








 , Shuqi Liang 2, Tianhao Qu 1, Shenyu Li 1, Ji Lei 1, Han Han 1, Yingying Guo 3, Man Xing 3, Xiaohong Wang 1,4, Shikun He 5, Mengyu Liao 1,4, Dongming Zhou 3,*
, Shuqi Liang 2, Tianhao Qu 1, Shenyu Li 1, Ji Lei 1, Han Han 1, Yingying Guo 3, Man Xing 3, Xiaohong Wang 1,4, Shikun He 5, Mengyu Liao 1,4, Dongming Zhou 3,* , Hua Yan 1,2,*
, Hua Yan 1,2,*1 Department of Ophthalmology, Tianjin Medical University General Hospital, International Joint Laboratory of Ocular Diseases (Ministry of Education), State Key Laboratory of Experimental Hematology, Tianjin Key Laboratory of Ocular Trauma, Laboratory of Molecular Ophthalmology, Tianjin Medical University, 300070 Tianjin, China
2 Department of Clinical Medicine, School of Medicine, Nankai University, 300071 Tianjin, China
3 School of Basic Medical Sciences, Tianjin Medical University, 300070 Tianjin, China
4 Department of Pharmacology, Tianjin Key Laboratory of Inflammation Biology, School of Basic Medical Sciences, Tianjin Medical University, 300070 Tianjin, China
5 Department of Pathology and USC Roski Eye Institute, Keck School of Medicine of the University of Southern California, Los Angeles, CA 90033, USA
Abstract
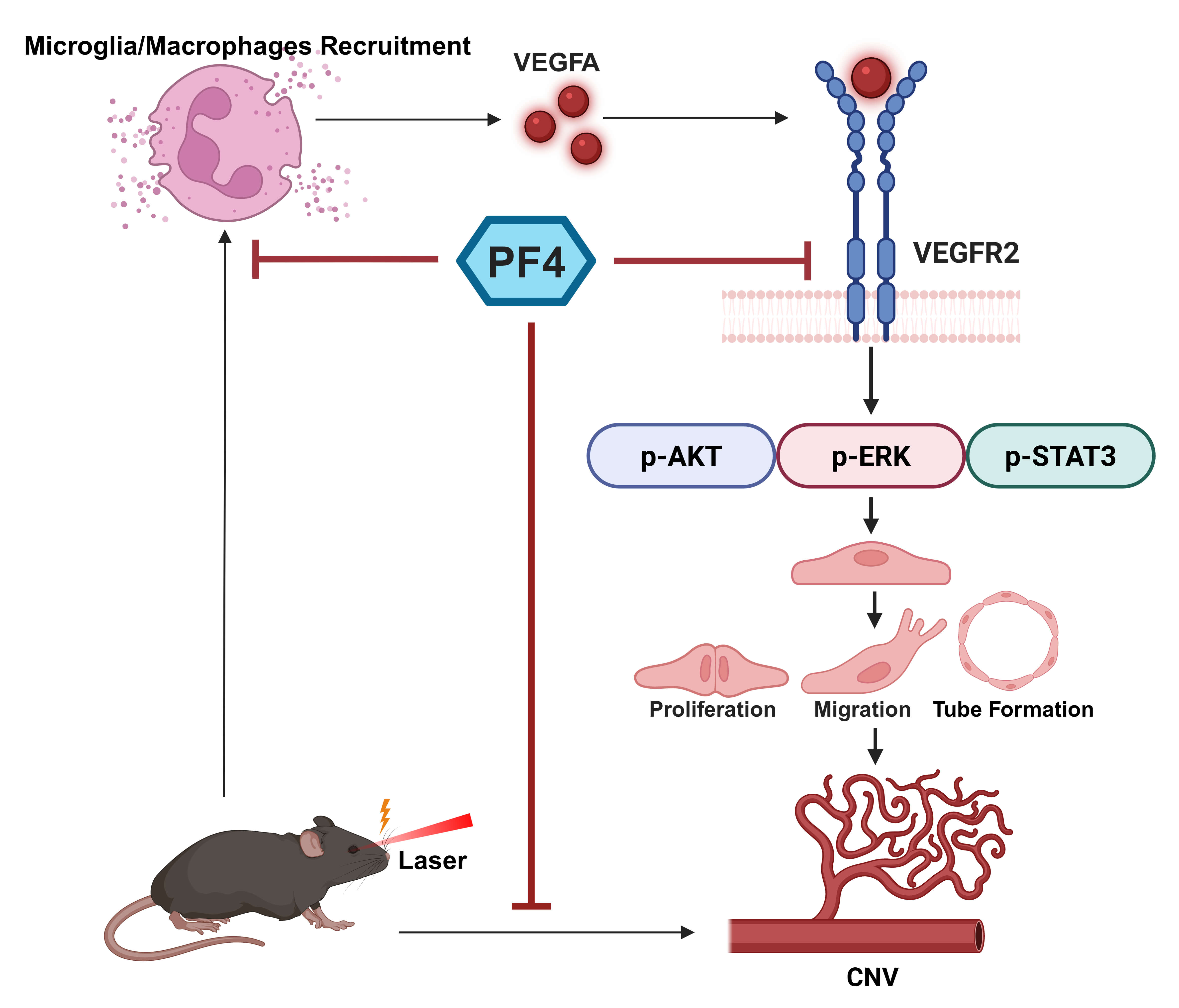
Platelet factor 4 (PF4/CXCL4) is a chemokine with reported anti-angiogenic and immunomodulatory properties; however, the role of PF4 in neovascular age-related macular degeneration (nAMD) remains unclear. Thus, this study aimed to evaluate the therapeutic potential of PF4 in experimental models of ocular pathological neovascularization and explored the underlying mechanisms.
PF4 expression was assessed in a laser-induced choroidal neovascularization (CNV) mouse model using quantitative real-time PCR (qRT-PCR), enzyme-linked immunosorbent assay (ELISA), and immunofluorescence. Recombinant PF4 was administered intravitreally in laser-induced CNV mice and very low-density lipoprotein receptor knockout (Vldlr⁻/⁻) mice, a model of spontaneous retinal neovascularization with retinal angiomatous proliferation (RAP)-like lesions. Pathological neovascularization and vascular leakage were quantified by fundus fluorescein angiography and choroidal/retinal flat-mount analyses. Immunofluorescence, qRT-PCR, and RNA sequencing were employed to evaluate inflammatory responses. Moreover, the effects of PF4 on vascular endothelial growth factor (VEGF)-induced proliferation, migration, and tube formation of human retinal microvascular endothelial cells were examined in vitro, and VEGF-mediated signaling was analyzed by Western blotting. Ocular safety was assessed by optical coherence tomography (OCT), electroretinography (ERG), hematoxylin and eosin (H&E) staining, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
Intravitreal PF4 significantly reduced pathological neovascularization and vascular leakage in both models and attenuated intraocular inflammation, as indicated by decreased expression of proinflammatory cytokines and reduced microglial/macrophage recruitment. PF4 inhibited VEGF-induced endothelial cell proliferation, migration, and tube formation in vitro. Mechanistically, PF4 downregulated VEGF expression in CNV lesions in vivo and suppressed VEGF-induced activation of vascular endothelial growth factor receptor 2 (VEGFR2) and downstream extracellular signal-regulated kinase (ERK), protein kinase B (AKT), and signal transducer and activator of transcription 3 (STAT3) signaling in vivo and in vitro. PF4 administration was well tolerated, with no detectable adverse effects on retinal structure or function.
PF4 effectively inhibits ocular pathological neovascularization and inflammation by modulating the VEGF/VEGFR2 signaling pathway. These findings support PF4 as a promising therapeutic candidate for nAMD and warrant further investigation.
Graphical Abstract
Keywords
- platelet factor 4
- wet macular degeneration
- neovascularization
- pathologic
- choroidal neovascularization
- inflammation
Age-related macular degeneration (AMD) is a leading cause of irreversible vision loss in the elderly worldwide [1]. It primarily manifests in two forms: the more prevalent dry (atrophic) type and the less common but visually more devastating wet (neovascular) form [2]. Neovascular AMD (nAMD) is characterized by pathological neovascularization; these aberrant vessels are prone to leakage, hemorrhage, and eventual fibrotic scarring, leading to rapid and severe central vision loss [3].
The pathogenesis of nAMD involves a complex interplay of genetic risk factors,
pro-angiogenic signaling, and chronic inflammation [4]. While several growth
factors, including FGF-2 and PDGF, contribute to this process [5], vascular
endothelial growth factor (VEGF) is established as the principal driver,
promoting endothelial cell proliferation, migration, and vascular permeability
[6]. Concurrently, innate immune activation, microglia/macrophages recruitment,
and the release of inflammatory cytokines such as IL-1
Current first-line therapy for nAMD relies on repeated intravitreal injections of anti-VEGF agents, which have revolutionized patient outcomes by effectively regressing neovascularization and reducing exudation [10, 11]. However, significant limitations remain, including incomplete or non-responsiveness in a subset of patients, the development of tachyphylaxis with repeated dosing, a substantial treatment burden, and the cumulative risks of endophthalmitis, geographic atrophy and fibrosis [12, 13]. Moreover, conventional anti-VEGF monotherapy does not adequately address the inflammatory component of the disease, highlighting the need for novel agents with broader mechanisms of action [14].
