







 , Donglan Piao 1,†, Jio Kang 1, Suh Jin Yoon 1,2, Hyun Ji Lee 1,2, Isoo Youn 1, Eun Kyoung Seo 1,*
, Donglan Piao 1,†, Jio Kang 1, Suh Jin Yoon 1,2, Hyun Ji Lee 1,2, Isoo Youn 1, Eun Kyoung Seo 1,* , Eun Sook Hwang 1,2,*
, Eun Sook Hwang 1,2,*1 College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, 03760 Seoul, Republic of Korea
2 Graduate Program in Innovative Biomaterials Convergence, Ewha Womans University, 03760 Seoul, Republic of Korea
†These authors contributed equally.
Abstract
Cordycepin (CDC), an adenosine (ADO) analog from Cordyceps mushrooms, exhibits potent anti-tumor and immunomodulatory activities. However, the precise mechanisms governing its effects on macrophage plasticity remain poorly understood. This study aimed to isolate CDC from a high-yielding Cordyceps cultivar, validate its systemic anti-tumor efficacy, and elucidate the mechanobiological cues regulating CDC-driven macrophage functions under varying cell-density states.
CDC (>98% purity) was isolated and structurally characterized via nuclear magnetic resonance (NMR) and high-resolution electrospray ionization mass spectrometry (HR-ESI-MS). Primary bone marrow-derived macrophages and RAW264.7 cells, cultured under sparse (~30%) or confluent (100%) conditions, were treated with CDC or ADO. We evaluated cell viability, pro-inflammatory cytokine expression, and Nuclear Factor kappa B (NF-κB) p65 signaling, phagocytosis, and migration. The therapeutic potential of CDC-primed macrophages was assessed via in vitro melanoma co-culture and in vivo intratumoral adoptive transfer in B16F10 tumor-bearing mice.
Systemic administration of CDC significantly inhibited melanoma growth in vivo, promoting apoptosis, enhancing macrophage infiltration. We discovered that CDC regulates macrophage functions via a density-dependent “switch”: while CDC induced cytotoxicity in sparse cultures, it significantly augmented M1-like cytokine production in confluent states without compromising viability. This density-dependent activation was mediated by the A2A adenosine receptor (A2AR), triggering the Akt–NF-κB p65 signaling axis. Furthermore, CDC upregulated migration- and phagocytosis-associated genes, enhancing tumor cell clearance. Notably, intratumoral injection of CDC-primed macrophages markedly reduced tumor volume and size in vivo.
CDC modulates macrophage activation through a unique mechanobiological switch. Under high-density conditions—mimicking the dense tumor microenvironment—CDC enhances A2AR-mediated NF-κB activation, boosting macrophage activation, recruitment, and phagocytosis to facilitate tumor regression. These findings establish CDC as a context-dependent immunomodulator capable of reprogramming macrophages toward a tumoricidal phenotype.
Graphical Abstract

Keywords
- cordycepin
- tumor microenvironment
- A2A adenosine receptor
- macrophage plasticity
- mechanobiology
Cordycepin (CDC), a bioactive nucleoside analog from Cordyceps spp., is widely recognized for its potent anti-diabetic, anti-tumor, and anti-metastatic activities [1, 2, 3]. Structurally analogous to adenosine (ADO) but lacking a 3ʹ-hydroxy group, CDC enters cells via nucleoside transporters [4] to disrupt RNA and DNA synthesis, thereby inhibiting tumor cell proliferation and inducing apoptosis [4, 5]. Beyond direct cytotoxicity, CDC modulates immune responses by interacting with extracellular ADO receptors (A2AR, A2BR, and A3R), which significantly influences tumor progression [6, 7, 8, 9, 10].
Recent paradigms in precision medicine emphasize the role of natural bioactive
compounds in fine-tuning immune signaling pathways, particularly the MAPK and
NF-
However, a functional paradox exists regarding the role of CDC in the tumor
microenvironment (TME). Effective tumor clearance often requires the robust
activation of M1-like macrophages—characterized by the secretion of
pro-inflammatory cytokines such as IL-1
In this study, we investigated the potential of CDC, isolated from a novel Cordyceps cultivar, to reprogram macrophage plasticity depending on environmental physical cues. We aimed to elucidate by which CDC modulates macrophage functional states to enhance anti-tumor immunity, thereby providing a strategic rationale for its application as a targeted immunotherapeutic agent within the complex tumor microenvironment.
A standard reference of CDC was purchased from Sigma-Aldrich (St. Louis, MO,
USA) for authentication. Recombinant cytokines were purchased from R&D Systems
(Minneapolis, MN, USA). LPS, MG132, 4′,6-diamidino-2-phenylindole (DAPI), and
ADO were sourced from Sigma-Aldrich or Cayman Chemical (Ann Arbor, MI, USA).
