







 , Jianjie Zhu 1,2,3,4,†, Yinhua Gong 5, Wenting Wu 1, Xinyu Zhang 1, Chang Li 1, Yang Yang 1, Yili Chen 1, Jianjun Li 1,2,3, Lei Gu 1,2,3, Jian-an Huang 1,2,3, Zeyi Liu 1,2,3, Dan Shen 1,*
, Jianjie Zhu 1,2,3,4,†, Yinhua Gong 5, Wenting Wu 1, Xinyu Zhang 1, Chang Li 1, Yang Yang 1, Yili Chen 1, Jianjun Li 1,2,3, Lei Gu 1,2,3, Jian-an Huang 1,2,3, Zeyi Liu 1,2,3, Dan Shen 1,* , Yuanyuan Zeng 1,2,3,*
, Yuanyuan Zeng 1,2,3,*1 Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Soochow University, 215006 Suzhou, Jiangsu, China
2 Institute of Respiratory Diseases, Soochow University, 215006 Suzhou, Jiangsu, China
3 Suzhou Key Laboratory for Respiratory Diseases, 215006 Suzhou, Jiangsu, China
4 Department of Cancer Diagnosis and Treatment Center, Affiliated Hospital of Jiangnan University, 214000 Wuxi, Jiangsu, China
5 Department of Pharmacy, The First Affiliated Hospital of Soochow University, 215006 Suzhou, Jiangsu, China
†These authors contributed equally.
Abstract
Lung cancer remains a major global public health challenge, with lung adenocarcinoma being the most prevalent histologic subtype. Rosuvastatin, a widely used lipid-lowering agent, has recently attracted attention for its potential antitumor properties. This study investigates the underlying mechanisms and therapeutic potential of rosuvastatin in lung cancer.
The effects of rosuvastatin were evaluated in lung adenocarcinoma cell lines using Cell Counting Kit-8 (CCK-8), 5-ethynyl-2′-deoxyuridine (EdU) incorporation, Transwell migration and invasion assays, wound-healing assays, and flow cytometry for apoptosis analysis. RNA sequencing identified cell-cycle signaling pathways as the primary targets of rosuvastatin. Analysis of survival curves and differential gene expression between tumor and adjacent non-tumor tissues using public databases, including the Human Protein Atlas, Gene Expression Profiling Interactive Analysis (GEPIA), and Tumor Immune Estimation Resource (TIMER), suggested that polo-like kinase 1 (PLK1) may be a key target mediating the antitumor effects of rosuvastatin. Western blotting and quantitative reverse transcription polymerase chain reaction (qRT-PCR) were used to confirm the differential expression of PLK1 and related cell-cycle proteins in lung adenocarcinoma cells following treatment with different doses of rosuvastatin. Furthermore, rescue experiments with PLK1 knockdown were performed to verify its role in the mechanism of rosuvastatin. A subcutaneous mouse xenograft model was established in vivo to assess the antitumor activity of rosuvastatin via PLK1 inhibition.
Rosuvastatin exerted significant antitumor effects against lung adenocarcinoma both in vitro and in vivo. Mechanistic studies indicated that its anticancer activity is mainly mediated by downregulating PLK1 expression.
By suppressing PLK1 expression, rosuvastatin inhibited cancer cell proliferation, migration, and invasion. These findings support the potential of rosuvastatin as a therapeutic agent for lung cancer, although further studies are needed to confirm its clinical utility.
Graphical Abstract
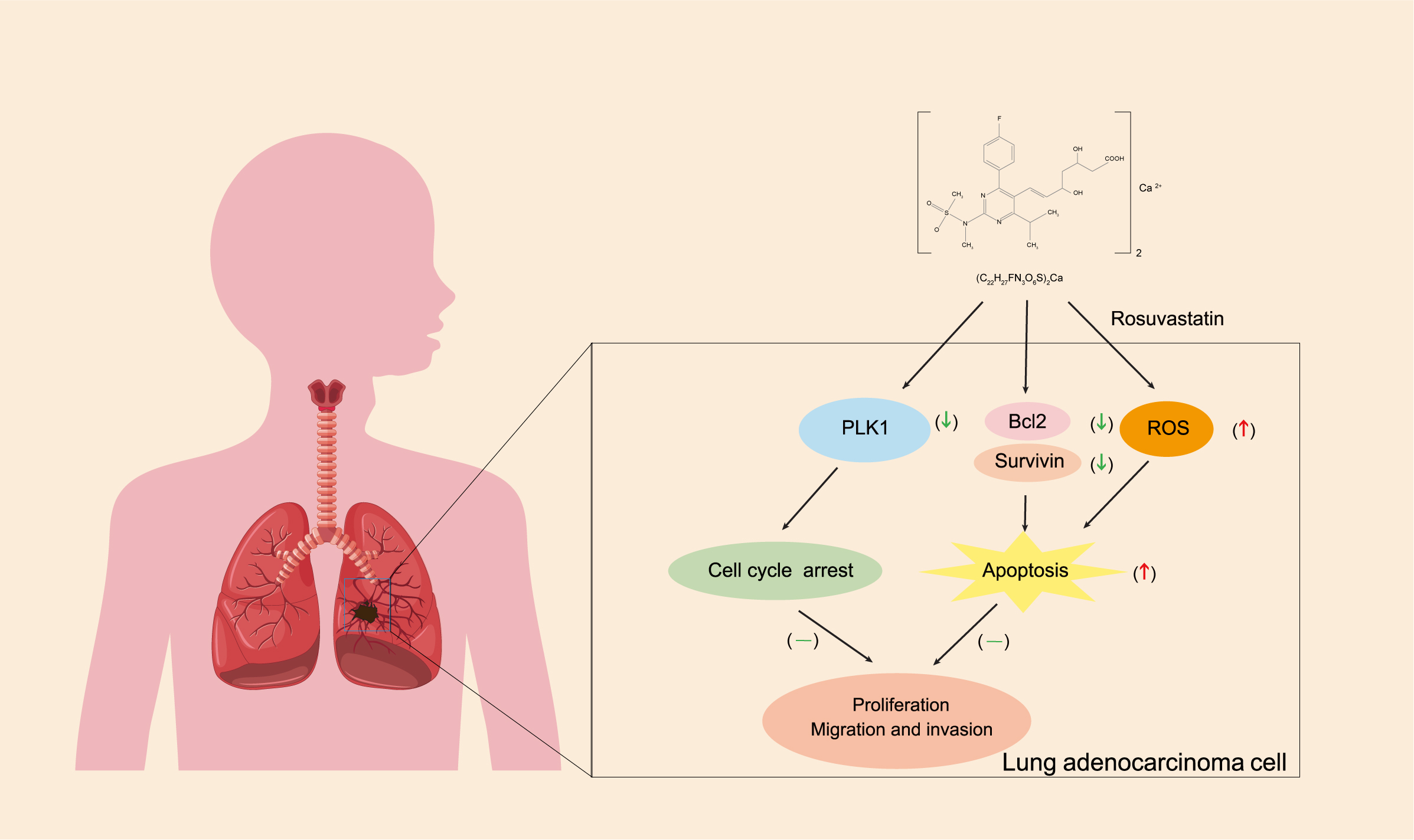
Keywords
- rosuvastatin
- lung adenocarcinoma
- polo-like kinase 1
- cell cycle
- apoptosis
Lung cancer is one of the most common malignancies worldwide and a leading cause of cancer-related mortality [1, 2, 3]. About 85% of instances of lung cancer are non-small cell lung cancer (NSCLC), with lung adenocarcinoma being the most prevalent subtype. The majority of patients are detected at an advanced stage of lung adenocarcinoma, even though surgical resection is an effective treatment for early-stage cases. With a 5-year survival rate of only 24% for advanced-stage lung adenocarcinoma, the outlook is still dismal despite advancements in targeted medicines and immunotherapies [2, 4, 5]. Thus, there is an urgent need to elucidate the mechanisms driving disease progression and explore new therapeutic strategies.
