






 , Jinyu Chang 2,†, Yi Liu 1,2,3, Yusang Shao 1, Yanyan Zhang 1, Yingru Zheng 2, Enfang Xiao 2, Yanqing Xie 1, Shuangshuang Tu 1, Haoxuan Lu 1, Wenming He 1,3,*
, Jinyu Chang 2,†, Yi Liu 1,2,3, Yusang Shao 1, Yanyan Zhang 1, Yingru Zheng 2, Enfang Xiao 2, Yanqing Xie 1, Shuangshuang Tu 1, Haoxuan Lu 1, Wenming He 1,3,*
1 Department of Cardiology, The First Affiliated Hospital of Ningbo University, 315010 Ningbo, Zhejiang, China
2 Department of Respiratory Disease, The First Affiliated Hospital, Jinzhou Medical University, 121000 Jinzhou, Liaoning, China
3 Institute of One Health Science, School of Civil & Environmental Engineering and Geography Science, State Key Laboratory for Quality and Safety of Agro-products, Ningbo University, 315211 Ningbo, Zhejiang, China
†These authors contributed equally.
Abstract
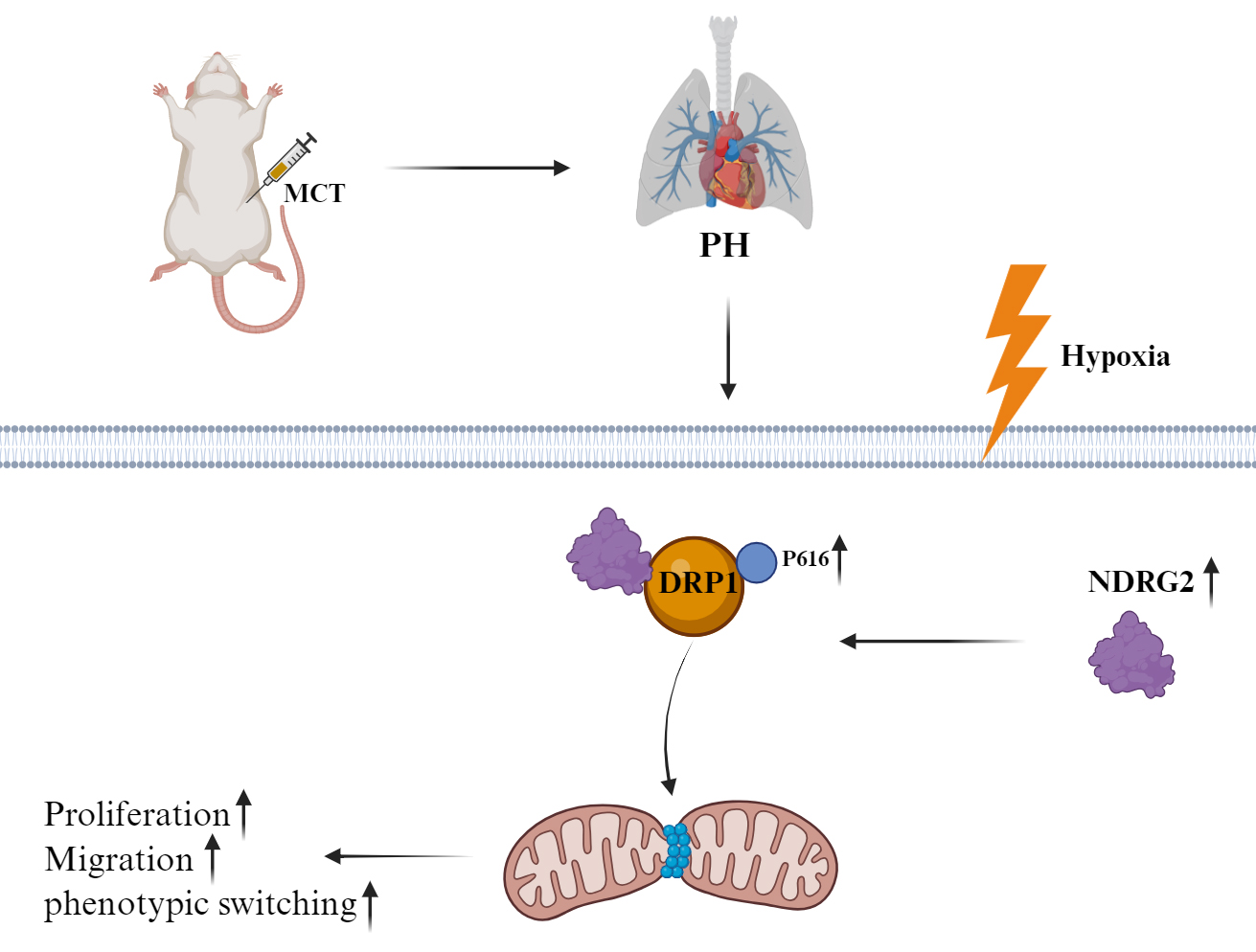
Pulmonary hypertension (PH) is a progressive disease characterized by obstructive pulmonary vascular remodeling, for which no curative therapies effectively reverse disease progression. This study investigated whether N-myc downstream-regulated gene 2 (NDRG2) drives PH pathogenesis by regulating mitochondrial dynamics.
