






 , Zhenjun Ji 1,2, Mi Wang 1,2, Zaixiao Tao 1,2, Xinxin Li 1,2, Rui Zhang 1,2, Junwei Xu 1,2,5,*
, Zhenjun Ji 1,2, Mi Wang 1,2, Zaixiao Tao 1,2, Xinxin Li 1,2, Rui Zhang 1,2, Junwei Xu 1,2,5,* , Genshan Ma 1,2,*
, Genshan Ma 1,2,*1 Department of Cardiology, Zhongda Hospital, 210009 Nanjing, Jiangsu, China
2 School of Medicine, Southeast University, 210009 Nanjing, Jiangsu, China
3 Department of Cardiology, Yancheng No. 1 People’s Hospital, Affiliated Hospital of Medical School, Nanjing University, 224000 Yancheng, Jiangsu, China
4 Department of Cardiology, The First people’s Hospital of Yancheng, 224000 Yancheng, Jiangsu, China
5 Department of Cardiology, Nanjing Chest Hospital, Affiliated Nanjing Brain Hospital, Nanjing Medical University, 210029 Nanjing, Jiangsu, China
Abstract
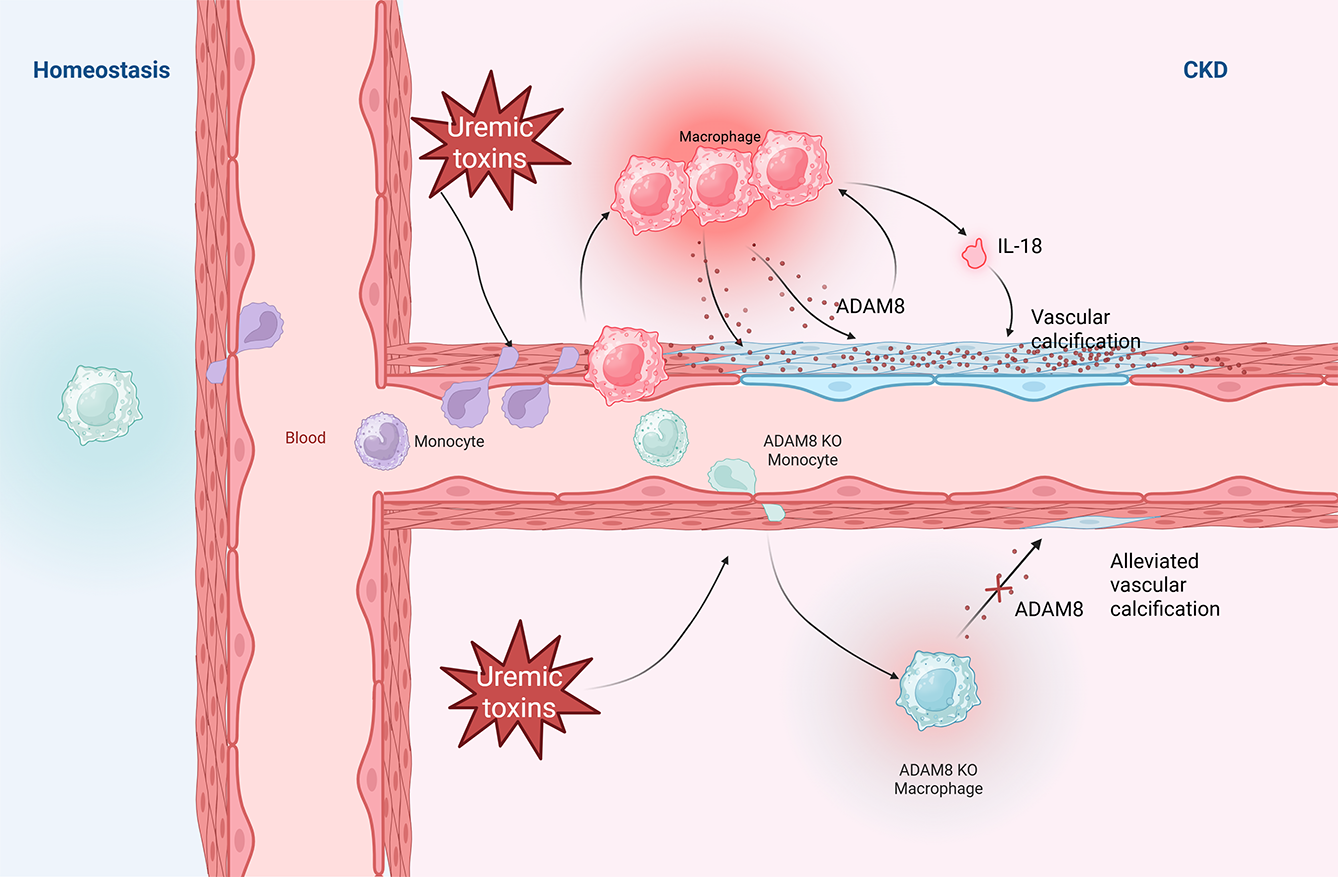
Vascular calcification (VC) is an inflammatory disease driven by aberrant cellular processes in which macrophages play an important role. The A disintegrin and metalloproteinase 8 (ADAM8) protein is an important regulatory factor in macrophages and contributes to the development of inflammatory diseases. However, the relationship between ADAM8 and VC remains unknown. Thus, this study aimed to investigate the role of macrophage ADAM8 in VC.
Plasma ADAM8 levels were compared between patients with and without aortic calcification. Immunofluorescence staining of human aortic tissue samples was performed to assess differences in ADAM8 expression between calcified and non-calcified tissues. Macrophage-specific Adam8 knockout mice (Adam8flox/flox, Lyz2-Cre; KO) and the corresponding control mice (Adam8flox/flox; Flox) were generated using CRISPR/Cas9 technology. Additionally, the adeno-associated virus AAV6-F4/80-Adam8 was employed to achieve macrophage-specific overexpression. The relationship between macrophage ADAM8 and VC was studied in a VC mouse model of chronic kidney disease (CKD) by comparing VC severity between groups.
Plasma ADAM8 levels were elevated in patients with aortic calcification. Immunofluorescence staining of human aortic samples suggested that VC was associated with ADAM8-positive macrophage infiltration and ADAM8 released by macrophages. In mice, macrophage-specific Adam8 knockout attenuated the development of CKD-associated VC, whereas macrophage-specific ADAM8 overexpression reversed the VC phenotype.
ADAM8 levels are elevated in patients with aortic calcification and are associated with macrophage infiltration into vascular tissue and ADAM8 release, leading to increased VC. These findings identify ADAM8 as a promising novel therapeutic target for preventing VC.
Graphical Abstract
Keywords
- A disintegrin and metalloproteinase 8 (ADAM8)
- macrophage
- vascular calcification
- inflammation
- chronic kidney disease (CKD)
Vascular calcification (VC) is characterized by the deposition of calcium phosphate on arterial walls and is an independent predictor of adverse cardiovascular events [1]. VC is commonly associated with aging [2], chronic kidney disease (CKD) [3], atherosclerosis [4], hypertension [5], and certain hereditary diseases. Due to population aging and other risk factors, the incidence of VC has increased in recent decades, constituting a threat to global public health. VC occurs insidiously, as it is clinically silent. It has a prolonged course, yet its progressive development is often overlooked, which can result in hemodynamic impairment associated with chronic ischemia in various organs. Ultimately, this can lead to adverse outcomes such as acute myocardial infarction, stroke, and peripheral arterial occlusion [6]. Due to the lack of effective preventative measures, most current treatments for VC, which are invasive and downstream modalities, target calcified vascular tissue. Although revascularization techniques can improve the survival rate of some vascular stenoses or occlusive diseases, the incidence of surgical complications and poor prognosis due to calcified vessels means that treating VC remains a formidable challenge.
