








1 Department of Biochemistry and Molecular Biology, School of Basic Medicine, Chongqing Medical University, 400016 Chongqing, China
2 Molecular Medicine and Cancer Research Center, Chongqing Medical University, 400016 Chongqing, China
3 Department of Laboratory Medicine, Xi’an No.1 Hospital, The First Affiliated Hospital of Northwest University, 710002 Xi’an, Shaanxi, China
4 The First Clinical College, Chongqing Medical University, 400016 Chongqing, China
†These authors contributed equally.
Abstract
Transfer RNA (tRNA)-derived small RNAs (tsRNAs) are an emerging class of small non-coding RNAs that play critical roles in tumor progression; however, the functions and underlying mechanisms of tsRNAs in lung cancer development remain poorly understood. Thus, this study aimed to elucidate the molecular mechanism through which alkB homolog 4, lysine demethylase (ALKBH4) influences non-small cell lung cancer (NSCLC) progression through 5′-tsRNAGlu, with a particular focus on the role of tsRNA in translational regulation and malignant phenotype formation.
Cell proliferation and cell cycle progression were assessed using Cell Counting Kit-8 (CCK-8), colony formation, and flow cytometry assays. Global translation efficiency was evaluated using nascent protein synthesis assays and polysome profiling. Northern blotting was used to confirm 5′-tsRNAGlu expression post-transfection, and downstream targets of 5′-tsRNAGlu were screened using a dual-luciferase reporter system. RNA pull-down and RNA immunoprecipitation (RIP) assays were employed to identify proteins that interacted with 5′-tsRNAGlu. Data were analyzed using Student’s t-test or one-way analysis of variance (ANOVA).
The demethylase ALKBH4 promoted the biogenesis of 5′-tsRNAGlu in NSCLC cells. Overexpression of ALKBH4 or 5′-tsRNAGlu inhibited cell proliferation, induced cell cycle arrest, and reduced global translational efficiency, whereas knockdown of 5′-tsRNAGlu attenuated the tumor-suppressive effects of ALKBH4. Mechanistically, 5′-tsRNAGlu did not function in a miRNA-like manner but instead interacted with ribosomal protein L17 (RPL17). Furthermore, 5′-tsRNAGlu was predominantly localized in the cytoplasm, and 5′-tsRNAGlu overexpression significantly increased the associated distribution in the 60S ribosomal subunit while diminishing the associated presence in the 80S monosome. Predictive modeling indicated that 5′-tsRNAGlu binds to the region of RPL17 spanning residues 137–142, the RPL17–rRNA interaction interface, suggesting that binding of 5′-tsRNAGlu to RPL17 interferes with RPL17–rRNA interaction, thereby disrupting the 80S ribosome assembly and global translational efficiency.
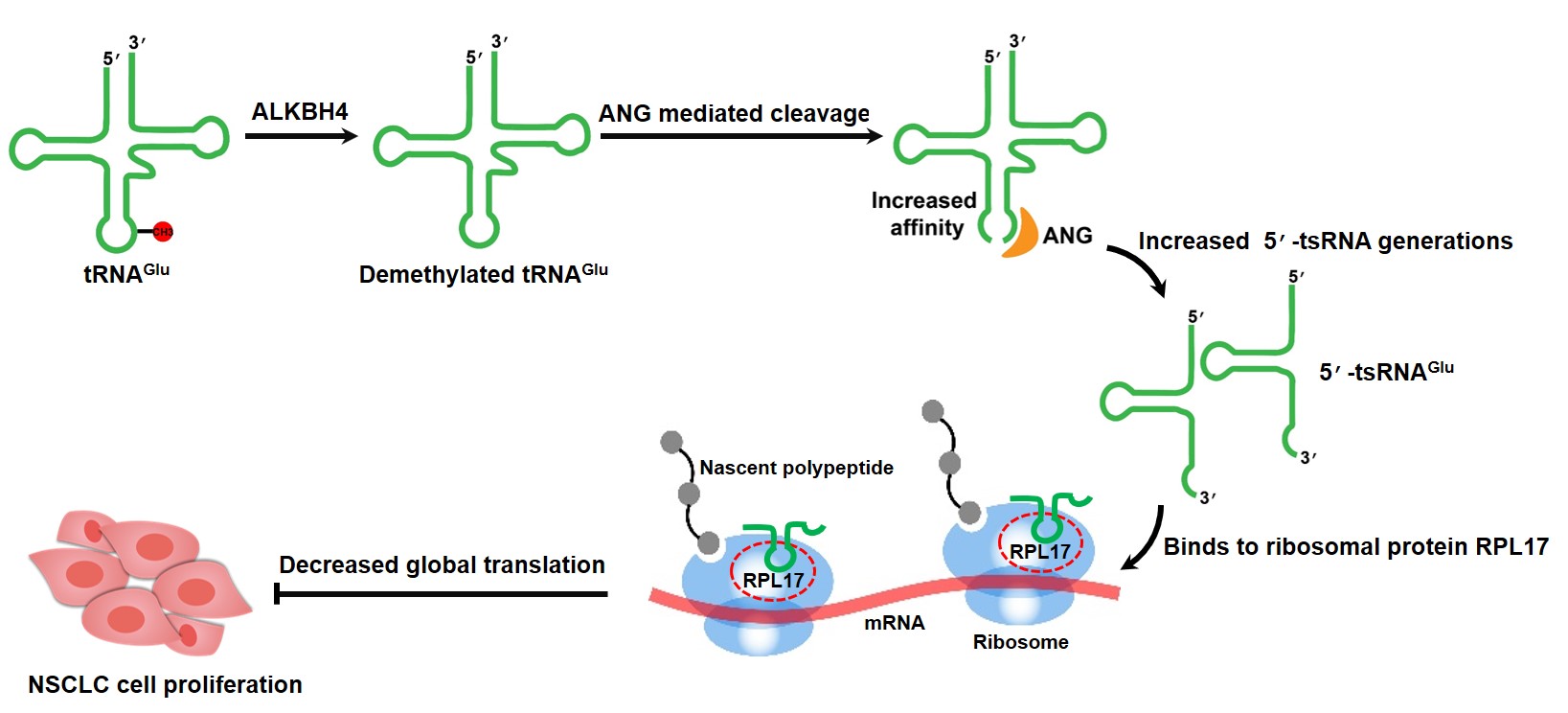
ALKBH4 suppresses malignant phenotypes in NSCLC cells via 5′-tsRNAGlu, likely through an interaction between 5′-tsRNAGlu and RPL17.
Graphical Abstract
Keywords
- non-small cell lung cancer
- non-coding RNA
- demethylase
- translation
Lung cancer is one of the most prevalent malignant tumors worldwide and ranks first in both incidence and mortality among all malignancies in China, showing no significant decline over the past decade [1]. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases, with lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LSCC) being the most common histological subtypes. The five-year survival rate of patients with NSCLC is approximately 15.9%, largely owing to challenges related to early detection and effective precision treatment regimens [2]. Therefore, elucidating the molecular mechanisms of NSCLC-related genes holds significant promise for the development of novel strategies for early diagnosis and targeted therapy.
tRNA-derived small RNAs (tsRNAs) constitute an emerging category of non-coding RNAs (ncRNAs) that are generated through the specific cleavage of mature or precursor tRNAs by ribonucleases. Fragments originating from the 5′- and 3′-ends of mature tRNAs are designated as 5′-tsRNAs and 3′-tsRNAs, respectively [3, 4, 5, 6]. As functional tRNA derivatives [7], tsRNAs have been implicated in a diverse range of biological processes, including stress responses, translational regulation, ribosome biogenesis, and transgenerational epigenetic inheritance [8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18]. For instance, under stress conditions, such as hypoxia, amino acid deprivation, oxidative stress, or ultraviolet (UV) irradiation, angiogenin (ANG) specifically cleaves tRNAs within the anticodon loop to produce tsRNAs, also known as tRNA-derived stress-induced RNAs (tiRNAs) [8, 19, 20]. tsRNAs are known to regulate translation via multiple mechanisms, such as inhibiting the initiation of translation by their unique RNA G-quadruplex (RG4) structures [10, 11, 12, 13, 21], suppressing the initiation of translation by specific tsRNA modifications [14, 15], repressing the translation of target mRNA in a miRNA-like manner [16], binding to ribosomes to either halt or promote translation [17], and altering mRNA secondary structures to enhance translational efficiency [18].
