







 , Yufeng Xing 1,2,3,†, Ming Wu 1,2,3, Dongping Chen 1,2,3, Feng Yang 1,2,3, Di Huang 1,2,3, Ayijiaken Kasimumali 4, Yijing Zhou 5, Chaoyang Ye 1,2,3,*
, Yufeng Xing 1,2,3,†, Ming Wu 1,2,3, Dongping Chen 1,2,3, Feng Yang 1,2,3, Di Huang 1,2,3, Ayijiaken Kasimumali 4, Yijing Zhou 5, Chaoyang Ye 1,2,3,*
1 Department of Nephrology, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, 201203 Shanghai, China
2 TCM Institute of Kidney Disease, Shanghai University of Traditional Chinese Medicine, 201203 Shanghai, China
3 Key Laboratory of Liver and Kidney Diseases (Shanghai University of Traditional Chinese Medicine), Ministry of Education, 201203 Shanghai, China
4 Department of Nephrology, The Second People’s Hospital of Kashgar Prefecture, 844000 Kashgar, Xinjiang Uygur Autonomous Region, China
5 Department of Nephrology, Jiaxing Hospital of TCM, 314100 Jiaxing, Zhejiang, China
†These authors contributed equally.
Abstract
As a predominant complication of chronic kidney disease (CKD), vascular calcification (VC) warrants investigation into its mechanisms. The epigenetic modulator Enhancer of Zeste Homologue 2 (EZH2) is implicated in various kidney disorders. This investigation aims to examine its function in the development of VC related to CKD.
Radial arteries collected from individuals with kidney failure (with or without calcification) following arterial venous fistula surgery were processed for Von Kossa and immunohistochemical (IHC) staining. A VC- associated CKD model was established by administering a diet containing adenine and phosphate (1.2%) and 3-deazaneplanocin A (3-DZNeP) was used to block EZH2 activity. To induce calcification, thoracic aortas isolated from SD rats were cultured ex vivo in a medium containing 2.6 mmol/L Pi. In parallel, an in vitro model of calcification was established by treating A7r5, a rat vascular smooth muscle cell line (VSMC), with a calcifying medium, and 3-DZNeP was used to inhibit EZH2. Adenovirus was used to overexpress EZH2 and siRNA was used to silence bone morphogenetic protein (BMP) 2.
EZH2 protein expression was higher in calcified arteries than in non-calcified arteries from individuals after the surgical creation of an arteriovenous fistula. In the mouse model, the upregulation of EZH2 and Runx2 at both the transcriptional and translational levels was demonstrated in calcified aortas. 3-DZNeP treatment decreased VC and transcriptional levels of EZH2. The ex vivo experiments revealed that high Pi stimulation significantly enhanced EZH2 expression and its histone mark, H3K27me3 (methylation of Histone H3 at lysine 27). This epigenetic alteration was accompanied by an elevation in osteogenic markers and a reduction in the expression of VSMC differentiation markers. In vitro, EZH2 and H3K27me3 were noticeably elevated in calcifying VSMCs. Mechanistically, overexpression of EZH2 aggravated the VSMCs’ calcification through upregulation of BMP2.
Our study demonstrated the elevated EZH2 expression in calcified vascular tissues from both CKD patients and preclinical models, including CKD mice, cultured rat aortas, and VSMCs calcification models. EZH2 may promote VC through activation of BMP2 signaling. Targeting EZH2 could be a therapeutic strategy against CKD associated VC.
Keywords
- vascular calcification
- kidney diseases
- enhancer of zeste homolog 2 protein
- bone morphogenetic protein 2
- osteogenesis
Chronic kidney disease (CKD) is a critical health issue with an overall prevalence of 13% [1]. Vascular calcification (VC) is a predominant complication associated with CKD, contributing to elevated cardiovascular morbidity and mortality [2]. The incidence of VC in patients with CKD reaches up to 65–70% [3].
VC, a complicated and active process, is distinguished by the ectopic deposition of calcium (Ca)-phosphate (Pi) crystals in the blood vessel wall that shares features with bone mineralization [4]. This process is driven by mechanisms such as hyperphosphatemia, osteogenic transformation of vascular smooth muscle cells (VSMCs), dysregulation of calcific inhibitors, and ultimately crystal deposition in the extracellular matrix [5]. Various biochemical signaling networks, including the bone morphogenetic protein (BMP) signaling [6] and the main transcription factor RUNX2 (Runt-related transcription factor 2), regulate trans-differentiation of VSMC [7]. However, there is a lack of unified clinical diagnostic criteria and effective drugs for early and middle stage VC in CKD [8].
Epigenetic reprogramming has a central function in driving the pathological progression of VC [9]. In CKD- associated calcification, elevated levels of Pi and indoxyl sulfate enhance DNA methylation, thereby dysregulating the genes of phenotypic switch in VC [10, 11]. In addition, epigenetic regulators such as non-Coding RNAs modulate the pathogenesis of arterial calcification through the miRNA sponging and interaction with transcription factors, thus altering signaling pathways that broadly influence cellular biology processes, like autophagy, senescence [12, 13]. CKD risk factors such as inflammation factors and high phosphorous, trigger VC via histone post-translational modifications, which promote VSMCs phenotypic switch by remodeling chromatin architecture and facilitating the transcriptional activation of osteogenic programs [14]. Enhancer of zeste homologue 2 (EZH2) is a vital epigenetic regulator [15, 16]. Accumulating evidence from animal studies shows that expression and activity of EZH2 are elevated in various animal models of kidney diseases [17]. Conversely, pharmacological or genetic inhibition of EZH2 provides reno-protective effects [18, 19]. Although suppression of EZH2 alleviated the calcification in models of atherosclerosis and medial calcification by suppressing the osteogenesis genes [20, 21]. The comprehensive understanding of the functions and mechanism of EZH2 exerted in CKD related VC is limited.
BMP2, is a key secreted ligand of the TGF-
Radial arteries in the presence or absence of calcification from individuals with kidney failure who experienced arterial venous fistular surgery (n = 3, respectively) were collected from Shuguang Hospital of Shanghai University of Traditional Chinese Medicine from September 2019 to January 2021. All procedures were conducted as per the Declaration of Helsinki and approved by the IRB of Shuguang Hospital affiliated with Shanghai University of TCM (approval number: 2018-575-04-01). Tissues were fixed in 4% paraformaldehyde-fixed and paraffin-embedded tissues (4 µm thick) and use them for further analysis.