Primary antibodies against pp65 (S536) (#3033), p65 (#8242), pI
CDC was isolated from Cordyceps militaris ARA301 (BIOARA Co., Ltd.,
Seoul, Korea), which was cultivated on a 100% edible insect medium. Briefly, 3 kg of C. militaris ARA301 was extracted using
methanol. The crude extract was subsequently partitioned with n-hexane,
ethyl acetate (EtOAc), and butanol (BuOH). The resulting fractions underwent
silica gel and Diaion HP-20 chromatography. CDC was further purified through
additional silica chromatography and crystallization, yielding 6.418 g of final
product. The chemical structure of CDC was confirmed via 1H/13C nuclear
magnetic resonance (NMR) and high-resolution electrospray ionization mass
spectrometry (HR-ESI-MS) ([M+H]+ m/z 252.1092, C10H13N5O3) [24].
Purity was validated to be
Bone marrow (BM) cells were isolated from male C57BL/6 mice (8–10 weeks old,
Orient Bio Inc., Gyeonggi-do, Korea) and plated at 2
BM-derived M1 macrophages or RAW264.7 cells were treated with CDC or ADO for 24 h. Cells were subsequently incubated with EZ-Cytox reagent for 30 min, according to the manufacturer’s instructions (EZ-Cytox Cell Viability Assay Kit, DoGenBio, Seoul, Korea). Cell viability was quantified by measuring optical density at 450 nm using a microplate reader (NFEC-2019-10-258101) (Molecular Devices, Sunnyvale, CA, USA) at the Ewha Fluorescence Imaging Core Center. Results were expressed relative to the vehicle-treated control.
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA),
and 2 µg of RNA were subjected to reverse transcription using MMLV Reverse
Transcriptase (Promega, Madison, WI, USA). qPCR was performed using Thunderbird
SYBR qPCR Mix (Toyobo, Osaka, Japan) on a StepOnePlus Real-Time PCR machine
(Applied Biosystems, Carlsbad, CA, USA). Relative transcript levels were
calculated using the comparative Ct (2-ΔΔCt) method, with
Cells on poly-L-lysine-coated coverslips or culture slides (Marienfeld,
Lauda-Königshofen, Germany) were treated with CDC, fixed, and stained with
antibodies against NF-
RAW264.7 or HEK293T cells were transfected with NF-
For phagocytosis, cells were incubated fluorescence-conjugated zymosan bioparticles (ab234053, Abcam, Cambridge, MA, USA) at a final concentration of 5 µg/mL for 30 min at 37 °C. Phagocytic activity was analyzed via flow cytometry (FACS Calibur, BD Biosciences, San Jose, CA, USA). For the migration assay, treated M1 macrophages were placed in the upper chamber of a 24-well Transwell insert (5 µm pore size, MERCK Millipore, Billerica, MA, USA), with B16F10 melanoma cells in the lower chamber. After 5 h of incubation, cells on the upper surface of the membrane were removed, and migrated cells were stained with 0.2% crystal violet, imaged, and quantified.
B16F10 melanoma tumor cells were labeled with carboxyfluorescein succinimidyl ester (CFSE, 5 µM) for 15 min. The CFSE-labeled tumor cells were co-cultured with macrophages pre-treated with CDC or ADO for 24 h. After co-culture, cells were stained with an anti-F4/80 antibody and analyzed by flow cytometry. Phagocytic macrophages and viable tumor cells were quantified using CellQuest software (BD Biosciences, San Jose, CA, USA).
Male C57BL6/J mice (10–12 weeks old) were anesthetized with isoflurane (3%)
using a small-animal inhalation anesthetic system (3–5 min) and subcutaneously
injected with B16F10 melanoma cells (5
Mice were euthanized by CO2 inhalation (at a displacement rate of 20% of the chamber volume per minute to ensure ethical and humane sacrifice), and tumors were harvested, fixed in 10% formalin, and embedded in paraffin. Tumor sections (4 µm thick) were stained for F4/80 and CD68 with fluorescent secondary antibodies and DAPI (1 µg/mL, Sigma-Aldrich) for nuclear counterstaining. Apoptotic cell death was evaluated by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, performed with a commercial kit (R&D Systems, 4810-30-K).
Data are presented as the mean
To obtain sufficient quantities of naturally occurring CDC, we utilized the high-yielding cultivar C. militaris ARA301. CDC was successfully isolated and purified from multiple fractions, yielding a total of 6.418 g from fractions XI-9 and BII (Fig. 1A). The identity and structural integrity of the purified compound were confirmed by 1H/13C NMR spectra, which were consistent with reported reference data [24]. High-resolution ESI-MS analysis confirmed the molecular formula as C10H13N5O3 (Fig. 1B). HPLC-UV analysis further verified a purity greater than 98% (Fig. 1C). To validate the in vivo anti-tumor efficacy of CDC, we administered the compound intraperitoneally to a melanoma mouse model. CDC treatment resulted in a significant inhibition of tumor growth, as evidenced by reduced tumor volume and size (Fig. 1D,E). Histological analysis of tumor tissues from CDC-treated mice revealed a marked increase in apoptotic cell death (TUNEL positive) and macrophage infiltration (F4/80 positive) (Fig. 1F). Furthermore, elevated systemic levels of pro-inflammatory cytokines were detected in the spleens of these mice, indicating a robust immune response (Fig. 1G). Collectively, these findings suggest that CDC not only inhibits tumor progression but also significantly enhances macrophage activation and recruitment within the TME, contributing to its overall anti-tumor effects.