tsRNAs represent a promising frontier for deciphering the complexity of malignancies. Through their distinct sequence characteristics, modification patterns, and structural configurations, tsRNAs exert context-dependent functions as either tumor suppressors or promoters [18, 20, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31]. To date, the precise function of tsRNAs in lung cancer remains largely unexplored. Specifically, tsRNA-46, tsRNA-47, and tsRNA-53 are shown to be downregulated in NSCLC samples and reduce the clonogenic capacity of NSCLC cells [23, 32]. Another study identified that a tsRNA (denoted as CAT1) is upregulated across multiple cancer types; CAT1 competitively binds to the RBPMS protein with NOTCH2 mRNA, thereby inhibiting the CCR4-NOT-mediated deadenylation of NOTCH2 mRNA, increasing its stability, and ultimately facilitating the proliferation and metastasis of NSCLC cells [33]. Collectively, these findings highlight the pivotal regulatory functions of tsRNAs in the progression of NSCLC. Consequently, a systematic investigation of tsRNAs will advance our understanding of the molecular mechanisms underlying NSCLC and support the development of innovative diagnostic and therapeutic approaches. In a previous study, we developed PANDORA-seq, a novel sequencing platform for small non-coding RNA, and discovered a 36-nt 5′-tsRNA derived from specific cleavage within the anticodon loop of tRNAGlu(CTC), termed 5′-tsRNAGlu. In addition, we found that 5′-tsRNAGlu positively regulated embryonic stem cell differentiation and attenuated translational efficiency [34]. From a comparative medical standpoint, it is likely that 5′-tsRNAGlu may act as a potential tumor suppressor, especially given that tumors often exhibit features of dedifferentiation and enhanced translation.
tRNAs are more extensively decorated with chemical modifications than other RNA
species, with methylation being the most abundant modification. The dynamic
regulation of tRNA methylation is orchestrated by tRNA methyltransferases and
demethylases [35]. Demethylation compromises the structural stability of tRNAs,
rendering them more susceptible to cleavage by ribonucleases, leading to the
production of tsRNAs [7, 36]. The ALKBH (alpha-ketoglutarate-dependent
dioxygenase alkB homolog) family is a class of Fe2⁺- and
In this study, we demonstrated that ALKBH4 promoted the generation of 5′-tsRNAGlu in NSCLC cells and that both ALKBH4 and 5′-tsRNAGlu suppressed their proliferative capabilities and global translational efficiency. These mechanisms involved the interaction between 5′-tsRNAGlu and ribosomal proteins.
Human NSCLC cell lines H1299 and A549 were obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All the cell lines were validated by STR profiling and tested negative for mycoplasma.
pCDH-Puro-3Flag was utilized as the vector to construct the ALKBH4 expression plasmid. Briefly, the ALKBH4 coding sequence was amplified using primers designed with homologous arms flanking insertion sites. Concurrently, the pCDH-Puro-3Flag vector was linearized through PCR amplification to eliminate the original coding sequence. Both PCR products were then subjected to in vitro recombination using the NovoRec® One-Step PCR Cloning Kit (Novoprotein, Suzhou, China) in a molar ratio-based reaction system. The ligation product was transformed into TransStbl3 competent cells and plated onto LB agar plates supplemented with ampicillin for colony growth. Positive clones were screened utilizing colony PCR. The ALKBH4 expression plasmid was subsequently verified by sequencing (Sangon Biotech, Shanghai, China).
For H2O2 treatment, H2O2 solution (Sigma-Aldrich, MO, USA) was diluted in complete medium and cells were treated with H2O2 solution at designated concentrations for 24 h. The cells were then collected and analyzed. For plasmid transfection, cells were transiently transfected with 2 µg of ALKBH4 plasmid or pCDH-Puro-3Flag plasmid using Neofect DNA Transfection Reagent (Neofect, Beijing, China) according to the manufacturer’s instructions. For RNA transfection, cells were transiently transfected with mimics, antisense oligonucleotides (ASO), or corresponding controls at a final concentration of 50 nM using LipofectamineTM RNAiMAX reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Cells were collected and analyzed 24 h after transfection. 5′-tsRNAGlu mimics and 5′-tsRNAGlu-targeting ASO were chemically synthesized (Sangon Biotech, Shanghai, China). The oligonucleotide sequences are listed in Supplementary Table 1.
For cell proliferation assays, cells were seeded in 96-well plates at a density of 1000 cells per well. Cell proliferation was subsequently measured using a Cell Counting Kit-8 (Bimake, TX, USA) according to the manufacturer’s instructions. The absorbance of each well was measured at 450 nm at each time point. For colony formation assays, cells were seeded in 6-well plates at a density of 1000 cells per well. After incubation at 37 °C for approximately 14 days, cells were washed three times with PBS, fixed with 4% paraformaldehyde for 30 min, and stained with 0.2% crystal violet solution for 30 min. After washing three times with PBS, each well was photographed, and the number of colonies was counted. Three independent experiments were performed, each in triplicate.
Cells were harvested and washed twice in PBS, suspended in 700 µL of 75% ethanol, mixed with 300 µL of PBS, and refrigerated at –20 °C overnight. Following centrifugation and washing twice with PBS, cells were resuspended, stained with propidium iodide (PI)/RNase Stain Binding Buffer, and placed at 4 °C for 30 min. DNA content was then measured using a FACSCalibur flow cytometer (BD Biosciences, CA, USA). Three independent experiments were performed, each in triplicate.
Experiments were performed using the Click-iT Plus OPP Protein Synthesis Assay Kit (Invitrogen, CA, USA). Briefly, cells in 6-well plates were incubated with 1 mL of medium containing 10 µM Click-iT Plus OPP working solution for 15 min. After centrifugation and washing with PBS, cells were fixed in 3.7% paraformaldehyde for 15 min, permeabilized in 0.5% Triton X-100 for 15 min, and then incubated in the dark with 100 µL of Click-iT Plus OPP reaction cocktail for 30 min. Subsequently, the cells were washed with the Click-iT reaction rinse buffer and stained with 4′,6-diamidino-2-phenylindole (DAPI). Samples were then analyzed using a FACSCanto II flow cytometer (BD Biosciences, CA, USA). Three independent experiments were performed, each in triplicate.
Cells were treated with 100 µg/mL cycloheximide (CHX) for 5 min, lysed with lysis buffer (20 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 150 mM NaCl, 100 µg/mL of CHX, 1 mM DTT, 100 U/mL of RNasein, 0.5% Triton X-100) on ice; subsequently, the cells were centrifuged and the supernatant was collected. The supernatant was layered on top of a 5–45% (w/v) sucrose gradient and centrifuged at 32,000 rpm at 4 °C for 1.5 h. Subsequently, the sample was fractionated using a Gradient Fractionator (Biocomp, Quebec, Canada) while continuously monitoring the absorbance at 260 nm. Fractions were categorized and used to isolate total RNA for subsequent RNA analysis. Three independent experiments were performed, each in triplicate.
The 5′-tsRNAGlu target sequence (reverse complement to the 5′-tsRNAGlu sequence) was subcloned into the pmirGLO vector (Tsingke, Beijing, China). Subsequently, the cells were co-transfected with the reporter plasmid and 5′-tsRNAGlu mimics or NC mimics using LipofectaminTM 3000 (Invitrogen, CA, USA). 24 h post-transfection, firefly and renilla luciferase activities were quantified using the Dual-Luciferase Reporter Assay System (Beyotime, Shanghai, China) following the manufacturer’s protocol. Three independent experiments were performed, each in triplicate.
Cells were washed with PBS and harvested by centrifugation. Nuclear and cytoplasmic extracts were then prepared using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China) following the manufacturer’s instructions. Briefly, cell pellets were resuspended in Reagent A, vortexed, and then incubated on ice. Following the addition of Reagent B and vortexing, the mixture was incubated on ice and centrifuged to separate the supernatant (containing cytoplasmic proteins) from the pellet (containing nuclear proteins). Cytoplasmic and nuclear RNAs were extracted from these fractions by adding TRIzolTM reagent (Invitrogen, CA, USA). Three independent experiments were performed, each in triplicate.
