










1 Department of Critical Care Medicine, Suzhou Hospital of Integrated Traditional Chinese and Western Medicine, 215101 Suzhou, Jiangsu, China
2 Department of Emergency, Suzhou Hospital of Integrated Traditional Chinese and Western Medicine, 215101 Suzhou, Jiangsu, China
3 Department of Central Laboratory, Suzhou Hospital of Integrated Traditional Chinese and Western Medicine, 215101 Suzhou, Jiangsu, China
†These authors contributed equally.
Abstract
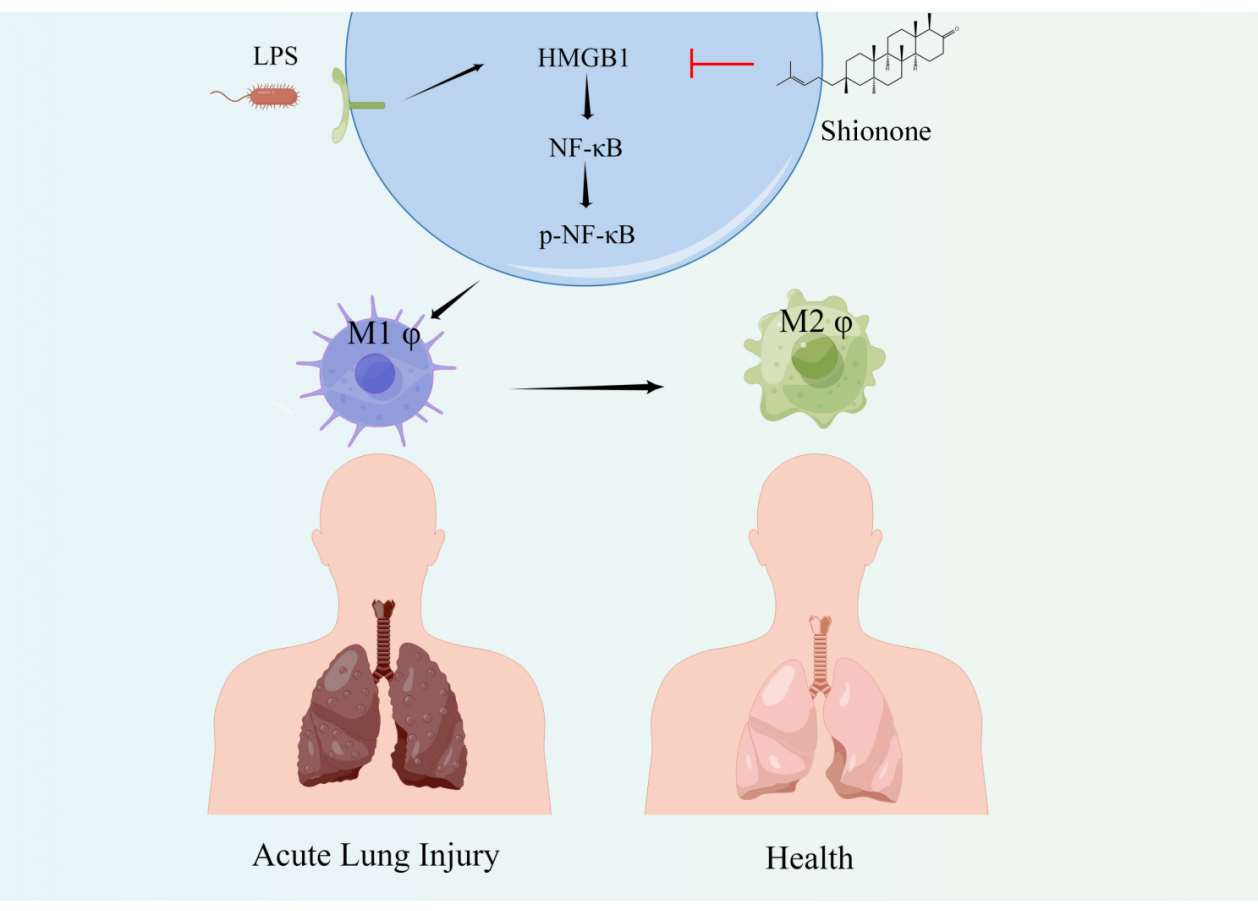
Sepsis-induced acute lung injury (ALI) poses a significant therapeutic challenge due to the lack of effective treatments. Shionone (SHI), a compound known for its anti-inflammatory properties, was investigated for its potential to mitigate ALI by modulating macrophage polarization, a key process in the inflammatory response. The underlying mechanism was also explored.
We established lipopolysaccharide (LPS)-induced models of ALI in mice and RAW264.7 cells. The protective effects of SHI were assessed in vivo using lung histopathology (hematoxylin and eosin [H&E] staining) and the lung wet-to-dry weight ratio. Cell viability was assessed using a Cell Counting Kit-8 (CCK-8) assay. The levels of inflammatory cytokines (interleukin-6 [IL-6], interleukin-1 beta [IL-1β], tumor necrosis factor-alpha [TNF-α], granulocyte-macrophage colony-stimulating factor [GM-CSF], Interleukin-10 [IL-10], transforming growth factor-beta 1 [TGF-β1]) and polarization markers (inducible nitric oxide synthase [iNOS], arginase-1 [Arg1]) were quantified by enzyme-linked immunosorbent assay [ELISA] and real-time quantitative PCR. The expression of key proteins in the high-mobility group box 1 (HMGB1)/nuclear factor κ B (NF-κB) pathway (HMGB1, toll-like receptor 4 [TLR4], myeloid differentiation primary response 88 [MyD88], NF-κB p65) was analyzed by western blot and immunofluorescence. The study used a small interfering RNA [siRNA] loss-of-function strategy to demonstrate that HMGB1 is a critical target of SHI.
SHI treatment significantly attenuated sepsis-induced ALI in mice, as evidenced by improved lung histology, lower lung injury scores, and reduced pulmonary edema. In both in vivo and in vitro models, SHI suppressed the production of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β) and the M1 macrophage marker iNOS, while enhancing the release of anti-inflammatory cytokines (GM-CSF, IL-10, TGF-β1) and the M2 marker Arg1. Mechanistically, SHI inhibited the activation of the HMGB1/NF-κB pathway by downregulating the expression of HMGB1, TLR4, MyD88, and NF-κB phosphorylation. The critical role of HMGB1 was further supported by the finding that siRNA-mediated knockdown of HMGB1 mimicked the anti-inflammatory and polarization-shifting effects induced by SHI.
Our findings demonstrate that SHI alleviates sepsis-induced ALI by reprogramming macrophage polarization from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype. This protective effect is primarily mediated through the inhibition of the HMGB1/NF-κB signaling pathway. Thus, SHI represents a potential therapeutic candidate for sepsis-associated lung injury.