Platelet factor 4 (PF4 or CXCL4), a cationic chemokine abundantly released from activated platelets, is a multifaceted regulator of vascular biology and immune homeostasis [15]. While PF4 is well- established for its potent heparin-neutralizing capacity, which localizes coagulation at injury sites, it also exhibits robust anti-angiogenic activity [16, 17]. This function is particularly evident in its inhibition of the VEGF–vascular endothelial growth factor receptor 2 (VEGFR2) signaling axis—the principal driver of angiogenesis [18]. Beyond these vascular functions, emerging evidence underscores PF4’s role as an immunomodulator. PF4 has been shown to attenuate neuroinflammation, notably by reducing inflammatory factor expression in the hippocampi of aged mice and in glaucomatous eyes, likely through direct or indirect regulation of microglia/macrophages activity [19, 20]. However, its specific expression, functional role, and therapeutic potential in the context of nAMD have remained largely unexplored.
In the current study, we hypothesized that PF4 serves as a regulator capable of concurrently suppressing pathological angiogenesis and mitigating intraocular inflammation—a dual mechanism that could offer a strategic advantage through a more comprehensive therapeutic approach. Accordingly, this study was designed to systematically evaluate the therapeutic efficacy and underlying molecular mechanisms of PF4 in experimental models of nAMD, focusing on its effects on pathological neovascularization, intraocular inflammation, VEGF signaling, and intraocular safety.
Male C57BL/6J mice (6–8 weeks old) were obtained from Beijing Vital River Laboratory Animal Technology (Beijing, China). Vldlr knockout (Vldlr⁻/⁻) mice were generated via CRISPR/Cas9-mediated deletion of exons 2–11 of the Vldlr 207 transcript. The mice were maintained under a 12-h light/dark cycle in a specific-pathogen-free (SPF) facility at Tianjin Medical University. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Tianjin Medical University (No. TMUaMEC 2024030) and conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. For euthanasia, mice were deeply anesthetized with 1.25% tribromoethanol (Avertin; Nanjing Aibei Biotechnology, Nanjing, China) administered intraperitoneally at a dose of 0.2 mL/10 g body weight, followed by cervical dislocation in accordance with the approved ethical guidelines.
The laser-induced choroidal neovascularization (CNV) model was established in C57BL/6J mice (6–8 weeks old) as previously described [21]. Briefly, mice were anesthetized, pupils were dilated with 1% tropicamide, and four laser spots (532 nm, 100 mW, 0.15 s, 100 µm spot size) were applied around the optic disc using a 532-nm photocoagulation system (OcuLight GL, Iridex, Mountain View, CA, USA). Formation of a cavitation bubble was taken as confirmation of Bruch’s membrane rupture. Eyes with retinal hemorrhage were excluded.
For intravitreal injections, the mice were anesthetized with 2%–3% isoflurane in oxygen at a flow rate of 0.5 L/min, and maintained under anesthesia for the duration of the injection procedure (approximately 5-10 min), and pupils were dilated with 1% tropicamide (Sinqi Pharmaceutical, Shenyang, Liaoning, China). A scleral entry site was created with a 30-gauge needle, and a 34-gauge Hamilton syringe was introduced into the vitreous cavity behind the limbus. Mice were randomly assigned to receive 1 µL recombinant human PF4 (0.3 µg/µL in 2% BSA/PBS; R&D Systems, Minneapolis, MN, USA) [22] or vehicle. Topical 0.5% levofloxacin (Sinqi Pharmaceutical, Shenyang, Liaoning, China) was applied before and after injection. Eyes with injection-related hemorrhage or lens injury were excluded from analysis.
FFA was performed at designated time points after laser using a Micron IV retinal imaging system (Phoenix Research Labs, Pleasanton, CA, USA). Mice were anesthetized and pupils dilated with 1% tropicamide. Fluorescein sodium (10%, 100 µL) was injected intraperitoneally, and fundus images were acquired later. Leakage area for each CNV lesion was measured in ImageJ (version 1.53t, National Institutes of Health, Bethesda, MD, USA) by outlining the hyperfluorescent region surrounding the laser spot as described [21]. The mean leakage area per mouse was calculated for statistical analysis.
For RPE–choroid flat mounts, mice were euthanized and eyes were enucleated and fixed in 4% paraformaldehyde for 1 h. Posterior eyecups were dissected, permeabilized in 1% Triton X‑100, and blocked in 3% BSA/0.3% Triton X‑100 in PBS. Tissues were incubated with anti-CD31 and anti-Iba1 primary antibodies, followed by fluorophore-conjugated secondary antibodies, and flat-mounted. Z-stack images of each laser lesion were acquired using a confocal microscope (LSM 800; Carl Zeiss, Oberkochen, Germany). CD31-positive neovascular volume and Iba1-positive microglia/macrophages volume were quantified using Imaris software (version 9.0.1, Bitplane AG, Zurich, Switzerland), and the mean value per mouse was used for analysis. The entire procedure was performed essentially as previously described [23]. Sources of antibodies are listed in Table 1.
| Antibody | Cat. No. | Company | Dilution Ratio |
| anti PF4 | 21157-1-AP | Proteintech | 1:200 |
| anti CD31 | MAB1398Z | Millipore | 1:150 |
| anti Iba1 | 019-19741 | Wako | 1:200 |
| anti VEGFA | 81323-2-RR | Proteintech | 1:200 |
PF4, platelet factor 4; CD31, cluster of differentiation 31; Iba1, ionized calcium-binding adapter molecule 1; VEGFA, vascular endothelial growth factor A.
For retinal cryosections, eyes were fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, embedded in Optical Coherence Tomography (OCT) (Sakura Finetek, Torrance, CA, USA), and sectioned at 20 µm. Sections containing laser lesions were permeabilized, blocked in 3% BSA containing 0.3% Triton X-100, and incubated overnight at 4 °C with primary antibodies against CD31, Iba1, vascular endothelial growth factor A (VEGFA), and PF4, followed by the application of fluorophore-conjugated secondary antibodies and DAPI. Sections were imaged by confocal microscopy. Sources of antibodies are listed in Table 1.
Apoptosis was assessed in retinal sections using a One-step TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) Apoptosis Assay Kit (Cat# C1086, Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, cryosections were equilibrated, permeabilized, and incubated with TUNEL reaction mixture, followed by DAPI counterstaining. Images were acquired using a confocal microscope (LSM 800; Carl Zeiss, Oberkochen, Germany).
Human retinal microvascular endothelial cells (HRMECs; Procell Life Science & Technology, Wuhan, China) were cultured in endothelial cell medium (ECM; ScienCell Research Laboratories, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement (ECGS), and 1% penicillin/streptomycin. Cells were maintained at 37 °C in a humidified 5% CO2 incubator, and passages 3–6 were used for the experiments. The HRMECs were validated by short tandem repeat (STR) profiling and tested negative for mycoplasma contamination by the supplier.
HRMEC viability in response to PF4 and VEGF was assessed using a CCK‑8 assay (NCM Biotech, Suzhou, China). Cells were treated with PF4 (0.25, 1, or 4 µg/mL) for 24 h in the presence or absence of VEGF (50 ng/mL). CCK‑8 reagent was added for 60 min at 37 °C, and absorbance at 450 nm was measured. Data were normalized to vehicle-treated or VEGF (Sino Biological, Beijing, China) + vehicle controls, as indicated, to identify a non‑toxic, effective PF4 concentration for subsequent functional assays.
HRMEC proliferation was evaluated using a 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay (BeyoClick™ EdU Cell Proliferation Kit; Cat# C0071S, Beyotime Biotechnology, Shanghai, China). Based on CCK‑8 assays showing that 1 µg/mL PF4 was the minimum concentration that significantly inhibited VEGF-induced HRMEC proliferation without reducing basal viability, this dose was used in subsequent in vitro assays. HRMECs were pretreated with PF4 (1 µg/mL) for 1 h and then stimulated with VEGF (50 ng/mL) for 24 h before EdU labeling and detection.
Cell migration was assessed using a scratch wound assay. HRMEC monolayers in 12-well plates were scratched with a 200-µL pipette tip and washed to remove debris. Cells were pretreated with PF4 (1 µg/mL) for 1 h and then stimulated with VEGF (50 ng/mL). Images were acquired at 0 and 12 h, and wound closure was quantified in ImageJ.
For tube formation, HRMECs were seeded onto growth factor-reduced Matrigel in 96-well plates. Cells were pretreated with PF4 (1 µg/mL) for 1 h and then exposed to VEGF (50 ng/mL). After 6 h, capillary-like networks were imaged, and total tube length and branch points were quantified using the Angiogenesis Analyzer plugin in ImageJ.
PF4 levels in retina–choroid complexes were determined using a commercial ELISA kit (Cat# ML037257, Shanghai Enzyme-linked Biotechnology, Shanghai, China). Tissues were homogenized in PBS and centrifuged, and supernatants were processed according to the manufacturer’s protocol. Absorbance at 450 nm was recorded, and PF4 concentrations were calculated from a standard curve and normalized to total protein.