Biotin-labeled 5′-tsRNAGlu and its antisense probe were synthesized by Sangon Biotechnology (Shanghai, China). RNA pull-down assays were then conducted using the PierceTM Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Briefly, 100 pmol of biotin-labeled 5′-tsRNAGlu or its antisense probe was added to 50 µL of streptavidin-coated magnetic beads and incubated for 30 min under constant agitation. The Master Mix containing pre-prepared cell lysates was then added to the RNA-bound beads and incubated at 4 °C for 60 min. Subsequently, the beads were washed three times with wash buffer, resuspended in loading buffer, followed by denaturation at 100 °C for 10 min. Eluted proteins were then separated by SDS-PAGE and visualized using Coomassie Blue staining. Protein bands within the molecular weight range of 20–150 kDa were excised and subjected to protein identification using LC-MS/MS on a Q-Exactive HF X tandem mass spectrometer (Thermo Fisher Scientific, CA, USA) at BGI (Shenzhen, China). The sequences of all RNA pull-down probes are provided in Supplementary Table 1. Three independent experiments of RNA pull-down assay were performed, each in triplicate.
RIP assays were conducted using an Immunoprecipitation Kit with Protein A+G Magnetic Beads (Beyotime, Shanghai, China), according to the manufacturer’s protocol. Briefly, cells were lysed in RIP lysis buffer containing protease inhibitors and RNase inhibitors. The lysate was centrifuged, and then the supernatant was collected and incubated with 0.5 µg of RPL17 antibody (14121-1-AP, Proteintech, IL, USA) or IgG antibody (A7016, Beyotime, Shanghai, China) under rotation at 4 °C for 24 h. Subsequently, 10 µL of Protein A+G magnetic beads were added and incubated with rotation at 4 °C for 16 h. RNA bound to the immunoprecipitated complexes was extracted using TRIzolTM reagent (Invitrogen, CA, USA) and subjected to quantitative RT-PCR analysis. Three independent experiments were performed, each in triplicate.
Total RNA was isolated from cell lines using TRIzolTM reagent (Invitrogen,
CA, USA), according to the manufacturer’s protocol. To detect small RNAs, cDNA
was reverse-transcribed from 500 ng of total RNA using the PrimeScripTM Ⅱ
1st Strand cDNA Synthesis Kit (Takara, Tokyo, Japan). For mRNA detection, cDNA
was synthesized from 500 ng of total RNA using a PrimeScriptTM RT Master Mix
Kit (Takara, Tokyo, Japan). Quantitative real-time PCR was performed using
2
Total RNAs were resolved on a 10% urea-PAGE gel and stained with SYBR Gold (Thermo Fisher Scientific, MA, USA), followed by immediate imaging. The separated RNAs were then transferred onto positively charged nylon membranes (Roche, Basel, Switzerland) and ultraviolet-crosslinked at an energy of 0.12 J. Subsequently, membranes were pre-hybridized with DIG Easy Hyb solution (Roche, Basel, Switzerland) for 1 h at 42 °C. After pre-hybridization, membranes were hybridized overnight at 42 °C with a DIG-labeled 5′-tsRNAGlu probe (Sangon Biotech, Shanghai, China). The probe sequences are listed in Supplementary Table 1. Subsequently, membranes were washed, blocked, and incubated with an anti-DIG-AP antibody (11093274910, 1:10,000, Roche, Basel, Switzerland) for 30 min. Signal detection was performed using the chemiluminescent substrate CSPD (Roche, Basel, Switzerland), and visualization was performed using a ChemiDocTM MP Imaging System (Bio-Rad, CA, USA).
Western blot analysis was performed according to a previously established
protocol [40]. The following primary antibodies were used: rabbit monoclonal
anti-ALKBH4 (ab195379, 1:1000, Abcam, Cambridge, UK), rabbit polyclonal
anti-Cyclin D1 (AF0126, 1:1000, Beyotime, Shanghai, China), rabbit polyclonal
anti-RPL17 (14121-1-AP, 1:1000, Proteintech, IL, USA), rabbit polyclonal
anti-Cyclin B1 (55004-1-AP, 1:1000, Proteintech, IL, USA), rabbit polyclonal
anti-Phospho-Histone-H3 (9701, 1:1000, Cell Signaling Technology, MA, USA),
rabbit monoclonal anti-Histone-H3 (4499T, 1:1000, Cell Signaling Technology, MA,
USA), rabbit polyclonal anti-GAPDH (AB-P-R001, 1:1000, Goodhere Biotech,
Hangzhou, China), and rabbit monoclonal anti-
H1299 cells were transiently transfected for 24 h with the pCDH plasmid, ALKBH4
expression plasmid, NC mimics, and 5′-tsRNAGlu mimics, as indicated.
Total RNA was extracted using TRIzolTM reagent (Invitrogen, CA, USA).
Following quantitative analysis and quality inspection, the RNA samples were
subjected to sequencing by BGI (Shenzhen, China). Libraries were constructed
using the Optimal Dual-mode mRNA Library Prep Kit (BGI, Shenzhen, China), and
sequenced on a BGIseq500 platform (BGI, Shenzhen, China). Raw sequencing data
were normalized for subsequent analysis. RNA-seq data are shown in
Supplementary Table 2. Differentially expressed genes were identified
using a threshold of
RNA sequencing data for both LUAD and LSCC tissues were retrieved from the TCGA
database [41]. Co-correlation patterns between ALKBH4 and other genes were
evaluated using Pearson’s correlation analysis [42]. Gene Set Enrichment Analysis
(GSEA) was performed with the GSEA portal
(https://www.gsea-msigdb.org/gsea/index.jsp) with the
“C5.go.bp.v7.5.symbols.gmt” gene set collection. The selection criteria were as
follows: nominal p-value
Statistical analyses were performed using SPSS software (version 22.0, SPSS
Inc., IL, USA). Comparisons between two groups were assessed using unpaired
two-tailed Student’s t-test, while comparisons among multiple groups
were performed using one-way ANOVA. p-value
5′-tsRNAGlu (36-nt in length), is produced from the specific cleavage of the anticodon loop of tRNAGlu(CTC). The type of 5′-tsRNA is also addressed by another name, i.e., tiRNA, in response to stress conditions. To test if 5′-tsRNAGlu was induced by oxidative stress, we compared the levels of 5′-tsRNAGlu in NSCLC cells responding to H2O2 treatment. H1299 cells subjected to H2O2 treatment exhibited a significant increase in the levels of 5′-tsRNAGlu, ALKBH4, and ANG (Fig. 1A,C). Similarly, their expression levels were remarkably upregulated in A549 cells following the H2O2 treatment (Fig. 1B,D). Oxidative stress increased the levels of 5′-tsRNAGlu in NSCLC cells. Under the same conditions, the expression of ALKBH4 and ANG was also elevated, suggesting that these factors are responsive to oxidative stress.