Graphical Abstract
Keywords
- acute respiratory distress syndrome
- shionone
- molecular mechanism
- macrophage polarization
- HMGB1
- traditional Chinese medicine
Acute lung injury (ALI) is a principal complication arising from sepsis [1]. Macrophage polarization, a key process in the development of sepsis, can lead to systemic decompensation, and exacerbate the inflammatory response, and have widespread effect during the development of sepsis. ALI involves dysregulated inflammation with an imbalance in macrophage polarization at its core. Excessive pro-inflammatory M1 macrophage activity promotes tissue damage, while deficient anti-inflammatory M2 macrophage function impairs repair. The specific signaling pathways governing this imbalance remain unclear, but elucidating these mechanisms is crucial for developing therapies to modulate macrophage responses and improve ALI outcomes [2]. Aster is a traditional medicine used to treat ALI, with shionone (SHI) as its main active component. Previous studies have reported that SHI improves ALI [3].
Macrophages, as central nodes of immune activity, exhibit strong plasticity and
versatility in response to changes in the body’s internal environment. Under
different stimulation conditions, macrophages can polarize into two distinct
phenotypes: M1 (pro-inflammatory) and M2 (anti-inflammatory), which play opposing
roles in inflammation, M1 pro-inflammatory type and M2 anti-inflammatory type,
which play different roles in inflammation. Their polarization functions are
almost antagonistic. M1 macrophages, induced by lipopolysaccharide (LPS), release
pro-inflammatory mediators, including interleukin-6 (IL-6), interleukin-1 beta
(IL-1
LPS activates nuclear factor
The Chinese herbal medicine Aster exhibits a protective effect against
respiratory diseases such as pharyngitis, cough, and asthma. Shionone, the
primary active ingredient extracted from Aster, possesses antiviral and
immunomodulatory properties. Previous study has reported that SHI can improve ALI
and inhibit the expression of inflammatory factors, potentially through the
regulation of macrophage polarization [3]. SHI exerts anti-inflammatory effects
through the NF-
SHI was purchased from Yuanye Bio-Technology (B21703, Shanghai, China) and was solubilized in 0.1% (w/v) DMSO. LPS was obtained from Sigma (L4516, St. Louis, United States). Dexamethasone (DXM) was purchased from Chenxin Drug Store (H37021969, Jining, China).
Male C57BL/6 mice (8 weeks old) were obtained from Jihui Co., Ltd. (Shanghai,
China). The mice were housed in a standard laboratory environment maintained a
temperature of 24
The lungs were removed from the sacrificed mice and weighed to obtain the wet weight (W). The lung tissues were then dried at 65 °C for 48 hours and weighed again to obtain the dry weight (D). The wet-to-dry weight (W/D) ratio was calculated to assess the degree of pulmonary edema.
Lung tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and
sectioned. After deparaffinization and rehydration, the sections were stained
with hematoxylin and eosin (H&E). Pulmonary histopathological changes were
observed under a CX53 microscope (Olympus, Tokyo, Japan). Lung injury scoring was
performed by an experienced investigator blinded to the treatment groups
(categorized as absent, mild, moderate, or severe with scores ranging from 0 to
3). The assessment was based on the presence of exudates, hyperemia, congestion,
neutrophilic infiltration, intra-alveolar hemorrhage, debris, and cellular
hyperplasia. Each of the following indicators was scored on a 0–4 scale: edema,
atelectasis, necrosis, alveolar and interstitial inflammation, hemorrhage, and
hyaline membrane formation. The scoring criteria were defined as follows: 0 = no
injury; 1 = 25% injury; 2 = 50% injury; 3 = 75% injury; 4 = 100% injury. For
each slide, the score of each injury indicator was evaluated in 10 randomly
selected fields at 200
Murine RAW264.7 macrophage cells were obtained from iCell (Shanghai, China) and
were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10%
heat-inactivated fetal bovine serum. Cells were maintained in a 37 °C
incubator with a humidified atmosphere of 5% CO2 and 95% air. Subculturing
was performed every 1–2 days, and only cells in the logarithmic growth phase
were used for experiments. Cells were seeded into six-well or 96-well plates at a
density of 5
HMGB1-siRNA was obtained from Hippobiotec Co., Ltd. (Huzhou, China). For cell
preparation, 1
Total RNA was isolated from cells using the MolPure Cell/Tissue Total RNA Kit
(Yeasen 19221ES50, Shanghai, China). Reverse transcription was performed using
with the HiScript II Q RT SuperMix Kit (Vazyme R223-01, Nanjing, China).
Quantitative real-time PCR (qRT-PCR) was then performed using SYBR qPCR Master
Mix (Medicalbio MR0321, Suzhou, China) according to the manufacturer’s protocols.
The 20 µL reaction system consisted of 1 µL cDNA template, 0.4
µL each of forward and reverse primers, 10 µL SYBR Premix Ex Taq, and
8.2 µL ddH2O.
| Primer name | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| GTGCTATGTTGCTCTAGACTTCG | ATGCCACAGGATTCCATACC | |
| High-mobility group box 1 | AGGCTGACAAGGCTCGTTATGAAAG | GGGCGGTACTCAGAACAGAACAAG |
| inducible nitric oxide synthase | ATCTTGGAGCGAGTTGTGGATTGTC | TAGGTGAGGGCTTGGCTGAGTG |
| Arginase-1 | AGACAGCAGAGGAGGTGAAGAGTAC | TGAGTTCCGAAGCAAGCCAAGG |
| Transforming growth factor-beta 1 | CAACAATTCCTGGCGTTACCTTGG | TGTATTCCGTCTCCTTGGTTCAGC |
| Interleukin-10 | TGGACAACATACTGCTAACCGACTC | GCCGCATCCTGAGGGTCTTC |
| Granulocyte-macrophage colony-stimulating factor | CCAGGAGATTCCACAACTCAGGTAG | TGAGAGGCTGTAGACCACAATGC |
| Interleukin-1 beta | TACAGGCTCCGAGATGAACAAC | TGCCGTCTTTCATTACACAGGA |
| Interleukin-6 | AGCCAGATCCTTCAGAGAGA | TGGTATTGGTCCTTAGCCAC |
| Tumor necrosis factor-alpha | CACAGAAAGCATGATCCGCG | ACTGATGAGAGGGAGGCCAT |
The concentrations of IL-1
Proteins were extracted from RAW264.7 cells using radio immunoprecipitation
assay (RIPA) buffer (Beyotime P0038, Shanghai, China), and their concentrations
were measured with a BCA kit (Beyotime P0009, Shanghai, China). After
denaturation, equal protein loads were resolved on 8–12% SDS-PAGE gels and
electrotransferred to PVDF membranes. Following a 1-hour block in 3% bovine
serum albumin (Servicebio GC305010, Wuhan, China), membranes were probed with
specific primary antibodies overnight at 4 °C. Primary antibodies against HMGB1
(ZenBio R22773 1:3000, Chengdu, China), p-NF-
Cells were fixed with 4% paraformaldehyde, then permeabilized and blocked with 3% BSA containing 0.1% Triton X-100. The cells were then incubated overnight at 4 °C with an anti-Arg1 primary antibody. After thorough washing, cells were incubated with an Alexa Fluor® 488-conjugated secondary antibody for 1 hour in the dark at room temperature. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 10 seconds. Finally, samples were mounted with an anti-fade mounting medium (Beyotime, Shanghai, China) and visualized under a CX53 microscope (Olympus, Tokyo, Japan). Fluorescence intensity was quantified using ImageJ software 2.1.4.7 (National Institutes of Health, Bethesda, MD, USA), and representative images were captured.