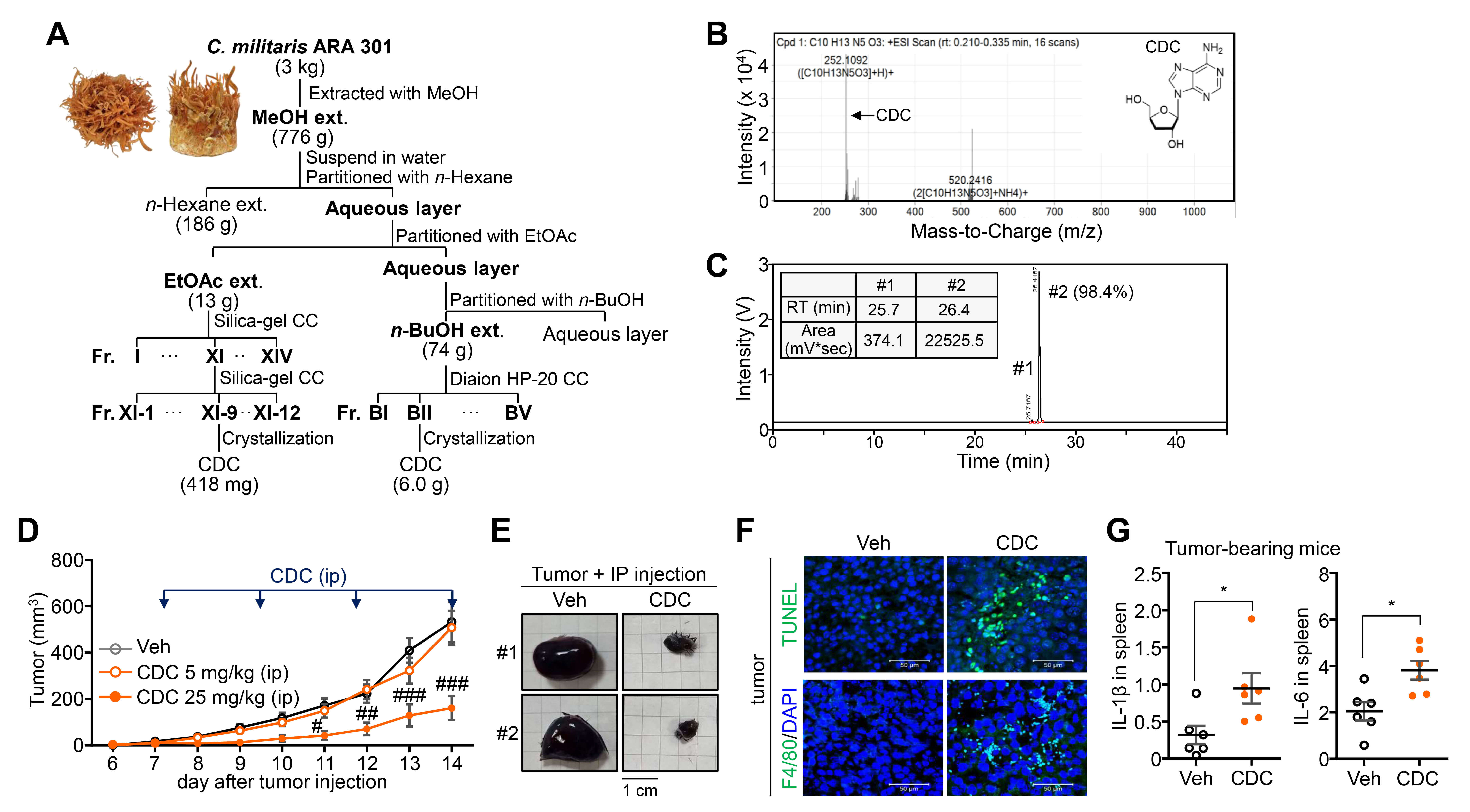 Fig. 1.
Fig. 1.
Identification and anti-tumor activity of Cordycepin (CDC)
isolated from a novel Cordyceps cultivar. (A) Isolation procedure and yield of
CDC from C. militaris ARA301 cultivar. (B) High-resolution electrospray
ionization mass spectrometry (HR-ESI-MS) analysis for the molecular weight
determination of isolated CDC. (C) High-performance liquid chromatography with
ultraviolet detection (HPLC-UV) chromatogram confirming the purity and identity
of the CDC fraction. (D) Inhibitory effect of CDC on in vivo B16F10 melanoma
tumor growth in a syngeneic mouse model. (E) Comparison of representative tumor
sizes and weights at the study endpoint. Scale bar = 1 cm. (F) Histological
analysis of tumor sections via terminal deoxynucleotidyl transferase dUTP
nick-end labeling (TUNEL) staining (apoptosis) and F4/80 immunohistochemistry
(macrophage infiltration). Scale bar = 50 µm. (G) qPCR analysis of
IL-1
Given prior reports indicating that CDC can reduce cell viability [16, 17, 25], we first evaluated its cytotoxic effects on BM-derived M1 macrophages across a range of concentrations. In confluent (100% density) cultures, CDC exhibited no significant cytotoxicity at concentrations up to 20 µM, with viability decreasing only at concentrations above 50 µM (Fig. 2A). As macrophage activation and functional states are known to be modulated by physical cues and mechanical forces [26, 27, 28], we examined whether cell density affects CDC’s efficacy. Notably, while CDC remained non-cytotoxic in confluent M1 macrophages, it significantly reduced cell numbers in low-density (approx. 30%) cultures at concentrations of 10–20 µM (Fig. 2B). A consistent pattern was observed in RAW264.7 macrophages, where CDC-induced cytotoxicity was strictly confined to low-density conditions (Fig. 2C). In contrast, the structural analog ADO resulted in no significant loss of viability in either M1 or RAW264.7 macrophages, regardless of cell density (Fig. 2D,E). These findings demonstrate that CDC-induced cytotoxicity is uniquely contingent upon cell density, suggesting that mechanically-driven changes in the microenvironment—such as altered cell-to-cell contact or cytoskeletal tension—fundamentally reconfigure the macrophage response to CDC.