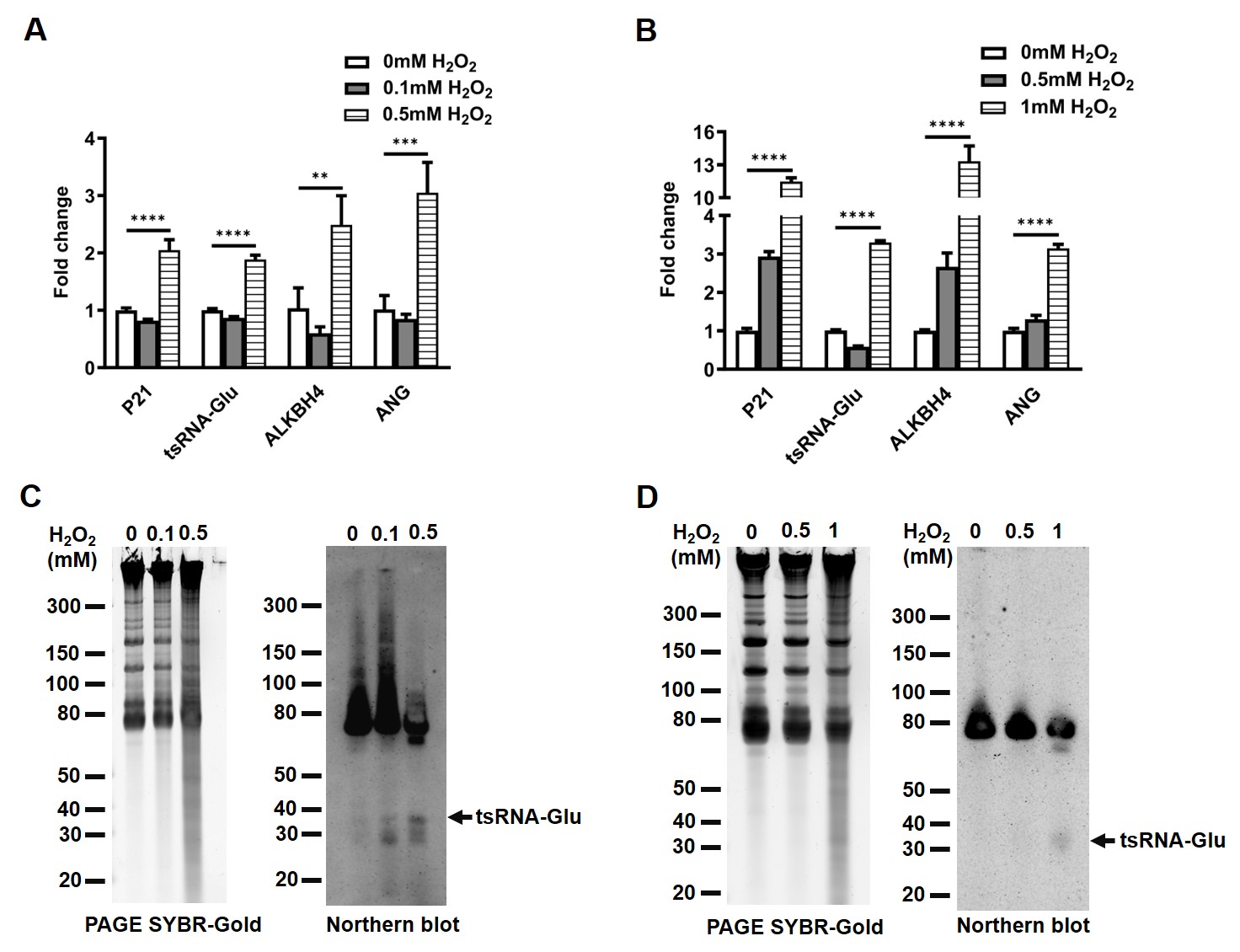 Fig. 1.
Fig. 1.
The levels of 5′-tsRNA𝐆𝐥𝐮, ALKBH4 and ANG in
NSCLC cells treated with H2O2. (A,B) The levels of
5′-tsRNAGlu, ALKBH4 and ANG in H1299 cells (A) and A549 cells (B)
treated with H2O2 were measured using quantitative RT-PCR, with U6 and
GAPDH as the loading controls, respectively. (C,D) The 5′-tsRNAGlu
levels in H1299 cells (C) and A549 cells (D) treated with H2O2 were
measured using Northern blot. The overall band intensity in PAGE served as the
loading control. Data are shown as the mean
RNA sequencing data for LUAD and LSCC were downloaded from the TCGA database and then used for gene expression correlation analysis, further creating a set of genes that were highly correlated with ALKBH4 expression. GSEA revealed that the gene set correlated with ALKBH4 was remarkably enriched in ‘tRNA processing’ and ‘RNA methylation’ (Supplementary Fig. 1), thus indicating that ALKBH4 might mediate the dynamic methylation of tRNAs.
Furthermore, NSCLC cells were transfected with the ALKBH4 plasmid (Fig. 2A,C). Both H1299 cells and A549 cells with ectopic expression of ALKBH4 showed a significant increase in 5′-tsRNAGlu levels (Fig. 2B,D). These results suggest that ALKBH4 may induce the biogenesis of 5′-tsRNAGlu in NSCLC cells, potentially through its tRNA demethylase activity. However, to determine whether ALKBH4 directly induces the biogenesis of 5′-tsRNAGlu, further studies will need to evaluate 5′-tsRNAGlu levels in ALKBH4 knockout cells.
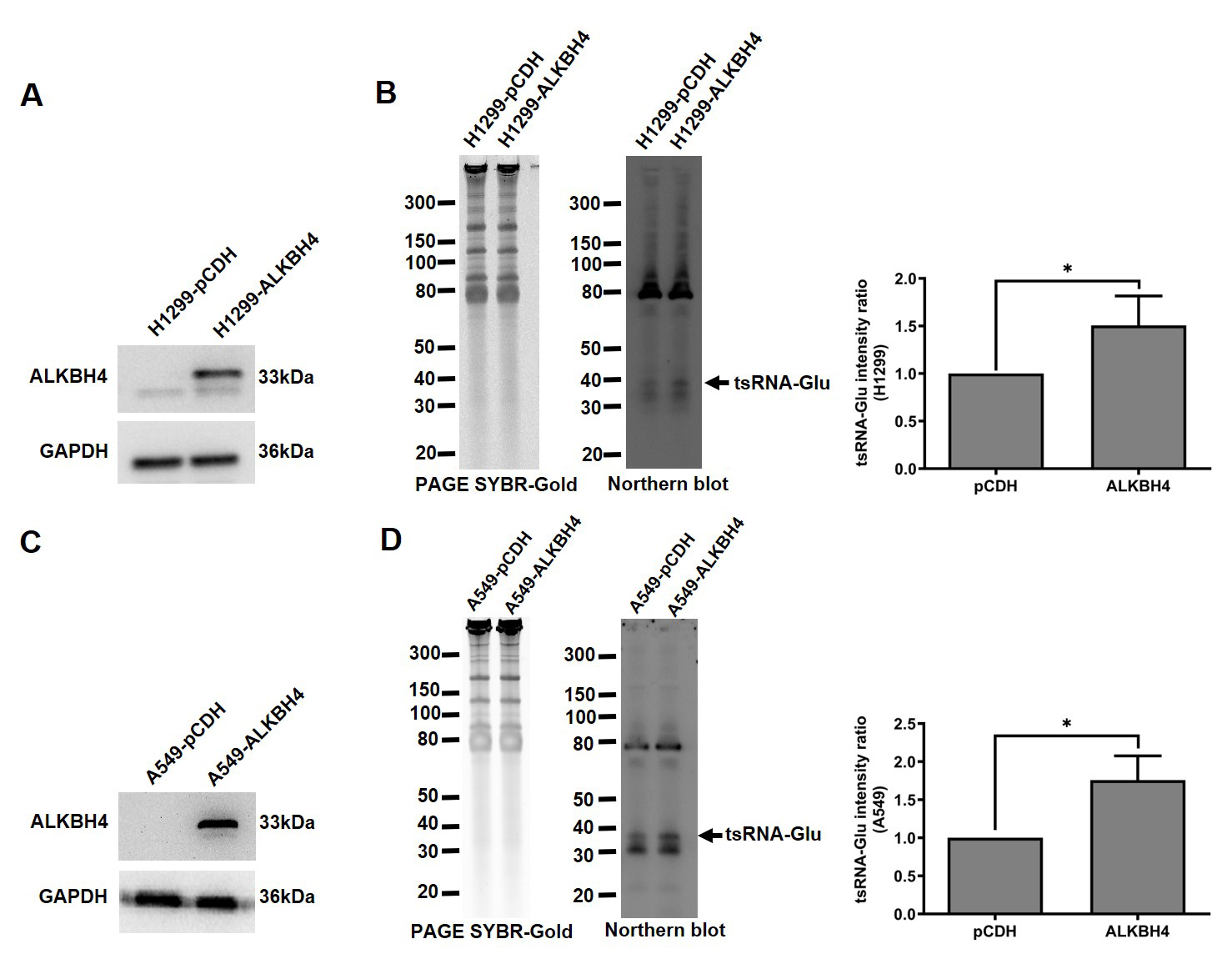 Fig. 2.
Fig. 2.