Data are expressed as mean
Our findings indicate that SHI can mitigate ALI in LPS-induced septic mice.
Histopathological changes were evaluated via H&E staining. As shown in Fig. 1A,
LPS challenge induced characteristic pathological features including inflammatory
cell infiltration, pulmonary edema, and alveolar wall thickening. Treatment with
SHI (100 mg/kg) or DXM markedly attenuated these changes, whereas SHI at 50 mg/kg
showed a lesser effect (Fig. 1A). Additionally, SHI significantly improved the
lung injury score (Fig. 1B). The LPS group exhibited a significantly elevated
wet-to-dry weight ratio compared to the control group (p
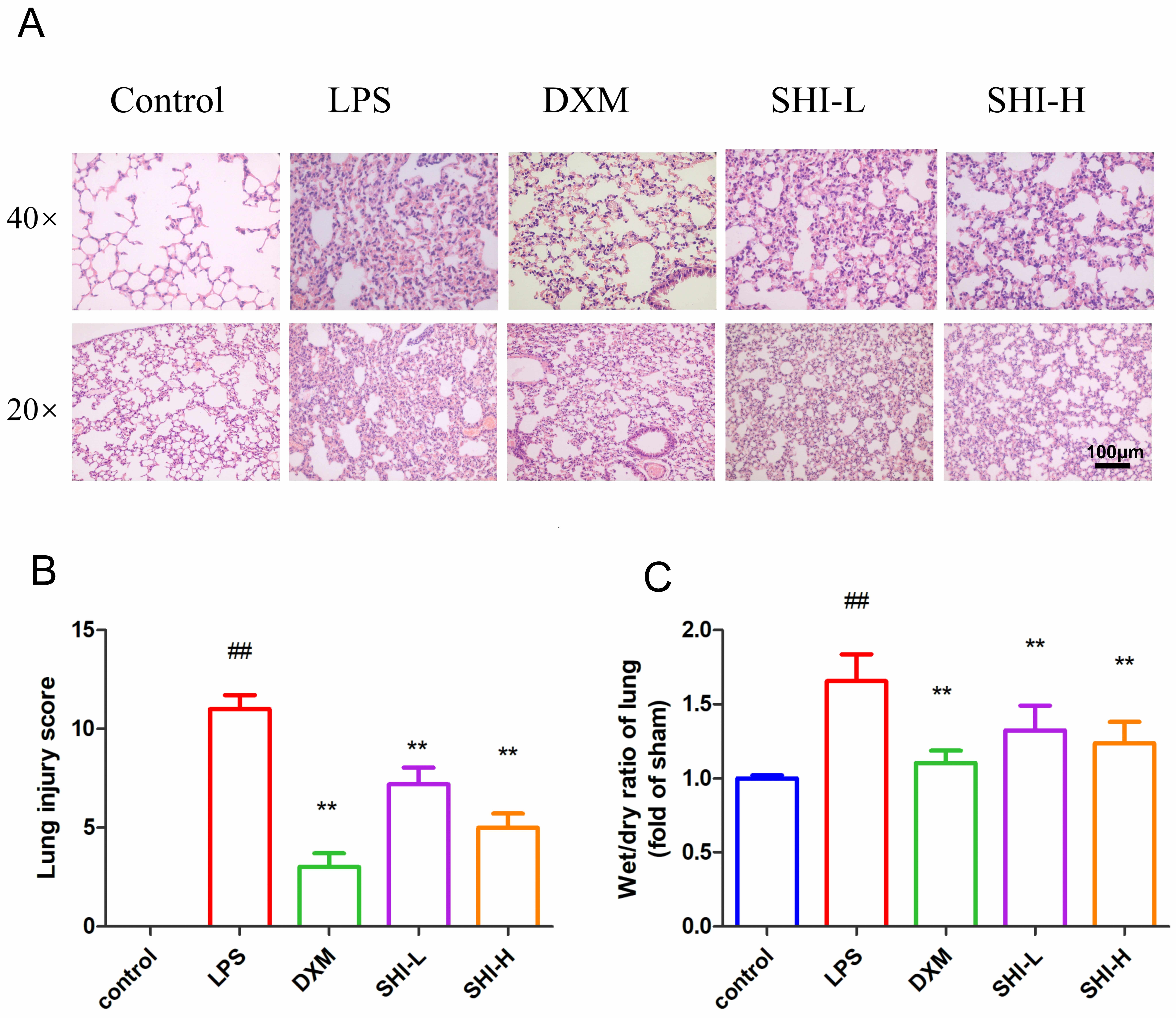 Fig. 1.
Fig. 1.