Ten-week-old male C57BL/6 mice (SPF grade) were bought from Shanghai SLAC Laboratory Animal Co., Ltd. The Animal Experimentation Ethics Committee of Shanghai University of Traditional Chinese Medicine (PZSHUTCM190920010) gave its approval for the protocol.
The adenine-induced CKD model was established over an eight-week period, following a previously described protocol with minor modifications (Supplementary Fig. 1). After a one-week acclimatization, ten-week-old male mice were fed with either the experimental diet or a standard control diet. The adenine diet was formulated with 10% casein to improve palatability and 1.2% phosphate to exacerbate VC [26, 27]. Seventeen mice were classified into two groups in a random manner: chow diet group (n = 8), and the adenine diet group (n = 9). Some mice were treated with 3-DZNeP (A8182, CAS: 120964-45-6, 98%, Ape Bio, Houston, TX, USA), a carbocyclic analog of adenosine used to block EZH2 activity (1 mg/kg/every 3 days) during the eight-week diet program. Eighteen mice were classified into three groups in a random manner: chow diet + vehicle (DMSO) group (n = 6), adenine diet + vehicle (DMSO) group (n = 6), adenine diet + 3-DZNep (1 mg/kg/per 3 days) group (n = 6).
Mice were euthanized by intraperitoneal injection of an overdose of pentobarbital sodium (100 mg/kg body weight; drug concentration: 8 mg/mL). Ca deposition was measured in the dissected thoracoabdominal aortas of mice. Plasma levels of blood urea nitrogen (BUN), creatinine (Cre), Calcium (Ca), alkaline phosphate (ALP), and Pi were estimated by automatic biochemical analyser (AU680, Beckman Coulter, Brea, CA, USA).
Thoracic aortas (from the arch to the iliac bifurcation) were dissected from male C57 mice weighing between 22–24 g or male Sprague-Dawley rats weighing between 180–200 g. Rat were euthanized by intraperitoneal injection of an overdose of pentobarbital sodium (150 mg/kg body weight; drug concentration: 50 mg/mL). After the removal the adventitia, vessels were sliced into 1–2 mm rings from mice or 3–5 mm rings from rats, which were cultured in HG-DMEM (11965092, Gbico, USA) medium containing 15% foetal bovine serum (FBS, 04-001-1ACS, Biological Industries, Israel) and 0.5% penicillin/streptomycin (P/S, C0222, Beyotime Biotech, Shanghai, China) at 37 °C, 5% CO2 atmosphere. Arota rings were incubated with a calcifying medium (pH 7.4) containing 2.6 mmol/L inorganic Pi, which is sodium phosphate (NaH2PO4/Na2HPO4) to induce calcification. Following incubation for indicated time points (2, 3, 5 and 7 days), the samples were harvested and analyzed. Each ex vivo experiment was conducted with at least three biological replicates.
The rat smooth muscle embryonic thoracic aorta cell line A7r5 was bought from
the National Collection of Authenticated Cell Cultures, Chinese Academy of
Medical Sciences (Cat. GNR 7). Cells were kept at 37 °C, 5% CO2
atmosphere using high-glucose DMEM (HG-DMEM; 11885084, Gibco, Waltham, MA, USA)
enriched with 10% FBS and 0.5% P/S. The cell line was authenticated by the
Chinese Academy of Medical Sciences and tested mycoplasma negative. Calcification
was induced by 2.6 mmol/L high Pi (Pi, NaH2PO4/Na2HPO4, PH =
7.4) in DMEM medium or 10 mmol/L
The adenovirus for EZH2 (Ad-EZH2) (packed in pADM-FH-GFP vector) were acquired
from WZ Biotech company (Shangdong, China). Cells cultured at
Rat-Scramble small interfering RNA (siRNA), and Rat-BMP2 siRNA were synthesized by GenePharma (Suzhou, China). Supplementary Table 1 displays the siRNA sequences. Cells were cultured in six-well plates and were transfected with 20 nmol/L siRNA per well via Lipofectamine 2000 Reagent (TL201, Vazyme, Nanjing, China).
VSMCs and aortic rings were treated for seven days in calcified medium with 2.6 mmol/L high Pi (NaH2PO4/Na2HPO4, PH = 7.4) to induce calcification. Following dissection from CKD animals, aortic tissues were homogenized, and VSMCs were isolated. These samples were then incubated in 0.6 mol/L HCl to facilitate Ca extraction. The Ca content in the HCl supernatant was analyzed via a commercial Kit (o-Cresolphthaleine complexone Assay) (20162400906, BioSino, Beijing, China) and normalized to protein concentration, which was detected via the Bradford assay (PA102, Tiangen, Beijing, China).
Following fixation with 4% paraformaldehyde, kidney samples, thoracic aortas from CKD models, and cultured aortic rings were embedded in paraffin. Each sample was sliced with a thickness of 4 µm. Masson’s trichrome staining was conducted on kidney slices according to the protocol of Trichrome stain kit (D026, Nanjing Jiancheng, Nanjing, China). IHC staining was performed on sections of thoracic aortas from CKD models and cultured aortic rings. The slices’ incubation was conducted with respective primary antibodies against Runx2 (1:200, 8486, CST, Danvers, MA, USA), EZH2 (1:200, 5246, CST, Danvers, MA, USA) and BMP2 (1:200, ER80602, HUABIO, Hangzhou, China). A microscope (80i, Nikon, Tokyo, Japan) was utilized to capture images.
For cell culture, VSMCs were grown in growth or calcifying mediums in six-well plates for 10 days, then the cells were PBS-rinsed three times and followed by fixation in 10% paraformaldehyde for 10 min. After another three times wash, cells were incubated with 1% Alizarin Red S for 30 min and PBS-rinsed three times again. Positively stained cells appeared red.
Von Kossa staining was performed on sections of thoracic aortas from CKD models and cultured aortic rings. The incubation of slices was first conducted in 5% silver nitrate (AgNO3) solution under ultraviolet light for 30 min and subsequently washed with 5% sodium thiosulfate (Na2S2O3). Hematoxylin was utilized to counterstain nuclei. Upon staining, the calcified nodules changed from brown to black. A microscope (80i, Nikon, Japan) was utilized to obtain images.