NDRG2 expression was examined in two rat models of PH (SuHx and MCT). Functional studies using NDRG2 knockdown and overexpression in PASMCs assessed phenotypic switching, proliferation, migration, mitochondrial morphology, and bioenergetics. The NDRG2-DRP1 interaction was investigated via co-immunoprecipitation and immunofluorescence. An in vivo rescue experiment was performed using intratracheal AAV9-shNDRG2 delivery in SuHx rats.
NDRG2 was significantly upregulated in PASMCs from PH rats, hypoxic human PASMCs, and patients with PH. NDRG2 knockdown attenuated, while its overexpression exacerbated, hypoxia-induced phenotypic switching, proliferation, and migration of PASMCs. Mechanistically, NDRG2 directly interacted with DRP1 and specifically promoted its activating phosphorylation at Ser616, leading to excessive mitochondrial fission, ATP depletion, and oxidative stress. These pathogenic effects were abolished by concurrent DRP1 knockdown. In vivo, NDRG2 knockdown ameliorated hemodynamic indices, right ventricular hypertrophy, pulmonary vascular remodeling, and exercise capacity in SuHx rats.
NDRG2 drives PH progression by promoting DRP1-mediated mitochondrial fission and vascular remodeling. The NDRG2-DRP1 axis represents a candidate pathway for therapeutic exploration in PH.
Graphical Abstract
Keywords
- NDRG2 protein
- dynamin-related protein 1
- hypertension
- pulmonary
- mitochondrial dynamics
- genetic vectors
- vascular remodeling
Pulmonary hypertension (PH) is a life-threatening pulmonary vascular disorder characterized by pathological pulmonary vascular remodeling and lumen obstruction, leading to a persistent increase in pulmonary vascular resistance and ultimately resulting in right-sided heart failure [1, 2, 3]. Although current therapies (e.g., endothelin receptor antagonists and prostacyclin analogues) can partially alleviate vasoconstriction, they generally fail to effectively halt or reverse structural vascular remodeling [4, 5, 6]. Therefore, exploring novel targets capable of directly reversing vascular remodeling is of great importance.
Excessive proliferation, migration, and phenotypic switching of pulmonary arterial smooth muscle cells (PASMCs) from a contractile to a synthetic secretory phenotype are considered central cellular mechanisms driving pulmonary vascular remodeling and lumen occlusion [7].
N-myc downstream-regulated gene 2 (NDRG2) is a member of the N-myc
downstream-regulated gene family, which possesses an
On the contrary, aberrant mitochondrial fragmentation has been identified as a significant contributor to PH, exacerbating vascular remodeling by promoting the proliferation, migration, and phenotypic switching of PASMCs, whereas the inhibition of this process confers therapeutic benefits [16, 17, 18]. This pathological mitochondrial fission event is primarily regulated by the post-translational modifications of dynamin-related protein 1 (DRP1): phosphorylation of DRP1 at serine 616 (Ser616) promotes its translocation to mitochondria and initiates fission, whereas the phosphorylation at serine 637 (Ser637) exerts an inhibitory effect [16, 19]. Given the central role of NDRG2 in regulating cellular signaling pathways, we hypothesize that NDRG2 may influence mitochondrial dynamics, and thereby contribute to PH progression, by perturbing the balance of DRP1 phosphorylation.