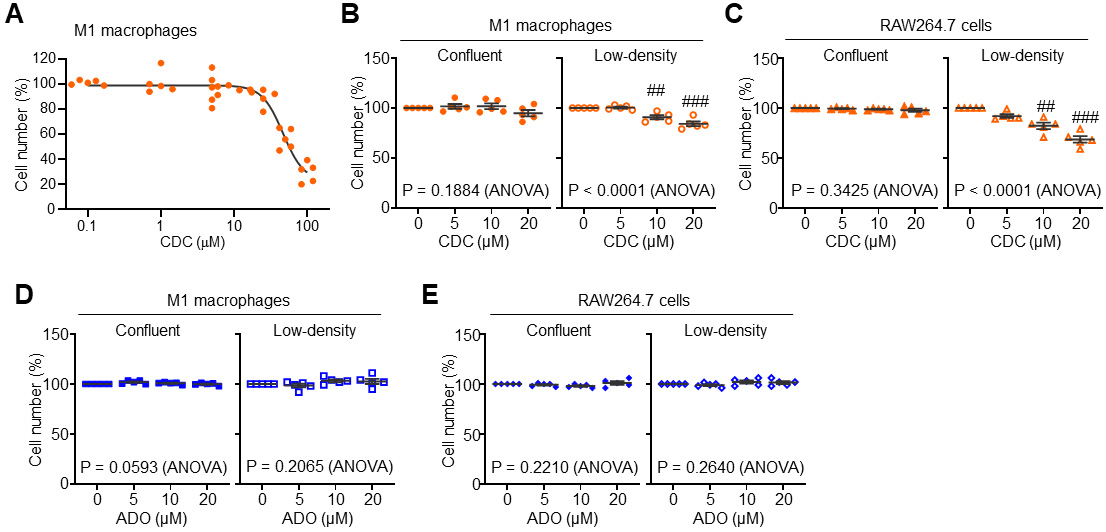 Fig. 2.
Fig. 2.
Context-dependent modulation of macrophage viability by CDC and
adenosine (ADO). (A) Dose-dependent effects of CDC on the viability of
BM-derived M1 macrophages across a range of concentrations
(0.1~100 µM). (B) Modulation of M1 macrophage
viability by CDC under varying cell densities (confluent vs. low-density). (C)
Cell viability modulation of RAW264.7 cells treated with CDC in confluent and
low-density cultures. (D) Effects of ADO on M1 macrophage viability in confluent
and low-density conditions. (E) Cell viability modulation of RAW264.7 cells
following ADO treatment under confluent and low-density conditions. Data are
presented as mean
We next measured inflammatory cytokine expression in both confluent and
low-density cultures of BM-derived M1 macrophages and RAW264.7 cells. In
confluent M1 macrophages, treatment with non-cytotoxic concentrations of CDC
(
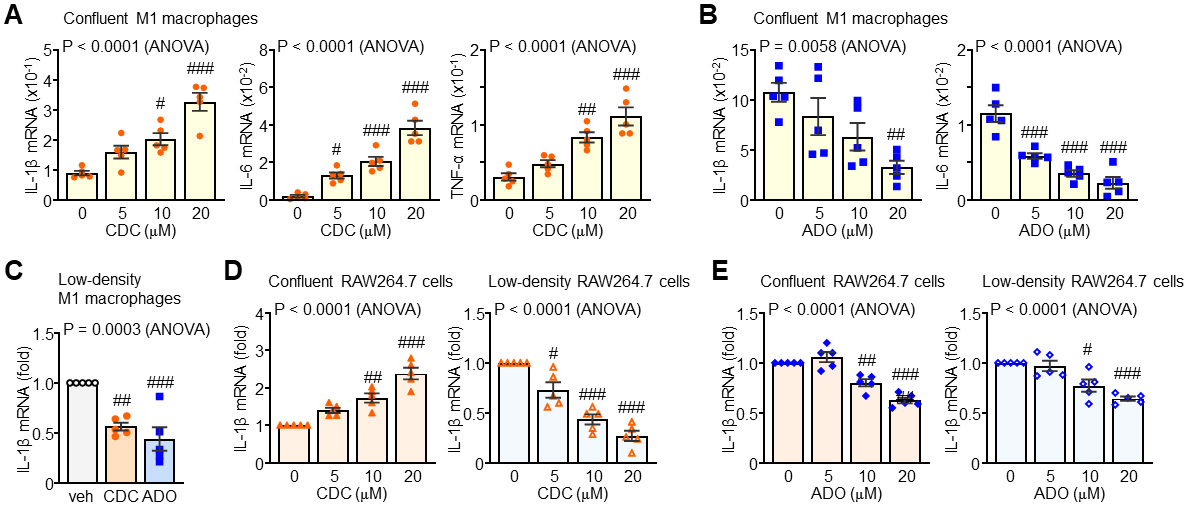 Fig. 3.