Protein lysates from rat primary VSMCs or cultured aortic rings were extracted
via RIPA lysis buffer (P0013B, Beyotime Biotech, Shanghai, China). Immunoblotting
was conducted as per previously published guidelines [28]. Primary antibodies are
Runx2 (1:1000, 8486, CST, USA), OPN (1:1000, BS1264, Bioworld, Dublin, OH, USA),
MSX2 (Msh homeobox 2) (1:1000, ab69058, abcam, Cambridge, UK), BMP2 (1:1000,
NBP1-19751, Novus, Littleton, CO, USA), a-SMA (1:10,000, ET1607-53, HUABIO,
Hangzhou, China), EZH2 (1:1000, 5246, CST, Danvers, MA, USA), H3k27me3 (Histone
H3 Lysine 27 Trimethylation) (1:1000, 9733, CST, Danvers, MA, USA), SM22
(1:10,000, A6760, abclonal, Wuhan, China), Calponin (1:10,000, ET1606-17, HUABIO,
Hangzhou, China),
TRIzol reagent (R401-1, Vazyme, China) was utilized to extract total RNA. RNA reverse-transcription to cDNA was conducted via HiScript II Q select RT SuperMix (R233-1, Vazyme, China). Hieff qPCR SYBR Green Master Mix (11203ES08, Yeasen, Shanghai, China) was employed per the manufacturer’s instructions. All amplification reactions were conducted over 40 cycles and were performed in triplicate using Real-Time PCR System (Step one plus, ABI, Carlsbad, CA, USA). Supplementary Table 2 displays the primer sequences.
Data are expressed as mean
An in vitro model of VC was established by using cell line A7r5
(VSMCs). A significant upregulation of Runx2 and Msx2 was observed in 10 mmol/L
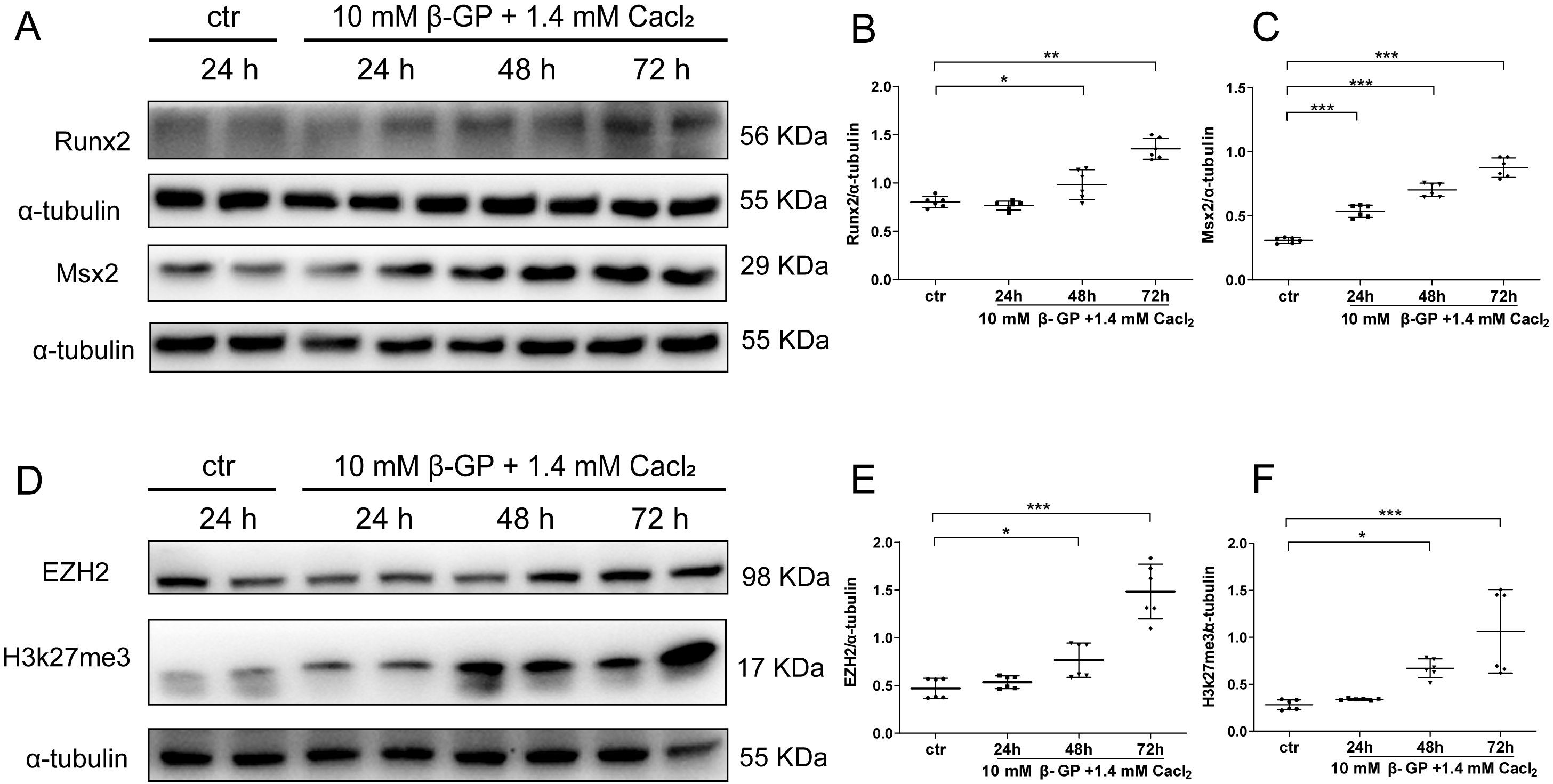 Fig. 1.
Fig. 1.
EZH2 is upregulated in calcified VSMC. Western blot analysis
and quantification of osteogenic markers Runx2 and Msx2 (A–C), and EZH2 and
H3k27me3 levels (D–F). Data are reported as mean
An ex vivo model of VC was established by culturing mouse thoracic aortas in high-phosphate (2.6 mmol/L Pi) HG/DMEM medium for 3, 5, and 7 days. A time-dependent increase in aortic Ca content was observed from day 3 to day 7. This model exhibited a time-dependent increase in Ca deposition (Fig. 2A), a result further verified by Von Kossa staining (Fig. 2B). In parallel, the expression of EZH2 was up-regulated in cultured aorta rings after Pi-stimulation as compared with the control group, as shown by immunostaining (Fig. 2C).