SHI ameliorated LPS-induced ALI in vivo. (A) The
histopathological alteration by H&E staining. Scale bar: 100 μm. (B) The lung injury score. (C) The
wet-to-dry ratio of the lungs. Compared with the control group, ##p
LPS stimulation significantly increased serum levels of IL-1
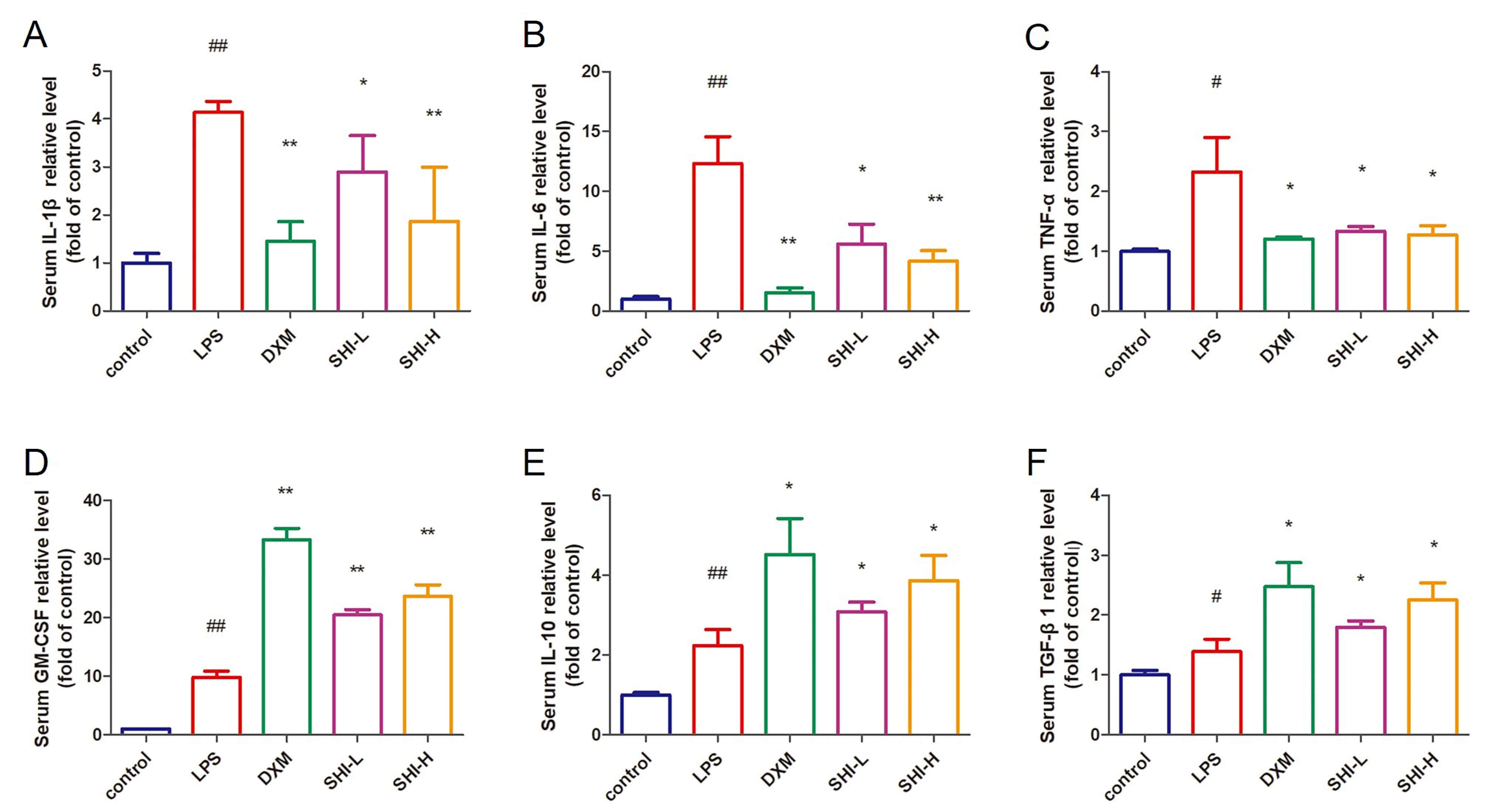 Fig. 2.
Fig. 2.
SHI inhibited the expression of serum inflammatory factors in
ALI models. (A–F) IL-1
The mRNA levels of IL-1
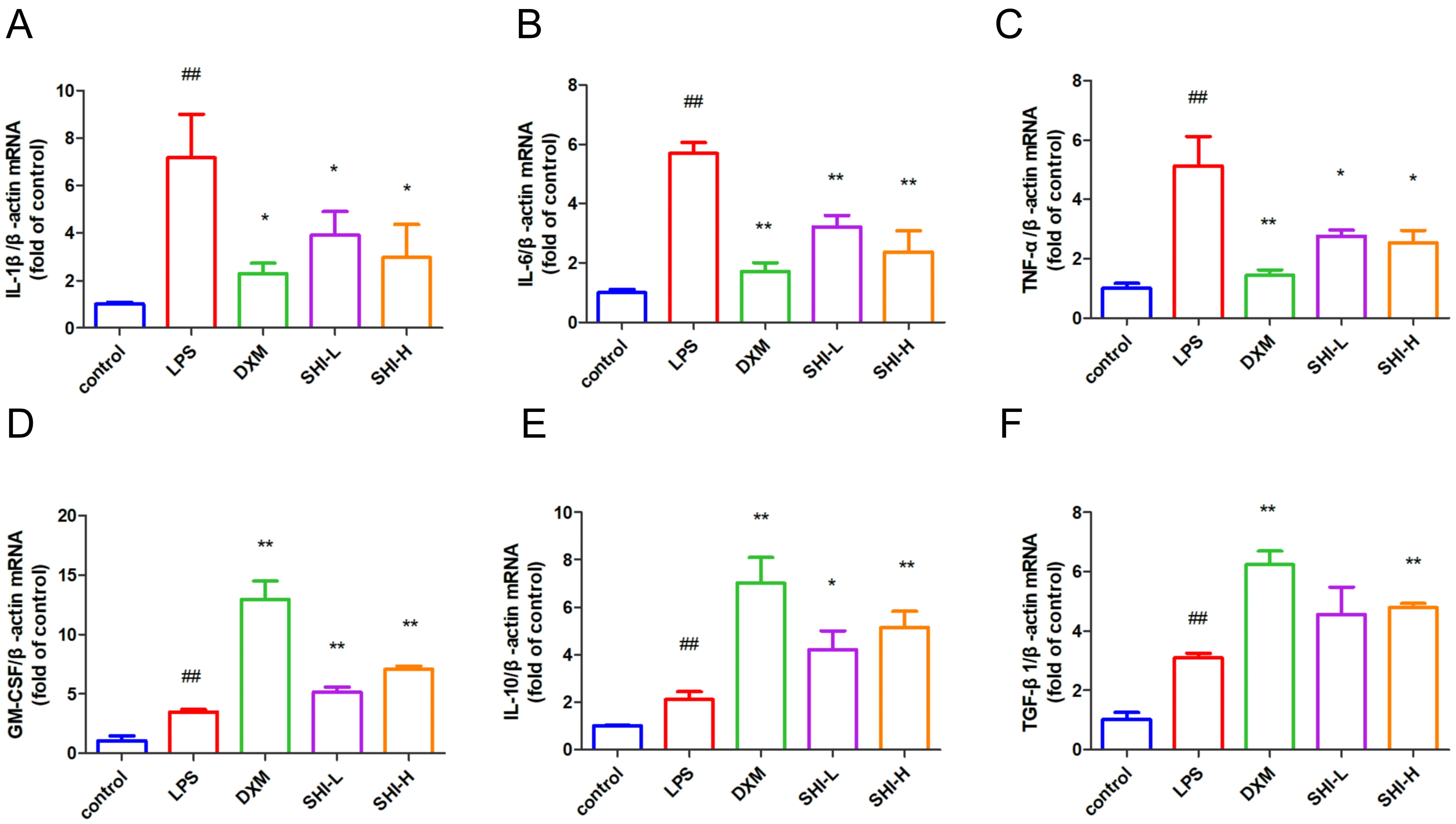 Fig. 3.