The 5′-tsRNA𝐆𝐥𝐮 levels in NSCLC cells
overexpressing ALKBH4. The ALKBH4 levels in H1299 cells (A) and A549 cells (C)
transiently transfected with the ALKBH4 plasmid were determined using Western
blot. GAPDH served as the loading control. The 5′-tsRNAGlu levels in
H1299 cells (B) and A549 cells (D) transiently transfected with the ALKBH4
plasmid were determined using Northern blot. The overall band intensity in PAGE
served as the loading control. The right panel shows the densitometric
quantification of 5′-tsRNAGlu levels, presented as fold change relative
to the control group. Data are shown as the mean
Next, we ivestigated the effects of ALKBH4 and 5′-tsRNAGlu on the proliferative ability of NSCLC cells. H1299 cells exhibiting ectopic ALKBH4 expression showed a remarkable reduction in cell proliferation, colony formation, and cell cycle arrest at the S-phase (Fig. 3). Likewise, H1299 cells transiently transfected with 5′-tsRNAGlu mimics revealed decreased capabilities for cell proliferation, colony formation, and cell cycle arrest at the G2/M phase (Fig. 4A–E). Conversely, the knockdown of 5′-tsRNAGlu using its specific ASO significantly promoted the cell proliferation and colony formation of H1299 cells (Supplementary Fig. 2). Furthermore, the reduction in cell proliferation due to ALKBH4 overexpression was rescued by the knockdown of 5′-tsRNAGlu (Fig. 4F). Collectively, these results suggest that ALKBH4 inhibited NSCLC cell proliferation probably via the induction of 5′-tsRNAGlu.
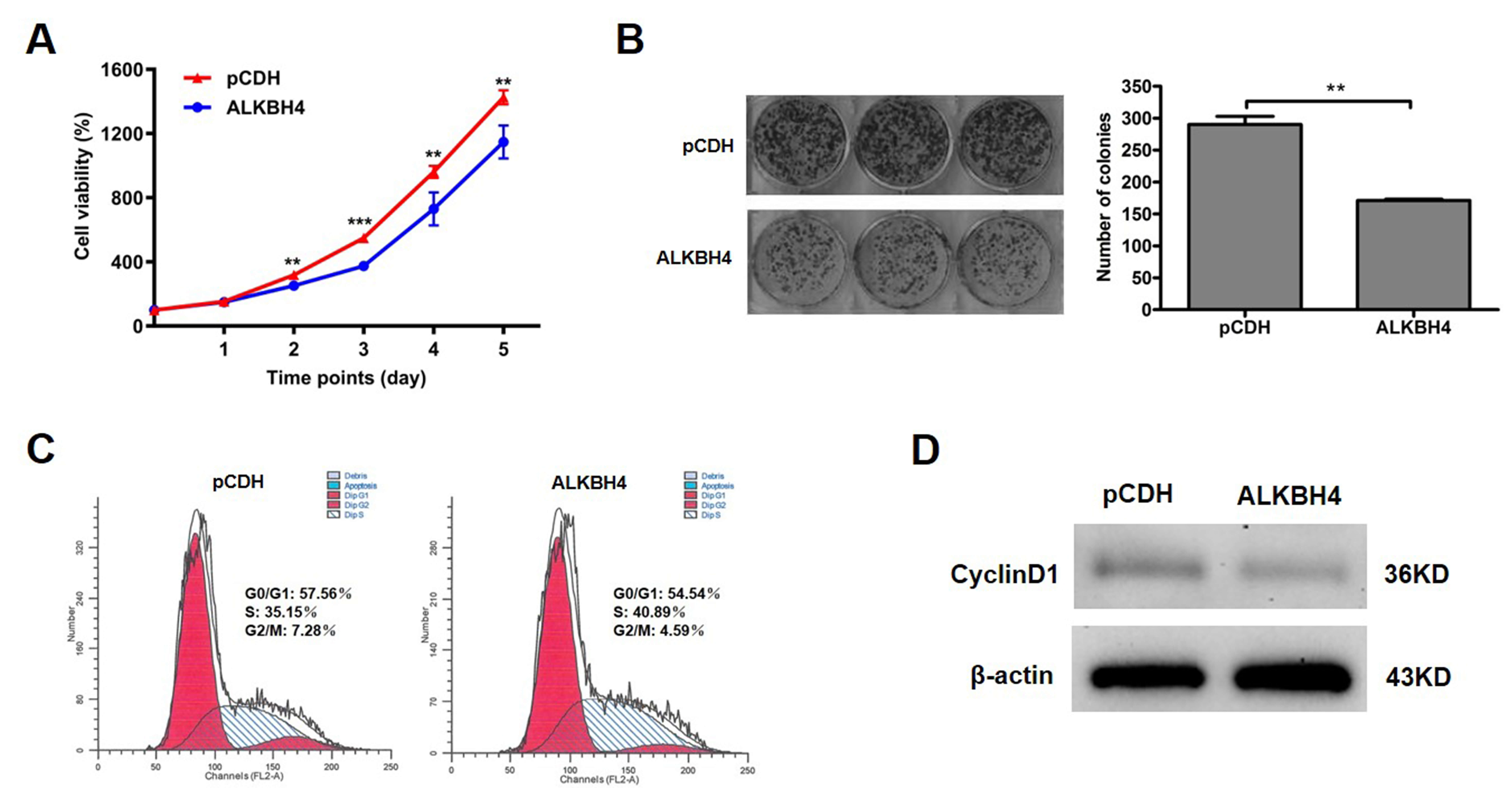 Fig. 3.
Fig. 3.
The effects of ALKBH4 on cell proliferation and cell cycle of
H1299 cells. (A) Proliferation curves of H1299 cells transiently transfected
with the ALKBH4 plasmid or the pCDH vector were monitored every 24 h, over 5
days. Three independent experiments were performed in triplicate. Data are shown
as the mean
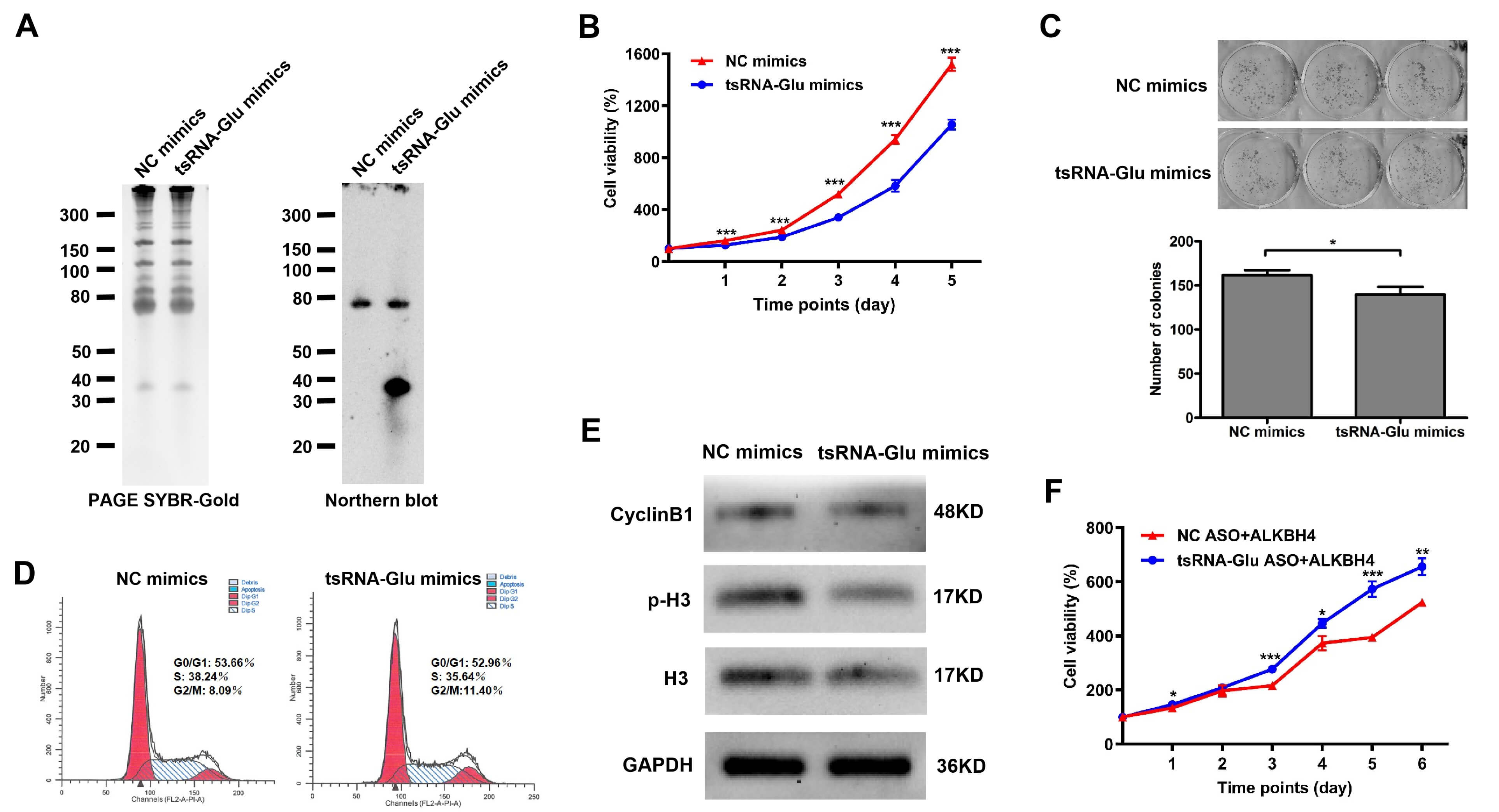 Fig. 4.