The development of new drugs is a lengthy, complex, and costly process, often spanning decades with increasing challenges and high failure rates. The success rate for cancer drug development remains remarkably low, at approximately 5% [6]. Consequently, the strategy of “drug repurposing”, identifying new therapeutic indications for existing drugs, has gained attention to overcome these limitations. This approach accelerates drug development, reduces costs and risks, and expands treatment options. Drug repurposing bypasses many early-stage research hurdles, making it one of the most efficient and cost-effective strategies for drug discovery. Notable success stories include disulfiram, a long-used alcohol deterrent, which was shown to reduce cancer-related mortality by 34% in a large study of 240,000 cancer patients in Denmark [7]. Similarly, Tan et al. [8] reported that desloratadine, a common antihistamine, significantly suppressed hepatocellular carcinoma growth in vitro and in vivo, suggesting its potential as an anti-cancer agent.
Rosuvastatin calcium, the calcium salt form of rosuvastatin, enhances the drug’s stability and solubility, thereby improving its bioavailability. Rosuvastatin is a third-generation hydrophilic statin with strong protein-binding affinity and minimal lipophilicity. Its primary method is to reduce endogenous cholesterol synthesis by competitively inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase [9]. Statins have been demonstrated to have extensive anti-inflammatory, immunomodulatory, and anti-tumor activities in addition to their everyday use as lipid-lowering medications. Research has shown that rosuvastatin inhibits several malignancies, including pancreatic, and liver cancer [10, 11]. However, its potential anti-cancer role in lung adenocarcinoma remains largely unexplored. Preliminary studies from our research group suggest that rosuvastatin may suppress the progression of lung adenocarcinoma, although the underlying mechanisms remain unclear.
Serine/threonine protein kinase polo-like kinase 1 (PLK1) is a crucial regulator of cell division and mitosis, according to mounting data [12]. PLK1 plays a major role in tumor growth and is aberrantly activated in several cancers, including lung cancer. Because of its function in promoting metastasis and carcinogenesis, it has emerged as a therapeutic target in numerous malignancies [13, 14, 15, 16]. The activation of the epithelial-mesenchymal transition (EMT) and the metastatic progression of NSCLC are linked to PLK1 signaling [17].
As a repurposed clinical drug, the precise anti-tumor mechanisms of rosuvastatin in lung adenocarcinoma remain undefined. Based on preliminary findings, we hypothesize that rosuvastatin exerts its anti-cancer effects by modulating PLK1 expression. The present study aimed to investigate this hypothesis systematically.
Procell Life Science & Technology Co., Ltd. (Wuhan, China) supplied the lung
cancer cell lines A549, H1299, and H838. All cell lines were validated by STR
profiling and tested negative for mycoplasma. These cells were cultured at 37
°C with 5% CO2 in RPMI 1640 (lot: WH0021A021, Procell, Wuhan,
China) supplemented with 10% fetal bovine serum (FBS; Lot. No. 2364724, Gibco,
Carlsbad, CA, USA) and 1% penicillin-streptomycin (Lot. No. 080421211115,
Beyotime, Shanghai, China). Cell viability was assessed using a Cell Counting
Kit-8 (CCK-8; Lot. No. 23002K1018, APExBIO, China). Rosuvastatin calcium was
obtained from Shanghai Yuanye Technology Co., Ltd. (Shanghai, China) with a
purity of
Each well of a 96-well plate received 100 µL of complete RPMI 1640
medium with 10% FBS, and each well was seeded with 3
An EdU kit was acquired from RiboBio (C10310-1, Lot. No. V1104, Guangzhou,
China). After 24 h of treatment with varying concentrations of rosuvastatin,
A549, H1299, and H838 cells were harvested. To ensure a density of 3
A 24-well Transwell plate was used for the migration assay. The lower chamber
received 800 µL of complete RPMI 1640 supplemented with 10% FBS,
while the upper chamber received serum-free RPMI 1640 medium (200
µL). After treating with rosuvastatin after 48 h, lung
adenocarcinoma cells (A549, H1299, or H838) were seeded in the upper chamber at 4
For the invasion assay, 50 µL of Matrigel diluted 1:4 with serum-free RPMI 1640 medium was added to the upper chamber, and allowed to solidify for 2 h at 37 °C. After that, 50 µL of serum-free RPMI 1640 was used to hydrate the Matrigel-coated chamber for 30 min. The subsequent procedures were identical to those described for the migration assay.
Using a sterile 10 µL pipette tip, a straight-line scratch was made
in a six-well plate once the cells attained approximately 80% confluence. After
aspirating the medium, unattached cells were removed from the wells by washing
them with phosphate-buffered saline (PBS). After adding 2 mL of fresh serum-free
medium, the plate was incubated for 15 min at 37 °C with 5% CO2.
Images were acquired at 200
Cell apoptosis was assessed using a Cell Apoptosis Detection Kit (C1065L, Lot.
No. 090921211105, Beyotime, China) according to the instructions outlined by the
manufacturer. Briefly, cells (4
A Reactive Oxygen Species Assay Kit (ID3130, Lot. No. 320A011, Solarbio, Beijing, China) was used to measure intracellular reactive oxygen species (ROS) levels. Cell seeding and drug treatment were performed using the same procedures as described for the apoptosis assay. After treatment, the culture medium were removed, and cells were incubated with 1 mL of serum-free medium containing DCFH-DA (diluted 1:1000, final concentration 10 µM) at 37 °C for 20 min in a CO2 incubator. Before flow cytometric analysis, the cells were washed three times with serum-free medium.
For 48 h, H838 cells were exposed to DMSO or 30 µM rosuvastatin. Following treatment, 1 mL of TRIzol reagent was used to lyse the samples. RNA sequencing was performed by Metware Biotechnology Co., Ltd. (Wuhan, China).
SiRNA sequences that target PLK1 were synthesized by GenePharma Co., Ltd. (Suzhou, China). Lipofectamine 2000 reagent (Invitrogen, MA, USA) was used for transfection according to the manufacturer’s instructions. Before transfection, A549, H1299, and H838 cells were seeded to achieve 50–60% confluence on the day of transfection. For transfection, two 1.5 mL Eppendorf tubes were prepared: 250 µL of Opti-MEMI and 5 µL of Lipofectamine 2000 were in Tube A, and 250 µL of Opti-MEMI and 5 µL of siRNA were in Tube B. After 5 min of room temperature incubation, both tubes were mixed and allowed to stand for a further 20 min. Next, 500 µL of the transfection mixture was added to 1.5 mL of medium (serum-free) and carefully mixed. After removing the culture medium from the six-well plate and performing two PBS washes, 2 mL of the prepared transfection mixture was added. Before the subsequent studies, the cells were cultured for an additional 48 h after the medium were changed to a complete serum-containing medium at 6 h. The specific interfering sequences employed for PLK1 knockdown are listed in Table 1.
| Interference sequence | (Sense 5′-3′) | (Anti-sense 5′-3′) |
| Si-NC | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
| Si-PLK1-1 | CCUCCGGAUCAAGAAGAAUTT | AUUCUUCUUGAUCCGGAGGTT |
| Si-PLK1-2 | GGUAUCAGCUCUGUGAUAATT | UUAUCACAGAGCUGAUACCTT |
| Si-PLK1-3 | GCGUGCAGAUCAACUUCUUTT | AAGAAGUUGAUCUGCACGCTT |
| Si-PLK1-4 | GGUGUAUCAUGUAUACCUUTT | AAGGUAUACAUGAUACACCTT |
Following a 48 h treatment with different doses of rosuvastatin in A549, H1299, and H838 cells, total protein was extracted, and Western blotting was performed. The following antibodies were employed in this investigation: anti-CDK1 (abs159273, rabbit, 1:1000, Absin); anti-CDK2 (10122-1-AP, rabbit, 1:1000, Proteintech); anti-cyclin B1 (55004-1-AP, rabbit, 1:5000, Proteintech); anti-cyclin A2 (18202-1-AP, rabbit, 1:5000, Proteintech); anti-GAPDH (60004-1-Ig, mouse, 1:5000, Proteintech); anti-cyclin D1 (AF0931, rabbit, 1:1000, Affinity); anti-P21 (S0B2351, rabbit, 1:1000, Starter); anti-PLK1 (S0B0035, rabbit, 1:10,000, Starter); anti-Bcl-2 (S0B2181, rabbit, 1:1000, Starter); anti-survivin (S0B6183, rabbit, 1:1000, Starter); anti-rabbit secondary antibody (98164S, goat, 1:5000, Cell Signaling Technology); anti-mouse secondary antibody (91186S, goat, 1:5000, Cell Signaling Technology).