Protein was extracted from HRMECs or retina–choroid complexes using RIPA buffer
with protease and phosphatase inhibitors. Protein concentrations were determined
by BCA assay. Equal amounts of protein were separated by SDS-PAGE and transferred
to PVDF membranes, blocked with 5% non-fat milk, and incubated overnight at 4
°C with primary antibodies against phospho-ERK (Cat# 9101S), total ERK
(Cat# 9102S), phospho-AKT (Cat# 2965T), total AKT (Cat# 4691T), phospho-STAT3
(Cat# 4113T), total STAT3 (Cat# 4904T), VEGFR2 (Cat# 9698), (all 1:1000; Cell
Signaling Technology, Danvers, MA, USA),
Total RNA from retina–choroid complexes was isolated using a commercial RNA extraction kit (Cat# AC0101, Sparkjade, Jinan, Shandong, China) and reverse-transcribed using Hifair III 1st Strand cDNA Synthesis SuperMix (Cat# 11141ES10, Yeasen, Shanghai, China). qPCR was performed with Hieff® qPCR SYBR Green Master Mix (Low Rox Plus) (Cat# 11202ES60, Yeasen, Shanghai, China) on a LightCycler 480 II system (Roche Diagnostics, Basel, Switzerland). The relative mRNA expression levels of target genes were normalized to the internal control Gapdh and quantified using the 2-ΔΔCt method. The primer sequences are listed in Table 2.
| Gene | Forward | Reverse |
| Gapdh | TGTGTCCGTCGTGGATCTGA | TTGCTGTTGAAGTCGCAGGAG |
| Vegfa | GCACATAGAGAGAATGAGCTTCC | CTCCGCTCTGAACAAGGCT |
| Tnf | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Il1b | CTTTCCCGTGGACCTTCCA | CTCGGAGCCTGTAGTGCAGTT |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| Nfkb1 | ATGGCAGACGATGATCCCTAC | CGGAATCGAAATCCCCTCTGTT |
Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Vegfa, vascular endothelial growth factor A; Tnf, tumor necrosis factor; Il1b, interleukin-1 beta; Il6, interleukin-6; Nfkb1, nuclear factor kappa-B subunit 1.
For transcriptomic analysis, retina–choroid complexes were collected from uninjured controls, laser-injured mice receiving vehicle, and laser-injured mice receiving PF4 (n = 3 per group) at day 7 after laser. Tissues were snap-frozen in liquid nitrogen and stored at –80 °C. RNA extraction, library preparation, sequencing, and bioinformatic analysis were performed by Majorbio (Shanghai, China).
Spectral‑domain OCT (Micron IV, Phoenix Research Labs) was used to assess retinal structure at designated time points after intravitreal injection. Mice were anesthetized and pupils dilated. Circular volume scans centered on the optic disc were obtained, and retinal thickness was measured on ImageJ by masked graders.
Full-field ERGs were recorded using a Celeris D430 system (Diagnosys, Lowell,
MA, USA) as described [25]. Mice were dark-adapted overnight, anesthetized under
dim red light, and pupils dilated with tropicamide. Corneal electrodes with
integrated stimulators were placed on lubricated corneas, and scotopic responses
were elicited at 0.01, 0.1, and 1 cd
To evaluate the potential retinal toxicity of PF4, C57BL/6J mice received a single intravitreal injection of PF4 (0.3 µg in 1 µL) or vehicle (PBS) on day 0. At day 7 post-injection, the mice were subjected to a series of safety assessments. First, fundus imaging and OCT were performed to examine the gross morphology and retinal thickness. Subsequently, retinal function was assessed using ERG as described in the previous section. Following functional testing, the mice were euthanized, and the eyes were harvested for histological analysis, including hematoxylin and eosin (H&E) staining to evaluate structural integrity and a TUNEL assay to detect cell apoptosis.
Data were analyzed using GraphPad Prism version 10 (GraphPad Software, San
Diego, CA, USA). All results are expressed as mean
To define PF4 expression in mouse CNV, we measured PF4 expression in a laser-induced CNV mouse model (Fig. 1A–C). Quantitative real-time PCR (qRT-PCR) showed that Pf4 mRNA levels were not significantly changed at day 7 after laser injury in posterior segment tissues compared with Normal control (i.e., untreated healthy mice) (Fig. 1A). Consistently, ELISA detected no significant difference in PF4 protein levels in posterior eye lysates at day 7 post‑laser (Fig. 1B). Immunofluorescence further revealed only weak PF4 signals in both Normal and laser-injured eyes, without an apparent increase within laser lesions (Fig. 1C).
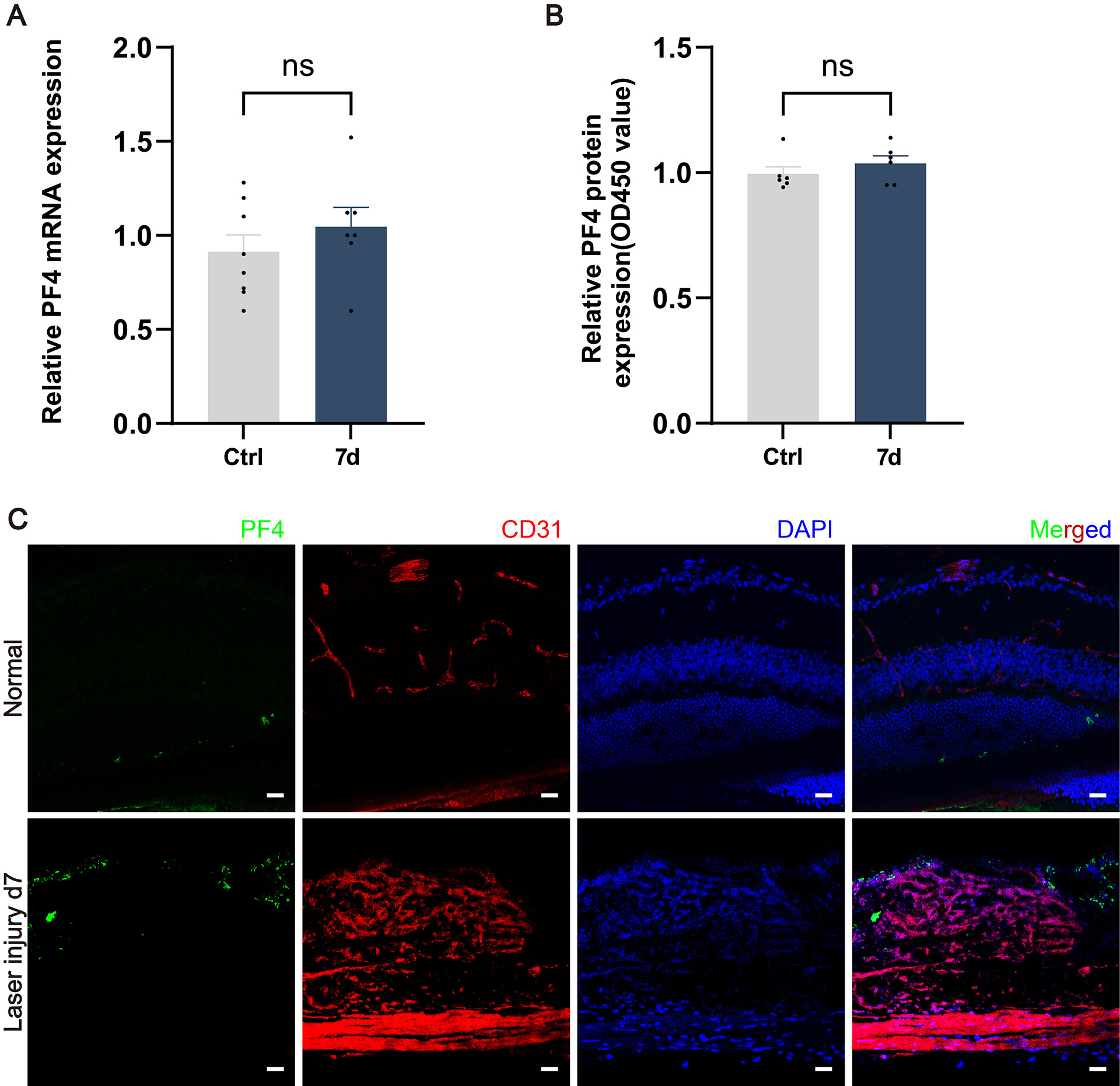 Fig. 1.
Fig. 1.
PF4 expression after laser application. (A) Pf4 mRNA
levels in the laser lesions of adult mice (6–8 weeks old) at the indicated time
points after laser injury, measured by quantitative real-time PCR (qRT-PCR). n =
7 eyes (from 4 mice) per group. (B) PF4 protein levels, measured by ELISA. n = 4
eyes (from 3 mice) per group. (C) Representative immunofluorescence images of PF4
(green) and CD31 (red) in cross-sections of the retinas from Normal control and
laser-induced CNV mice at day 7 post-injury; nuclei are counterstained with DAPI
(blue). Scale bar = 20 µm. Data are shown as mean
To investigate the role of PF4 in pathological neovascularization, we initially utilized a laser-induced mouse model of CNV. Six-week-old mice underwent laser induction, followed by intravitreal injection of PF4 protein (Fig. 2A). We first assessed the hallmark feature of CNV, vascular leakage, by FFA on day 3 post-laser. FFA images demonstrated that dye leakage was markedly reduced in PF4-treated mice at days 3 and 7 after laser injury (Fig. 2B). Quantitative analysis further confirmed this robust inhibitory effect, showing a significant reduction in leakage area in the PF4-treated group at both time points (Fig. 2C). We then evaluated the extent of neovascularization by performing CD31 immunofluorescence staining on RPE–choroid flat mounts at day 7 post-laser. Representative confocal images revealed that CNV lesions were noticeably smaller in PF4-treated mice than in Vehicle (i.e., mice injected with the carrier solution without PF4) (Fig. 2D). Quantification of CNV volume corroborated these findings, demonstrating a significant decrease in lesion volume in the PF4-treated group (Fig. 2E). To validate these results histologically, we performed H&E staining on retinal cross-sections. H&E images showed a clear reduction of CNV lesions in PF4-treated eyes (Fig. 2F).