Fig. 4.
The effects of 5′-tsRNA𝐆𝐥𝐮 on cell
proliferation and cell cycle of H1299 cells. (A) The 5′-tsRNAGlu level
in H1299 cells transiently transfected with 5′-tsRNAGlu mimics was
determined using Northern blot. The overall band intensity in PAGE served as the
loading control. (B) Proliferation curves of H1299 cells transiently transfected
with 5′-tsRNAGlu mimics or NC mimics were monitored every 24 h, over 5
days. Three independent experiments were performed in triplicate. Data are shown
as the mean
We further evaluated the effects of ALKBH4 and 5′-tsRNAGlu on protein synthesis in NSCLC cells. OPP protein synthesis assays showed that the overexpression of ALKBH4 decreased nascent protein synthesis in H1299 cells (Fig. 5A). Furthermore, the overexpression of ALKBH4 reduced 80S monosome and polysome contents in polysome profiling (Fig. 5B). Consistently, the overexpression of 5′-tsRNAGlu receded nascent protein synthesis as well as 80S monosome and polysome contents in H1299 cells (Fig. 5C,D). Overall, these results indicate that both ALKBH4 and 5′-tsRNAGlu contributed to the downregulation of global translation efficiency in NSCLC cells.
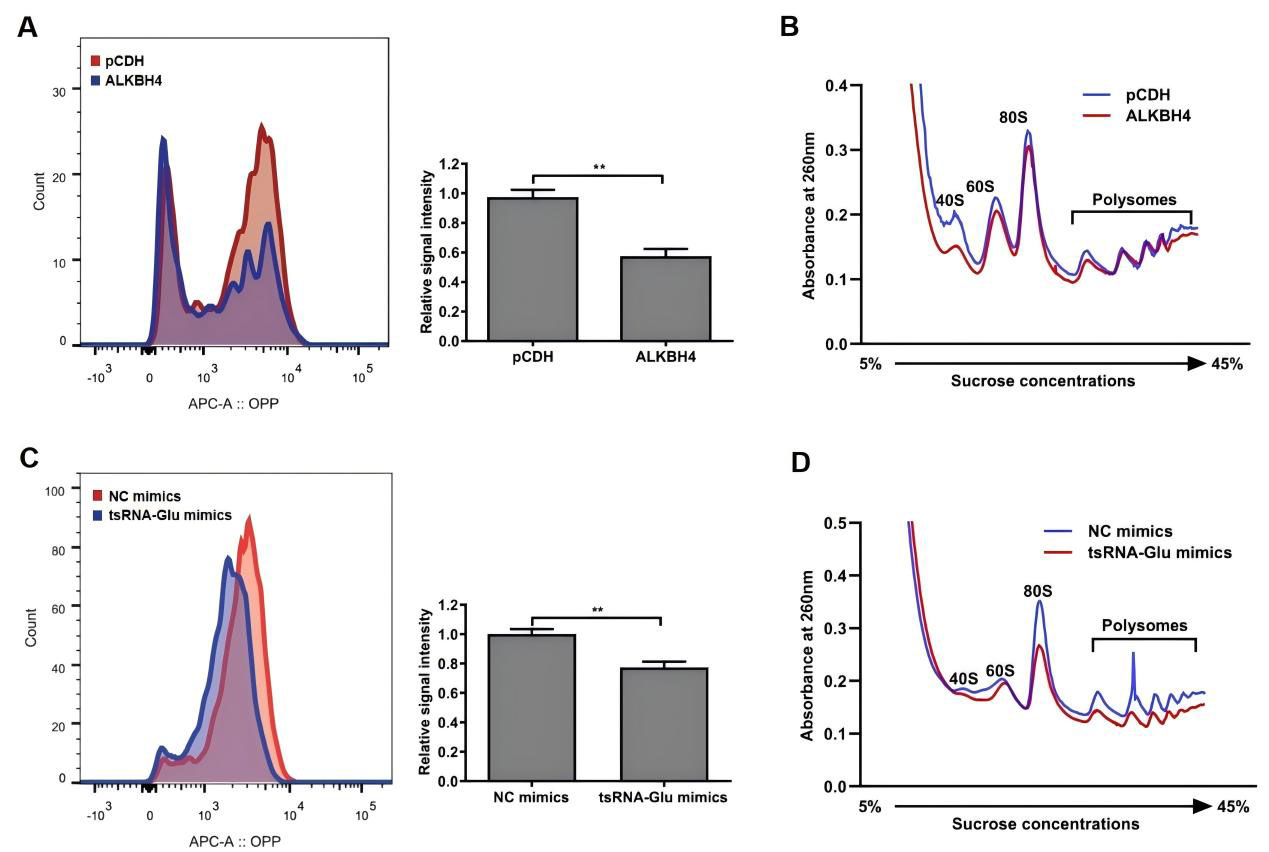 Fig. 5.
Fig. 5.
The effects of ALKBH4 and 5′-tsRNA𝐆𝐥𝐮 on the
global translational efficiency of H1299 cells. (A) The nascent protein
synthesis of H1299 cells transiently transfected with the ALKBH4 plasmid or the
pCDH vector was determined using OPP protein synthesis assay. Their relative
fluorescence intensities were quantified in the right section. (B) The polysome
profiling of H1299 cells transiently transfected with the ALKBH4 plasmid or the
pCDH vector was analyzed. (C) The nascent protein synthesis of H1299 cells
transiently transfected with 5′-tsRNAGlu mimics or NC mimics was
determined using OPP protein synthesis assay. Their relative fluorescence
intensities were quantified in the right section. (D) The polysome profiling of
H1299 cells transiently transfected with 5′-tsRNAGlu mimics or NC mimics
was analyzed. Three independent experiments were performed in triplicate. Data
are shown as the mean
Previous studies have established that tsRNAs can inhibit the translation of mRNAs via binding complementarily with the 3′UTR of these mRNAs, in a miRNA-like manner [22, 24, 28, 44, 45, 46, 47]. Based on this, we proceeded to screen the target mRNAs of 5′-tsRNAGlu. The mRNA expression profiles of NSCLC cells transiently transfected with the ALKBH4 plasmid (termed the ALKBH4 profile) and those with 5′-tsRNAGlu mimics (termed the tsRNA-Glu profile) were detected using RNA sequencing. GSEA revealed that the biological process categories enriched in the ALKBH4 profile were translational termination, ribosome biogenesis, ncRNA processing, etc. (Supplementary Fig. 3A). The biological process categories enriched in the tsRNA-Glu profile were the regulation of response to extracellular stimulus, the negative regulation of epithelial cell proliferation, and drug catabolic process, among others (Supplementary Fig. 3B). It displayed 11 differentially expressed genes with the same trend in the ALKBH4 profile and the tsRNA-Glu profile (Supplementary Table 3). Of these, three genes that strongly associated with tumor progression, i.e., XAF1, EIF2S3B and REC8, were selected for further validation using quantitative RT-PCR. It showed that EIF2S3B and REC8 were significantly upregulated in both profiles, consistent with the RNA sequencing results, whereas XAF1 expression displayed the contrary tendency in both profiles (Supplementary Fig. 4A,B). Subsequently, we analyzed the expression patterns of XAF1 and REC8 in NSCLC tumor tissues compared to normal lung tissues, based on the TCGA and GTEx database (EIF2S3B data were unavailable). The expression levels of XAF1 and REC8 in LUAD and LSCC tissues were markedly lower than those in normal lung tissues (Supplementary Fig. 4C,D). These findings imply that REC8 mRNA may represent a downstream target of 5′-tsRNAGlu. Nevertheless, sequence alignment analysis failed to identify complementary pairing between 5′-tsRNAGlu and all regions of the REC8 mRNA, thus indicating that REC8 mRNA is probably not the direct target of 5′-tsRNAGlu.