Based on this background, this study was conducted to systematically address three core questions: (1) to determine whether NDRG2 is involved in the pathogenesis and progression of PH; (2) to investigate the molecular mechanism by which NDRG2 promotes vascular remodeling, specifically examining whether it acts by regulating DRP1 phosphorylation-mediated mitochondrial fragmentation; and (3) to evaluate the potential therapeutic value of targeting NDRG2 for ameliorating pathological vascular remodeling.
All experimental procedures involving animals were approved by the Animal Ethics
Committee of Ningbo University (Approval No.: AEWC-NBU20250371). Twenty-four male
Sprague–Dawley (SD) rats (aged 6–8 weeks and weighing 180–220 g) were housed
under standard laboratory conditions and randomly allocated to four experimental
groups (n = 6 per group): normoxia control, normoxia + AAV9-shNDRG2,
hypoxia + Sugen 5416 (SuHx), and SuHx + AAV9-shNDRG2. For genetic knockdown, the
rats received an intratracheal instillation of adeno-associated virus 9 (AAV9)
encoding short hairpin RNA against NDRG2 (AAV9-shNDRG2; 1.75
The PH model was subsequently induced. The rats in the SuHx and SuHx+AAV9-shNDRG2 groups were housed in a hypoxic chamber (10% O2, Oxycycler A84XOV; BioSpherix, Parish, NY, USA), with CO2 maintained below 3% using soda lime. On the first day of hypoxia, these rats received a single intraperitoneal injection of Sugen 5416 (20 mg/kg; Bio-Techne, America) dissolved in vehicle. The rats in the control groups were maintained under normoxia (21% O2) throughout the experiment. The body weight and health status were monitored weekly. At the study endpoint, all rats were euthanized under sodium pentobarbital anesthesia (150 mg/kg, i.p.).
The exercise capacity was evaluated using a motorized treadmill. After acclimatization, the test commenced at 10 m/min, with speed increasing by 5 m/min every 5 min until exhaustion or a maximum of 30 min. The total running distance was recorded [21].
Following euthanasia, the heart and lung tissues were harvested. The right
ventricle (RV) was dissected and weighed separately from the left ventricle plus
septum (LV + S) to calculate the RV hypertrophy index (RVHI = RV/[LV + S]). The
right lung was snap-frozen for molecular analysis. The left lung was
inflation-fixed with 4% paraformaldehyde (PFA, P0099-3L, Beyotime, Shanghai,
China), processed for paraffin embedding, and sectioned (5 µm). Hematoxylin
and eosin (H&E)-stained sections were used to quantify the wall thickness (%)
of pulmonary arterioles in vessels with an external diameter of 50–150 µm,
calculated as (medial wall thickness/external diameter)
RV hemodynamics were assessed via direct catheterization before euthanasia. Under anesthesia (sodium pentobarbital, 40 mg/kg, i.p.), the right external jugular vein was cannulated with a heparinized PE-50 catheter (Taimeng, Chengdu, China). The catheter was advanced into the RV under continuous-pressure monitoring (BL-420N; Taimeng, Chengdu, China). RV systolic pressure (RVSP) was recorded in real time and used as a surrogate for pulmonary arterial systolic pressure.