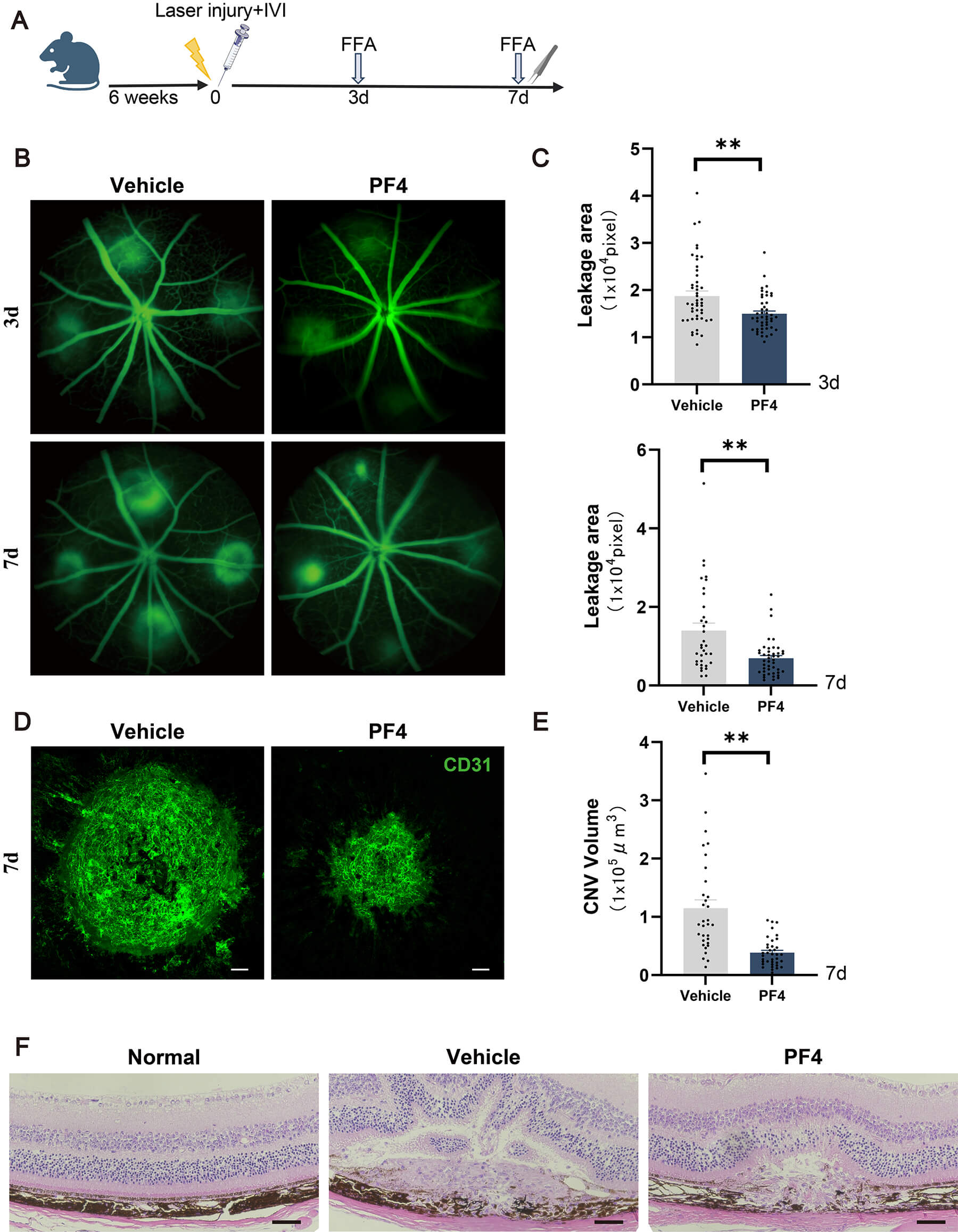 Fig. 2.
Fig. 2.
PF4 administration attenuates vascular leakage and choroidal
neovascularization in a laser-induced mouse model of CNV. (A) Schematic of the
laser injury, intravitreal injection, and analysis schedule. (B) Representative
longitudinal paired FFA images showing vascular leakage in the same eyes at days
3 and 7 after laser injury in vehicle- and PF4-treated mice. (C) Quantification
of leakage area at days 3 and 7 is shown in (B). n = 30 lesions from 5 mice per
group. (D) Representative confocal images of RPE–choroid flat mounts at day 7
after laser injury from vehicle- and PF4-treated mice, stained for CD31 (green)
to visualize CNV lesions. Scale bar = 50 µm. (E) Quantification of CNV
volume shown in (D), assessed as CD31-positive fluorescence volume. n = 32
lesions from 5 mice per group. (F) Representative retinal sections stained with
H&E from Normal, vehicle-treated, and PF4-treated mice. Scale bar = 50 µm.
Data are shown as mean
To further evaluate the protective role of PF4, we employed the Vldlr⁻/⁻ mouse model, which recapitulates key features of human neovascular retinal diseases [26]. Vldlr⁻/⁻ mice develop pathological retinal neovascularization (PRNV) from postnatal day 11 (P11), accompanied by progressive retinal ischemia, breakdown of the outer blood–retinal barrier, and functional decline. This pathology stems from impaired RPE lipid transport, resulting in HIF-driven upregulation of proangiogenic factors such as VEGF, mirroring the pathogenesis of proliferative diabetic retinopathy and nAMD [27].
We intravitreally injected PF4 or vehicle into Vldlr⁻/⁻ mice at P11 and analyzed retinal angiogenesis at P18. CD31 immunostaining of retinal flat mounts revealed marked reduction in neovascular lesions in PF4-treated eyes compared with Vehicle, with higher-magnification and three-dimensional (3D)-reconstructed images further demonstrating attenuated aberrant vascular growth (Fig. 3A). Quantitative analysis confirmed that PF4 significantly decreased both the number and area of neovascular lesions (Fig. 3B,C). Similarly, in adult Vldlr⁻/⁻ mice, PF4 injection reduced vascular leakage, as evaluated by FFA at baseline (i.e., the pre-injection time point, day 0) and 7 days post-injection (Fig. 3D). Quantification showed a significant improvement in vascular leakage following PF4 treatment (Fig. 3E).
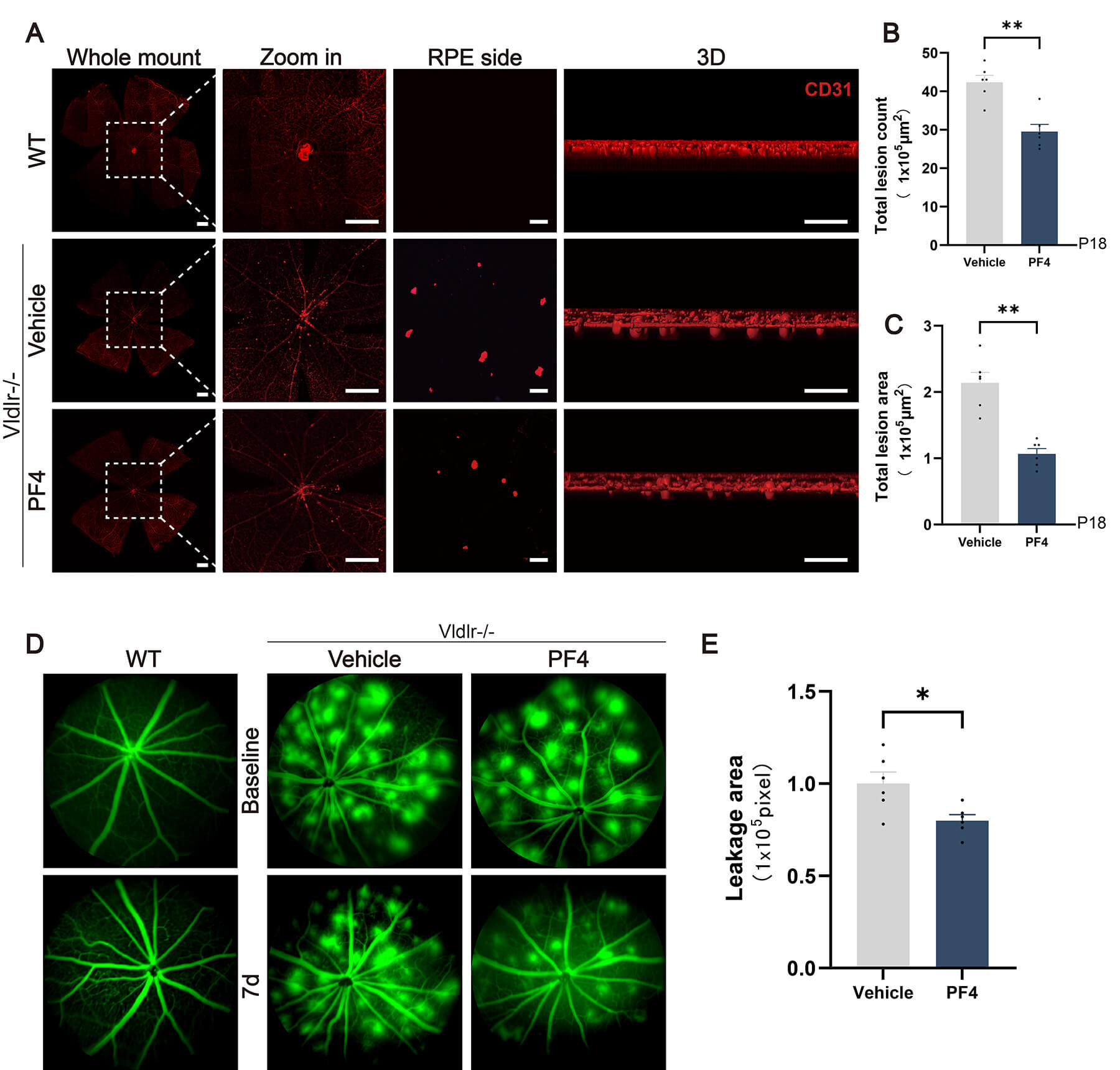 Fig. 3.
