts.
Inhibition of NF-
Recent research focusing on the transcriptomic and proteomic profiling of human
donor eyes and animal models affected by glaucoma has identified an early
increase in the expression of numerous molecules and pathways involved in
inflammatory signaling, such as pattern recognition receptors, TLRs, NLRs,
Myeloid Differentiation Primary Response 88 (MyD88), Mitogen-Activated Protein
Kinases (MAPKs) and NF-
In an experimental glaucoma model, the conditional deletion of
I
Lebrun-Julien and colleagues [110] demonstrated that the activation of the
NF-
Targeting the NF-
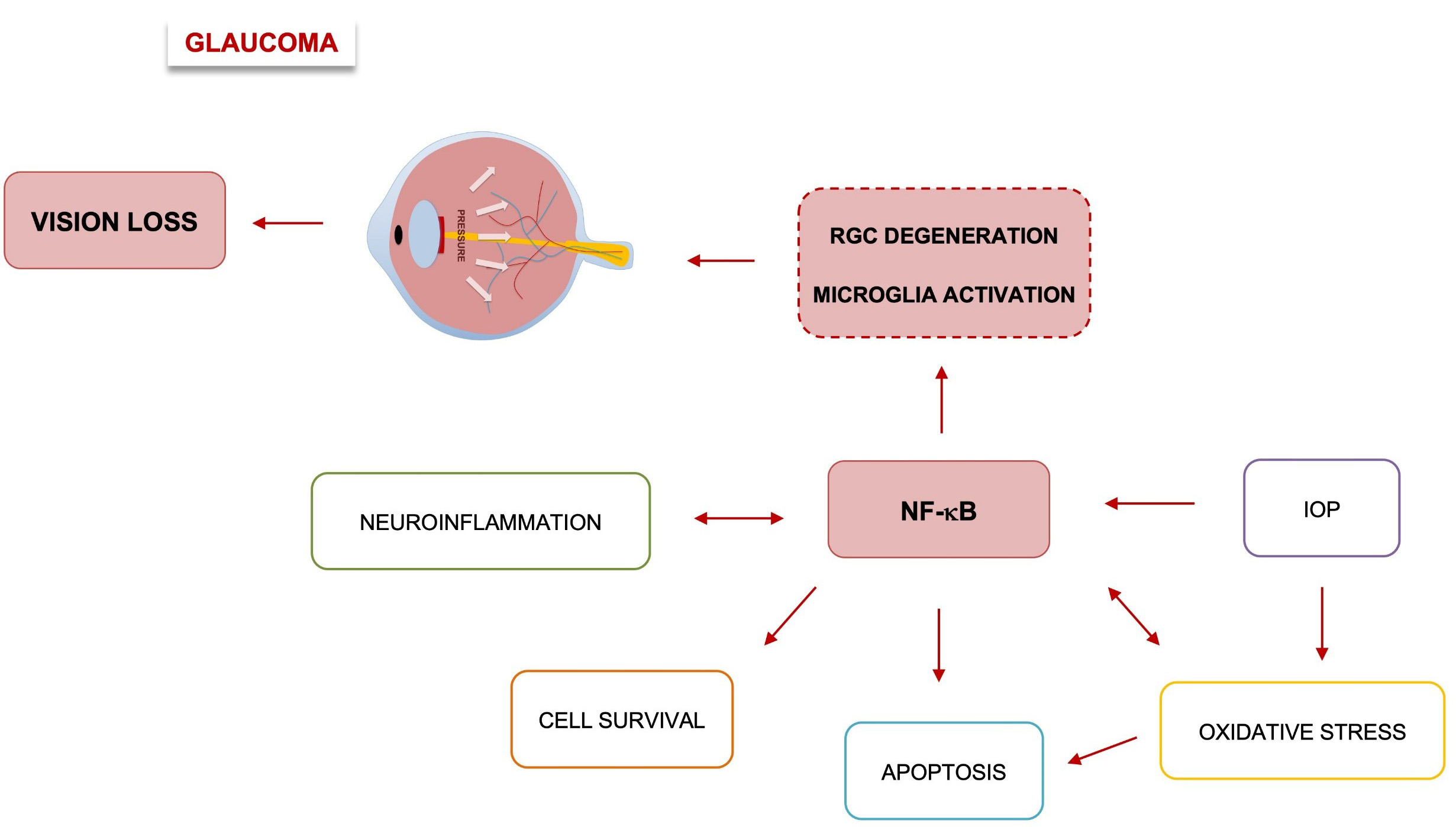 Fig. 3.
Fig. 3.
Role of NF-
Reported evidence suggests that NF-
A clear pathologic description centered on NF-
Understanding these molecular dynamics is crucial for developing targeted
therapies aimed at mitigating glaucomatous damage and preserving vision. Research
in this area may lead to the development of novel therapeutic approaches that
modulate NF-
In summary, the role of NF-
FV and MDVN drafting the manuscript prepared the figure and performed bibliographic search. IF, FDA, FDG and DVer performed bibliographic search and formatting; DC, AA, EA gives a fundamental contributes to the design of the study and also support the revision process. DVec and FZ conceived the project and reviewed the final version of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was funded by intramural “DISCAB GRANT 2022 code 07_DG_2022_24” awarded by the Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, by Progetto di Ricerca di Ateneo per l’avvio alla Ricerca code 07_PROGETTO_RICERCA_ATENEO_2023_VECCHIOTTI to DVec. awarded by University of L’Aquila and by grant number F/310074/01-02/X56 CUP B19J23000180005 by Ministry of Enterprises and Made in Italy to EA and FZ. FV is by supported by the L’Aquila University Ph.D. program in Experimental CUP E11I23000130001, CODICE BORSA 39-411-55-DOT13SR6G7-7917.
The authors declare no conflict of interest.
Search Methodology is provided in the supplementary materials. Supplementary material associated with this article can be found in the online version, at https://doi.org/10.31083/FBL45644.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.