Male BALB/c nude mice (3–5 weeks old, weighing 18–20 g) were purchased and
kept in a specified pathogen-free (SPF) environment. A549 cells (2.5
The Human Protein Atlas, GEPIA, and TIMER databases were used to validate differential PLK1 expression between tumor and adjacent normal tissues in patients with human lung adenocarcinoma. Additionally, survival analysis was performed using these platforms.
Every experiment was conducted three times. The statistical significance between
the two groups was evaluated using an unpaired Student’s t-test. The
mean
The chemical structure of rosuvastatin is shown in Fig. 1A. A549, H1299, and H838 lung adenocarcinoma cells were treated with rosuvastatin at various doses, and cell viability, cytotoxicity, and growth inhibition were assessed using the CCK-8 assay. The results demonstrated that rosuvastatin reduced cell viability in a dose-dependent way. Following treatment for 72 h, the IC50 values for A549, H1299, and H838 cells were 159.1 µM, 150.4 µM, and 47.5 µM, respectively (Fig. 1B). To further evaluate its effect on proliferation, an EdU assay was performed. The results showed that rosuvastatin treatment significantly decreased the number of EdU-positive A549, H1299, and H838 cells (Fig. 1C–E). Collectively, these findings indicate that rosuvastatin effectively suppresses the proliferation of lung adenocarcinoma cells.
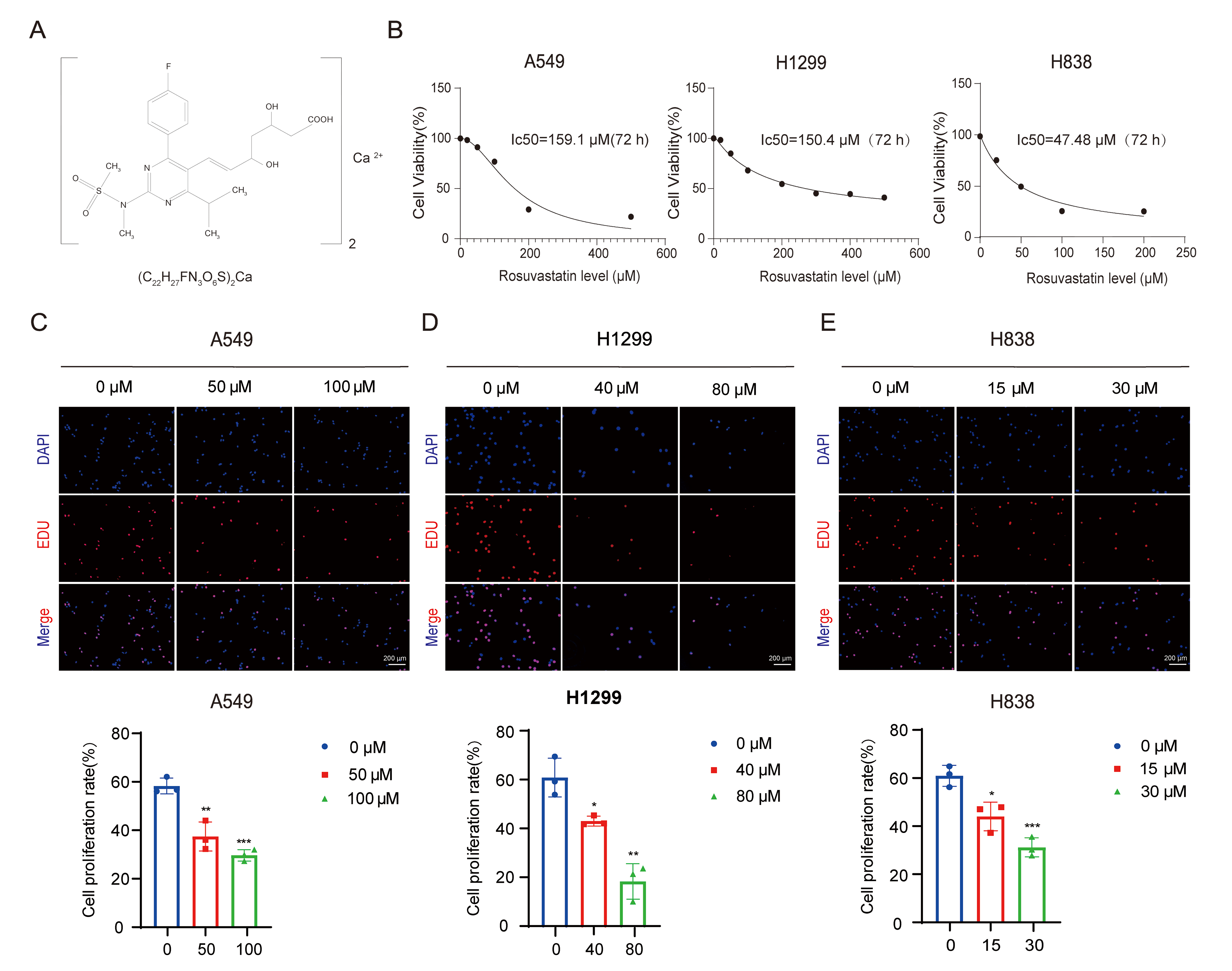 Fig. 1.
Fig. 1.
Rosuvastatin inhibits the proliferation of lung
adenocarcinoma cells. (A) Chemical structure of rosuvastatin. (B) The effect of
rosuvastatin on the proliferation of the A549, H1299, and H838 human lung
adenocarcinoma cell lines was assessed using the CCK-8 assay. (C–E) EdU uptake
was used to assess the frequency of EdU-positive cells with or without
rosuvastatin treatment. The drug concentrations for each cell line were as
follows: A549 (0, 50, and 100 µM); H1299 (0, 40, and 80 µM); and H838
(0, 15, and 30 µM). A 48-hour treatment time point was used for all cell
lines. Scale bar = 200 µm. *p
Transwell migration and invasion assays revealed that rosuvastatin significantly suppressed the migratory and invasive abilities of A549, H1299, and H838 cells in a dose-dependent manner (Fig. 2A–C). The wound-healing assay demonstrated that as the drug concentration increased, wound closure was significantly delayed in A549, H1299, and H838 cells, further confirming the anti-migratory effects of rosuvastatin (Fig. 2D–F). These results show that rosuvastatin suppressed lung adenocarcinoma cell invasion and migration.