Fig. 3.
PF4 administration attenuates vascular leakage retinal
neovascularization in Vldlr⁻/⁻ mice. (A) Representative confocal images of
retinal flat mounts from wild-type (WT), vehicle-treated Vldlr⁻/⁻, and
PF4-treated Vldlr⁻/⁻ mice following CD31 (red) immunostaining. Whole-mount views,
magnified regions (zoomed in), images from the RPE side, and corresponding
three-dimensional (3D) reconstructions illustrating abnormal vascular growth are
shown. Scale bar = 500 µm (whole mount), 167 µm (zoomed in), 50
µm (RPE side), and 50 µm (3D). (B,C) Quantifications of the lesion
number and lesion area are shown in (A). n = 6 eyes (from 3 mice) per group. (D)
Adult Vldlr⁻/⁻ mice received intravitreal injections of the indicated treatments
(Vehicle or PF4) and were analyzed on day 7. Representative FFA images show
vascular leakage at baseline (day 0, pre-injection) and at day 7 post-injection
in Vehicle-treated Vldlr⁻/⁻ and PF4-treated Vldlr⁻/⁻ mice, compared with
age-matched healthy WT controls. (The two WT images are biological replicates
from independent mice, demonstrating the consistent absence of vascular leakage
in healthy retinas.) (E) Quantification of leakage area post-treatment is shown
in (D). n = 6 eyes (from 3 mice) per group. Data are shown as mean
Because inflammation contributes to CNV, we examined whether PF4 modulates laser-induced inflammatory responses. Compared with vehicle, PF4 significantly reduced mRNA levels of Il1b, Il6, Tnf, and Nfkb1 in posterior segment retinal tissues at day 7 post-laser injury (Fig. 4A). We next assessed microglia/macrophages recruitment at lesion sites. Immunostaining showed fewer Iba1+ microglia/macrophages with reduced aggregation in PF4-treated eyes (Fig. 4B), and quantitative analysis confirmed reduced Iba1+ microglia/macrophages volume within lesions (Fig. 4C).
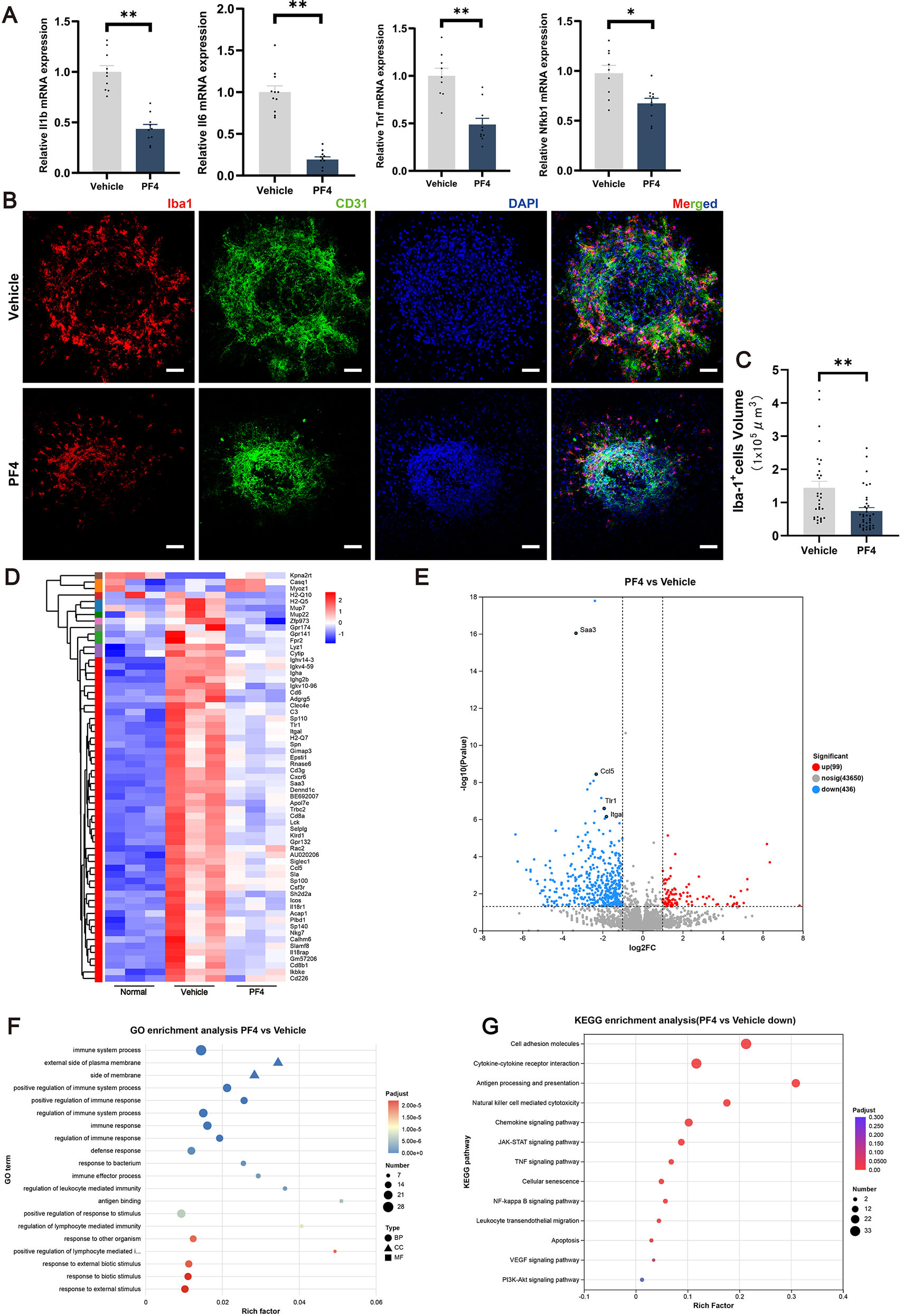 Fig. 4.
Fig. 4.
PF4 suppresses intraocular inflammation induced by laser injury.
(A) Relative mRNA expression of Il1b, Il6, Tnf, and Nfkb1 in the posterior
segment 7 days after laser photocoagulation in vehicle- and PF4-treated eyes. (B)
Representative confocal images of laser lesions from vehicle- and PF4-treated
retinas immunostained for Iba1 (red) and CD31 (green), with nuclei counterstained
with DAPI (blue). Scale bar = 50 µm. (C) Quantification of Iba1+
microglia/macrophages volume within the laser lesions shown in (B). n = 32
lesions (from 5 eyes) per group. (D) Heatmap of differentially expressed genes in
retina–choroid complexes from Normal, vehicle-treated, and PF4-treated mice. (E)
Volcano plot of differentially expressed genes in PF4-treated versus
vehicle-treated groups. (F) GO enrichment analysis of differentially expressed
genes between PF4- and vehicle-treated groups. (G) KEGG pathway enrichment
analysis of differentially expressed genes between PF4- and vehicle-treated
groups. n = 3 eyes (from 3 mice) per group for RNA sequencing. Data are shown as
mean
To comprehensively characterize PF4-mediated transcriptomic changes, we
performed bulk RNA sequencing of retina–choroid complexes from Normal,
vehicle-treated, and PF4-treated mice. Hierarchical clustering of differentially
expressed genes (DEGs) across the three groups revealed clearly segregated
expression profiles (Fig. 4D). Vehicle-treated samples displayed a pronounced
proinflammatory signature, whereas PF4-treated samples formed a distinct cluster
whose expression pattern partially shifted toward that of Normal controls,
suggesting that PF4 counteracts injury-induced inflammatory reprogramming.
Differential expression analysis between the Vehicle and PF4 groups showed that
PF4 induced widespread transcriptional remodeling (Fig. 4E). PF4 significantly
downregulated a broad set of inflammation-related genes, including chemokines,
cytokines, adhesion molecules, and genes associated with leukocyte trafficking
and microglia/macrophages activation, while genes involved in tissue homeostasis
were relatively preserved. We interrogated the biological processes associated
with PF4-regulated genes using GO enrichment analysis. PF4-downregulated DEGs
were predominantly enriched in terms related to innate immune responses, cytokine
production, regulation of inflammatory responses, and responses to external
stimuli (Fig. 4F), indicating broad suppression of inflammatory activation
programs. We then examined pathway changes using KEGG enrichment analysis, which
revealed that PF4 strongly inhibited signaling cascades implicated in retinal
inflammation and leukocyte recruitment, including the NF-
Given that inflammatory mediators can induce VEGFA expression [28, 29, 30, 31, 32, 33], we tested whether PF4 affects VEGFA levels in laser-induced CNV. Compared with the Normal control group, laser photocoagulation markedly increased VEGFA protein expression in the RPE–choroid complex, whereas PF4 treatment significantly attenuated this upregulation (Fig. 5A,B). Consistent with the western blot results, immunofluorescence staining of sections through the laser lesions revealed strong immunoreactivity of VEGFA around CD31-positive neovessels in the vehicle group, which was markedly reduced in PF4-treated eyes (Fig. 5C). These findings suggest that PF4 may inhibit CNV formation at least in part by suppressing inflammation-driven upregulation of VEGFA.