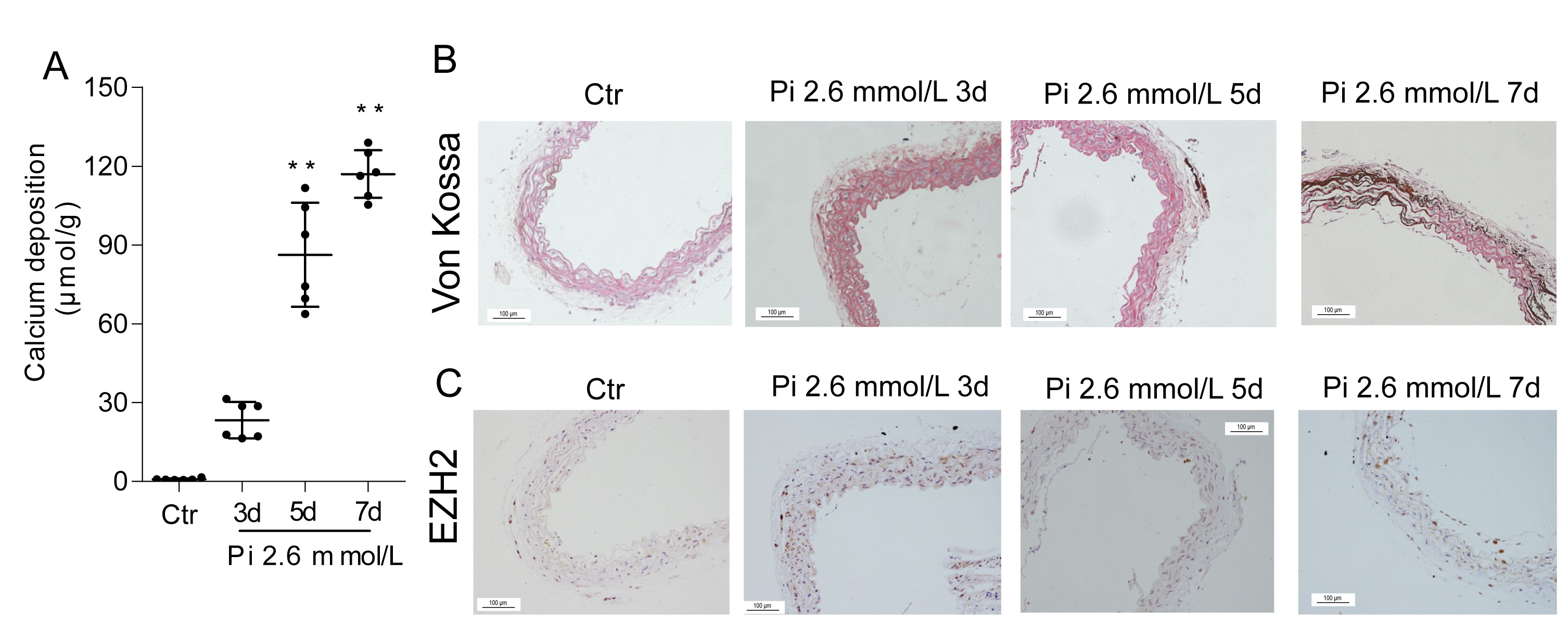 Fig. 2.
Fig. 2.
EZH2 is upregulated in calcified mouse aortas. Mouse aortas were
incubated with medium containing 2.6 mmol/L Pi for 3, 5 and 7 days. (A)
Quantification of calcium (Ca) deposition. (B) Von Kossa staining. (C)
Immunohistochemical staining of EZH2. Scale bar = 100 µm. Data are mean
We further established an ex vivo model of VC using SD rats. Fig. 3A
illustrates that the aortic Ca deposition of rat thoracic aortas was
significantly increased upon Pi stimulation from day 5 to day 7. The enhanced
calcification of elastic lamellae in rat thoracic aortas in calcifying medium for
7 days was further shown by Von Kossa staining (Fig. 3B). Western blotting
analysis demonstrated that Pi stimulation induced elevation of osteogenic markers
(Fig. 3C–F), while repressing the expression of VSMC differentiation marker
Calponin (CNN),
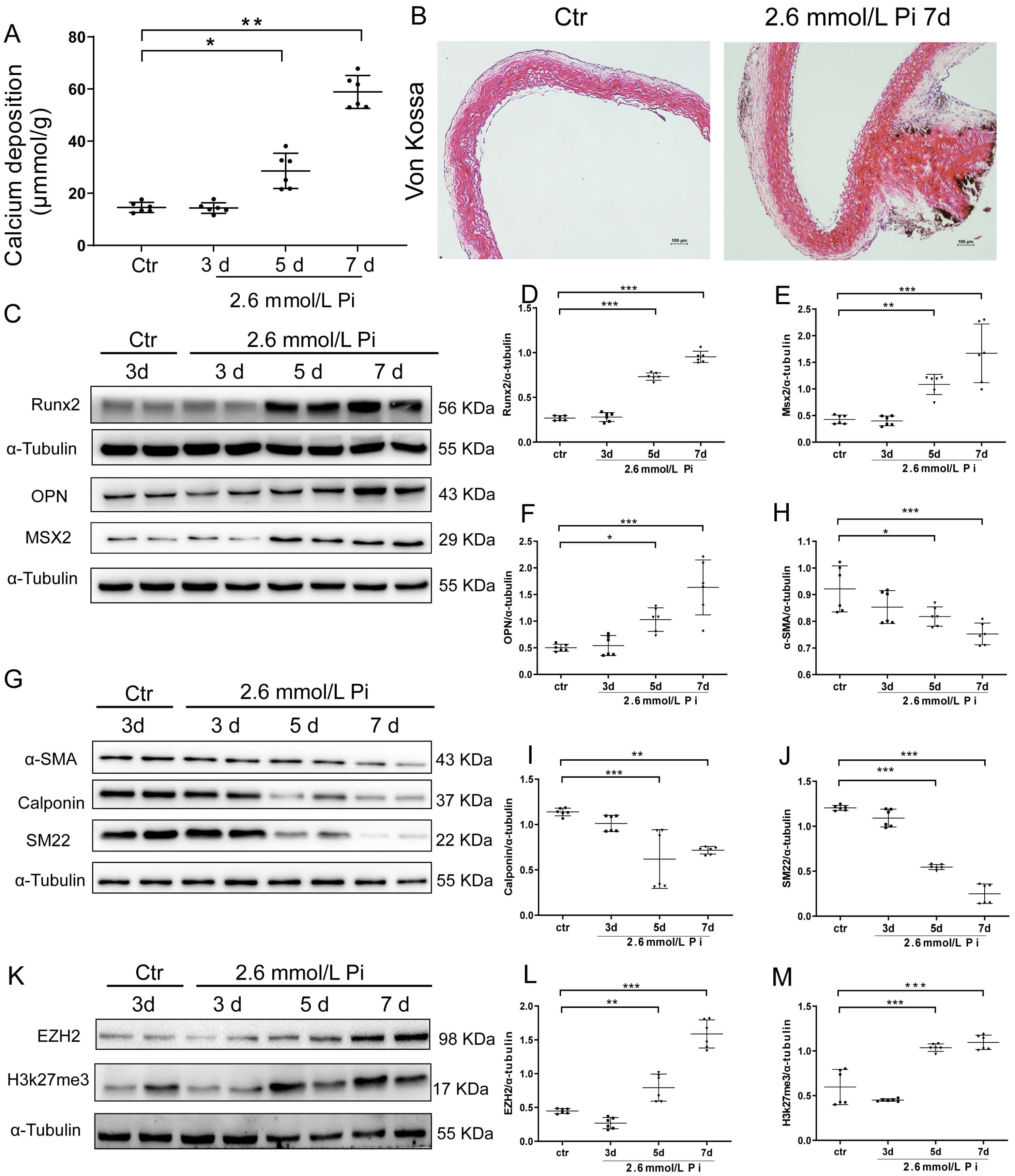 Fig. 3.