For immunofluorescence analysis, 5-µm lung sections were deparaffinized,
rehydrated, and subjected to antigen retrieval. The sections were blocked with
5% bovine serum albumin (BSA) and incubated overnight at 4 °C with
primary antibodies against
Human pulmonary arterial smooth muscle cells (HPASMCs; #3110, Sciencell, Carlsbad, CA, USA) were cultured in smooth muscle cell medium (#1101; ScienCell) at 37 °C with 5% CO2. Following is the example of the statement for cell lines: All cell lines were validated by STR profiling and tested negative for mycoplasma. Experiments used cells at passages 2–6. For stable NDRG2 knockdown, the cells were transduced with lentiviral vectors encoding short hairpin RNA (shRNA) against NDRG2 (shNDRG2; GeneChem). The targeting sequence was as follows: 5′-CCGGGAGGACATGCAGGAAATCATTCTCGAGAATGATTTCCTGCATGTCCTCTTTTTG-3′. A nontargeting shRNA (shRandom) served as the control. The cells were infected at a multiplicity of infection of 20 in the presence of 8 µg/mL polybrene for 24 h, followed by selection with 2 µg/mL puromycin for 72 h. For hypoxia treatment, the transduced cells were exposed to 1% O2 in a tri-gas incubator for 24 h. This condition was selected based on previous study showing that it robustly induced PASMC dysfunction [22].
The fixed and permeabilized HPASMCs were blocked with 5% BSA (ST023-50g, Beyotime, Shanghai, China) and incubated with primary antibodies against the translocase of outer membrane 20 (TOMM20; 1:200; ab186735, Abcam) or co-incubated with anti-NDRG2 and anti-dynamin-related protein 1 (DRP1; 1:200; ab184247, Abcam) antibodies. After incubation with fluorophore-conjugated secondary antibodies, the nuclei were stained with DAPI. The confocal images were captured using a Zeiss LSM 880 microscope (Carl Zeiss AG, Oberkochen, Germany). Specificity was confirmed using isotype and secondary antibody–only controls.
Cell proliferation was assessed using 5-ethynyl-2′-deoxyuridine (EdU) incorporation and cell counting kit-8 (CCK-8) assays. For the EdU assay, the cells were pulsed with 10 µM EdU for 2 h, fixed, and detected using a Click-iT kit (K1076l; APExbio, Houston, TX, USA). EdU-positive nuclei were counted. For the CCK-8 assay, the cells were seeded in 96-well plates, and the absorbance at 450 nm was measured 2 h after adding CCK-8 reagent at the indicated time points.
Cell migration was evaluated using Transwell chambers. HPASMCs (1
Cell lysates were prepared using RIPA buffer. Proteins (20 µg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and probed with specific primary antibodies (Table 1), followed by horseradish peroxidase–conjugated secondary antibodies. The signals were detected by enhanced chemiluminescence and quantified with ImageJ (version 1.52, National Institutes of Health, Bethesda, MD, USA).
| Antibody | Cat. No. | Producer | Dilution ratio |
| Anti-NDRG2 | Ab174850 | Abcam | 1:1000 |
| Anti-DRP1 | Ab184247 | Abcam | 1:2000 |
| Anti-p-DRP1 (Ser616) | Ab614755 | Abcam | 1:2000 |
| Anti-p-DRP1 (Ser637) | Ab193216 | Abcam | 1:2000 |
| Anti-MFN1 | Ab221661 | Abcam | 1:2000 |
| Anti-MFN2 | Ab124773 | Abcam | 1:1000 |
| Anti-OPA1 | Ab157457 | Abcam | 1:1000 |
| Anti-FN1 | Ab45688 | Abcam | 1:1000 |
| Anti-Col I | Ab6308 | Abcam | 1:2000 |
| Anti- |
#19245 | CST | 1:5000 |
| Anti- |
AF7018 | Affinity | 1:5000 |
HPASMC lysates (500 µg) from hypoxic cells were incubated with anti-NDRG2 antibody overnight, followed by incubation with Protein A/G beads. The immunocomplexes were washed, eluted, and analyzed by Western blotting. Control immunoglobulin G was used to confirm specificity.
The data were presented as mean
We established two widely used rat models to investigate the role of NDRG2 in PH: the SuHx model and the monocrotaline (MCT)-induced model. The model rats developed the hallmark features of PH, including significant right ventricular hypertrophy, cardiomyocyte enlargement, and pronounced pulmonary vascular remodeling and occlusion (Fig. 1A,D,E). These pathological changes were accompanied by elevated RVSP and RVHI (Fig. 1B,C), as well as impaired exercise tolerance (Fig. 1F), confirming successful establishment of the model.