Moving beyond theories of passive calcium phosphate deposition, VC is now
understood to be an active, cell-driven process. This process is tightly
controlled by various inducers and inhibitors, sharing fundamental cellular
mechanisms, regulatory pathways, and structural characteristics with bone
formation [7, 8, 9, 10, 11]. A key step in the development of VC is the
osteogenic differentiation of vascular smooth muscle cells (VSMCs) [12]. Although
the drivers and mechanisms underpinning the development of VC vary in different
conditions, the pathological features of phenotypic transition are essentially
the same. Fundamentally, these phenotypic shifts involve the downregulation of
contractile markers, including smooth muscle protein 22-alpha (SM22
Robust evidence suggests that VC is a chronic, sterile inflammatory disease. During aging or CKD, inflammation can lead to the uptake of inorganic phosphate, which promotes the osteogenic differentiation of VSMCs. Therefore, inhibiting this inflammatory response is a key means of antagonizing VC. Furthermore, macrophage infiltration is one of the key steps in VC development. Macrophages are recognized to play a dynamic and multifaceted role throughout the course of VC, including initiation, progression, and potential regression [19]. Macrophages can be secondarily recruited to inflammation or microcalcification, which, in turn, induces the release of inflammatory factors, the expression of osteogenic genes, or the formation of apoptotic vesicles. On the one hand, this can continue to drive macrophage recruitment. However, on the other hand, some inflammatory mediators can further induce the osteogenic differentiation of VSMCs. Therefore, an in-depth study of the characteristics of infiltrating macrophages at the onset of VC and the potentially targetable upstream pro-calcification mediators is essential for the clinical management of VC.
A disintegrin and metalloprotease 8 (ADAM8), also known as MS2 or CD156, is a member of the ADAM protein family. Adam8 was initially identified through cloning mouse macrophage cDNA from a library. The gene is preferentially expressed in lymphoid tissues, immune cells, and tumor cells. Structurally, ADAM8 contains the characteristic domains shared by the ADAM protein family: a prodomain (Pro), a metalloproteinase (MP) domain, a disintegrin (DIS) domain, a cysteine-rich domain (Cys), an epidermal growth factor (EGF)-like domain, a transmembrane domain (TM), and a cytoplasmic domain (CD) [20]. ADAM8 possesses protein hydrolytic activity, which is mainly responsible for the cleavage of cell membrane proteins. It is critically involved in diverse physiological and pathological processes, such as cell adhesion, differentiation, fertilization, inflammation, and tumorigenesis.
Evidence from cardiovascular disease studies suggests that plasma ADAM8 levels correlate with the severity of atherosclerosis [21]. Evidence also suggests that ADAM8 enhances osteoclast formation both in vivo and ex vivo and that it mediates bone resorption during inflammatory processes [22, 23]. A strong association between osteoporosis and VC has been widely reported, and the role of ADAM8 in both inflammation and bone homeostasis suggests that it may also be involved in the pathogenesis of VC. However, the relationship between ADAM8 and VC has not been described.
Thus, the aim of this study was to investigate the relationship between macrophage ADAM8 and VC. We found that VC was associated with the macrophage infiltration of vascular tissues and the release of ADAM8. Macrophage ADAM8 in CKD-background mice promoted VC through inflammatory mechanisms, whereas the knockout of macrophage Adam8 attenuated VC in the CKD background, providing a rationale for targeted interventions against VC.
Eighty-five CKD patients and 44 controls were enrolled from the Cardiology Department of our hospital from July 2023 to January 2024 for the aortic calcification study. This study complied with the Declaration of Helsinki and was approved by the Ethics Committee of Southeast University-affiliated Zhongda Hospital (Number: 2023ZDSYLL298-P01). Blood was collected from the patients along with basic clinical data (Supplementary Table 1). The inclusion criteria were: (1) Willing participants and signing the informed consent form. (2) Individuals aged 18 years or older, excluding pregnant women. (3) Case group: patients with stage III–IV CKD; control group: individuals with normal renal function, no history of hematuria, proteinuria, or CKD, or patients with stage I CKD. The exclusion criteria were: (1) Individuals with a history of ischemic stroke within the past week, intracranial or gastrointestinal hemorrhage within the last 6 months, or major surgery within the previous 30 days. (2) Those with severe respiratory conditions, including but not limited to chronic obstructive pulmonary disease (COPD) or asthma. (3) Severe liver disease (hepatic insufficiency), with aspartate aminotransferase/alanine aminotransferase (AST/ALT) levels exceeding three times the upper limit of normal due to non-cardiac causes, or history of cirrhosis. (4) Severe infectious diseases, or patients with severe dyspnea due to conditions such as pneumonia or COPD. (5) The presence of other severe comorbid conditions that limit life expectancy to less than 6 months. (6) Cardiac conduction disorders, including sick sinus syndrome, advanced atrioventricular block, or bradycardia-induced syncope. (7) Pregnant or lactating women. (8) Participants deemed by the investigator to be otherwise unsuitable for the study.
Eight patients who had undergone aortic replacement were enrolled in this study from the cardiothoracic surgery department of Zhongda Hospital from May 2024 to July 2024 and were included in the immunofluorescence imaging of human aortic tissues. This study complied with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Zhongda Hospital, affiliated with Southeast University (Number: 2024ZDSYLL150-P01). Aortic tissues were collected from the patients. The inclusion criteria were: (1) Patients requiring aortic replacement. (2) Provided written informed consent and voluntarily agreed to take part in the study. (3) Males or nonpregnant females over 18 years of age. The exclusion criteria were: (1) Patients with an autoimmune disease. (2) Pregnant or lactating women. (3) Any other status or condition that would make participation in the study inappropriate.
All animal experiments were approved by the Animal Care and Welfare Committee (Number: 20200326014) and were performed in adherence to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the ARRIVE guidelines. All animal procedures were conducted in strict compliance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition, 2011). Efforts were made to reduce the number of animals used and to minimize animal pain and discomfort. Macrophage-specific Adam8 KO mice (Cyagen Biosciences, Suzhou, China) on a C57BL/6 background were generated using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system [24] (Supplementary Fig. 1). Mice were housed under controlled conditions at the Southeast University Laboratory Animal Center. They were maintained in a specific-pathogen-free environment within ventilated cages under a 12-hour light/dark cycle, with ad libitum access to food and water. Male mice aged 6–10 weeks were utilized in the current study. The study employed randomization with allocation concealment, and the operators were blinded to group assignment. Mice (n = 28) were fed a 0.25% adenine diet (Macklin, Shanghai, China) for 4 weeks to establish the CKD model. Subsequently, the CKD model mice were switched to a high-phosphorus diet (1.8% phosphorus; Xie Tong Sheng Wu) and received intramuscular injections of calcitriol (1 µg/kg; Sigma-Aldrich, Darmstadt, Germany) twice weekly to induce VC. A control group (n = 12) was maintained on a standard normal diet (ND) group (Xie Tong Sheng Wu, Nanjing, China) and received saline injections in parallel. Tail-tip blood sampling was performed at the start of the experiment (0 W), the fourth week (4 W), and the tenth week (10 W) without anesthesia. Prior to blood collection, the tails were immersed in warm water at approximately 50 °C and disinfected. After blood collection, hemostasis was applied, and the mice were adequately comforted. After 16 weeks, the mice were euthanized via an intraperitoneal injection of sodium pentobarbital (concentration 15 mg/mL, 200 mg/kg), and the aorta and blood were collected. At the conclusion of the experiment, surviving animals were transferred to other approved protocols within the institution for continued use.
Human plasma was processed by centrifugation (1000 rpm, 20 minutes, 4 °C) to pellet cellular debris, and the resulting supernatant was collected for subsequent analysis. Enzyme-linked immunosorbent assays (ELISAs) were performed according to the manufacturer’s instructions (Elabscience Biotechnology Co., Ltd., Wuhan, China). Optical density (OD) was measured at a wavelength of 450 nm.