Moreover, utilizing the TargetRank database, we predicted PPM1F, MECP2 and C14orf172 as the targets of 5′-tsRNAGlu. However, subsequent validation disproved these predictions (Supplementary Fig. 5A,B). To directly test the gene-silencing activity of 5′-tsRNAGlu, we next constructed a dual-luciferase reporter that has the complementary sequence of 5′-tsRNAGlu at the 3′UTR of the Renilla luciferase gene (Supplementary Fig. 5C,D). The dual-luciferase reporter gene assay revealed that 5′-tsRNAGlu had no effect on the relative luciferase activity of the reporter gene containing the complementary sequence of 5′-tsRNAGlu (Supplementary Fig. 5E–H). Taking the inhibitory effects of 5′-tsRNAGlu on global translation efficiency into consideration, we propose that 5′-tsRNAGlu interacts with specific proteins rather than specific mRNAs, to suppress the global translation efficiency of NSCLC cells and thus their malignant phenotypes.
To verify the hypothesis that 5′-tsRNAGlu could suppress global translational efficiency via binding target proteins, we next employed RNA pull-down coupled with mass spectrometry to identify proteins that interacted with 5′-tsRNAGlu (Fig. 6A). Based on the results of two replicates, we identified seven overlapping proteins, including MYEF2, RPS5, RPL17, DEK, NUDT21, SRSF10, and SRSF5 (Fig. 6B). Subsequently, the interaction probabilities between 5′-tsRNAGlu and the candidate proteins were predicted using the RPIseq database; RPL17 exhibited the highest interaction probability among these candidate proteins (Fig. 6C). Considering that RPL17 is a ribosomal large subunit protein closely associated with translation, we selected RPL17 for further validation. Independent RNA pull-down assays and WB assays confirmed the interaction of 5′-tsRNAGlu and RPL17 in both H1299 and A549 cells (Fig. 6D). Consistently, RIP assays also proved that 5′-tsRNAGlu was enriched by the RPL17 antibody group, compared with the IgG antibody group (Fig. 6E). In addition, 5′-tsRNAGlu did not affect the expression of RPL17 (Supplementary Fig. 6). To gain insight into the specific binding pattern between 5′-tsRNAGlu and RPL17, we utilized the online database HDOCK to predict the detailed binding sites of 5′-tsRNAGlu to RPL17 (Fig. 6F). It showed that 5′-tsRNAGlu bound to the region spanning residues 137-142 of RPL17, which corresponds to the protein-rRNA interaction interface. To determine the subcellular localization of 5′-tsRNAGlu, we next performed nucleoplasmic separation assays and found that over 80% of 5′-tsRNAGlu localized in the cytoplasm of both H1299 cells and A549 cells (Fig. 6G). Furthermore, we explored the distribution of 5′-tsRNAGlu in the 40S, 60S, and 80S ribosomal fractions. The overexpression of 5′-tsRNAGlu led to an increased ratio of 5′-tsRNAGlu in the 60S but a decreased ratio of 5′-tsRNAGlu in the 80S ribosomal fraction (Fig. 6H). Given these findings, we speculate that the binding of 5′-tsRNAGlu to RPL17 disrupts the binding of RPL17 to rRNAs, and thus 80S ribosome assembly.
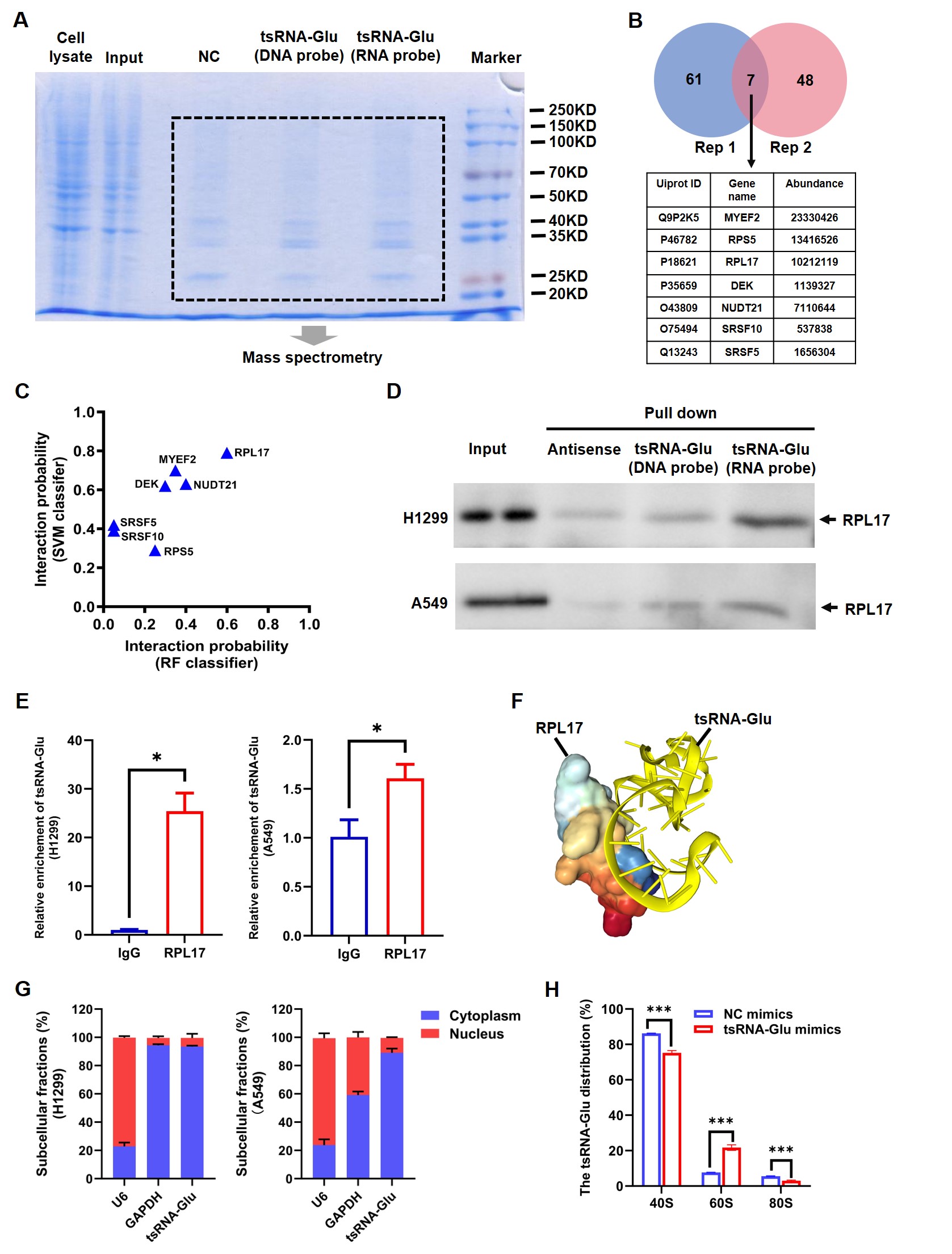 Fig. 6.
Fig. 6.
5′-tsRNA𝐆𝐥𝐮 selectively interacts with RPL17.
(A) SDS-PAGE and then Coomassie staining of RNA pull-down samples from H1299
cells. tsGlu (RNA Probe): 5′-tsRNAGlu; tsGlu (DNA Probe): the
corresponding DNA sequence of 5′-tsRNAGlu; NC: the reverse complementary
DNA sequence of 5′-tsRNAGlu. (B) The counts of candidate
5′-tsRNAGlu interacted proteins identified in two biological replicates
as well as these seven overlapping proteins. (C) The interaction probability of
5′-tsRNAGlu and candidate proteins predicted by RPIseq database. The
horizontal axis represents the interaction probability using RF classifier, and
the vertical axis represents the interaction probability using SVM classifier.
(D) Validation of the binding of 5′-tsRNAGlu to RPL17 using RNA
pull-down assays. 5′-Biotinylated synthetic oligonucleotides were used for
RNA pull-down assays. The antisense sequence of 5′-tsRNAGlu was used as
the negative control. Three independent experiments were performed in triplicate.
(E) Validation of the binding of 5′-tsRNAGlu to RPL17 using RIP assays.