Fig. 3.
Context-dependent modulation of inflammatory cytokine
transcripts by CDC and ADO. (A) Regulatory effects of CDC on pro-inflammatory
cytokine mRNA expression in confluent M1 macrophages. (B) Inhibitory effects of
ADO on pro-inflammatory cytokine expression in confluent M1 macrophages. (C)
Comparative modulation of cytokine profiles by CDC or ADO in low-density M1
macrophages. (D) Density-dependent effects of CDC on pro-inflammatory cytokine
expression in RAW264.7 cells (confluent vs. low-density). (E) Suppression of
pro-inflammatory cytokines by ADO in RAW264.7 cells across varying culture
densities. Statistical significance: #p
To elucidate the molecular mechanisms by which CDC induces immunomodulatory
cytokine production, we examined the activation of the NF-
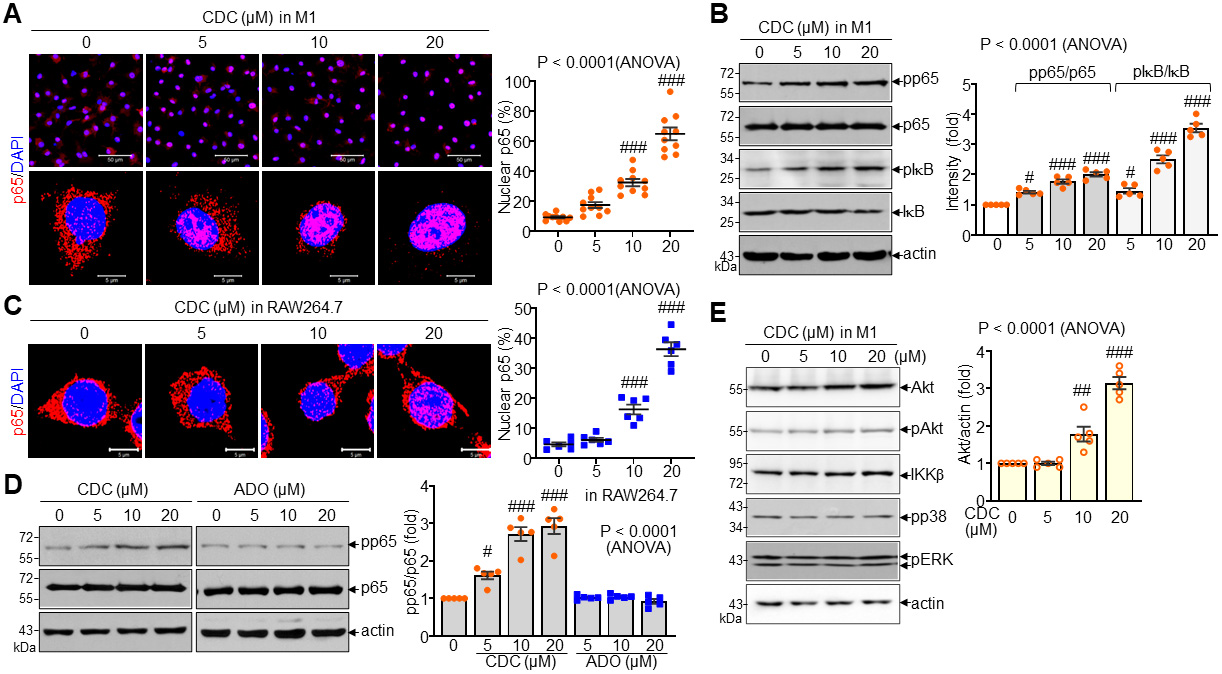 Fig. 4.
Fig. 4.