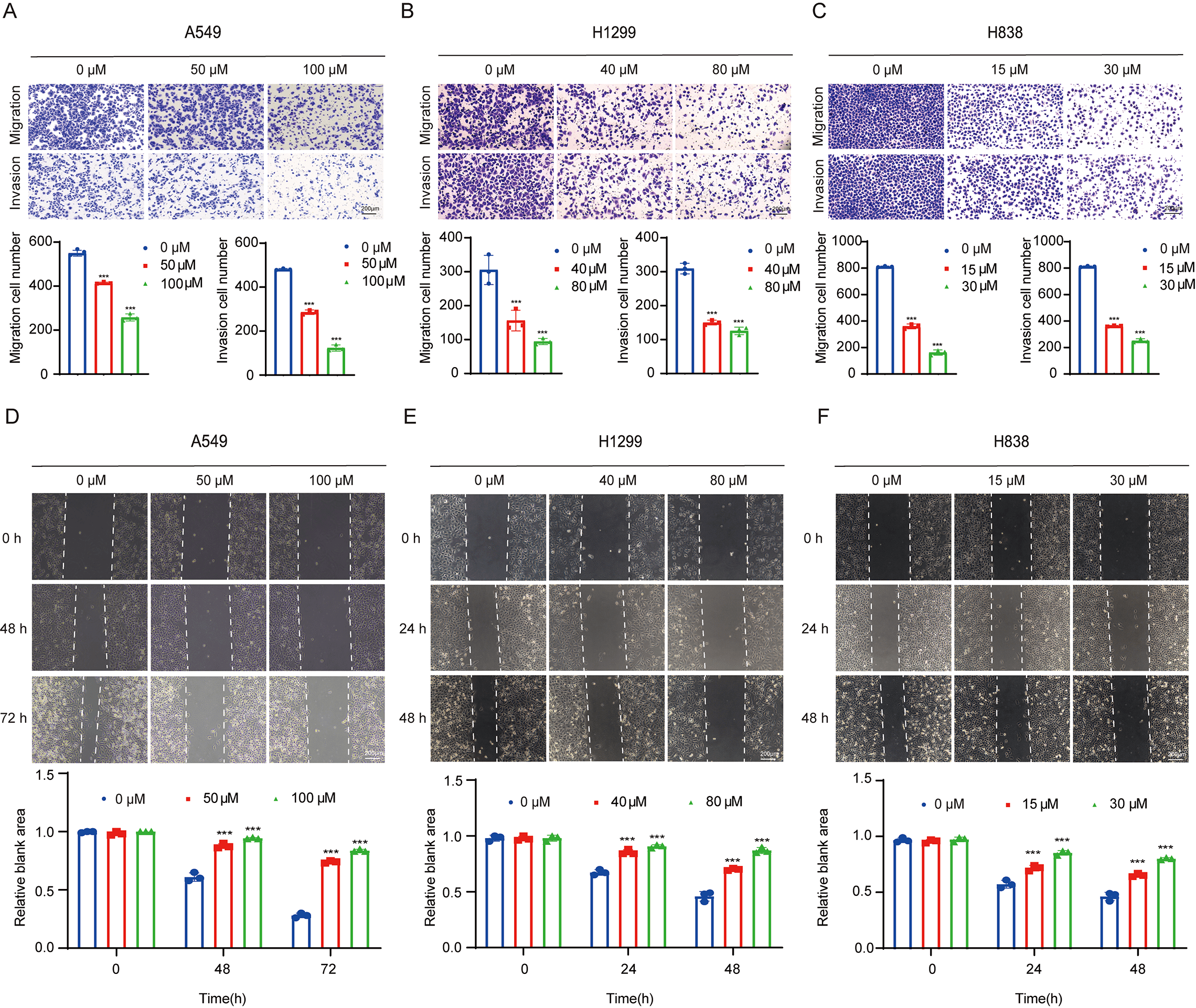 Fig. 2.
Fig. 2.
Rosuvastatin inhibits the migration and invasion of lung
adenocarcinoma cells. (A–C) Transwell migration and invasion assays assessing
the changes in the migration and invasion abilities of A549, H1299, and H838
cells after treatment with different concentrations of rosuvastatin. The drug
concentrations for each cell line were as follows: A549 (0, 50, and 100
µM); H1299 (0, 40, and 80 µM); and H838 (0, 15, and 30 µM). A
48-hour treatment time point was used for all cell lines. Scale bar = 200
µm. (D–F) Wound-healing assay-based evaluation of the effects of different
concentrations of rosuvastatin on the migration and invasion of A549, H1299, and
H838 cells. The drug concentrations and treatment timing were the same as in
(A–C). Scratch wounds were imaged and measured at defined time points for each
cell line: A549 (0, 48, and 72 h); H1299 (0, 24, and 48 h); and H838 (0, 24, and
48 h). Scale bar = 200 µm. ***p
The pro-apoptotic effects of rosuvastatin were evaluated by flow cytometry (Fig. 3A–C), and Western blotting was used to assess apoptosis-related protein expression. The findings demonstrated that in A549, H1299, and H838 cells, rosuvastatin administration resulted in the dose-dependent downregulation of Bcl-2 and survivin (Fig. 3D–F). Moreover, rosuvastatin treatment significantly increased ROS levels in these cells (Fig. 3G–I). This suggests that rosuvastatin may induce apoptosis in lung adenocarcinoma cells, at least in part, by promoting oxidative stress. Overall, our data show that rosuvastatin can trigger apoptosis and inhibit the progression of lung adenocarcinoma.
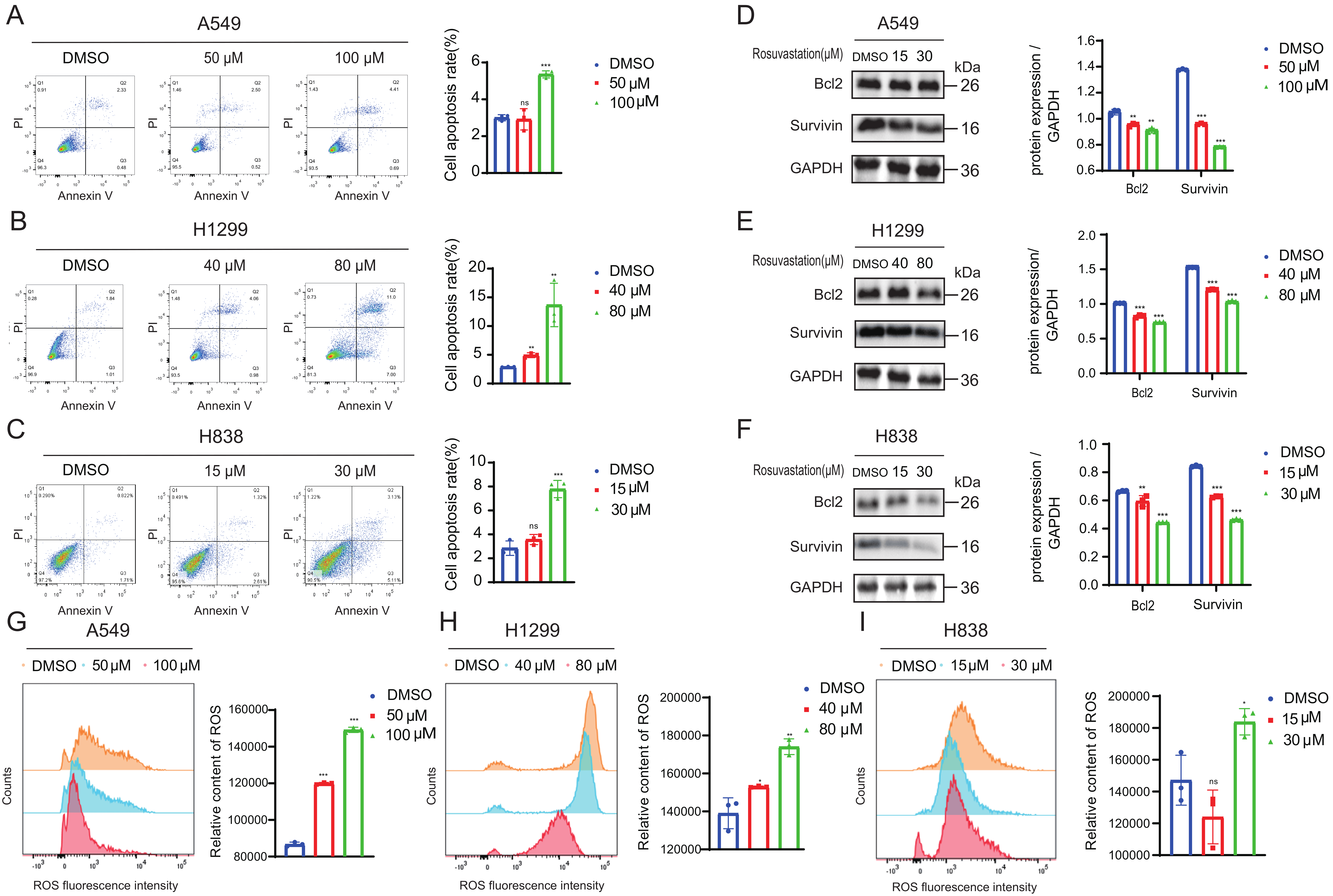 Fig. 3.
Fig. 3.