Fig. 3.
EZH2 is upregulated in calcified rat aortas. Rat aortas were
cultivated with calcifying medium with 2.6 mmol/L Pi for 3, 5 and 7 days. (A)
Calcium deposition was quantified by calcium assay. (B) Calcified area was shown
by Von Kossa staining (Magnification
We next created mouse model of VC associated with CKD by feeding mice with adenine and a high-Pi diet (Supplementary Fig. 1). Blood levels of creatinine (CREA), blood urea nitrogen (BUN), uric acid (UA), phosphate (Pi), and alkaline phosphatase (ALP) were elevated in CKD mice (Fig. 4A–E) (Supplementary Tables 3,4). Elevated calcium deposition (Fig. 4F) and intensified Von Kossa staining (Fig. 4G upper) in the aortic tissue confirmed successful VC induction in the adenine-fed CKD mice. Fig. 4G middle and Fig. 4H shows that EZH2 was upregulated in calcified aortas of CKD mice, as shown by IHC staining and qRT-PCR, which was associated with elevated osteogenic marker Runx2 (Fig. 4G lower and Fig. 4I).
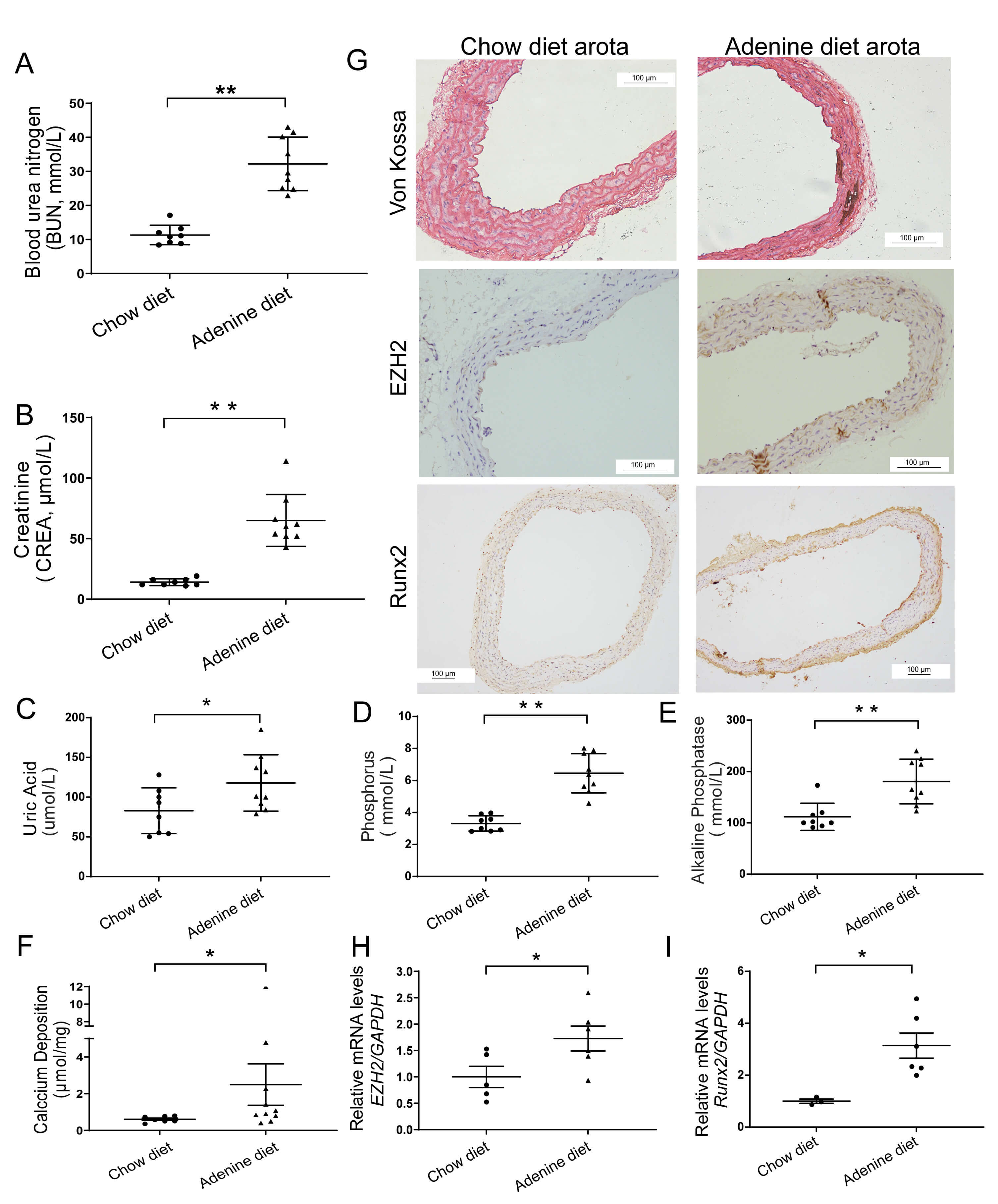 Fig. 4.
Fig. 4.
Vascular EZH2 is upregulated in mice with CKD-associated VC. In
eight-week adenine-triggered renal failure model. Plasma levels of renal function
indices were measured (A–E). Aortic calcification was assessed by calcium
content quantification (F; chow diet, n = 8 and adenine diet, n = 9) and Von
Kossa staining, with EZH2 expression (Magnification
We further measured the expression of EZH2 in vascular tissues of CKD patients. VC was revealed by Von Kossa assays in radial arteries from individuals with kidney failure who underwent arterial venous fistular operation. A significantly higher expression EZH2 in tunica media of calcified arteries was revealed by IHC, supporting the positive correlation of EZH2 expression and VC in patients with CKD (Fig. 5).