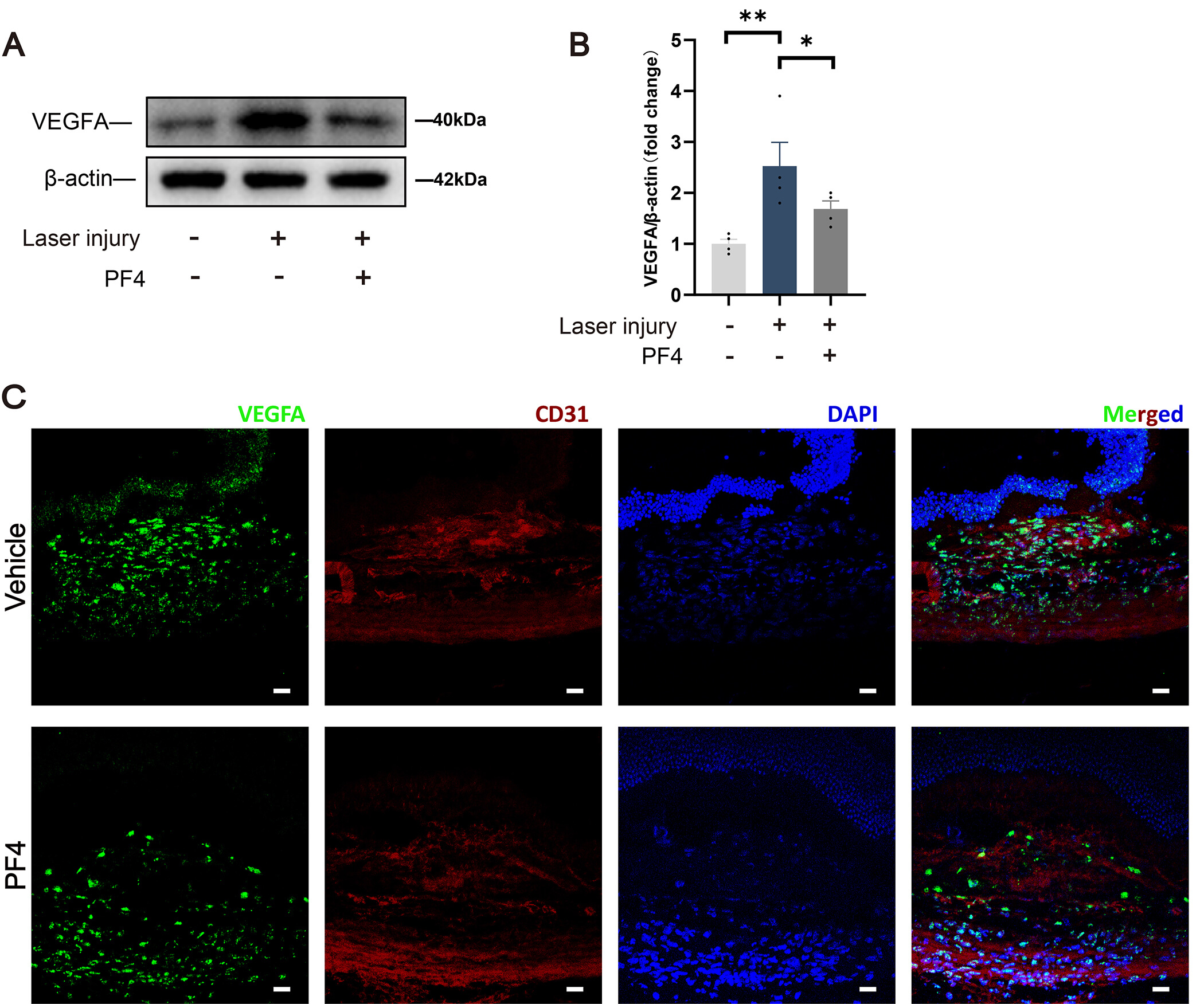 Fig. 5.
Fig. 5.
PF4 attenuates VEGFA expression in laser‑induced CNV lesions. (A) Representative immunoblot of VEGFA expression in Normal (no laser),
laser‑injured + vehicle, and laser‑injured + PF4–treated eyes.
In addition to suppressing inflammation-driven upregulation of VEGFA in vivo, we sought to investigate whether PF4 could counteract VEGF-induced angiogenic responses in endothelial cells in vitro. We initially investigated the impact of this factor on key angiogenic processes in vitro using HRMECs—a type of vascular endothelial cell that highly expresses VEGFR2 and is highly sensitive to VEGF stimulation [34, 35, 36, 37].
The results showed that the number of proliferating HRMECs was markedly reduced in the PF4-pretreated group compared with the group stimulated with VEGF alone (Fig. 6A). Quantitative analysis further revealed a significant decrease in the percentage of EdU-positive cells (Fig. 6B). We next assessed cell migration capacity using a scratch wound-healing assay, which revealed that PF4 pretreatment significantly suppressed VEGF-stimulated migration of HRMECs (Fig. 6C). The scratch closure rate was significantly lower in the PF4-treated group than in the control group (Fig. 6D). Furthermore, we examined the effect of PF4 on the ability of HRMECs to form capillary-like tubular structures, a critical step in the angiogenesis process. The results demonstrated that PF4 pretreatment disrupted VEGF-driven tube formation in HRMECs (Fig. 6E). The number of branch points was reduced in the PF4-treated group compared with the VEGF control group (Fig. 6F).
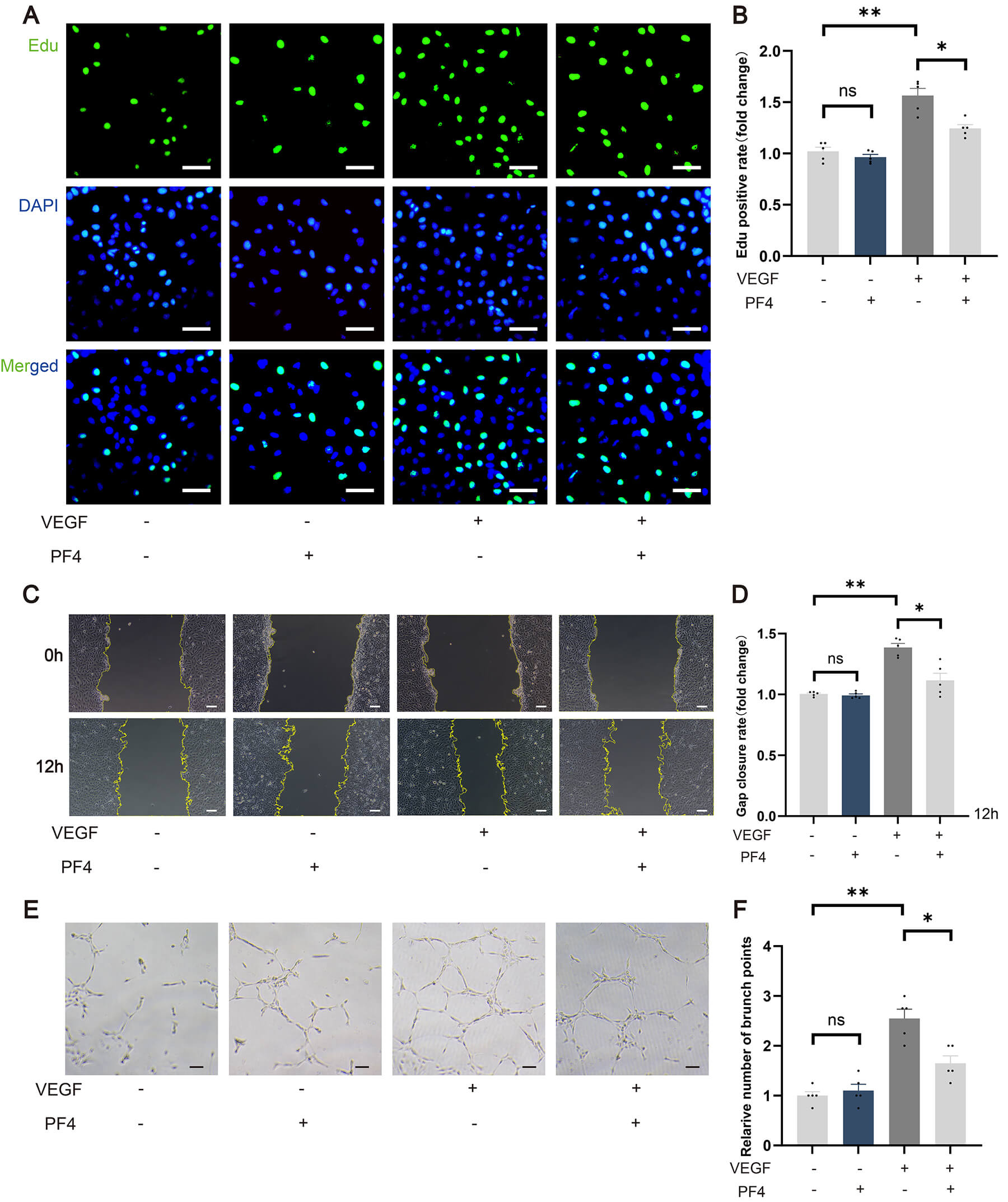 Fig. 6.
Fig. 6.
PF4 inhibits VEGF-induced proliferation, migration, and tube
formation of human retinal microvascular endothelial cells. (A) Representative
EdU staining of HRMECs after PF4 pretreatment and VEGF stimulation, stained for
EdU (green) and DAPI (blue). Scale bar = 50 µm. (B) Quantification of
EdU-positive cells as a percentage of total cells in (A). n = 5 per group. (C)
Representative images of HRMEC migration in a scratch-wound assay at 0 and 12
hours after PF4 pretreatment and VEGF stimulation. Scale bar = 50 µm. (D)
Quantification of gap closure in (C). n = 5 per group. (E) Representative images
of tube formation by HRMECs following PF4 pretreatment and VEGF stimulation.