Rosuvastatin promotes apoptosis in lung adenocarcinoma cells. (A–C) Flow cytometry-based quantification of the frequencies of apoptotic A549,
H1299, and H838 cells following treatment with various rosuvastatin
concentrations for 48 h. (D–F) Changes in apoptosis-related Bcl-2 and survivin
protein expression in lung adenocarcinoma cells following treatment with various
concentrations of rosuvastatin for 48 h, as detected by Western blotting. (G–I)
Effects of treatment with different concentrations of rosuvastatin for 48 h on
reactive oxygen species (ROS) levels in A549, H1299, and H838 cells. ns, no
significance; *p
RNA sequencing (RNA-seq) analysis of H838 cells treated with 30 µM
rosuvastatin for 48 h was conducted further to explore the molecular mechanisms
underlying rosuvastatin’s effects. A total of 1700 differentially expressed genes
(DEGs) were discovered using a criterion of FDR
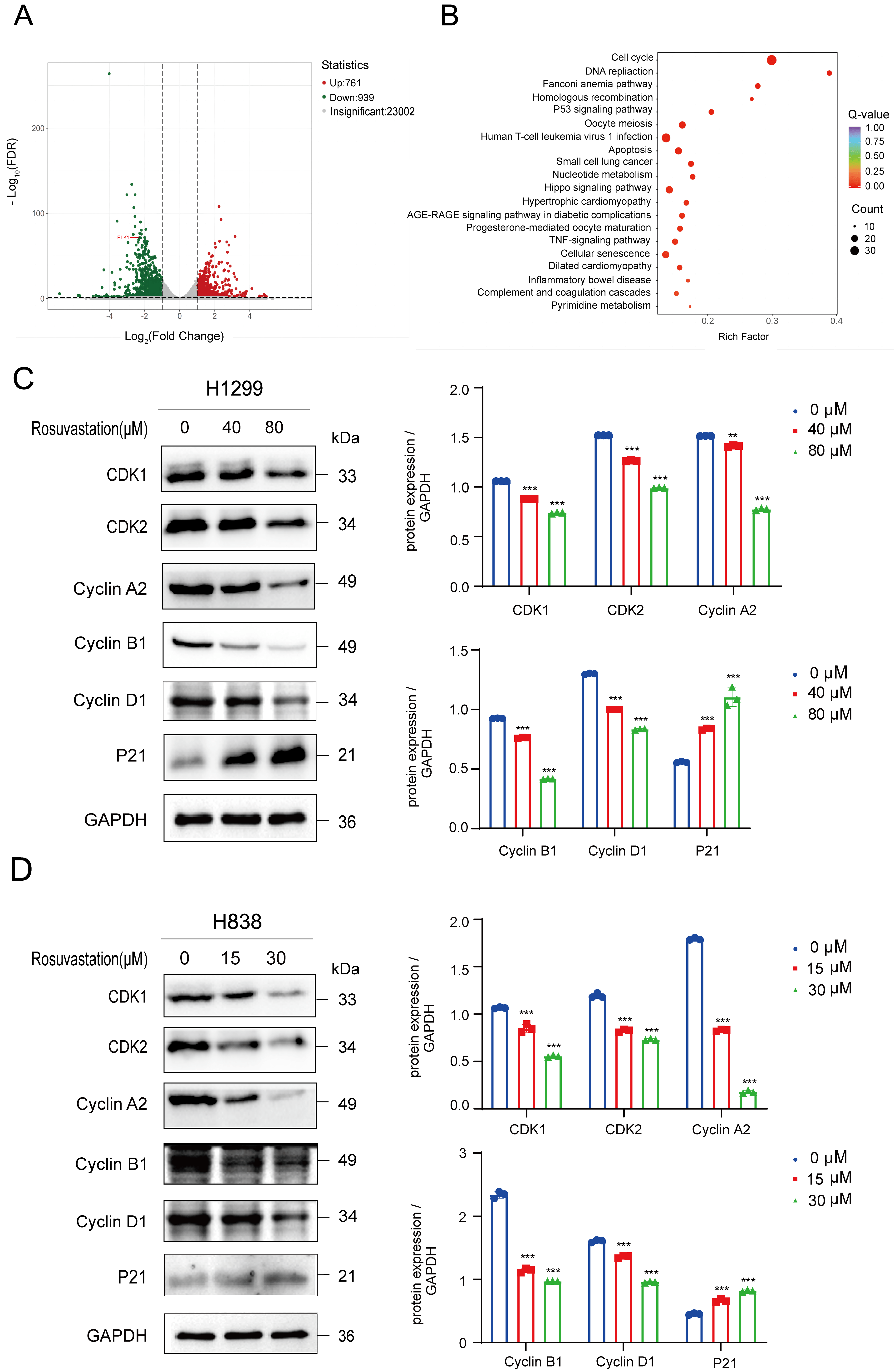 Fig. 4.
Fig. 4.
Rosuvastatin modulates the cell cycle in lung adenocarcinoma
cells to exert anti-tumor effects. (A) A volcano plot highlighting the
differentially expressed genes (DEGs) identified by RNA sequencing (RNA-seq) in
the transcriptional profiling of rosuvastatin-treated H838 cells. (B) Gene
ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional
enrichment analyses of DEGs identified by RNA-seq. (C,D) CDK1, CDK2, cyclin A2,
cyclin B1, cyclin D1, and P21 protein levels were detected in H1299 and H838
cells by Western blotting before and after rosuvastatin treatment. **p
We then examined the transcriptional patterns of rosuvastatin-treated H838 cells using these RNA-seq results to find DEGs associated with the cell cycle. Fig. 5A displays the heatmap that was produced. This conclusion was further supported by immunohistochemical data from the Human Protein Atlas, which showed elevated PLK1 expression levels in lung cancer tissues (Fig. 5B). Furthermore, analysis using the GEPIA and TIMER databases revealed that PLK1 was significantly overexpressed in lung adenocarcinoma tissues and negatively correlated with patient survival (Fig. 5C–E). We treated A549, H1299, and H838 cells with varying concentrations of rosuvastatin for 48 h to further explore its impact on PLK1. Next, we collected protein and total RNA for Western blot and qRT-PCR analysis, respectively. The findings demonstrated that rosuvastatin significantly reduced PLK1 expression at the mRNA and protein levels (Fig. 5F–I).
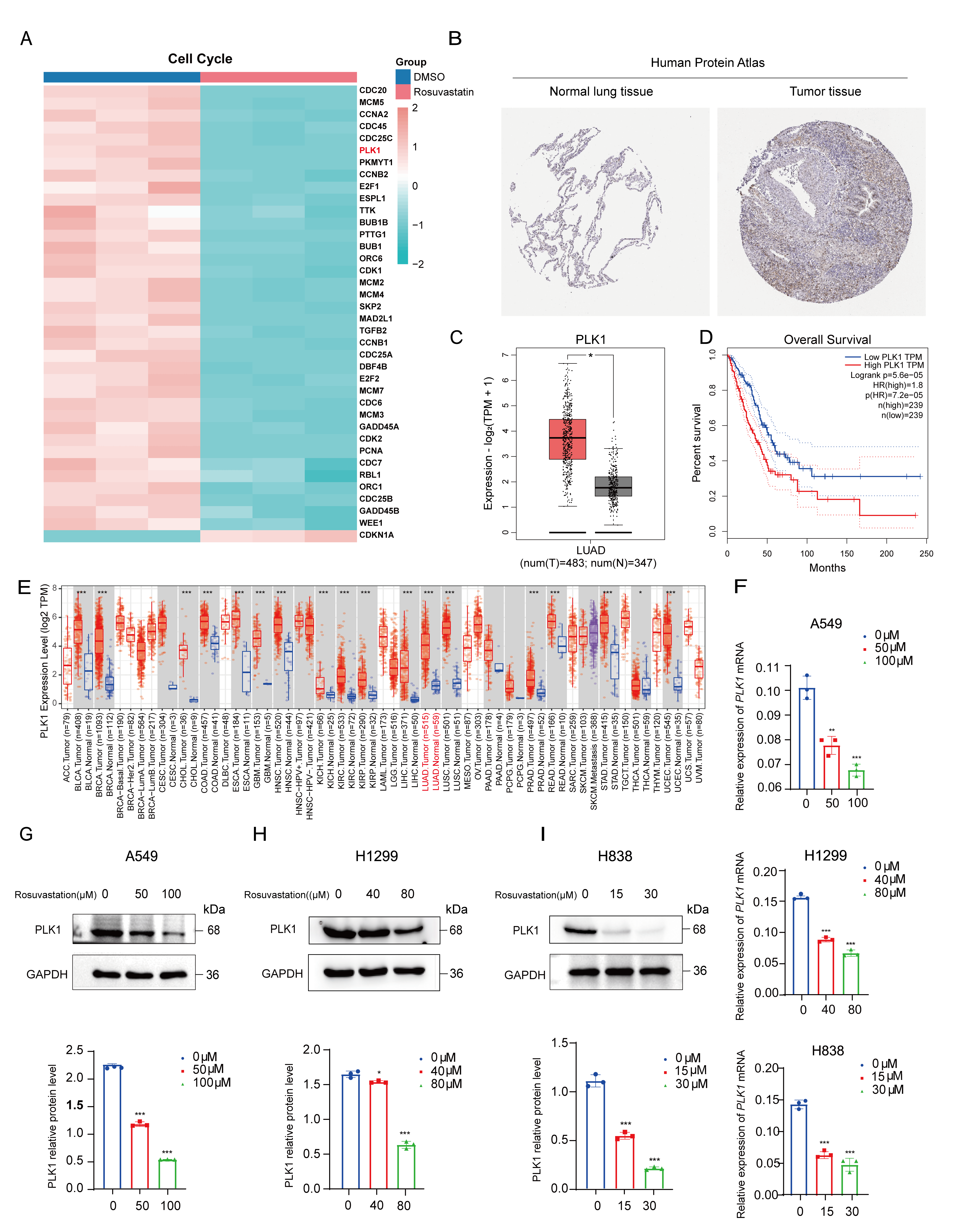 Fig. 5.