Enhancement of p65 nuclear localization and phosphorylation by
CDC. (A) Representative immunofluorescence images and quantitative analysis of
p65 nuclear localization in M1 macrophages following CDC treatment. Scale bar =
50 µm (top) and 5 µm (bottom). (B) Immunoblot analysis and
densitometric quantification of phosphorylated p65 (pp65), total p65,
pI
Consistent with its effect on NF-
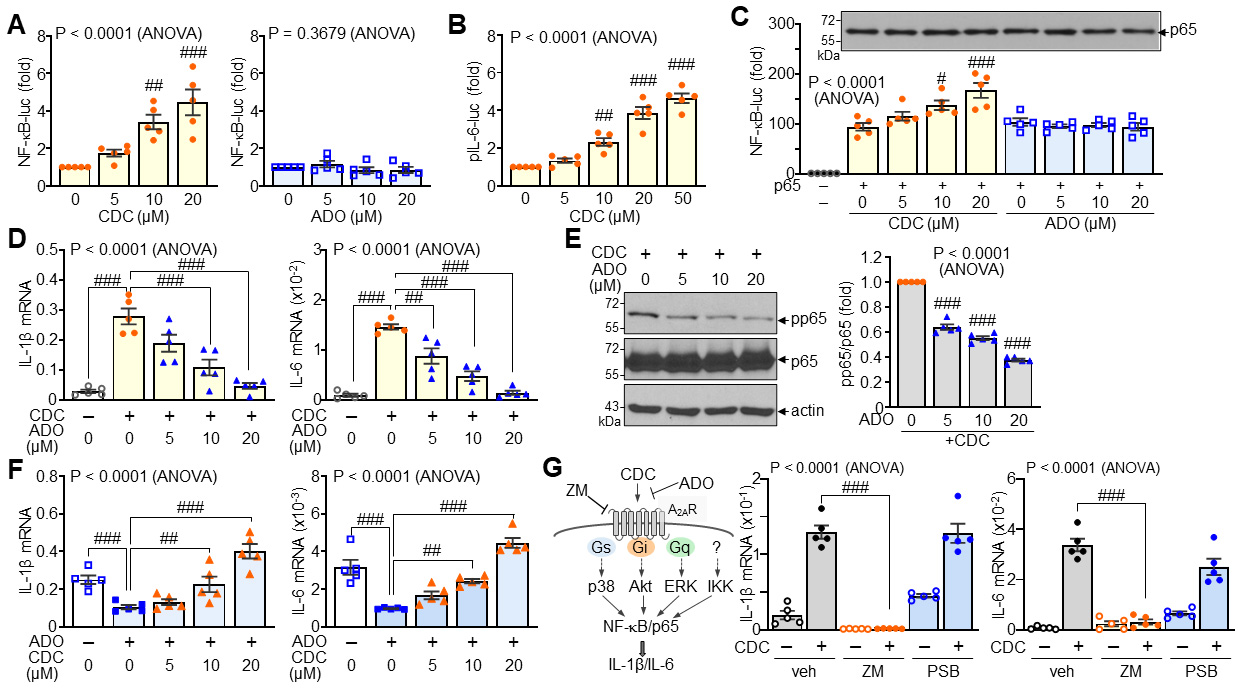 Fig. 5.
Fig. 5.
Activation of A2A adenosine receptor (A2AR)-mediated signaling
by CDC. (A) Reporter assay measuring NF-
Building on the engagement of A2AR–Akt–NF-
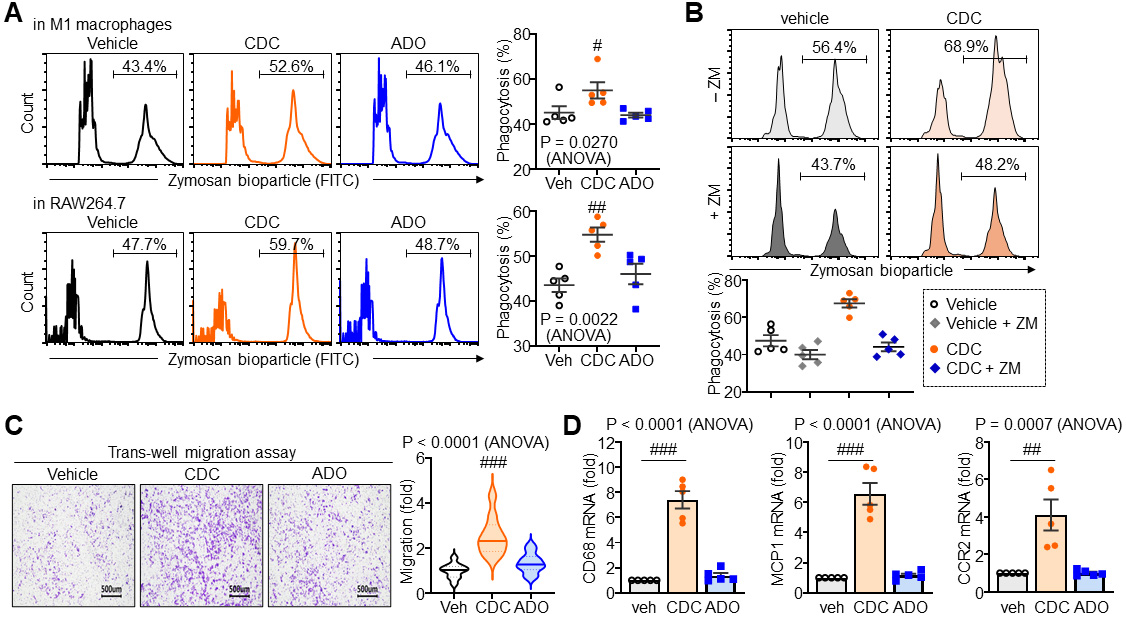 Fig. 6.
Fig. 6.