Scale bar = 100 µm. (F) Quantification of branch points in (E). n = 5 per
group. Data are shown as mean
To elucidate the molecular basis underlying these anti-angiogenic effects of PF4, we next examined whether PF4 modulates VEGF/VEGFR2 signaling and its key downstream pathways [38, 39]. In the laser-induced CNV mouse model, PF4 administration markedly reduced VEGFR2 expression, as well as the phosphorylation of ERK and STAT3 in choroidal tissue (Fig. 7A,B), indicating that PF4 attenuates VEGF/VEGFR2-related signal transduction in vivo. Consistent with these findings, PF4 significantly inhibited VEGF-induced phosphorylation of AKT, ERK, and STAT3 in cultured HRMECs (Fig. 7C,D), further supporting its direct suppression of VEGF/VEGFR2-driven pro-angiogenic signaling.
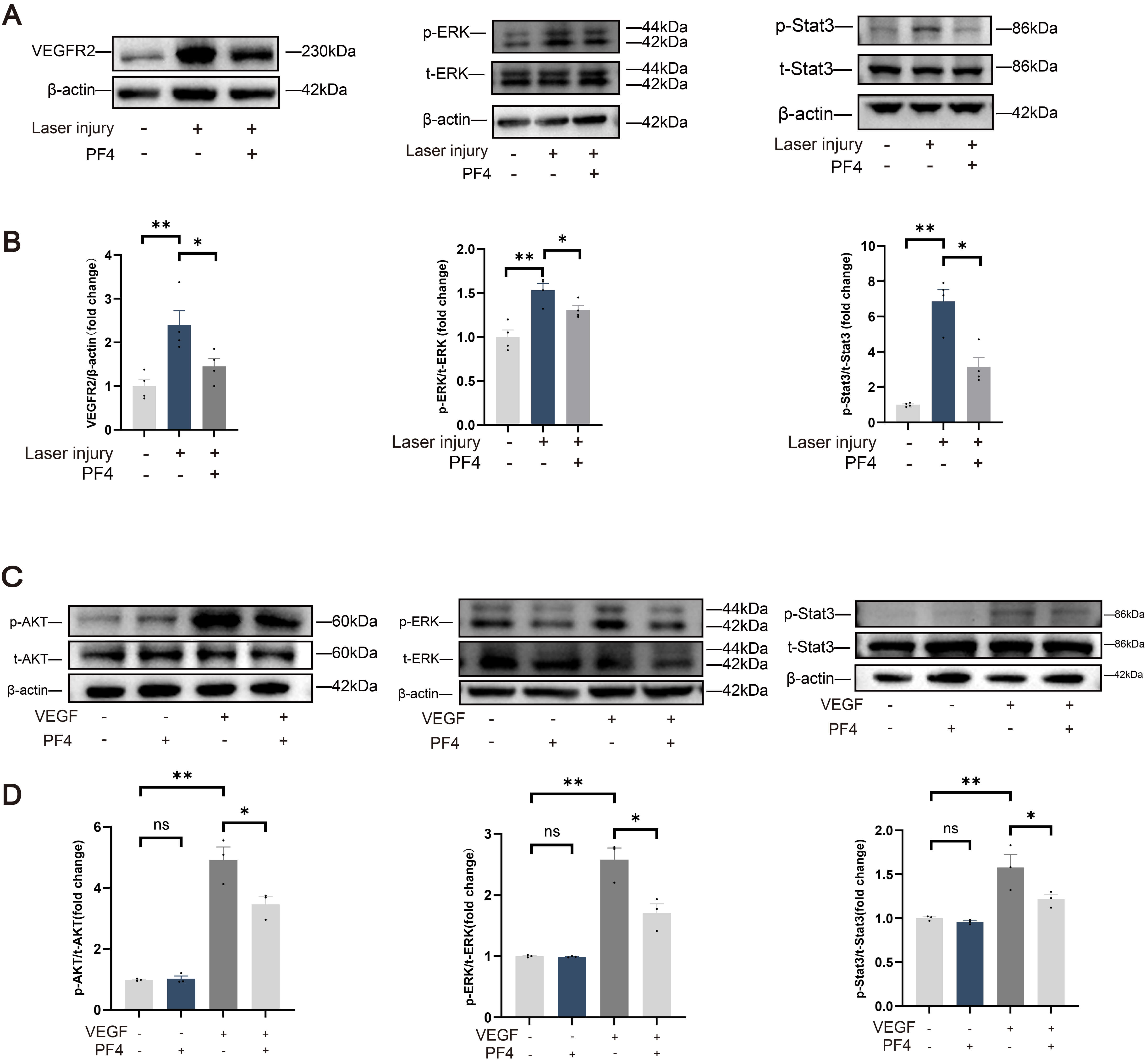 Fig. 7.
Fig. 7.
PF4 attenuates VEGF/VEGFR2-driven ERK, AKT, and STAT3 signaling
in vivo and in vitro.(A) Representative immunoblots of
VEGFR2, phosphorylated (p-ERK) and total (t-ERK) ERK, and phosphorylated
(p-STAT3) and total (t-STAT3) STAT3 in RPE–choroid–sclera complex tissue from
sham (no laser), laser-injured + vehicle, and laser-injured + PF4–treated mice.
We next evaluated the retinal safety of intraocular PF4 administration in mice at day 7 post-injection. OCT imaging showed that retinal morphology and thickness were preserved following intraocular injection of either vehicle or PF4 (Fig. 8A,B). Consistently, ERG revealed no significant reduction in a-wave amplitudes (originating primarily from photoreceptor rods and cones) or b-wave amplitudes (reflecting inner retinal activity, mainly Müller and bipolar cells) in PF4-treated eyes compared with vehicle-treated controls (Fig. 8C,D). Histological examination further confirmed the structural integrity of the retina (Fig. 8E), and TUNEL staining demonstrated an absence of appreciable cell death (Fig. 8F). Taken together, these data indicate that a single intraocular administration of PF4 (0.3 µg/eye) was well tolerated and exhibited a favorable retinal safety profile in mice.
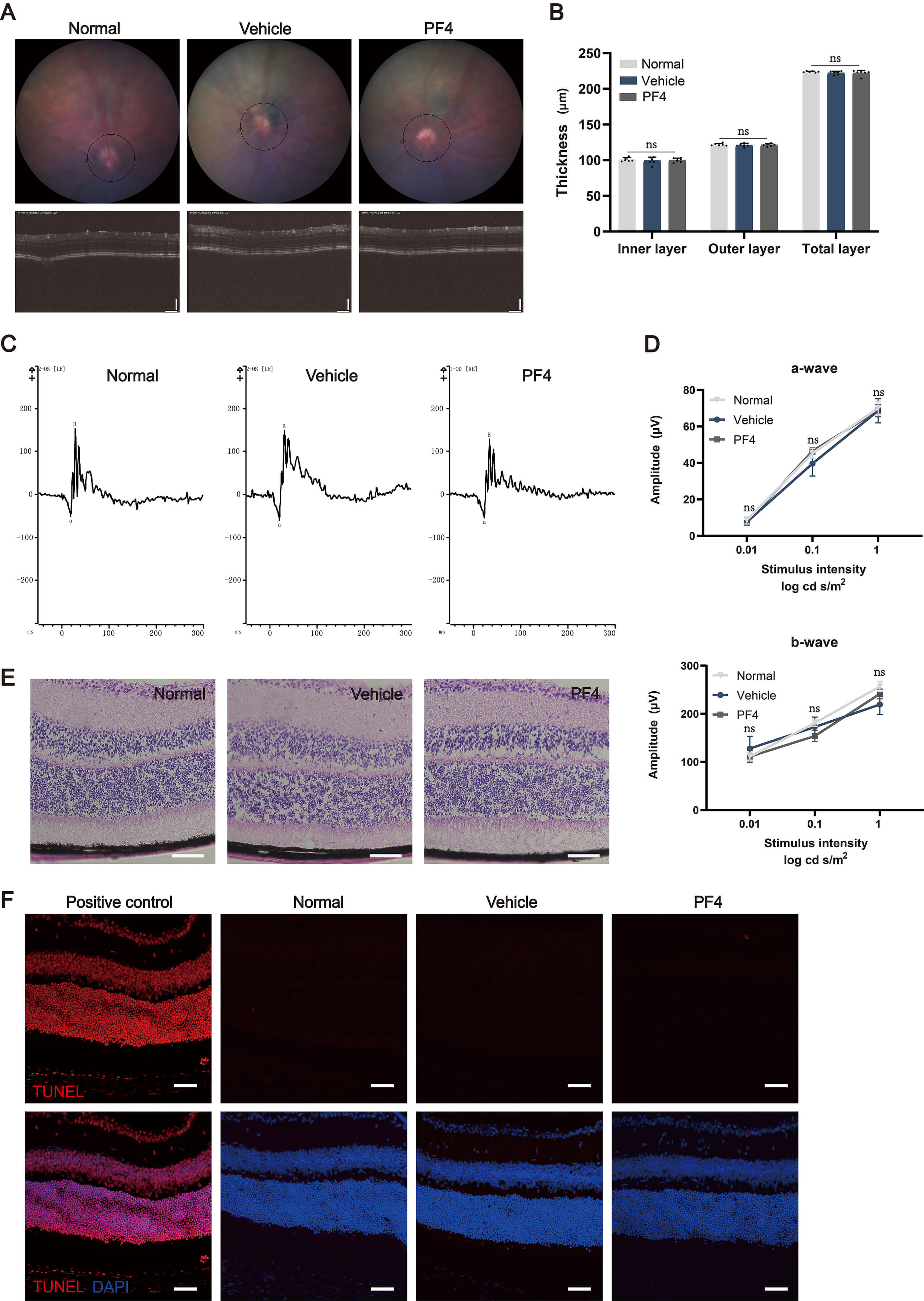 Fig. 8.