Fig. 3.
SHI inhibited the expression of inflammatory factors in ALI
model lung tissue. (A–F) IL-1
The expression of the polarization markers iNOS and Arg1 mRNA was examined in
the lung tissues of mice stimulated by LPS for 24 hours. The results showed that
the iNOS mRNA level in ALI lung tissues significantly increased after LPS
stimulation for 24 hours (p
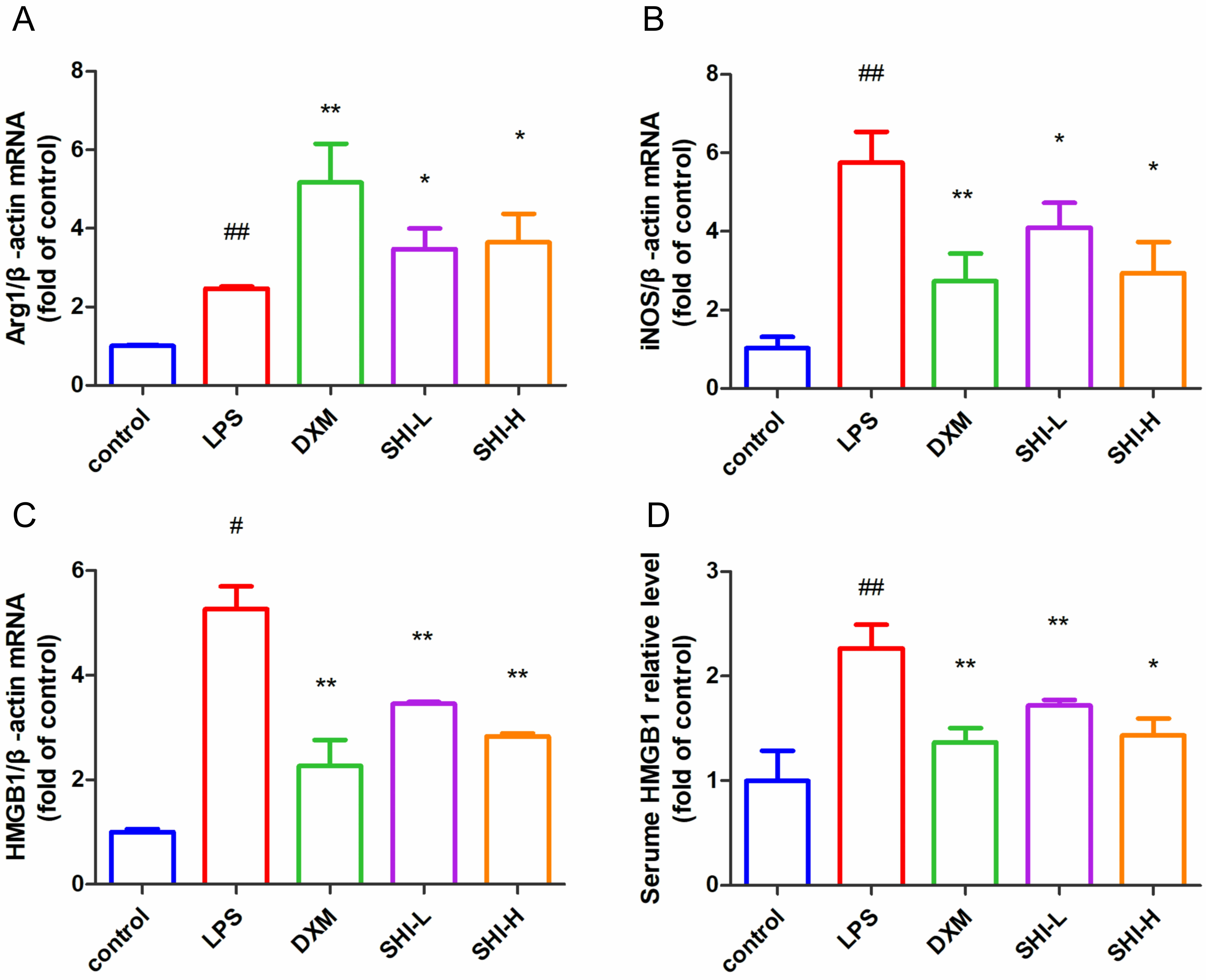 Fig. 4.
Fig. 4.
SHI affects the polarization and inhibits HMGB1 expression in
ALI model lung tissue. (A,B) mRNA expression of Arg1 and iNOS in lung tissues.