Fig. 5.
PLK1 may be a key target for rosuvastatin’s anti-cancer
effects. (A) Heatmap showing DEGs related to the cell cycle identified by
RNA-seq analysis of H838 cells treated with rosuvastatin. (B) Immunohistochemical
staining from the Human Protein Atlas showing PLK1 expression in lung
adenocarcinoma and adjacent normal tissues. (C,D) GEPIA database analysis
results show that PLK1 is significantly overexpressed in lung adenocarcinoma
patients (C) and is negatively correlated with survival (D). (E) TIMER database
analysis results show differential PLK1 expression between lung adenocarcinoma
tissues and adjacent normal tissues. (F) qRT-PCR analysis of PLK1 mRNA levels in
A549, H1299, and H838 cells treated with increasing concentrations of
rosuvastatin for 48 h. The concentrations used were 0, 50, and 100 µM for
A549; 0, 40, and 80 µM for H1299; and 0, 15, and 30 µM for H838.
(G–I) Western blot analysis of PLK1 protein expression in A549 (G), H1299 (H),
and H838 (I) cells treated with the indicated concentrations of rosuvastatin for
48 h. *p
To investigate whether PLK1 is a key mediator of rosuvastatin’s anti-cancer actions, we used RNA interference to inhibit PLK1 expression in A549 and H838 cells. Western blotting and qRT-PCR were used to confirm the knockdown’s effectiveness (Fig. 6A–D). Two highly effective siRNA sequences were selected for PLK1 knockdown in A549 and H838 cells. PLK1 knockdown significantly reduced the growth of these lung adenocarcinoma cells, as shown by EdU staining. Furthermore, rosuvastatin and PLK1 knockdown showed synergistic effects, resulting in an even greater reduction in proliferation (Fig. 6E, F). Compared with the control group, the PLK1-silenced lung adenocarcinoma cells showed a significant decrease in their ability to migrate and invade in Transwell assays. Although rosuvastatin treatment further inhibited migration and invasion, its effects were partially attenuated in PLK1-knockdown cells, suggesting a potential additive interaction (Fig. 6G,H). Taken together, these findings demonstrate rosuvastatin inhibits lung adenocarcinoma cell proliferation, migration, and invasion by downregulating PLK1. This demonstrates that PLK1 is an essential mediator of rosuvastatin’s anti-cancer effects.
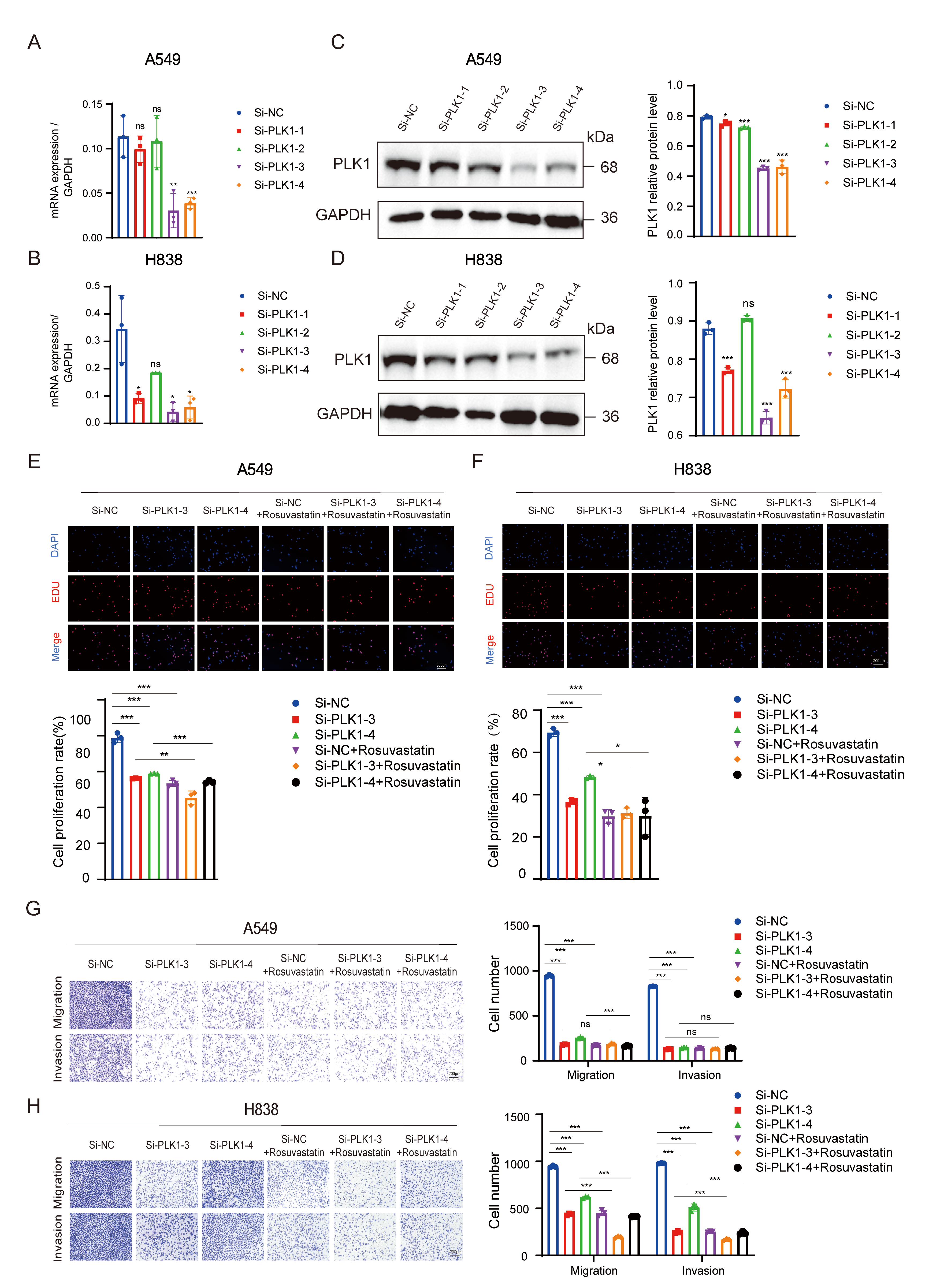 Fig. 6.
Fig. 6.