Following standard protocols, mouse aortas were paraffin-embedded and sectioned. For Von Kossa staining, sections were deparaffinized and rehydrated, then immersed in silver nitrate solution under ultraviolet light for 2 hours. After rinsing in phosphate-buffered saline (PBS), the sections were subjected to hematoxylin and eosin staining. Finally, all sections were dehydrated, mounted, and scanned using a digital slide scanner (Olympus VS200, Tokyo, Japan).
Paraffin sections of human aortic tissue were prepared, dewaxed, and subjected
to antigen retrieval. The tissue areas were outlined with a PAP Pen to prevent
antibody dispersion. Sections were then incubated with the corresponding primary
antibody at 4 °C overnight in a humidified chamber. After washing with
PBS on a shaker, a secondary antibody was applied and incubated for 1 hour at
room temperature. Nuclei were counterstained with
4′,6-diamidino-2-phenylindole. Following the application of an anti-fade
mounting medium, the slides were coverslipped and imaged with a digital slide
scanner (Olympus VS200). For quantitative analysis, five distinct regions of
interest per sample were randomly selected at a higher magnification. The mean
fluorescence intensity of ADAM8 and
The serum levels of creatinine, phosphorus, urea, calcium, ALP, as well as aortic calcium content, were measured using commercially available assay kits (Elabscience, Wuhan, China). Following the manufacturer’s protocol, aortic tissue segments were lysed in the designated buffer. Calcium ions in the lysate were then chelated with methyl thymol blue under alkaline conditions to form a blue complex. The absorbance of the colored product was measured to quantify calcium content. The results were normalized to the total protein concentration determined with a BCA protein assay kit.
The AAV vector for macrophage-specific Adam8 OE was generated by
subcloning the mouse Adam8 coding sequence (NM_007403) downstream of an
EGFP-FT2A cassette into a GV650 backbone, under the control of a
macrophage-specific F4/80 promoter (sequence provided in Supplementary
Information). Briefly, the 3Flag sequence of GV650 was removed to generate a
pAAV-F4/80p-EGFP-FT2A-NM_007403-SV40 PolyA construct. The Adam8
fragment was amplified and inserted into the vector via NheI/EcoRI cloning sites.
Following plasmid construction, recombinant AAV was packaged, purified, and its
titer was determined, and cryopreserved at –80 °C (Genechem Co., Ltd.,
Shanghai, China). For in vivo delivery, 4 to 6-week-old male mice were
administered approximately 8
The aortic tissues were homogenized in RIPA lysis buffer supplemented with protease and phosphatase inhibitors (KeyGen Bio TECH, Nanjing, China). Equal amounts of total protein were loaded onto a pre-cast gel and separated by electrophoresis at a constant voltage of 120 V. Once the bromophenol blue dye front reached the bottom of the gel, proteins were transferred to polyvinylidene membranes using a rapid transfer system at 400 mA for 30 minutes. Following transfer, the membranes were blocked with 5% non-fat milk for 1 hour at room temperature. After blocking, membranes were trimmed and incubated overnight at 4 °C with the indicated primary antibodies. The next day, the membranes were washed and incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature with gentle shaking. Protein bands were visualized using an automated chemiluminescence imaging system (Tanon 5200, Shanghai, China). Band intensities were quantified with ImageJ software (version 2; National Institutes of Health, Bethesda, MD, USA), and statistical analysis was performed accordingly. Details of the antibodies used are provided in Supplementary Table 2.
The sample sizes for both the clinical cohort and the mouse experiments were determined based on previously published studies with similar designs. Strict exclusion criteria were applied to the clinical cohort to minimize confounding factors and enhance the robustness of the results. Data analyses were performed using SPSS 26.0 (IBM, Armonk, NY, USA) and GraphPad Prism 9.5.0 software (GraphPad Software, Inc. San Diego, CA, USA).
Continuous data are presented as mean
Aortic calcification was used as the target lesion to explore the relationship
between ADAM8 and VC. Ultimately, a total of 129 patients were enrolled for this
segment of the research, including 85 CKD patients and 44 controls. Plasma
samples were collected, and thoracoabdominal computed tomography (CT) scans were
performed on all participants. The demographic and clinical characteristics of
patients in both groups are presented in Supplementary Table 1.
Patients in the CKD group exhibited higher neutrophil counts (5.21
The chest and abdominal CT findings were used to categorize the participants
into a calcified group (n = 97) and a non-calcified group (n = 32). Patients with
aortic calcification were older (67.26
| Characteristics | Non-VC group (n = 32) | VC group (n = 97) | p-value | |
| Age (years) | 53.91 |
67.26 |
||
| Sex (male, n %) | 20 (63%) | 59 (61%) | 0.867 | |
| Laboratory tests | ||||
| WBCs, ×109/L | 6.56 |
6.98 |
0.370 | |
| RBCs, ×1012/L | 4.11 |
3.89 |
0.173 | |
| PLTs, ×109/L | 217.91 |
186.77 |
0.077 | |
| Neutrophils, ×109/L | 4.44 |
5.00 |
0.232 | |
| Lymphocytes, ×109/L | 1.51 |
1.17 |
0.002** | |
| Monocytes, ×109/L | 0.41 |
0.46 |
0.240 | |
| ALT, U/L | 20.13 |
19.25 |
0.819 | |
| AST, U/L | 19.25 |
22.83 |
0.179 | |
| BUN, mmol/L | 8.81 |
12.67 |
0.026* | |
| Cr, mmol/L | 225.97 |
247.45 |
0.685 | |
| UA, µmol/L | 356.53 |
362.84 |
0.830 | |
| eGFR, mL/min/1.73 m2 | 69.83 |
48.18 |
0.008** | |
| TC, mmol/L | 4.18 |
3.49 |
0.007** | |
| TG, mmol/L | 1.94 |
1.51 |
0.240 | |
| HDL-C, mmol/L | 1.10 |
1.01 |
0.226 | |
| LDL-C, mmol/L | 2.23 |
1.86 |
0.037* | |
| HbA1c (%) | 5.93 |
6.50 |
0.065 | |
| ADAM8, pg/mL | 746.87 |
1030.98 |
0.008** | |
| Indexes | ||||
| PIV | 315.14 |
494.25 |
0.025* | |
| SII | 719.66 |
986.23 |
0.045* | |
Values are presented as mean
We collected eight aortic tissue samples from patients who underwent aortic
replacement to further analyze the relationship between ADAM8 and VC. The Von
Kossa staining results were used to classify the samples into the aortic
calcification group (AC) and the non-aortic calcification group (non-AC). The
baseline demographic and clinical characteristics of the two groups are presented
in Table 2. Patients in the AC group exhibited significantly higher uric acid
(UA) levels (270.25
| Characteristics | AC (n = 4) | Non AC (n = 4) | p-value |
| Age (years) | 67 |
60.75 |
0.374 |
| Sex (male, n %) | 3 (75%) | 3 (75%) | 1.000 |
| WBCs, ×109/L | 8.56 |
8.98 |
0.876 |
| RBCs, ×1012/L | 3.46 |
3.71 |
0.690 |
| PLTs, ×109/L | 202.50 |
144.75 |
0.448 |
| Neutrophils, ×109/L | 6.88 |
7.41 |
0.843 |
| Lymphocytes, ×109/L | 1.08 |
1.37 |
0.530 |
| Monocytes, ×109/L | 0.55 |
0.83 |
0.212 |
| ALT, U/L | 64.25 |
34.00 |
0.225 |
| AST, U/L | 49.75 |
33.00 |
0.500 |
| BUN, mmol/L | 7.73 |
6.33 |
0.204 |
| Cr, mmol/L | 94.50 |
64.75 |
0.056 |
| UA, µmol/L | 270.25 |
185.75 |
0.040* |
| eGFR, mL/min/1.73 m2 | 66.51 |
103.72 |
0.012* |
| TC, mmol/L | 2.95 |
3.43 |
0.470 |
| TG, mmol/L | 1.46 |
1.49 |
0.960 |
| HDL-C, mmol/L | 0.90 |
0.97 |
0.655 |
| LDL-C, mmol/L | 1.61 |
1.94 |
0.456 |
| FBG, mmol/L | 7.36 |
5.79 |
0.204 |
Values are presented as mean
Three adjacent sections from the same sample were respectively subjected to Von
Kossa staining, immunofluorescence staining for ADAM8 and CD68 and
immunofluorescence staining for ADAM8 and
Through comparative analysis, we observed that the calcified regions were co-localized with ADAM8, demonstrating significant ADAM8 expression (Fig. 1), and the expression level of ADAM8 was positively correlated with the degree of calcification (Supplementary Fig. 2). In the non-calcified regions of the CG, scattered ADAM8 expression was also detected, whereas no ADAM8 expression was observed in the tunica media in the NG (Fig. 1). In the CG, macrophage infiltration was evident around the calcified regions, termed the macrophage infiltration region, predominantly located within the medial layer. This region showed high ADAM8 expression in macrophages. In contrast, the macrophage region in the NG exhibited low ADAM8 expression, with a small number of macrophages, and this region was primarily localized at the junction between the intima and media (Fig. 2).