(C,D) HMGB1 mRNA expression in lung tissues and serum level. Compared with the
control group, #p
In this study, RAW264.7 cells were stimulated with lipopolysaccharide (LPS) as a
model, and then treated with low (2 µg/mL) or high (4
µg/mL) doses of shionone (SHI). Shi proliferation was assessed using
CCK-8 assays. Compared to the control group, LPS group was significantly reduced
RAW264.7 cell proliferation (p
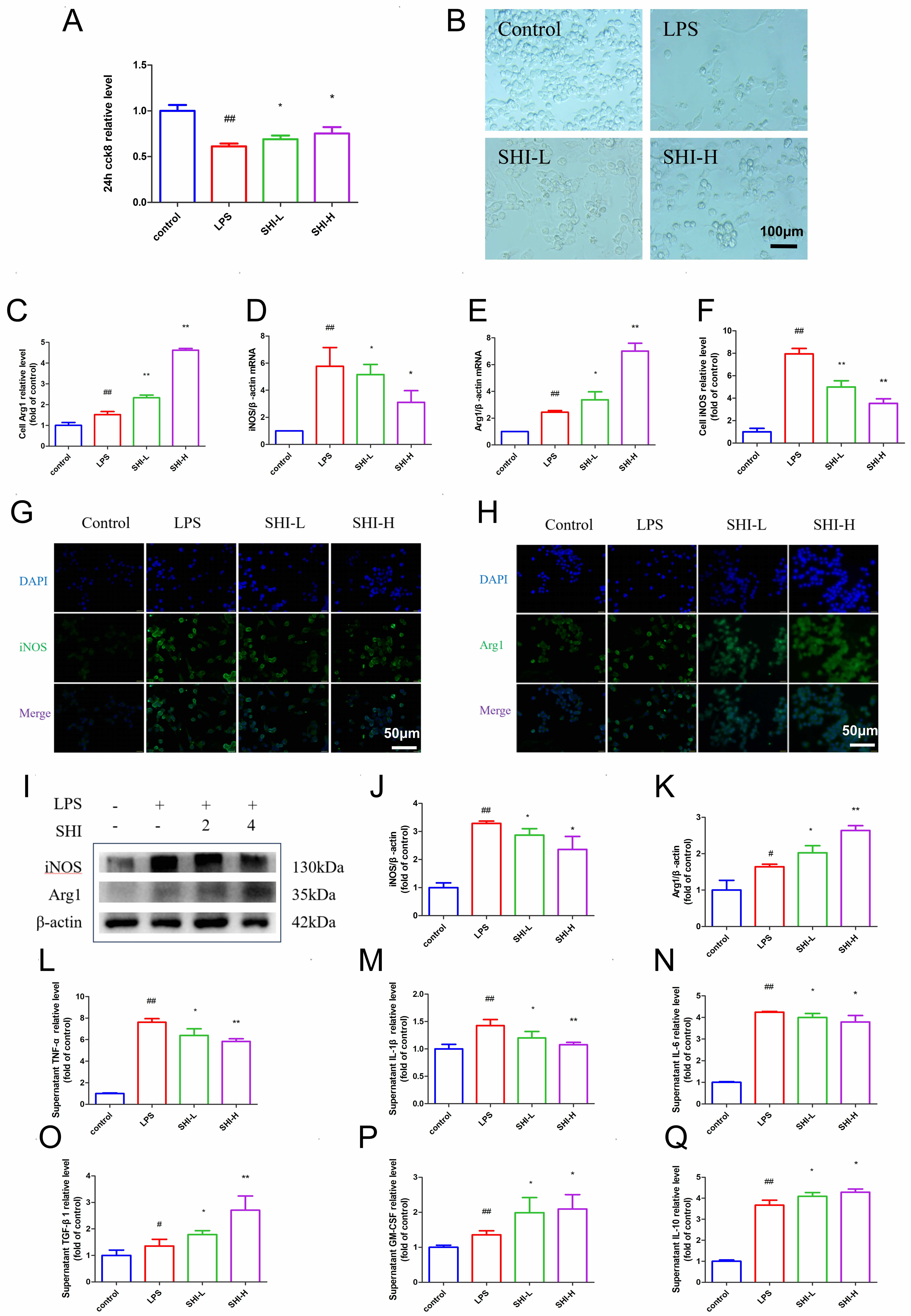 Fig. 5.
Fig. 5.
The effect of SHI on macrophage polarization. (A) Cell
Proliferation after 24 hours (CCK-8 assay). (B) Microscopic images. Scale bar: 100 μm. (C,D) Arg1
and iNOS mRNA expression in RAW264.7 cells. (E–H) Arg1
and iNOS protein
expression in RAW264.7 cells by Immunofluorescence. Scale bar: 50 μm. (I–K) iNOS and Arg1 protein
expression in RAW264.7 cells by Western Blot. (L–Q) TNF-
To investigate whether SHI mediates macrophage polarization by regulating the
HMGB1, p-NF-
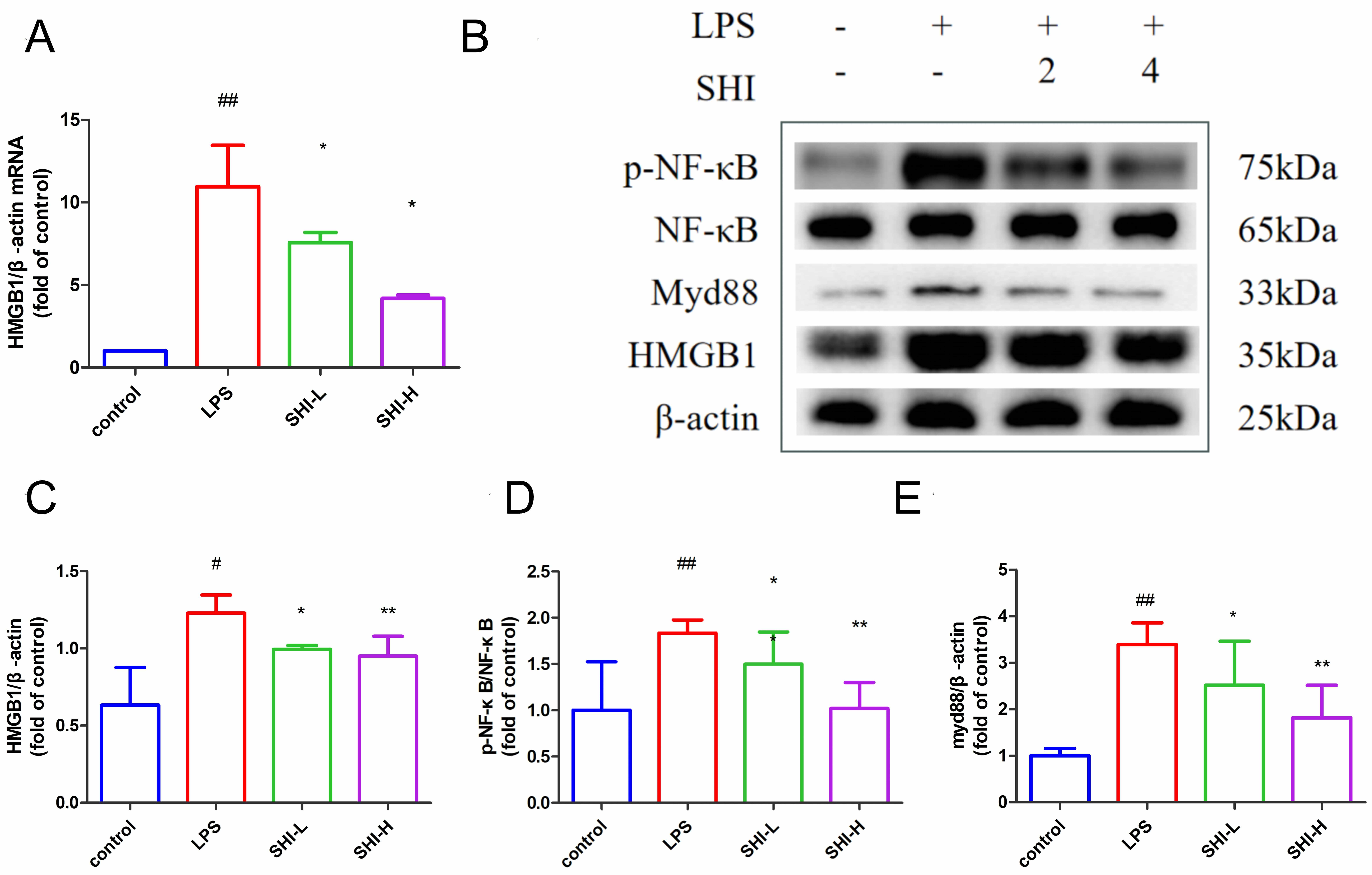 Fig. 6.