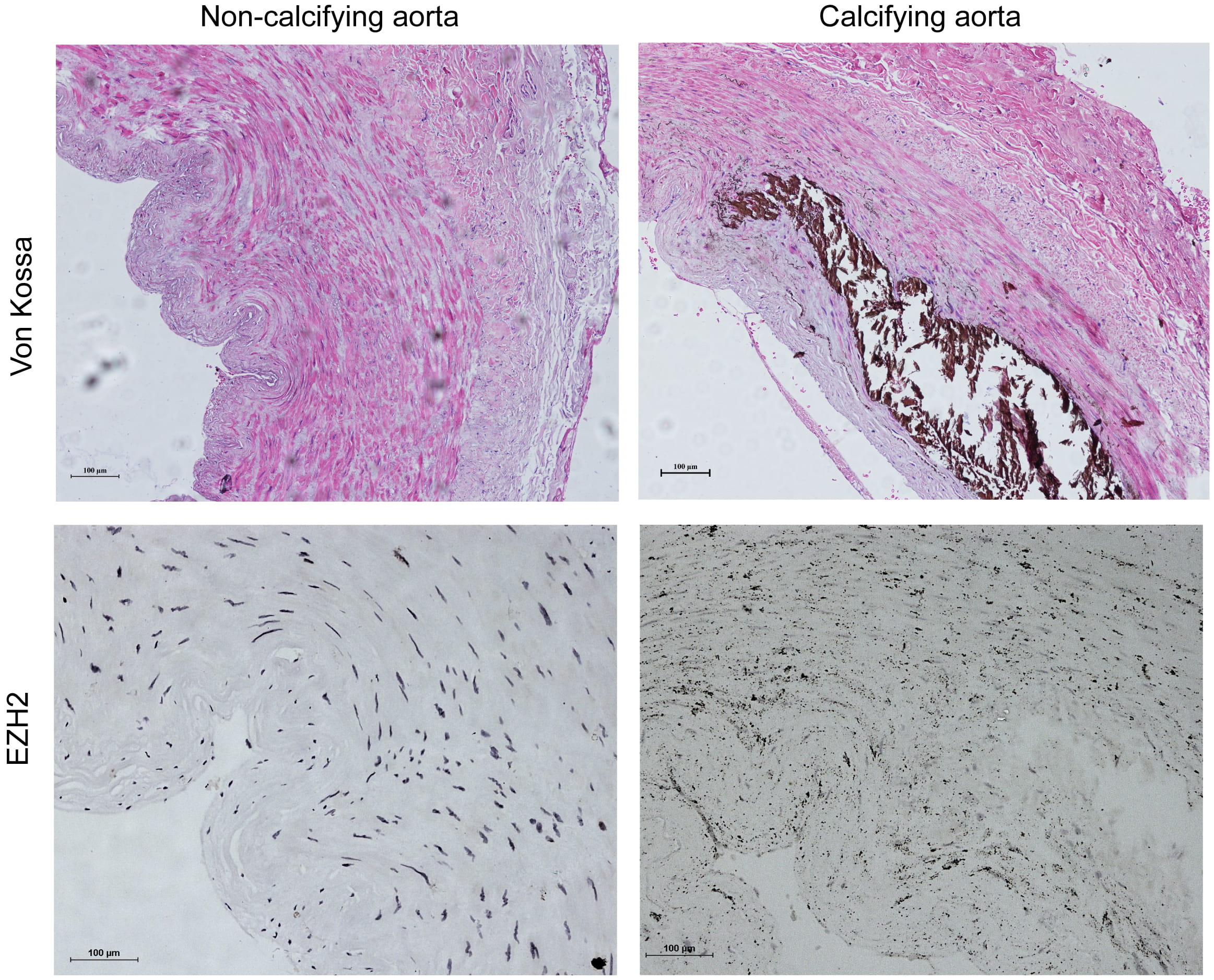 Fig. 5.
Fig. 5.
EZH2 upregulation in human Calcifying arteries associated with
CKD. Von Kossa staining (upper) in radial arteries in the presence of
calcification from CKD individual who experienced an arterial venous fistula
operation. Immunohistology analysis of EZH2 expression (Magnification
We next examined the potential function of EZH2 in VC in vitro. VSMC Ca
deposition and calcifying nodules were increased in A7r5 cells after incubation
with high Pi calcifying medium (2.6 mmol/L Pi) for 3, 5, 7 or 10 days
(Supplementary Fig. 2A,B). CCK-8 assessment was conducted to observe the
optimal concentration of 3-DZNeP on A7r5 cells (Fig. 6A). Compared to the control
group, two days of treatment with 3-DZNeP did not reduce cell viability below
100% at any concentrations from 1 to 20 µmol/L. Dose-dependent treatment
with 3-DZNeP (2, 10 and 20 µM) for seven days reduced Ca deposition in the
presence of calcifying culture medium (Fig. 6B). VSMCs were treated with 3-DZNeP
(20 µmol/L) under
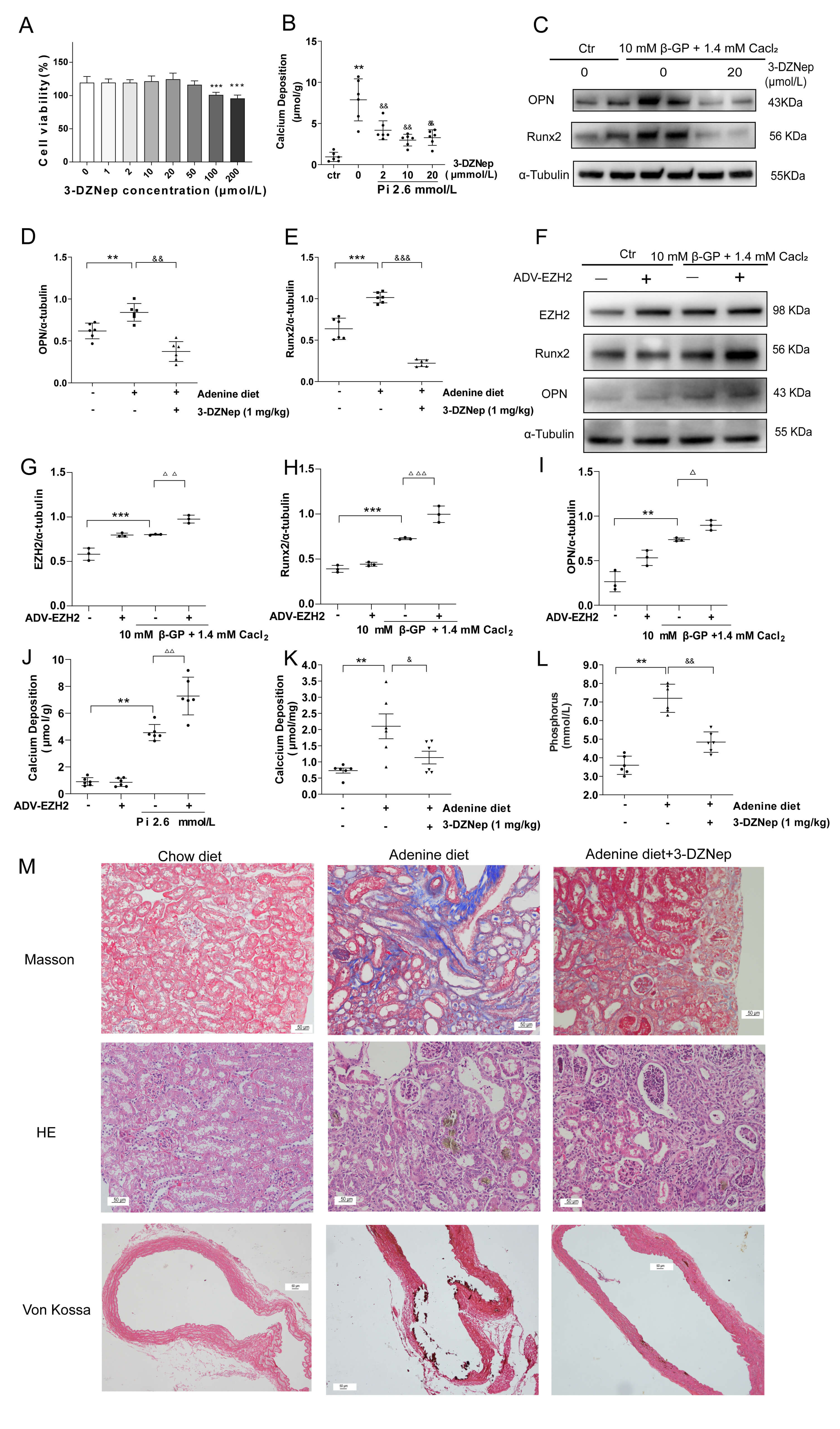 Fig. 6.
Fig. 6.
EZH2 suppression reduces VC in vitro and in
vivo. (A) Cytotoxicity of the EZH2 inhibitor 3-DZNeP (1–200
µmol/L) was assessed in VSMCs (n = 6 per group). (B) Dose
dependent effects of 3-DZNeP (2, 10, 20 µmol/L) on calcium deposition in
VSMC calcification model for 7 days were evaluated (n = 2 per group).
(C–E) Under
Next, we assessed the 3-DZNeP effect on disease progression in the mouse model of VC associated with CKD. CKD mice demonstrated a progressive interstitial fibrosis and adenine crystal deposition in the kidney as shown by Masson and HE staining, which