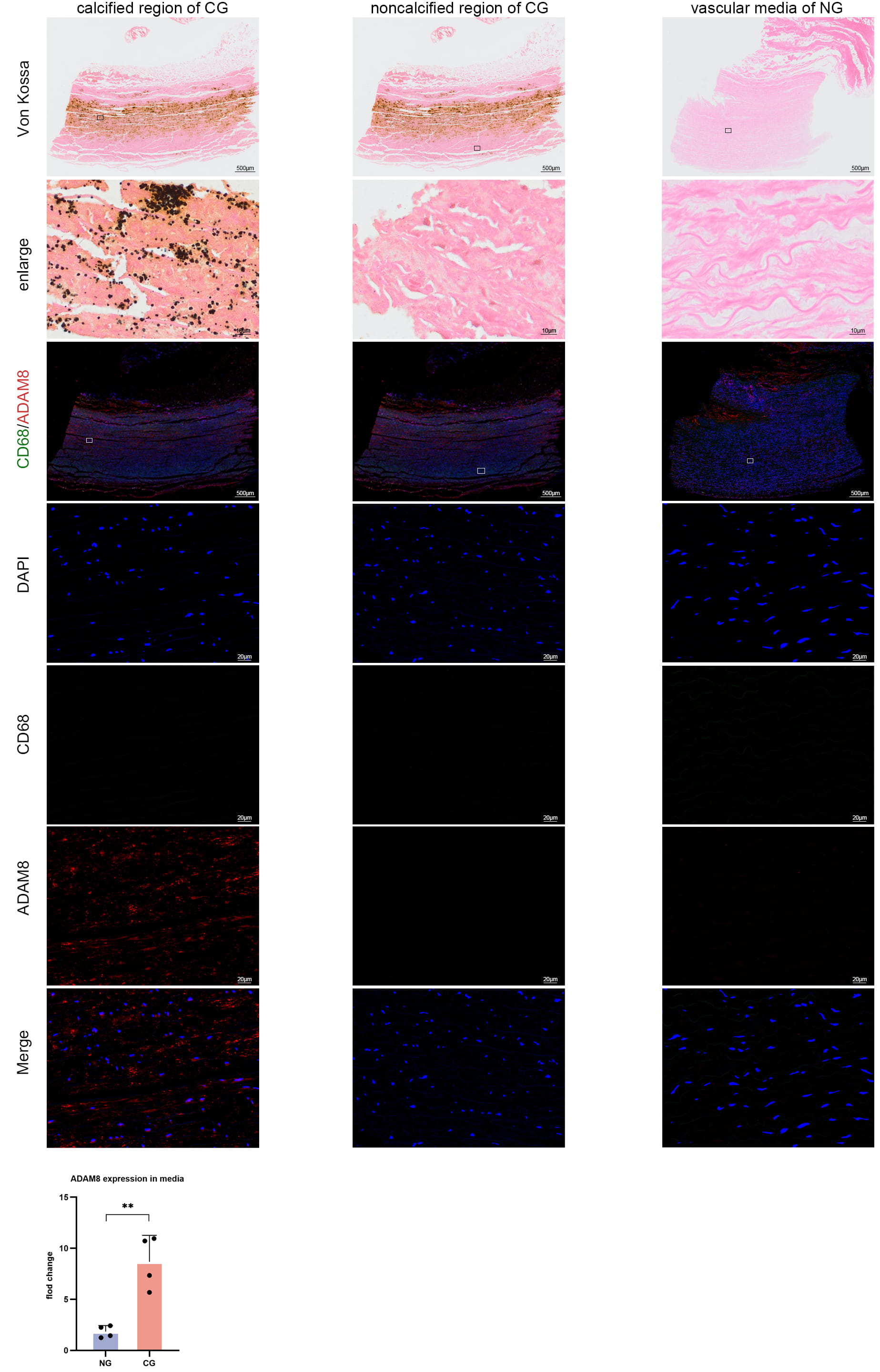 Fig. 1.
Fig. 1.
Von Kossa staining images and immunofluorescence staining images
of CD68 and ADAM8 in human aortic tissues. ADAM8 expression was co-localized
with the calcified region, while hardly any ADAM8 expression was observed in
non-calcified regions (n = 4) (p = 0.0038, unpaired t-test).
**p
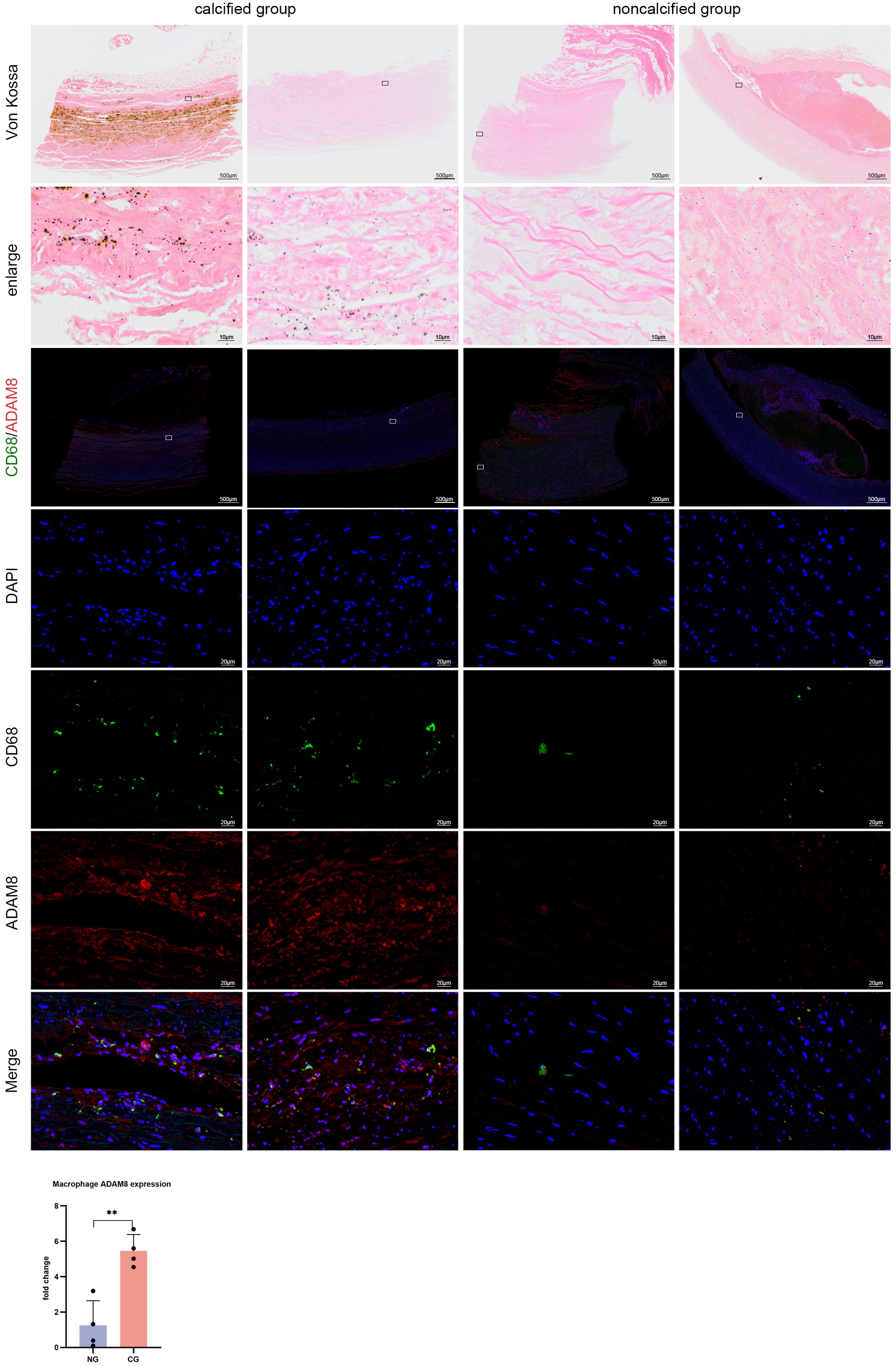 Fig. 2.