Fig. 6.
The effect of SHI on HMGB1/NF-
To further investigate the role of SHI in HMGB1-mediated polarization of
macrophages, we used HMGB1-siRNA to reduce HMGB1 expression in cells. We then
detected the mRNA expression and protein levels of the polarization markers iNOS
and Arg1 in RAW264.7 cells after 24 h of LPS stimulation. As shown in Fig. 7A–E,
knocking down HMGB1 (LPS+HMGB1-siRNA group) significantly inhibited the
expression of iNOS (p
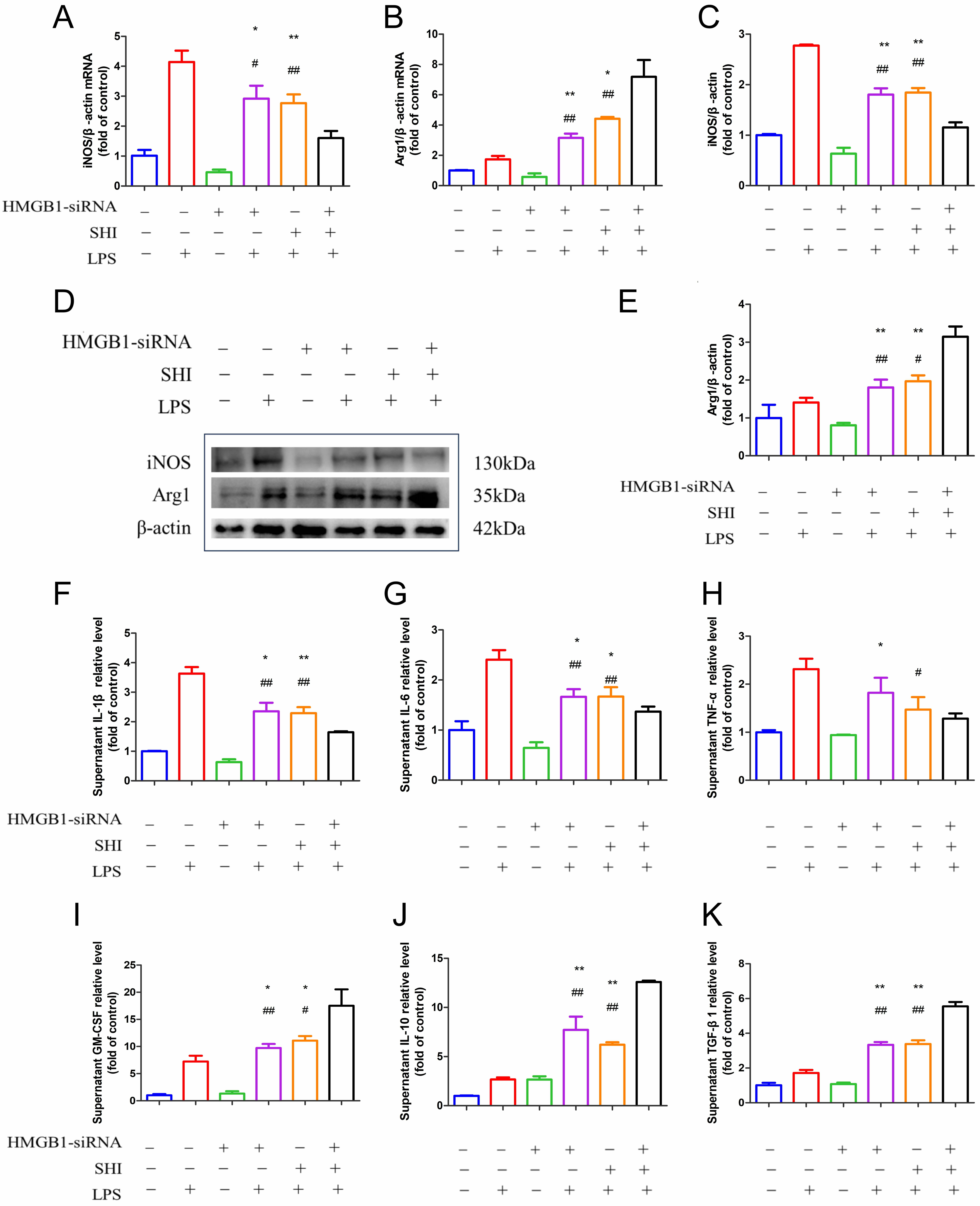 Fig. 7.
Fig. 7.