were attenuated by 3-DZNeP (Fig. 6M upper and middle). The level of serum Pi (Fig. 6K) was enhanced in CKD mice and reduced by 3-DZNeP treatment. Notably, Von Kossa staining (Fig. 6M lower) and calcium content assay (Fig. 6L) revealed an increased Ca deposition in aorta tissue in CKD mice, which was significantly reduced by treatment with 3-DZNeP (Supplementary Table 4).
To examine the mechanism of EZH2 underlying its role in VC, the expression of BMP2, a key inducer of osteoblast differentiation was examined. BMP2 protein level was significantly upregulated in high Pi-stimulated VSMCs in a time dependent manner (Fig. 7A,B). Moreover, overexpression of EZH2 by adenovirus increased the expression of BMP2 (Fig. 7C–E). Furthermore, 3-DZNeP treatment diminished the overexpression of BMP2 in high Pi medium stimulated VSMCs, which was associated with down-expression of EZH2 and H3K27me3 (Supplementary Fig. 3A–D). IHC staining result showed that BMP2 was upregulated in calcified cultured aorta rings of mouse (Fig. 7F). The expression of EZH2 and BMP2 was increased, as shown by IHC staining (Fig. 7G), which was in paralleled with the up-regulation of Ezh2 mRNA expression in aorta tissues of CKD mice (Fig. 7H). Mechanistically, BMP2 silencing counteracted the aggravated Ca deposition induced by EZH2-overexpressing adenovirus in calcifying culture medium (Fig. 7I). Moreover, knockdown of BMP2 (Fig. 7J–L) antagonized the upregulation of Runx2 caused by EZH2 overexpression (Fig. 7M,N). Taken together, the data suggest that the progression of VC driven by EZH2 is related to the up-regulation of BMP2.
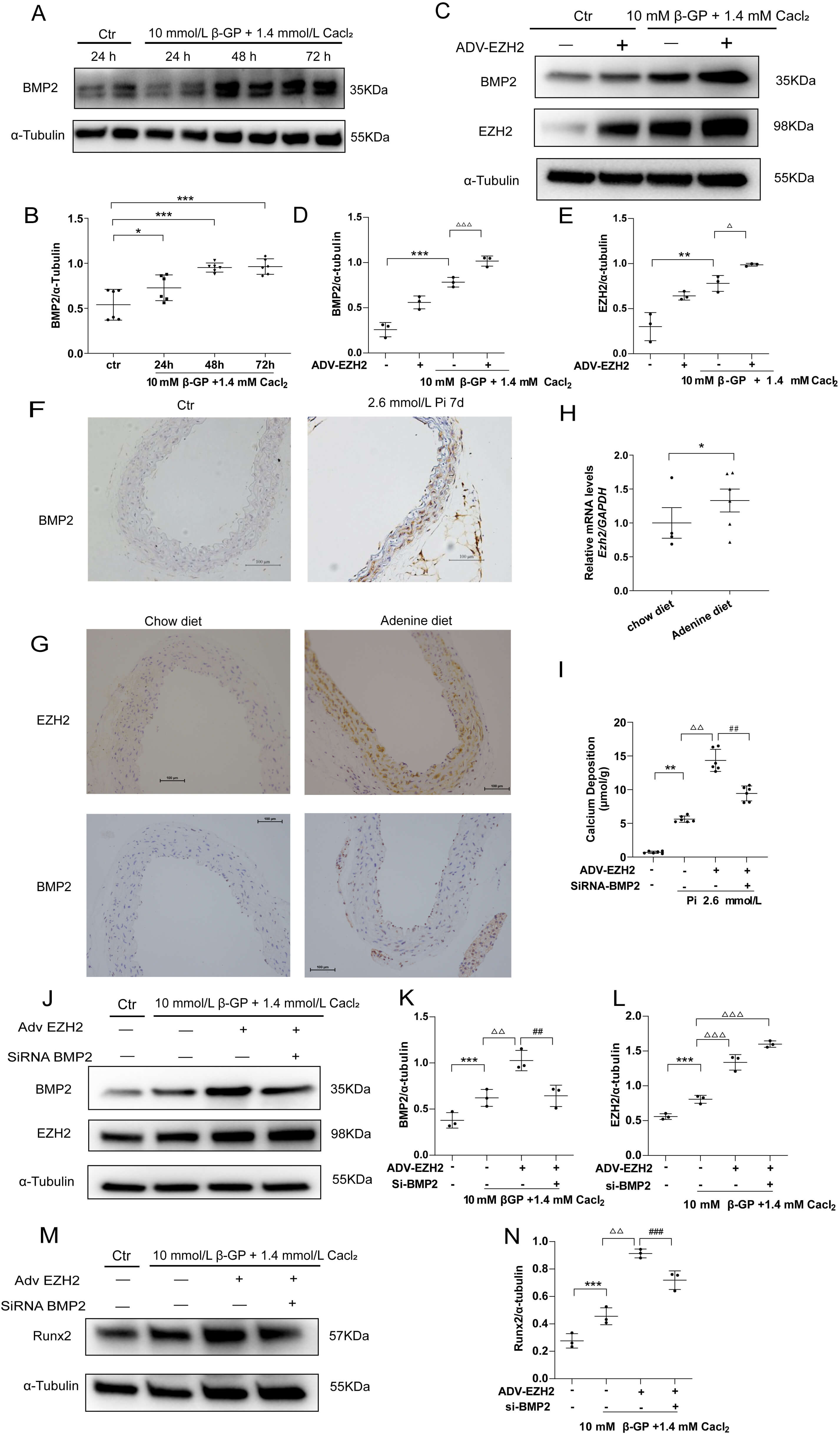 Fig. 7.