Fig. 2.
Von Kossa staining images and CD68 and ADAM8 immunofluorescence
staining images in human aortic samples. Comparison of Von Kossa staining and
CD68 and ADAM8 immunofluorescence images between macrophage infiltration regions
in the calcified group and macrophage regions in the non-calcified group revealed
abundant ADAM8 expression in macrophage infiltration regions in the calcified
group, while no such phenomenon was observed in the non-calcified group (n = 4)
(p = 0.0024, unpaired t-test). **p
Immunofluorescence staining for both
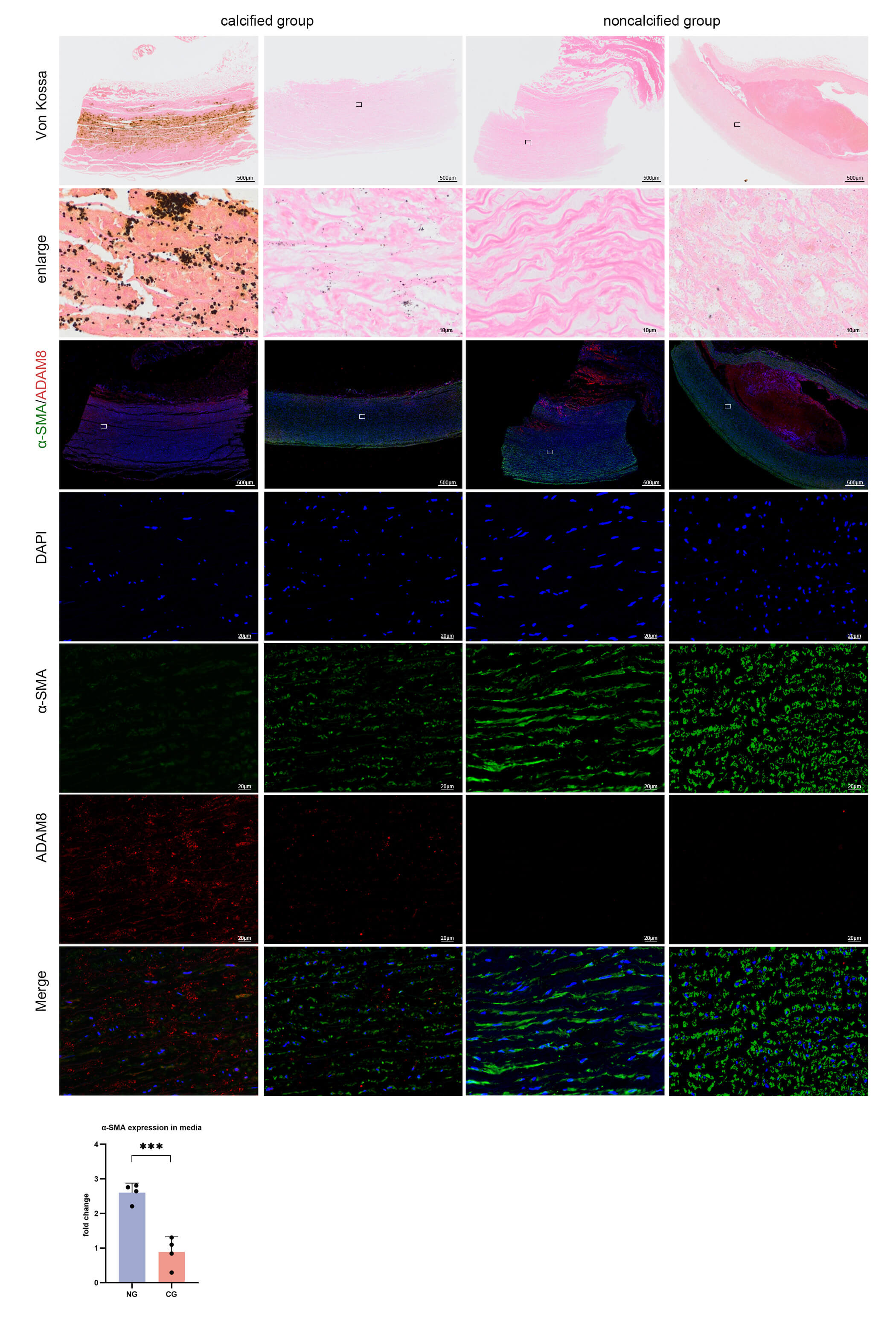 Fig. 3.
Fig. 3.
Von Kossa staining images and immunofluorescence staining images
of
We established macrophage-specific Adam8 KO mice. Knocking out Adam8 did not cause phenotypic changes in the mice. As shown in Fig. 4A, we established an in vivo CKD model using Adam8 KO mice and control mice from the same litter. Knocking out macrophage Adam8 KO did not affect the survival of mice in either sub-model group (Fig. 4B). As renal function deteriorated, the weight of the mice in both model groups decreased equally (Fig. 4C). At the beginning of the experiment (0 weeks), as well as at the fourth and tenth weeks, we evaluated renal function impairment. The results showed that serum phosphorus (Fig. 4D), ALP (Fig. 4E), creatinine (Fig. 4F), and urea (Fig. 4G) levels were significantly elevated in both model groups, with no statistically significant differences observed between the two groups. No significant differences in serum calcium levels were observed among the four groups (Fig. 4H), suggesting that the adenine diet was successful in inducing renal injury and that the KO of macrophage-specific Adam8 did not alter renal function. The aortic calcium content in both CKD groups was increased compared with the ND groups and was higher in the CKD-Flox group than in the CKD-KO group (Fig. 4I).