SHI affects macrophage polarization through HMGB1. (A,B) iNOS
and Arg1 mRNA expression in the RAW264.7 cells. (C–E) iNOS and Arg1 protein
expression in the RAW264.7 by WB. (F–K) IL-1
We detected the expression of HMGB1, TLR4, MyD88, NF-
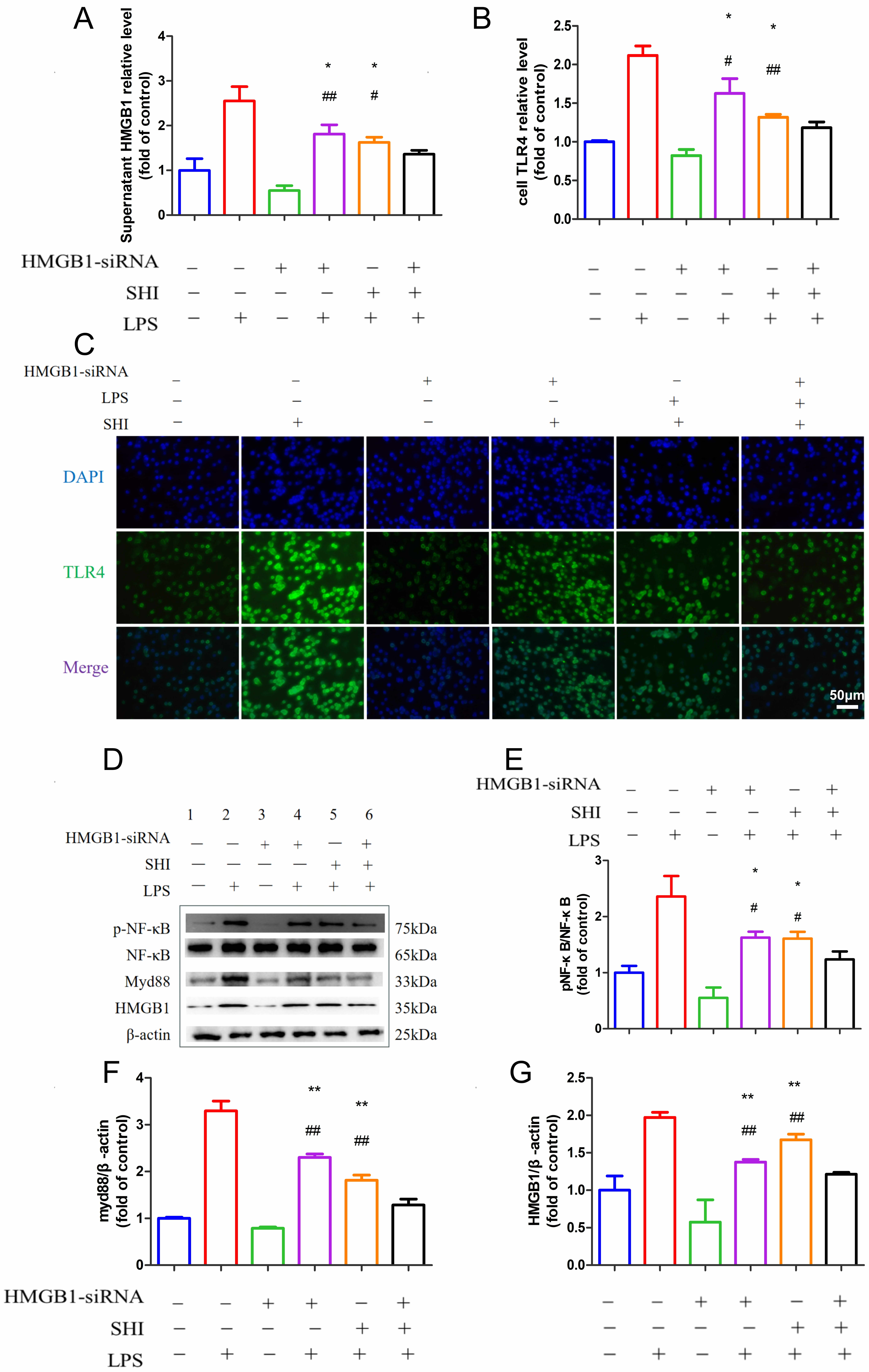 Fig. 8.
Fig. 8.
SHI affects macrophage polarization through the regulation of
the TLR4/NF-
This study investigates the therapeutic mechanism by which SHI alleviates
sepsis-induced ALI by regulating macrophage polarization via the
HMGB1/NF-
Sepsis frequently leads to multiple organ damage, with the development of a “systemic inflammatory response syndrome” impacting both humans and animals [17]. Acute lung injury, a major complication of sepsis, typically results in lung inflammation and pathological injury, significantly affecting lung pathophysiology. Sepsis can induce high concentrations of pro-inflammatory cytokines in serum and lung tissue, creating an “inflammatory storm” that further exacerbates lung damage [18].Previous studies have reported that ECM1 serves as a critical factor required for driving M1 macrophage polarization in inflammatory bowel disease (IBD) upon LPS stimulation. [19]. This model revealed changes in lung histopathology, including inflammatory cell infiltration, alveolar wall thickening, and pulmonary edema. Drugs intervention improved these pulmonary inflammatory manifestations, indicating that SHI can be effectively reverse sepsis-induced ALI.
Sepsis alters the body’s immune environment and activates macrophages, which are
crucial phagocytes in the immune system distributed throughout various tissues,
including the lung. macrophages perform various functions, including pathogen
phagocytosis and elimination, removal of damaged or dead cells, regulation of
inflammatory responses, and promotion of tissue regeneration. Macrophage
polarization is associated with the occurrence and development of immune
disorders. Polarization of macrophages can lead to immune system decompensation,
exacerbate the inflammatory response, and have a wide impact on the body during
sepsis development [2]. Under LPS stimulation, macrophages can exhibit both
pro-inflammatory and anti-inflammatory properties [20]. In the early stages of
the inflammatory response, M1 macrophages activated by the classical activation
via pathway, respond rapidly to infection and tissue damage by secreting various
pro-inflammatory factors such as TNF-
Upon detecting danger signals, such as microbial stimulation or tissue damage,
macrophages rapidly reshape themselves and activate the transcription of key
downstream genes in a spatiotemporal dependent manner. This allows them to
quickly and effectively perform functions such as endocytosis, phagocytosis,
cytokines secretion and immune response regulation. Many studies have
demonstrated that transcription regulation is crucial for macrophages
polarization, and several transcription regulatory factors have been identified,
including NF-
HMGB1 typically resides in the nucleus. Upon proper stimulation HMGB1 is either
actively secreted by immune cells or passively released from necrotic or damaged
cells. It translocates to the cytoplasm and acts as an initiator of the
inflammatory cascade, participating in the progression of inflammatory reactions
[9], exacerbating HMGB1 further exacerbates the inflammatory response, worsening
patient conditions, and affecting prognosis [12]. HMGB1 binds to Toll-like
receptors (TLR4,9) to activate NF-
This study is limited by its focus on macrophage-mediated mechanisms; future investigations should evaluate SHI’s effects on other immune cell populations and explore potential crosstalk between macrophage polarization and metabolic reprogramming in sepsis.
Shionone mitigates sepsis-induced acute lung injury by shifting macrophage
polarization from a pro-inflammatory M1 to an anti-inflammatory M2 phenotype.
This therapeutic effect is achieved by suppressing the HMGB1/NF-
All data supporting the findings of this study are available within the article and are also accessible from the lead contact on request.
QW, BZ, HJ and YS designed the research study. QW and GX performed the research. QW, JZ and YL analyzed the data. QW and BZ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was conducted in accordance with the ARRIVE guidelines. The research protocol was approved by the Ethics Committee of Suzhou Hospital of Integrated Traditional Chinese and Western Medicine (Ethic Approval Number: 2023030).
We gratefully acknowledge the assistance and instruction from Dr. Xin Wang and professor Weiwei Tao.
This research was supported by Jiangsu Provincial Association of Chinese Medicine Project No. CYTF2024037. Jiangsu Province Traditional Chinese Medicine Technology Development Project No. MS2023037. Suzhou Applied Basic Research (Medical and Health) Science and Technology Innovation Project No. SYWD2024333. Medical and Health Project on Suzhou Wuzhong District Science and Technology Plan Youth Project No. WZYW2023017. Applied Basic Research on Special Research of Wumenyipai with Shicaixuepai of Suzhou No. SC2023002.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.