PLK1 knockdown reduces the proliferation, migration, and
invasion of lung adenocarcinoma cells. (A–D) qRT-PCR and Western blot
validation of PLK1 knockdown efficiency in A549 and H838 cells transfected with
PLK1-targeting siRNA. (E,F) An EdU assay-based analysis of the effect of PLK1
knockdown and rosuvastatin treatment on the proliferation of lung adenocarcinoma
cells. Scale bar = 200 µm. (G,H) Transwell migration and invasion assays
of A549 (G) and H838 (H) cells after PLK1 knockdown, rosuvastatin treatment, or
their combination. Scale bar = 200 µm. ns, no significance; *p
To evaluate rosuvastatin’s in vivo anti-tumor activities, a BALB/c nude mouse xenograft model was established (Fig. 7A). After the tumors grew to a volume of around 200 mm3, the mice were randomly divided into three groups (n = 5 per group) and given daily intraperitoneal injections of vehicle control, 10 mg/kg rosuvastatin, or 20 mg/kg rosuvastatin. The xenograft tumors were removed, and the mice were euthanized after 12 days of treatment. The results demonstrated that rosuvastatin significantly inhibited lung adenocarcinoma cell growth in vivo (Fig. 7B–D), with no significant changes in body weight observed among the treated mice (Fig. 7E). Western blotting and immunohistochemical staining analyses of the excised tumor tissues were conducted to further investigate the effect of rosuvastatin on PLK1 expression in xenograft tumors. Both studies confirmed the significant downregulation of PLK1 at the protein level following rosuvastatin treatment (Fig. 7F,G).
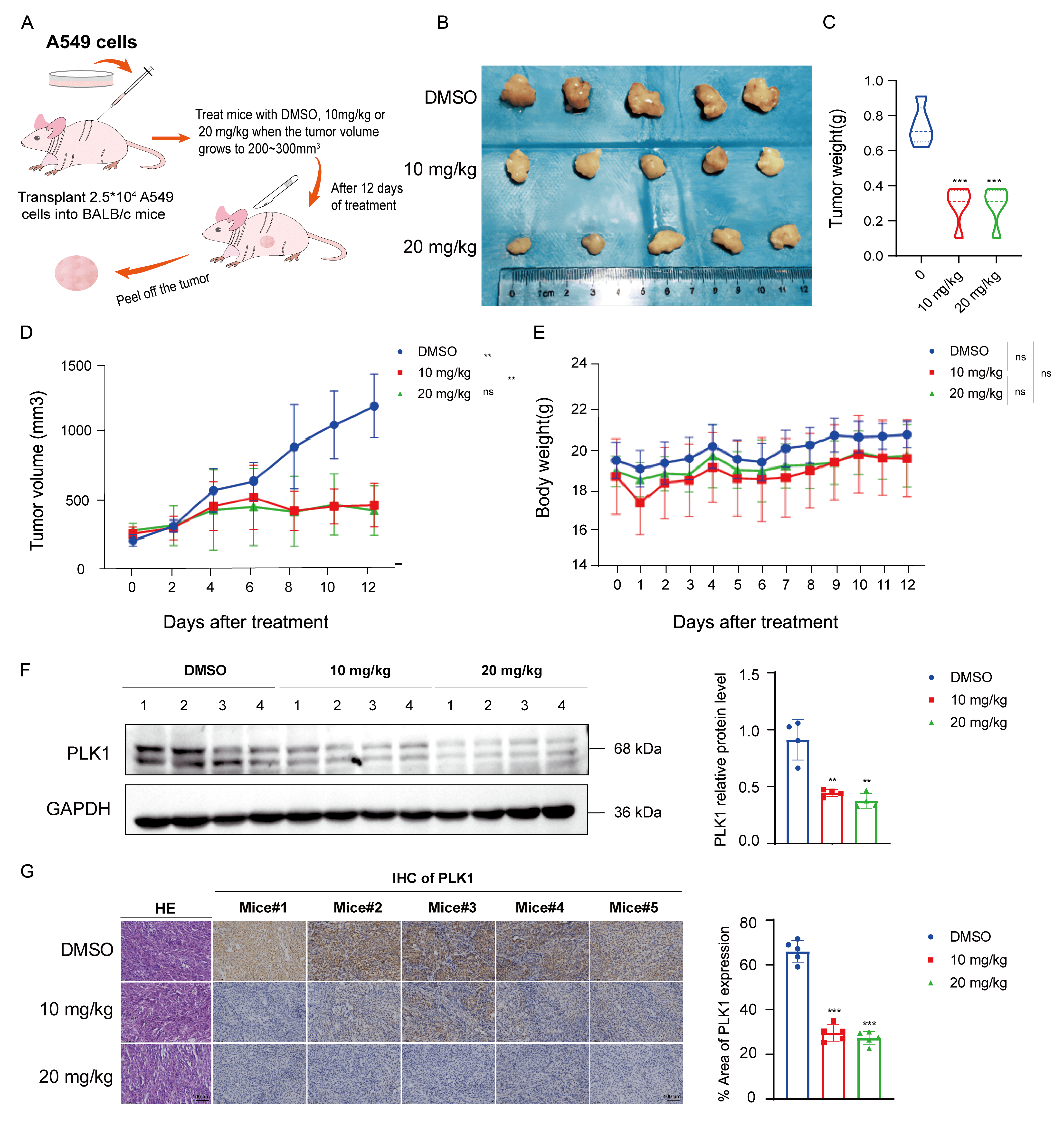 Fig. 7.
Fig. 7.
Rosuvastatin inhibits lung adenocarcinoma tumor growth
in vivo. (A) Schematic overview of the establishment of a xenograft
tumor model in nude mice. Created using Adobe Illustrator 2024. (B) Images of
excised tumors after treatment (n = 5). (C) Endpoint tumor weights. (D) Tumor
volume was monitored every 2 days during the treatment period. (E) Changes in
murine body weight were measured every 2 days during the treatment period. (F)
Western blotting analysis of PLK1 protein expression levels in tumors from the
control (DMSO), low-dose treatment (rosuvastatin, 10 mg/kg), and high-dose
treatment (rosuvastatin, 20 mg/kg) groups. (G) Immunohistochemical staining of
PLK1 protein expression in tumors from different treatment groups and hematoxylin
and eosin (H&E) staining of lung adenocarcinoma tissue. Scale bar = 100
µm. ns, no significance. **p
Lung adenocarcinoma is a primary source of cancer-related morbidity and mortality, despite the availability of many therapeutic methods, including chemotherapy, molecular targeted therapy, surgical resection, radiation, and immunotherapy. More efficient treatment options are desperately needed to enhance patient outcomes because of their high prevalence and poor prognosis.
Rosuvastatin, a commonly used lipid-lowering drug, has revealed potential anti-cancer benefits in several malignancies. Clinical studies have reported that rosuvastatin enhances survival outcomes when used concurrently with chemoradiotherapy or as postoperative treatment for cancers such as nasopharyngeal carcinoma [18], breast cancer [19], and esophageal squamous cell carcinoma [20]. Furthermore, rosuvastatin has been shown to inhibit lung adenocarcinoma progression by downregulating cyclin A2 and enhancing sensitivity to chemotherapy drugs such as cisplatin and gemcitabine [21].
The results of our study demonstrated that rosuvastatin effectively inhibited lung adenocarcinoma cell proliferation, migration, and invasion in vitro while also promoting apoptosis by increasing ROS levels and downregulating Bcl-2 and survivin expression. In addition, rosuvastatin inhibited tumor growth in xenograft models without causing significant adverse effects, according to in vivo studies. These findings reveal that rosuvastatin may be a useful treatment for lung cancer, although the underlying mechanisms remain unclear. PLK1, PLK2, PLK3, PLK4, and PLK5 are members of the highly conserved serine/threonine kinase family, of which PLK1 has been the subject of the most research. PLK1 demonstrates cell cycle-dependent expression and activity and regulates mitosis. In the G0, G1, and S phases, PLK1 expression is comparatively low; it rises in the G2 phase and peaks in the M phase [22, 23, 24]. PLK1 is abnormally regulated in several human malignancies, and its overexpression is frequently linked to a poor prognosis for patients. For example, PLK1 increased cancer stemness and induced EMT in colorectal cancer, according to Poyil et al. [25]. Similarly, Wang et al. [26] demonstrated that inhibiting PLK1 expression in hepatocellular carcinoma cells induced G2/M phase arrest and promoted tumor cell apoptosis.