Fig. 8.
Evaluation of retinal toxicity following the intravitreal
administration of PF4 in mice. (A) Representative fundus imaging and OCT imaging
of normal mice and mice treated with vehicle or PF4. The circular bounding box in
the OCT images indicates the standardized region scanned and used for layer
thickness measurement. (B) Thickness of total retinal layers. n = 6 eyes (from 3
mice) per group. (C) Representative electroretinogram imaging of normal mice and
mice treated with vehicle or PF4, where “a” and “B” denote the a-wave and b-wave
amplitudes, respectively. The y-axis represents response amplitude in microvolts
(µV). (D) Electroretinogram and quantification of normal mice and mice
treated with vehicle or PF4. n = 6 eyes (from 3 mice) per group. (E) H&E
staining of eyeball sections from normal mice and mice treated with vehicle or
PF4. Scale bar = 100 µm. (F) TUNEL assay of retinal sections from normal
mice and mice treated with vehicle or PF4. Scale bar = 50 µm. Data are
shown as mean
In this study, we evaluated the therapeutic potential of PF4 in experimental animal models of nAMD. Our results demonstrated that intravitreal PF4 injection significantly reduced vascular leakage and CNV lesion size in the laser-induced mouse CNV model. These findings were further supported by results obtained in Vldlr⁻/⁻ mice, in which retinal neovascularization was suppressed. In parallel with the in vivo studies, we showed that PF4 inhibited VEGF-induced proliferation, migration, and tube formation in HRMECs, indicating a consistent anti-angiogenic effect across experimental systems.
A key finding from our work is the absence of significant upregulation of endogenous Pf4 mRNA or protein in laser-induced CNV lesions at the time point the specimens were collected (Fig. 1A–C), which provides critical context for subsequent experiments. Despite PF4’s established anti-angiogenic and immunomodulatory properties in systemic models [38, 39] (e.g., tumor angiogenesis, atherosclerosis), the lack of a reported response to laser injury in the retina suggests a “therapeutic gap” in the ocular inflammatory-angiogenic cascade. This observation justifies the use of exogenous PF4 supplementation, as endogenous levels are insufficient to counteract pathological neovascularization or inflammation. Notably, weak PF4 immunoreactivity in injured eyes (Fig. 1C) further supports the need for targeted delivery.
Building on this rationale, we validated PF4’s anti-angiogenic efficacy across two distinct mouse models of neovascular retinal disease: laser-induced CNV (mimicking nAMD) and Vldlr⁻/⁻ mice (recapitulating retinal ischemia and PRNV, as seen in retinopathy of prematurity or diabetic retinopathy) [40, 41]. In the laser-induced CNV model, PF4 significantly reduced vascular leakage (FFA; Fig. 2B,C) and CNV lesions, as shown by CD31 and H&E staining (Fig. 2D–F), demonstrating attenuation of both functional and structural hallmarks of pathological angiogenesis. In Vldlr⁻/⁻ mice, PF4 similarly reduced neovascular lesion number and area (P18; Fig. 3A–C) and vascular leakage (adult mice; Fig. 3D,E). Although the Vldlr⁻/⁻ model is primarily driven by metabolic fuel shortages, its pathological progression involves secondary inflammatory cell infiltration. The consistent efficacy of PF4 across both laser-induced (acute/inflammatory) and Vldlr⁻/⁻ (chronic/metabolic) models suggests potential utility across diverse pathological contexts [42]. These cross-model results are particularly impactful, as they confirm PF4’s efficacy in the inhibition of retinal angiogenesis regardless of the initial trigger (laser-induced choroidal ischemia vs. developmental retinal ischemia in Vldlr⁻/⁻ mice). This versatility suggests that PF4 may act on pathways common to pathological angiogenesis, warranting further investigation across multiple neovascular retinal diseases. Importantly, the concordance between functional (FFA) and structural (immunostaining and H&E) outcomes strengthens the conclusion that PF4 suppresses neovessel formation and vascular leakage, rather than merely masking functional deficits.
Our results demonstrate that PF4 suppresses laser-induced inflammation at both
molecular and cellular levels: (1) reduced mRNA expression of proinflammatory
mediators (Il1b, Il6, Tnf, and Nfkb1; Fig. 4A); (2) decreased recruitment of
activated Iba1+ microglia/macrophages to lesion sites (Fig. 4B,C); and (3)
broad transcriptional remodeling of inflammation-related pathways (RNA-seq; Fig. 4D–G). Bulk RNA-seq analysis further reveals that PF4 inhibits injury-induced
inflammatory reprogramming: PF4-treated samples cluster closer to uninjured
controls (Fig. 4D) and downregulate genes involved in innate immunity, cytokine
production, and leukocyte trafficking (GO enrichment; Fig. 4F). KEGG pathway
analysis identifies key suppressed signaling cascades, including NF-
Given the central role of VEGF in ocular neovascularization, we further
investigated whether PF4 targets VEGF-driven pathways, uncovering a dual
mechanism of action. In laser-induced CNV, PF4 significantly attenuated
laser-induced VEGF upregulation (western blot and immunofluorescence; Fig. 5A–C), likely via suppression of inflammation (since IL-1
This study provides a preclinical proof-of-concept validating the therapeutic potential of PF4 for CNV. However, several limitations should be acknowledged when interpreting our findings, and addressing these gaps will be essential to advance PF4 from a therapeutic concept to a viable drug candidate.
First, while the laser-induced CNV model is a standard for testing anti-angiogenic therapies, it is fundamentally a wound-healing model triggered by acute thermal injury, which differs from the chronic, multifactorial process of human age-related macular degeneration (AMD). Second, due to technical constraints in sampling the minute vitreous volume in mice, a precise intraocular pharmacokinetic (PK) profile for exogenous PF4 was not established. Future studies in larger animal models, such as rabbits, are required to accurately map its clearance dynamics. Furthermore, while our 7-day assessments demonstrated a favorable retinal safety profile, the long-term effects and potential immunogenicity (e.g., anti-drug antibodies) of repeated dosing must be rigorously evaluated in future translational studies. Third, although we frame PF4 as a potential complementary agent to existing anti-VEGF therapies, this study did not include direct head-to-head comparisons or combination studies with anti-VEGF agents. Such studies will be essential to establish its relative efficacy and potential synergistic benefits. Finally, although we characterized the tissue-level localization of PF4, its precise subcellular trafficking following intravitreal injection requires further investigation. Addressing these gaps in future studies will be essential for translating PF4 toward clinical application.
In conclusion, intravitreal PF4 markedly reduced neovascular lesion formation and vascular leakage in the laser-induced CNV model, while attenuating retinal inflammatory responses, decreasing VEGFA and total VEGFR2 expression, and suppressing ERK/STAT3 activation, with good structural and functional tolerability in the eye. In addition, PF4 reduced PRNV in Vldlr⁻/⁻ mice, providing complementary in vivo support for its anti-angiogenic activity. In vitro, PF4 inhibited VEGF-induced angiogenic behaviors of HRMECs (proliferation, migration, and tube formation) and reduced VEGF-triggered AKT/ERK/STAT3 phosphorylation. Together, these findings demonstrate that PF4 exerts both anti-angiogenic and anti-inflammatory effects in preclinical models, supporting further investigation of PF4 for the treatment of nAMD and related neovascular retinal diseases.
nAMD, neovascular age-related macular degeneration; CNV, choroidal neovascularization; PF4, platelet factor 4; VEGF, vascular endothelial growth factor; VEGFA, vascular endothelial growth factor A; VEGFR2, vascular endothelial growth factor receptor 2; ERK, extracellular signal-regulated kinase; STAT3, signal transducer and activator of transcription 3; AKT, protein kinase B; HRMECs, human retinal microvascular endothelial cells; Vldlr⁻/⁻, very low‑density lipoprotein receptor knockout.
All data reported in this paper will also be shared by the lead contact upon request.
XK was the primary lead for experiments and data collection and drafted the original manuscript. SLiang, TQ, SLi, JL, and HH participated in data collection. XW, SH, DZ, and HY contributed to overall study conception and top-level design. YG provided key input on study conceptualization (including defining experimental groups) and interpreted histological data; MX designed critical experimental protocols and analyzed and interpreted molecular data; ML developed the detailed methodological framework and analyzed and interpreted functional outcomes. YG, MX, ML, and HY critically reviewed the manuscript and provided final manuscript oversight. HY also secured funding. All authors contributed to editorial changes in the manuscript, read and approved the final version of the manuscript for publication, and all authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (TMUaMEC 2024030) and conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, as well as the ARRIVE guidelines.
The authors thank the clinical and research staff at Tianjin Medical University General Hospital and Tianjin Medical University for their valuable assistance with data collection and technical support.
This work is supported by grants from the National Natural Science Foundation of China (82530032, 82330031) to HY, the National Natural Science Foundation of China (82401296) to ML, the Natural Science Foundation of Tianjin (25JCZDJC00390) to HY, the Natural Science Foundation of Tianjin (25JCQNJC00680) to ML, the Natural Science Foundation of Tianjin (23JCQNJC01180) to HH, and the Tianjin Key Medical Discipline Construction Project (NO. TJYXZDXK-3-004A) to HY.
The authors declare no conflict of interest. Given his role as the Editorial Board member, Shikun He had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Dario Rusciano.
During the preparation of this work the authors used ChatGPT-3.5 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.