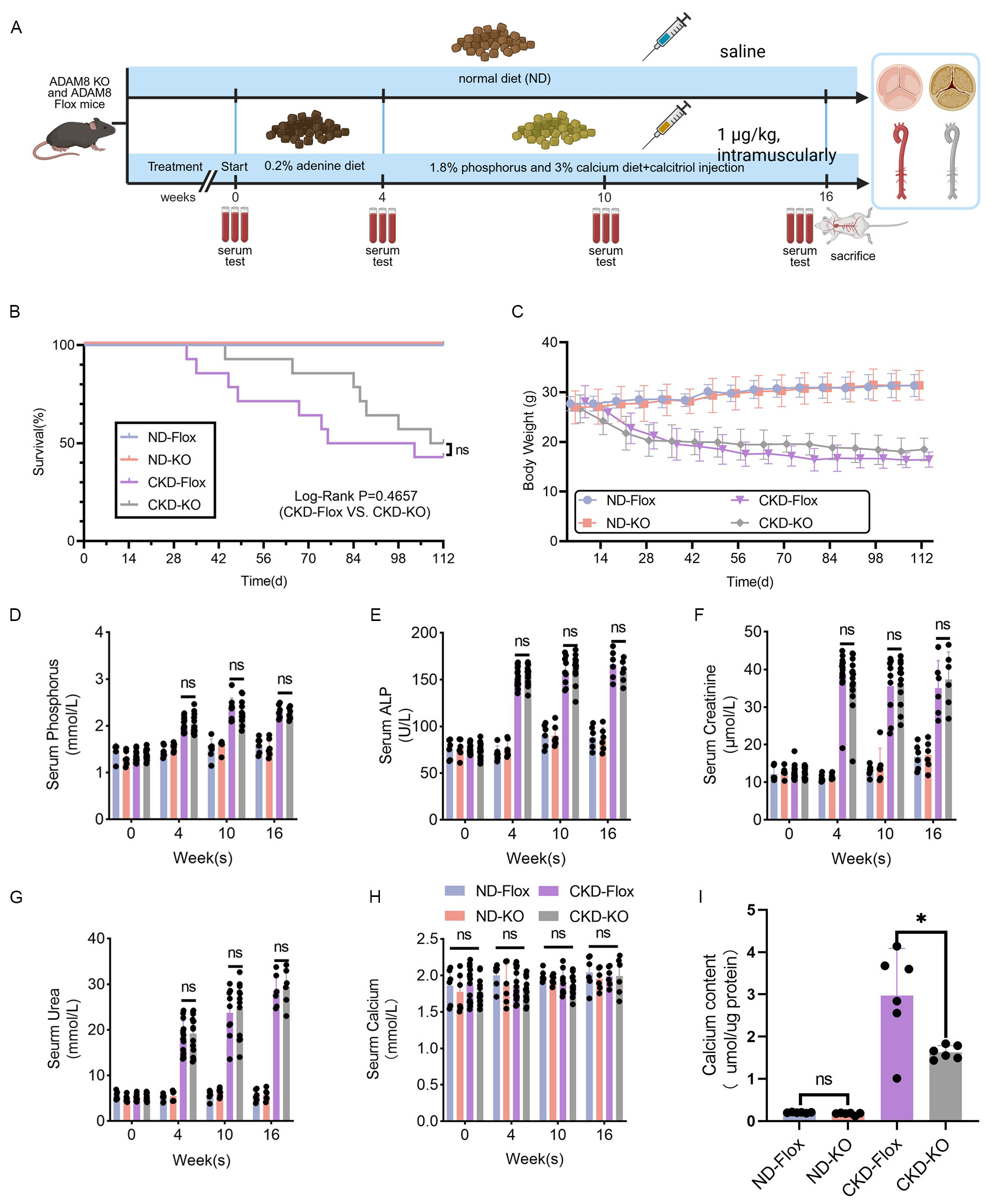 Fig. 4.
Fig. 4.
Adam8 KO has no protective effect on renal function in
CKD mice. (A) Development of a mouse model for CKD-associated vascular
calcification. Created in BioRender. Dong, R. (2025).
https://BioRender.com/u16l265. (B) Survival curves. Cumulative mortality in the
four groups of mice. ND-Flox mice (n = 6), ND-KO mice (n = 6), CKD-Flox mice (n =
14), and CKD-KO mice (n = 14). (C) Body weights (n = 6–14). (D) Biochemical
analyses of serum phosphorus, serum alkaline phosphatase (ALP) (E), creatinine
(F), urea (G), and calcium concentrations (H) at 0, 4, and 10 weeks during modeling. (I) Quantification of
aortic tissue calcium content (n = 6–14). (p = 0.015, unpaired
t-test). Notes: “n value” refers to the number of biological
replicates. *p
The extent of VC in the aortic media was assessed by Von Kossa staining and
measuring the calcium content. We dissected mice that died during the experiment
and compared the calcification of the aortic valve and aorta in the two sub-model
groups. Calcification occurred earlier in the aortic valves than in the aortic
vessels. Furthermore, the valvular calcification was more severe in the CKD-Flox
group compared to the CKD-KO group (Supplementary Fig. 5). The CKD-Flox
group displayed more severe vascular calcification than the CKD-KO group (Fig. 5A). Western blots (Fig. 5B and Supplementary Figs. 6,7) showed that
Runx2 expression was increased in vascular tissue in the CKD groups (Fig. 5E)
Sm22
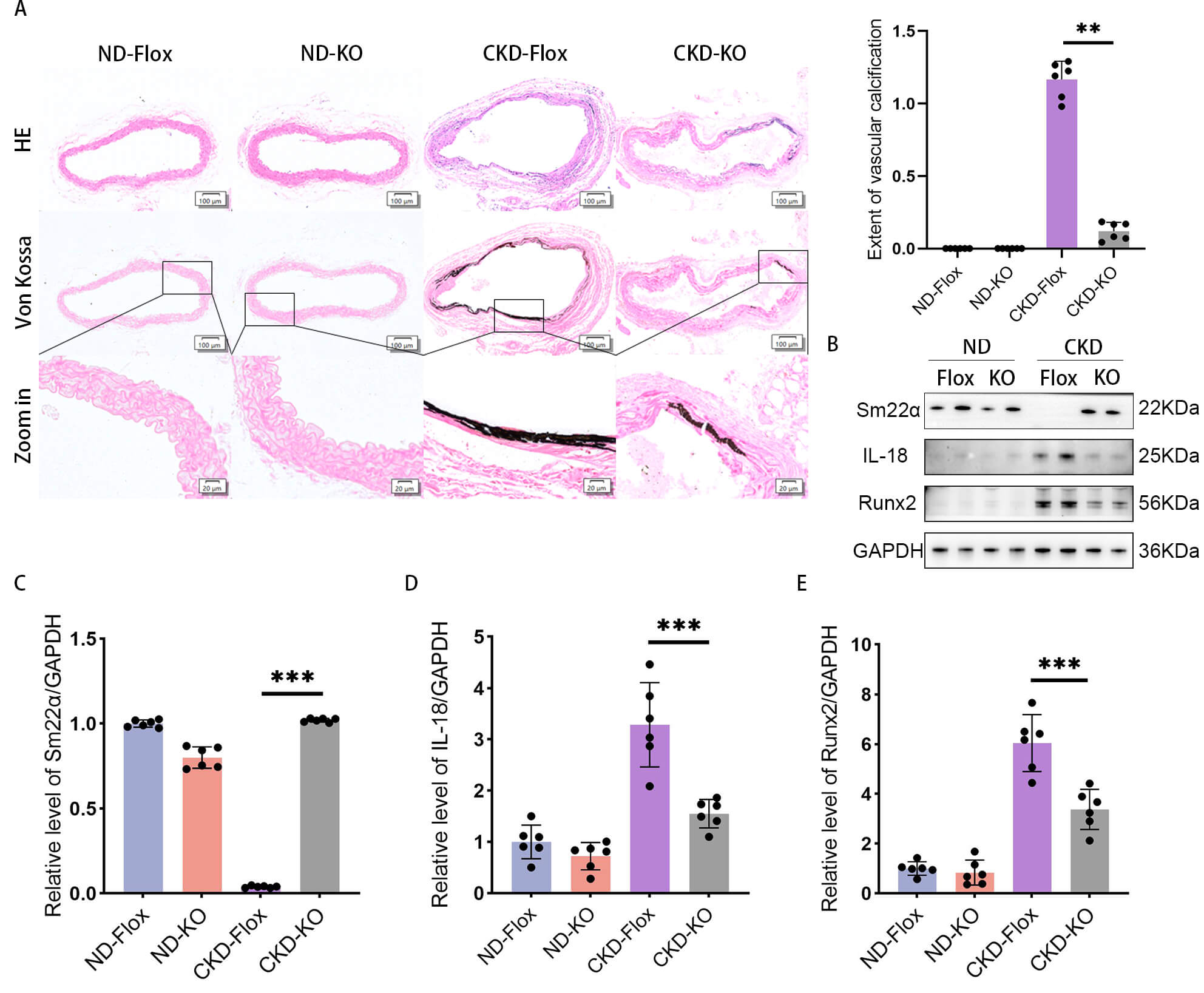 Fig. 5.