Fig. 7.
EZH2 may promote VC through up-regulation of BMP2. (A,B) BMP2
expression in VSMCs stimulated with
Herein, we found that histone methylation modification promotes VC. Throughout in vitro and ex vivo trials, we observed that EZH2 was highly expressed in calcified VSMCs and aorta culture models. In vivo, by using an adenine with high Pi diet induced VC-associated CKD model, we observed EZH2 upregulation in calcified arteries. The increased EZH2 expression was further validated in calcified radial artery samples from individuals with kidney failure who experienced arteriovenous fistula operation. We further showed that EZH2 inhibition attenuated VC in vivo and alleviated calcification of VSMCs in vitro. The specific effect of EZH2 on calcification was confirmed in VSMCs by pharmacological inhibition and genetic evidence. Finally, we showed that the pro-calcific effect of EZH2 was related to BMP2 up-regulation.
Based on our findings and previous studies, it can be proposed that EZH2 promotes VC in CKD through several epigenetic mechanisms. Firstly, EZH2 promotes osteogenic epientiation by silencing key inhibitors of the BMP signaling. EZH2 repress extracellular BMP antagonists such as Noggin [30], leading to enhanced BMP2-driven osteogenic trans-differentiation [31]. Secondly, EZH2 probably removes physiological barriers against calcification by epigenetically silencing genes associated with cytoprotective and homeostatic maintenance processes. Recent studies have demonstrated that EZH2 enhances osteogenic differentiation of VSMCs through epigenetic silencing of senescence-related genes (P16, TIMP3 and P21) [21]. Inhibiting EZH2 with GSK343 reduced the VSMCs’ calcification by activating autophagy [20].
EZH2 is up-regulated and promotes disease progression in various type of CKD [19, 32]. Our data extend the role of EZH2 in CKD-associated complications. In this study, inhibition of EZH2 may not only directly attenuate the osteogenic transformation of VSMCs and indirectly mitigate VC by delaying the progression of CKD for improving Ca/Pi homeostasis. Indeed, we observed improved renal fibrosis and reduced Pi concentration in 3-DZNeP-treated CKD mice in this study. Thus, EZH2 could be a multi-organ therapeutic target for intervention in CKD-associated VC.
However, there are several limitations in our study. On one hand, the mechanistic link between EZH2 and BMP2 is unclear, which warrants further epigenetic investigation. In the next step of study, approaches such as ChIP, CUT&Tag assays would be employed to elucidate EZH2’s epigenetic regulatory effects on downstream targets. On the other hand, the analysis of human vascular samples was based on a relatively small cohort, which was exploratory in nature. Although the findings align with our laboratory results, the limited sample size may affect the statistical power and generalizability of the outcomes. Upcoming investigations with larger, multi-center cohorts are required to verify these observations.
Building on the observation of elevated EZH2 levels in calcified vessels from CKD individuals to various preclinical calcification models (CKD mice, rat aorta culture and VSMCs), EZH2 was defined as a key driver of VC in CKD , and its inhibition by 3-DZNeP reduced calcification in VSMCs. Mechanistically, EZH2 probably promotes VC through increasing BMP2 expression. This study not only identifies EZH2 as a critcal regulator in the VC development associated with CKD but also broadens our understanding of the importance of epigenetic regulation in VC. These evidences underline the targeting of EZH2 as an innovative therapeutic strategy for treating VC.
All data supporting the conclusions of this article are presented in the paper, but raw data will be available by request to the corresponding author.
PL: Writing—original draft, Investigation. YX: Investigation, Data curation. DC: Investigation. FY: Formal analysis. DH: Data curation. AK: Visualization. YZ: Writing—review & editing, Conceptualization. MW: Writing—review & editing, Formal analysis, Investigation. CY: Supervision, Conceptualization, Writing—review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was conducted in accordance with the Declaration of Helsinki and approved by the ethics Committees of Shuguang Hospital of Traditional Chinese Medicine University of Shanghai (approval number: 2018-575-04-01). Written informed consent was obtained from the patient before surgery. The animal experimental protocol was approved by the Animal Experimentation Ethics Committee of Shanghai University of Traditional Chinese Medicine (PZSHUTCM190920010). All experiments were performed in accordance with the guidelines set forth in the Guidance Suggestions for the Humane Treatment of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China and the national standard’Laboratory Animal—Guideline for Ethical Review of Animal Welfare (GB/T 35892-2018).
Not applicable.
This study was supported by National Natural Foundation of China [Grant numbers 82170747, 82305095] and the joint funding of the Kashgar Workstation of the State Key Laboratory of Causes and Prevention of High Incidence in Central Asia [Grant numbers SKL-HIDCA2024-KS].
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL48723.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.