Enhancement of macrophage phagocytosis and migratory capacity by
CDC. (A) Representative images and quantitative analysis of phagocytic activity
in M1 macrophages and RAW264.7 cells following treatment with CDC (20 µM)
or ADO (20 µM). (B) Phagocytosis assay and corresponding quantification of
M1 macrophages in the presence or absence of the A2AR antagonist ZM241385 (ZM, 10
µM). (C) Transwell migration assay and quantification of M1 macrophages
following treatment with 20 µM CDC or ADO. Scale bar = 500 µm. (D)
qPCR analysis of migration-related markers (CD68, monocyte chemoattractant
protein 1 (MCP-1), and CC chemokine receptor 2 (CCR2)) in M1 macrophages treated
with CDC. Statistical significance: #p
Having established that CDC enhances macrophage migration and phagocytosis, we evaluated its anti-tumor effects in co-culture and syngeneic mouse models. CFSE-labelled B16F10 melanoma cells were co-cultured with M1 macrophages pre-treated with either CDC or ADO. CDC-treated M1 macrophages exhibited a significantly increased phagocytic uptake of tumor cells but ADO did not (Fig. 7A). Consequently, tumor cell viability was markedly reduced in co-cultures with CDC-pre-treated M1 macrophages, and this tumoricidal effect was further enhanced when macrophages were primed with CDC. However, ADO treatment failed to produce any significant enhancement in tumor killing (Fig. 7B,C). To assess in vivo efficacy, mice bearing subcutaneous melanomas received intratumoral injections of either vehicle-treated or CDC-treated M1 macrophages. CDC-activated M1 macrophages significantly suppressed tumor growth, resulting in a substantial reduction in both tumor volume and weight compared to vehicle-treated macrophage controls (Fig. 7D,E). Histological analysis of the tumor tissues confirmed the therapeutic mechanism: tumors receiving CDC-primed macrophages showed a marked increase in apoptotic cell death (TUNEL positive) and a higher density of CD68-positive macrophage infiltration (Fig. 7F). These findings demonstrate that CDC functionally re-programs M1 macrophages into a potent tumoricidal phenotype, enhancing their ability to infiltrate the tumor mass and execute tumor cell clearance, thereby suppressing tumor progression in vivo.
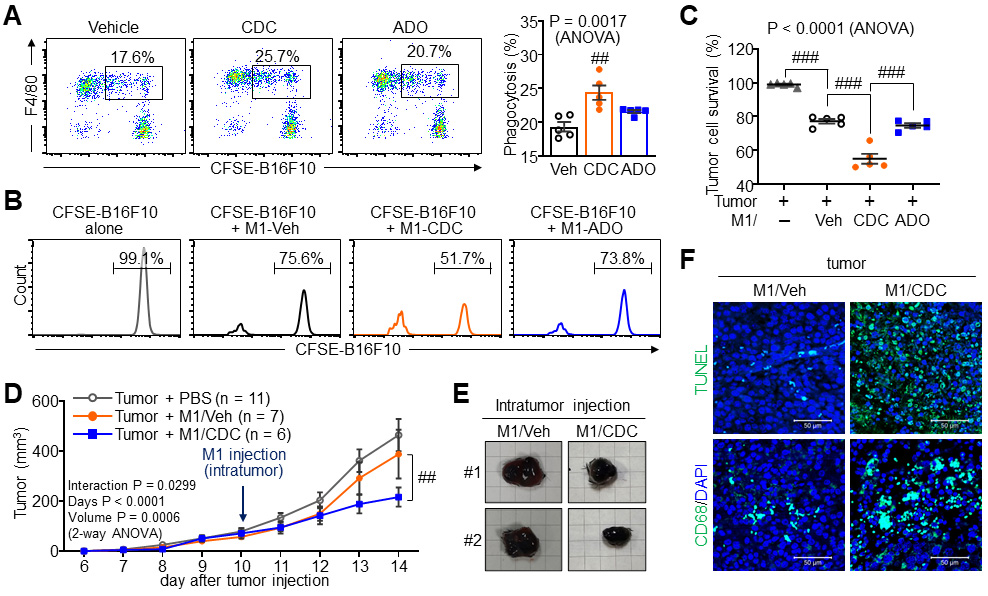 Fig. 7.
Fig. 7.
Anti-tumor efficacy of CDC-activated M1 macrophages. (A) Tumor
phagocytosis assay of M1 macrophages treated with 20 µM CDC or ADO
co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled B16F10
melanoma cells. (B,C) Tumor cell survival assay in the presence of M1
macrophages. Survival of live B16F10 cells was analyzed by flow cytometry (B) and
corresponding quantification (C). (D–F) Therapeutic evaluation in a syngeneic
B16F10 tumor model (C57BL/6 mice) following intratumoral injection of vehicle- or
CDC-pre-treated M1 macrophages. (D) Longitudinal tumor growth curves over the
experimental period. (E) Comparison of terminal tumor size and mass. Each grid
square is 0.5 cm. (F) Representative images of TUNEL staining (apoptosis) and CD68
staining (macrophage infiltration) in tumor sections. Sacle bar = 50 µm.