Fig. 5.
Aortic calcification is attenuated by Adam8 KO in CKD
mice. (A) Representative Von Kossa staining images of vascular tissue from the
four groups (positive staining: black) (n = 6). (B) Western blots of (C)
SM22
The association between macrophage-derived ADAM8 and VC was investigated using
macrophage-specific Adam8 OE. This was achieved by injecting the tail
veins of 4-week-old Adam8 KO mice with either AAV6-F4/80-Adam8 or a
control virus. Following viral delivery, CKD-associated VC was induced in these
animals. The results showed that the mice successfully overexpressed
ADAM8 after viral injection (Fig. 6B,C and Supplementary Fig.
8). VC was significantly aggravated in the CKD+KO+OE group compared to the
CKD+KO+NC group (Fig. 6A). Consistent with this phenotypic enhancement, Western
blot analysis revealed a concurrent upregulation of osteogenic (Runx2) and
inflammatory (IL-18) markers, and a downregulation of Sm22
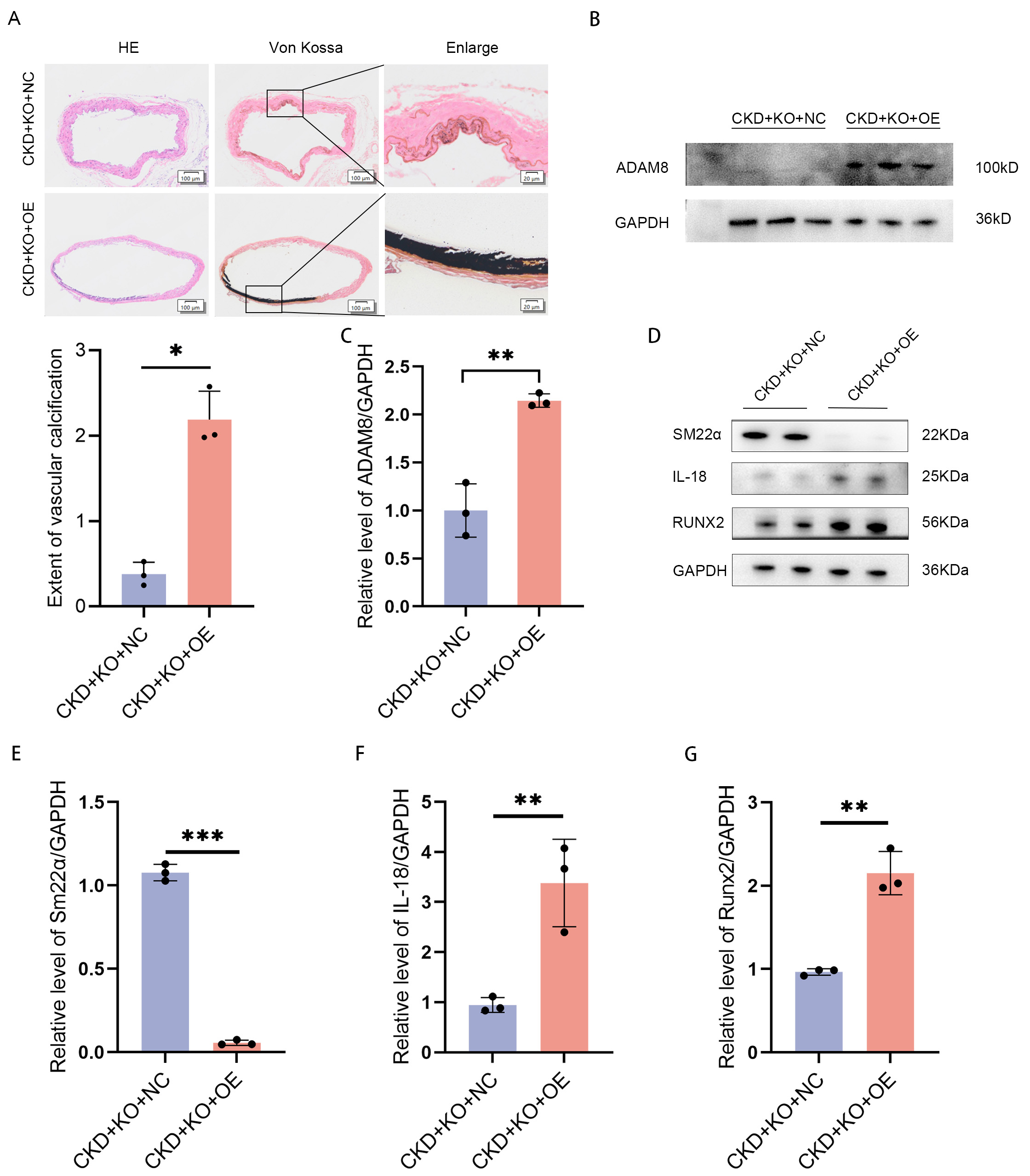 Fig. 6.
Fig. 6.
Adam8 OE exacerbates aortic calcification in
Adam8 KO mice under CKD conditions. (A) Representative images of Von
Kossa staining in the aortic tissues of mice from the two CKD groups (positive
staining: black). (B) Western blot analysis and (C) quantification of ADAM8
protein levels (p
Vascular calcification is very common in elderly people and CKD populations, and has become a potential killer in seniors due to a lack of sufficient attention and effective prevention and treatment [25, 26]. VC has also been associated with cognitive decline [27, 28]. The dual threat of VC to long-term health and quality of life has increased its social burden in the context of an aging society and population. Deciphering the mechanisms of VC can lay a foundation for its prevention and targeted treatment. Here, our study demonstrated a relationship between macrophage ADAM8 and VC, and macrophage-specific Adam8 KO attenuated VC in CKD model mice. These findings extend beyond identifying a regulatory role for ADAM8 in VC to provide novel evidence for the macrophage-mediated regulation of this pathological process.
ADAM8 is a shedding enzyme that is involved in a series of physiological and pathological processes, such as ovulation, inflammation, tumor metastasis, and progression. ADAM8 is not essential for homeostasis, and it is expressed at very low levels in normal conditions. It can be found in either a transmembrane location or in a soluble form. Previous studies have shown that serum ADAM8 levels are dramatically increased in various inflammatory diseases such as asthma, COPD [22, 29], gingivitis [30], periprosthetic inflammation of the hip joint [31], and neoplastic diseases [20]. In addition, serum or tissue fluid levels of ADAM8 have been shown to correlate with disease severity. Although inhibiting or knocking down Adam8 was shown to decrease inflammatory responses or slow tumor metastasis [32, 33], it is not clear whether ADAM8 deficiency inhibits VC. Considering that VC is an inflammatory disease and ADAM8 is associated with inflammation, we hypothesized that ADAM8 promotes VC.