The 5′-tsRNAGlu level in the product obtained from RIP was detected
using quantitative RT-PCR. Three independent experiments were performed in
triplicate. (F) The binding pattern of 5′-tsRNAGlu and RPL17 protein
predicted by HDOCK database. 5′-tsRNAGlu is shown in yellow, which is
predicted to bind to the protein-rRNA interface (spanning residues 137–142) of
RPL17. (G) The levels of 5′-tsRNAGlu in the cytoplasm and the nucleus of
H1299 cells (left) and A549 cells (right) were determined using quantitative
RT-PCR following subcellular RNA fractionation. GAPDH and U6 served as the
controls for the cytoplasmic fraction and the nuclear fraction, respectively.
Three independent experiments were performed in triplicate. Data are shown as the
mean
ALKBH4 was initially characterized as a catalyst for actin demethylation that
modulates the contraction of the actomyosin network [48, 49, 50]. Subsequent studies
have revealed its potential role in regulating chromatin states and gene
expression [51, 52]. Although studies focused on the potential role of ALKBH4 in
oncology are relatively scarce, current evidence suggests that it may function as
a tumor suppressor. In a previous study, Shen et al. [53] demonstrated
that ALKBH4 suppressed the metastasis of colorectal cancer by inhibiting
miR-21 expression and epithelial-mesenchymal transition (EMT). This
functionality was attributed to its competitive binding to the methyltransferase
WDR5, which is responsible for H3K4me3 modification, leading to reduced H3K4me3
levels in the chromatin region associated with the WDR5 target gene
miR-21 [53]. Another study found that among the ALKBH family, only
ALKBH4 was downregulated by hypoxia-inducible factor HIF-1
Our study further demonstrated that the inhibitory effect of ALKBH4 on the proliferation of NSCLC cells was dependent on 5′-tsRNAGlu. Specifically, ALKBH4 induced the biogenesis of 5′-tsRNAGlu in NSCLC cells (Fig. 2). 5′-tsRNAGlu markedly suppressed the proliferation of NSCLC cells, arrested cell cycle arrest at the G2/M phase (Fig. 4), and attenuated global translational efficiency by decreasing the abundance of 80S ribosomes and polysomes (Fig. 5). Rescue assays further confirmed that ALKBH4 inhibited the proliferation of NSCLC cells via a 5′-tsRNAGlu-mediated mechanism (Fig. 4F). It is plausible that the ALKBH4-induced production of 5′-tsRNAGlu involves RNA modification. RNA molecules carry over 100 types of functional chemical modifications [35], whose dynamic and reversible regulation, along with their functional consequences, are coordinated by “writer”, “eraser” and “reader” proteins [36]. tRNAs harbor more chemical modifications than other RNA species, with an average of 13 modifications per molecule [57]; these modifications modulate tRNA stability and function through structural influences [58]. Methylation is the most abundant category of tRNA chemical modifications [59, 60], including m1A, m1G, m3C, m5C, and m22G [61]. tRNA methyltransferases (“writers”) act in concert with tRNA demethylases (“erasers”) to dynamically regulate tRNA methylation. Reduced tRNA methylation can destabilize tRNAs, rendering them more susceptible to ribonuclease cleavage and leading to the biogenesis of tsRNA [36]. The ALKBH family comprises nine members (ALKBH1-ALKBH8 and FTO), many of which exhibit demethylase activity. For instance, Chen et al. [38] discovered that ALKBH3 catalyzed the demethylation of m1A and m3C in tRNAs. m1A-demethylated tRNAs, in particular, were more prone to cleavage by ANG, predominantly yielding 5′-tsRNAs. ALKBH3 was shown to facilitate tumor cell proliferation, invasion, chemotherapy resistance, and tumorigenesis in vivo via specific tsRNAs such as 5′-tDR-GlyGCC [38]. Although ALKBH4 has not yet been definitely characterized as a tRNA demethylase, one study indicated that it could modulate translational efficiency through regulating uridine modifications in tRNAs [56]. To test the hypothesis that ALKBH4 catalyzes demethylation at specific sites of tRNAGlu, thereby promoting its cleavage to generate 5′-tsRNAGlu, future studies will employ liquid chromatography-tandem mass spectrometry (LC-MS/MS) to analyze the impact of ALKBH4 on nucleotide methylation levels at specific positions of tRNAGlu.
Despite the growing interest in tsRNAs, the involvement of tsRNAs in lung cancer
develpment remains poorly characterized. Early investigations revealed that
tsRNA-46, tsRNA-47, and tsRNA-53 impaired the clonogenic potential of NSCLC cells
via unknown mechanisms [23, 32]. Subsequent work by Shao et al. [62]
demonstrated that silencing a tiRNA derived from tRNALeu-CAG suppressed NSCLC
cell proliferation and induced G0/G1 cell cycle arrest, putatively through
downregulation of AURKA. Expanding on this, Young and colleagues uncovered that
AS-tDR-007333 drove both proliferation and metastasis in NSCLC cells via the
HSPB1-MED29 and ELK4-MED29 signaling axes [63]. In a distinct mechanistic
paradigm, Yu et al. [33] identified CAT1, a pan-cancer-upregulated
tsRNA, which stabilized NOTCH2 mRNA by binding RBPMS and subsequently abrogating
CCR4-NOT-mediated deadenylation, thus facilitating lung cancer progression. More
recently, Zhao et al. [64] found that tumor-derived exosomal
3′-tsRNA-AlaCGC activated the TGF-
While our study uncovers a previously unrecognized role of the ALKBH4/5′-tsRNAGlu axis in modulating translational efficiency and cellular proliferation in NSCLC, several limitations warrant consideration. First, the precise molecular mechanism by which 5′-tsRNAGlu binds to RPL17 to suppress translation and proliferation remains incompletely elucidated. Computational predications indicate that 5′-tsRNAGlu interacts with a region spanning residues 137–142 of RPL17, corresponding to the protein-rRNA interaction interface, further studies employing systematic truncations of RPL17 are necessary to delineate the minimal binding domain and identify specific amino acid residues essential for 5′-tsRNAGlu recognition. Furthermore, well-designed rescue experiments, wherein wild-type and mutant RPL17 constructs will be introduced to confirm whether the 5′-tsRNAGlu-RPL17 interaction is directly responsible for the defects in 60S ribosomal subunit assembly, global translation efficiency, and proliferative capacity. Second, the diagnostic and therapeutic potential of the ALKBH4/5′-tsRNAGlu axis has yet to be systematically evaluated. Future investigations should focus on collecting adequate cohorts of clinical peripheral blood specimens to assess the utility of ALKBH4 and 5′-tsRNAGlu as non-invasive biomarkers for NSCLC diagnosis. In addition, in vivo xenograft models is imperative to determine whether targeting ALKBH4/5′-tsRNAGlu represents a viable therapeutic target in NSCLC.
Our findings establish the ALKBH4/5′-tsRNAGlu axis as a critical pathway suppressing the progression of NSCLC. ALKBH4 promotes the biogenesis of 5′-tsRNAGlu, which in turn exerts tumor-suppressive effects that are unlike miRNA, but rather via direct interaction with the ribosomal protein RPL17. This interaction is proposed to destabilize the RPL17-rRNA interface, thereby impeding assembly of the 80S ribosome, thus attenuating global translational efficiency, and ultimately inhibiting cell proliferation. Therefore, this study implies both ALKBH4 and 5′-tsRNAGlu represent promising candidates for the diagnosis and treatment of NSCLC.
tsRNAs, tRNA-derived small RNAs; ALKBH, alpha-ketoglutarate-dependent dioxygenase alkB homolog; NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; LSCC, lung squamous cell carcinoma; ncRNAs, non-coding RNAs; UV, ultraviolet; ANG, angiogenin; tiRNAs, tRNA-derived stress-induced RNAs; RG4, RNA G-quadruplexes; ASO, antisense oligonucleotide; PI, propidium iodide; CHX, cycloheximide; LC-MS/MS, liquid chromatography-tandem mass spectrometry; RIP, RNA immunoprecipitation; FDR, false discovery rate.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
CS: Investigation, Methodology, Writing – original draft. HC: Investigation, Methodology, Writing – original draft. YL: Investigation, Methodology. DX: Investigation, Methodology. WD: Data curation, Software, Validation. MP: Data curation, Software, Validation. LS: Data curation, Software, Validation. JT: Validation. YZ: Conceptualization, Funding acquisition, Project administration, Writing – review and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by Chongqing Natural Science Foundation (CSTB2022NSCQ-MSX0123).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL50109.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.