Statistical significance: ##p
Our study demonstrates that CDC activates M1 macrophages and enhances anti-tumor
functions by engaging the A2AR‒Akt‒NF-
While CDC is often recognized for its anti-inflammatory effects, our findings
highlight its broader, more nuanced role in modulating macrophage plasticity
within the TME. Although pro-inflammatory cytokines such as IL-1
CDC, a nucleoside analog structurally similar to ADO, exerts its effects through AR-dependent signaling. Our comparative analysis with ADO and the use of the AR-selective antagonists confirms that CDC’s pro-inflammatory effects are specifically mediated through the A2AR axis. Because CDC and ADO share structural motifs and compete for similar receptor populations, their functional relationship is intrinsically linked. However, CDC possesses a unique, density-dependent “switch” that is absent in standard ADO signaling, allowing it to bypass the typical immunosuppressive cues of the TME.
Previous studies indicated that CDC suppresses NF-
Notably, CDC-induced functional reprogramming extends beyond phagocytosis to a significantly enhanced migratory capacity. We confirmed that CDC upregulates not only CD68 and MCP-1 but also CCR2, effectively priming macrophages for active tumor infiltration. This density-dependent transition—from a cytotoxic agent in sparse conditions to a potent immunostimulator in confluent states—provides a novel mechanobiological insight into how CDC reconfigures the inflammatory landscape based on the physical architecture of the tumor.
Mechanistically, CDC was found to enhance total Akt expression, providing a larger signaling reservoir for p65 phosphorylation. While ADO remained functionally inert regarding migration and recruitment markers, CDC’s ability to integrate mechanical cues allows it to overcome the restrictive conditions that typically favor M2 exhaustion. In conclusion, CDC promotes macrophage activation and anti-tumor activity associated with enhanced migration and phagocytosis, driven by a unique sensitivity to the cellular microenvironment. This study establishes CDC as a promising candidate for reprogramming the TME, warranting further clinical characterization of its context-dependent therapeutic applications.
Despite the significant findings of this study, several limitations should be acknowledged. First, while we demonstrated the density-dependent “switch” of CDC in vitro using primary BMDMs and RAW264.7 cells, the precise mechanical sensors (such as Piezo1 or integrin-mediated signaling) that interact with the A2AR-Akt axis remain to be fully identified. Second, although the B16F10 melanoma model provided a robust platform for validating systemic and adoptive transfer efficacy, further validation in diverse syngeneic or orthotopic tumor models is required to generalize the immunomodulatory role of CDC across different tumor architectures. Lastly, the long-term systemic effects of CDC-primed macrophage therapy on other organ systems were not extensively evaluated in this study, which may impact the clinical translation of this approach regarding potential off-target inflammatory responses.
In conclusion, this study demonstrates that Cordycepin (CDC) functions as a
context-dependent immunomodulator that reconfigures macrophage plasticity through
a mechanobiological switch. Unlike its endogenous analog adenosine, CDC promotes
an M1-like tumoricidal phenotype specifically under high-density
conditions—characteristic of the dense tumor microenvironment—by activating
the A2AR–Akt–NF-
ADO, Adenosine; ANOVA, Analysis of Variance; AR, Adenosine receptor; BM, Bone Marrow; BMDMs, Bone Marrow-Derived Macrophages; CCR2, CC chemokine receptor 2; CDC, Cordycepin; CFSE, Carboxyfluorescein succinimidyl ester; DAPI, 4′,6-Diamidino-2-Phenylindole; HPLC, High-Performance Liquid Chromatography; HR-ESI-MS, High-Resolution Electrospray Ionization Mass Spectrometry; IACUC, Institutional Animal Care and Use Committee; I
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
JL: Formal Analysis, Investigation, Methodology, Visualization, Writing – Original Draft Preparation. DP: Investigation, Methodology, Resources. JK: Formal Analysis, Investigation, Methodology, Visualization. SJY: Investigation, Validation, Data Curation. HJL: Investigation, Methodology, Validation. IY: Methodology, Software, Writing – Original Draft Preparation. EKS: Conceptualization, Resources, Supervision, Writing – Reviewing and Editing. ESH: Conceptualization, Funding Acquisition, Supervision, Visualization, Writing – Original Draft Preparation, Writing – Reviewing and Editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were conducted in strict compliance with the NIH Guide for the Care and Use of Laboratory Animals and the national guidelines provided by the Ministry of Food and Drug Safety of Korea. The study protocols were formally reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Ewha Womans University (Approval Nos. IACUC 19-031 and IACUC 24-046). All efforts were made to minimize animal suffering during the experimental procedures. Animal experiments should adhere to the 3Rs principle: substitution, reduction, and optimization.
Not applicable.
This work was supported by the National Research Foundation of Korea [RS-2025-00558072 and RS-2021-NF000578], funded by the Ministry of Science and ICT.
The authors declare no conflicts of interest. Given the role as the Guest Editor and Editorial Board member, the corresponding author Eun Sook Hwang had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
During the preparation of this work, the author(s) used Gemini (Google) to refine the manuscript’s English language and to assist in creating the graphical abstract. The author(s) reviewed and edited the AI-generated output and take(s) full responsibility for the final content.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.