First, we included CKD patients and controls. We found that elevated plasma ADAM8 levels were associated with poorer renal function. The participants were categorized into VC and non-VC groups according to the CT scan findings. A comparison of the two groups found that aortic calcification was associated with age and plasma ADAM8 levels. Previous studies have identified a correlation between plasma ADAM8 levels and aortic calcification. Notably, patients with vascular calcification accompanied by renal dysfunction exhibited significantly elevated plasma ADAM8 concentrations.
Our previous work found that plasma ADAM8 is associated with inflammation, renal
function, and aortic calcification, and various risk factors, including aging,
smoking, diabetes, and hypertension, can lead to VC. As a result of its
inflammatory nature, the chronic progression of VC leads to a state of sustained,
low-grade innate immune activation. This persistent signaling ultimately drives
maladaptive tissue responses [34]. Closely linked and critically involved in VC
[34], macrophages play essential roles in its onset, progression, and regression
[19]. Furthermore, alterations in the tissue microenvironment can lead to
adaptive responses in macrophages, with different polarities, phenotypes, or
secretome profiles. Given that the infiltration of monocytes/macrophages into
vascular tissues is a key step in the formation of VC, through a variety of
mechanisms, we chose to perform Von Kossa staining and immunofluorescence
staining of CD68, ADAM8, and
The staining results showed that the calcified vascular intima-media tissue area co-localized with ADAM8 and there was a high prevalence of ADAM8-positive macrophages infiltrating around the calcified vascular tissues (Fig. 1). On the one hand, this finding corroborated that VC is associated with macrophage infiltration, and on the other hand, we found that the infiltration of macrophages into the vascular intima-media might promote VC by releasing ADAM8. A large amount of ADAM8 expression was found in the vicinity of atherosclerotic lipid plaques, accompanied by high macrophage infiltration. Both inflammatory diseases exhibited the release of ADAM8, but the sites where macrophages infiltrated and released ADAM8 differed.
The immunofluorescence imaging of
Whether ADAM8 is a participant or a bystander in cardiovascular disease is still under debate. Because ADAM8 is widely involved in inflammatory responses and exists in both transmembrane and soluble forms, studies of one form of ADAM8 do not reflect its complete function. Second, ADAM8 contains multiple structural domains, each of which can exercise different functions [38]. Nevertheless, the general consensus is that serum ADAM8 levels correlate with the severity of cardiovascular disease [21]. Theodorou et al. [39] found that ADAM8 was associated with plaque progression in humans, but systemic and hematopoietic ADAM8 deficiency did not affect the development of advanced atherosclerotic lesions. In addition, ADAM8 expression was most prominent in the shoulder region of human atherosclerotic lesions [39]. Elevated serum ADAM8 levels were associated with postoperative organ dysfunction in coronary artery disease patients [21].
In this study, we found a correlation between plasma ADAM8 levels and aortic calcification in patients with CKD. Furthermore, tissue samples showed that ADAM8 was enriched in human calcified arterial tissue, and there was high infiltration of ADAM8-positive macrophages around calcified vascular tissues, strongly suggesting that macrophage ADAM8 may contribute to VC development.
Although ADAM8 is essential in homeostasis, ADAM8 deficiency does not cause a
phenotypic shift, and Adam8 KO mice show only mild developmental
defects, as well as increased neovascularization in a model of retinopathy under
oxygen-induced conditions [40]. Therefore, we constructed macrophage-specific
Adam8 KO mice to further investigate the relationship between ADAM8 and
VC. We chose the CKD model, an accelerated model of aging [41, 42, 43], to
validate the relationship between ADAM8 and VC. The CKD groups of mice were first
subjected to renal function impairment. The hematological index tests during
modeling suggested that the renal function of mice in the model groups was
impaired to similar extents. Adam8 KO had no effect on renal function
impairment, body weight change, or survival rates during modeling. Autopsies of
the mice that died during modeling revealed that mice in the CKD-Flox group first
developed aortic valve calcification. Von Kossa staining of the aorta in all four
groups of mice suggested that VC was more severe in the CKD-Flox group than in
the CKD-KO group, while tissue Western blotting suggested that calcification
proteins (such as Runx2) were increased and contractile markers (such as
Sm22
Finally, we performed rescue experiments by overexpressing macrophage
ADAM8 by injecting AAV virus into the tail vein of Adam8 KO
mice. Four weeks later, CKD-related VC was induced in the mice. Mice in the
CKD+KO+OE group exhibited significantly aggravated VC compared to the CKD+KO+NC
group (Fig. 6A). Furthermore, the Western blotting data suggested that the
expression of Runx2 and IL-18 was elevated in arterial tissues, and
Sm22
The present study still has shortcomings. For example, aging and VC development have long-term chronic courses, and our model was different from the normal aging process. Additionally, there was a limitation in the design of our rescue experiment. We employed the F4/80 promoter to drive macrophage-specific ADAM8 expression. Although F4/80 is a valuable pan-macrophage marker, its broad activity in various tissue-resident macrophages, particularly the abundant Kupffer cells in the liver, introduces a potentially confounding factor. For instance, inducing ADAM8 expression in Kupffer cells may alter their functions, such as cytokine secretion, the clearance of circulating metabolites, and overall liver homeostasis. This hepatic alteration presents a plausible indirect mechanism that could influence our primary observation—the modulation of VC. An impaired or activated liver may systemically release a cascade of factors, including pro-inflammatory cytokines, uremic toxins in the context of renal impairment, or dysregulated calcification mediators such as Fetuin-A or Matrix Gla Protein, all of which are known to profoundly affect the VC process [46]. Therefore, although our data robustly demonstrated that ADAM8 re-expression rescued the VC phenotype in CKD, it failed to further distinguish the direct local effects of macrophage ADAM8 from the indirect effects mediated by systemic signals originating from the liver. Future studies utilizing more spatially restricted macrophage-specific promoters, such as those targeting plaque macrophages, or bone marrow transplantation models, combined with the direct analysis of liver function and systemic cytokine and adipokine profiles, will be essential to dissecting local versus systemic mechanisms and fully elucidating the role of ADAM8 in this complex pathology.
The present study revealed the relationship between macrophage ADAM8 and VC, as well as the potential role of macrophage ADAM8 in the pathogenesis of VC. Macrophage ADAM8 deletion slowed the CKD-induced VC. ADAM8 deletion might attenuate VC by decreasing the expression of IL-18. Combined with these results, ADAM8 inhibition or KO may have some preventive potential in VC.
The original data of the study are appended to the article/Supplementary Material. All data reported in this paper will also be shared by the lead contact upon request.
RD, ZJJ and GSM designed the research study. RD, MW, ZXT and XXL performed the research. ZXT, XXL and JWX participated in the acquisition of date. RD and RZ analyzed the data. RD and ZJJ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was conducted in accordance with the Declaration of Helsinki. The research protocol was approved by the Ethics Committee of Southeast University affiliated Zhongda Hospital (Number: 2023ZDSYLL298-P01) (Number: 2024ZDSYLL150-P01). All of the participants provided signed informed consent. Besides, all animal experiments were performed in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition, 2011). All animal experiments conformed to animal experimental ethics rules and were approved by the Animal Care & Welfare Committee of Southeast University (Number: 20200326014).
We are grateful to the staff in Biobank of Zhongda Hospital, School of Medicine, Southeast University for technical assistance. We thank Cyagen Biosciences Co., Ltd. (Suzhou, China) for technical assistance in generation of macrophage-specific Adam8-KO mice. Graphical protocols for modeling are Created with BioRender.com (Agreement number: YZ27OEY0B4). We thank the contribution of BioRender in help us creating Graphical Protocols. We greatly appreciate the patients who willingly participated in this study.
This report was funded by Jiangsu Provincial Key Research and Development Program (BE2022852) and Research Personnel Cultivation Program of Zhongda Hospital Southeast University (CZXM-GSP-RC167).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL49495.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.