Furthermore, PLK1 has been identified as a crucial factor in lung adenocarcinoma development through bioinformatics and machine learning analyses [27]. PLK1 is highly expressed in lung cancer and has a negative correlation with patient prognosis, according to our analysis of the Human Protein Atlas, GEPIA, and TIMER public databases. According to our experiments, rosuvastatin reduces PLK1 mRNA and protein levels in the cell lines A549, H1299, and H838. To confirm PLK1’s functional role as a mediator of rosuvastatin’s anti-cancer actions, we performed PLK1 knockdown experiments in A549 and H838 cells. The findings showed that in both cell lines, PLK1 knockdown significantly lowered invasion, migration, and proliferation. These results provide compelling evidence that PLK1 mediates the therapeutic effects of rosuvastatin in lung cancer.
The cell cycle signaling pathway was the most significantly enriched pathway by GO and KEGG enrichment analyses of our RNA-seq data. This suggests that rosuvastatin primarily exerts its effects on lung adenocarcinoma cells by disrupting cell cycle progression. Cyclin-dependent kinases (CDKs) and cyclins play direct regulatory roles in cell cycle control. Their abnormal expression can disrupt the initiation, progression, and termination of the cell cycle, potentially leading to excessive cell proliferation and reduced apoptosis, thereby accelerating tumorigenesis. CDKs are key regulators that drive the eukaryotic cell cycle in a precise and orderly manner. Among them, CDK1 has shown to be an important regulator of the G2/M phase transition, and its overexpression has been shown to promote G2/M progression and accelerate tumor cell growth [28, 29]. CDK2, another essential member of the CDK family, regulates the G1/S and S/G2 phase transitions. However, CDK2 itself is not intrinsically active; its protein levels remain relatively stable throughout the cell cycle, and the periodic fluctuations in cyclin levels control its activity. CDK2 interacts with cyclin E to facilitate G1/S transition and with cyclin A to promote entry into the G2 phase [30].
Cyclin A2 is a highly conserved cyclin expressed in nearly all human tissues [31]. According to research, cyclin A2 is overexpressed in several malignancies and could be a useful prognostic and diagnostic indicator for breast, lung, esophageal, and colorectal cancers (CRC). It is believed to bind to CDK2, controlling the G2/M transition and S-phase progression, thereby promoting tumor cell growth [32, 33, 34]. Similarly, cyclin B1 overexpression enhances the G2/M transition, which can lead to uncontrolled cell proliferation and malignant transformation [35]. Yuan et al. [36] reported that inhibiting cyclin B1 expression induced G2/M phase arrest, suppressed tumor growth, and promoted apoptosis. Cyclin D1 is another critical positive regulator of the cell cycle. It interacts with CDKs to drive G1/S transition by forming a cyclin D1-CDK4 complex, which regulates CDK4-mediated phosphorylation of the retinoblastoma protein. This phosphorylation event releases E2F transcription factors, which enter the nucleus and activate the transcription of genes required for cell cycle progression. Dysregulation of this pathway contributes to uncontrolled cell proliferation and malignant transformation, playing a significant role in tumor development [37, 38, 39, 40, 41]. In addition to its role as a cell cycle inhibitor, P21 functions as a broad-spectrum regulator [42] and is involved in senescence induction, tumor suppression, apoptosis, differentiation, DNA repair, transcriptional regulation, and cell migration [43]. P21 induction has been implicated in both cancer cell proliferation and tumorigenesis [44]. To learn more about rosuvastatin’s effects on cell cycle regulation, we conducted Western blot analysis on H1299 and H838 cells treated with different concentrations of the drug for 48 h. The findings showed that P21 expression was significantly elevated, while CDK1, CDK2, cyclin A2, cyclin B1, and cyclin D1 expression levels were significantly decreased. These results suggest that rosuvastatin inhibits the growth of lung cancer cells by disrupting their cell cycle.
To assess rosuvastatin’s anti-tumor properties in vivo, a subcutaneous tumor model in male BALB/c nude mice was established while employing A549 cells. The findings showed that the tumor weight and size in the rosuvastatin-treated groups were considerably lower than those in the control group. In particular, during treatment, there were no significant differences in body weight between the treated and control groups, indicating that rosuvastatin did not have any harmful side effects. PLK1 protein levels were considerably lower in the rosuvastatin-treated group, according to Western blot examination of PLK1 expression in tumor tissues from the control and rosuvastatin-treated groups. Moreover, the reduction in PLK1 expression was dose-dependent, with higher drug concentrations leading to a more pronounced decrease. Immunohistochemical staining further confirmed these findings, supporting the hypothesis that rosuvastatin exerts its anti-tumor effects by downregulating PLK1 expression.
Several limitations of this study should be noted for the correct interpretation of the results. First, although our findings confirmed that rosuvastatin exerts anti-tumor effects by downregulating PLK1 and interfering with cell cycle progression, the present study only focused on the PLK1–cell cycle signaling axis as the core mechanism. As a multi-functional repurposed drug, rosuvastatin also exerts important regulatory effects on lipid metabolism, inflammation, oxidative stress, and tumor immune microenvironment. Whether these biological processes are also involved in its anti-lung adenocarcinoma activity and whether there is crosstalk between different pathways remain unclear. Therefore, the comprehensive molecular network underlying the anti-tumor effects of rosuvastatin needs to be further explored and supplemented. Second, this study lacks systematic pharmacokinetic and pharmacodynamic (PK/PD) evaluations in vivo. Consequently, the translatability of these preclinical doses to clinical medication regimens remains uncertain, and the optimal therapeutic window and safety threshold of rosuvastatin in the treatment of lung adenocarcinoma have not been established. Third, the present study only explored the monotherapy effect of rosuvastatin, without evaluating its combination value with mainstream clinical treatments for lung adenocarcinoma. At present, advanced lung adenocarcinoma is mainly treated with targeted therapy, immune checkpoint inhibitors, and chemotherapy. Whether rosuvastatin can synergize with these therapies to enhance anti-tumor efficacy, reduce toxic side effects, or overcome drug resistance has not been investigated. This missing evidence limits the formulation of clinical combination strategies and the practical application prospects of rosuvastatin as a repurposed drug. Taken together, these limitations suggest that further mechanism exploration, standardized PK/PD research, and combination therapy verification are needed to promote the clinical transformation and application of rosuvastatin in lung adenocarcinoma.
In conclusion, this study provides initial evidence of rosuvastatin’s anti-lung adenocarcinoma properties in both in vitro and in vivo models. Our findings reveal, for the first time, that rosuvastatin suppresses the proliferation, migration, and invasion of lung adenocarcinoma cells by downregulating PLK1 expression. Furthermore, by inducing cell cycle arrest and promoting apoptosis, rosuvastatin effectively inhibited the growth of lung adenocarcinoma. Collectively, these results suggest that rosuvastatin may be a therapeutic option for patients with lung adenocarcinoma. To fully understand the molecular mechanisms underlying its anti-cancer properties and to investigate its potential for clinical applicability, further research is needed.
CDKs, cyclin-dependent kinases; DMSO, dimethyl sulfoxide; EMT, epithelial-mesenchymal transition; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; IHC, immunohistochemistry; NSCLC, non-small cell lung cancer; PLKs, polo-like kinases; ROS, reactive oxygen species.
Raw data will be made available on reasonable request.
JY: Designed the research, writing—original draft, and data curation. JZ: Designed the research, investigation, writing—review & editing, and funding acquisition. YG: Designed the research, investigation, writing—review. WW: Performed the research. XZ: Writing—review & editing and data curation. CL: Writing—review & editing and data curation. YY: Performed the research, and analyzed the data. YC: Performed the research, and analyzed the data. JL: Data curation. LG: Participated in animal experiments. JH: Participated in designing the research study, and funding acquisition. ZL: Participated in designing the research study. DS: Designed the research, supervision, and writing—review & editing. YZ: Designed the research, Supervision, writing—review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were performed in compliance with the ARRIVE guidelines and approved by the Animal Ethics Committee of Soochow University (registration number: 202312A016, ethical approval date: 2023–12–05).
Not applicable.
This work was supported by grants from the Science and Technology Plan Project of Suzhou (SKY2023160) and Jiangsu Provincial Medical Key Discipline (ZDXK202201).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.