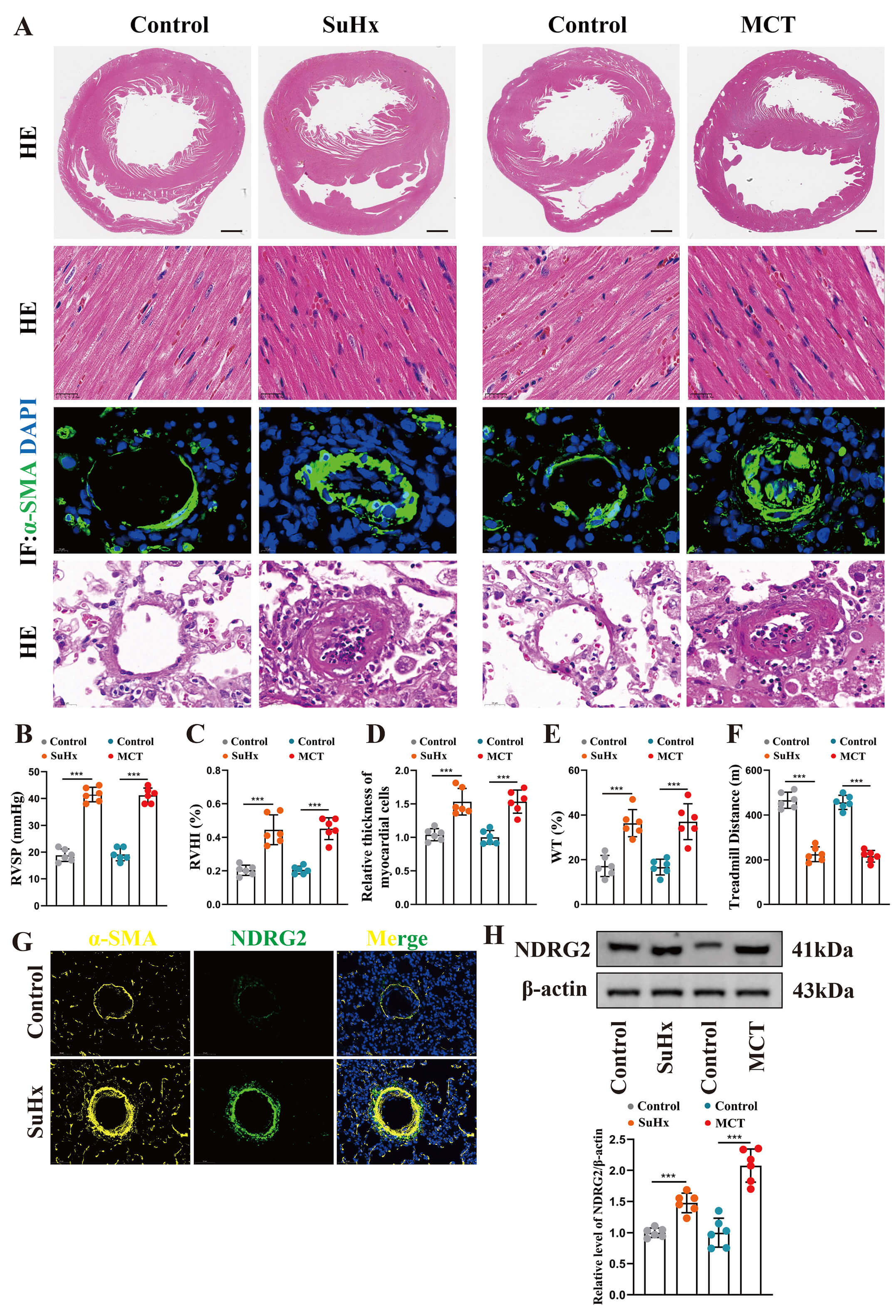 Fig. 1.
Fig. 1.
NDRG2 was upregulated in experimental pulmonary hypertension rat
models. (A) Representative images of heart and lung tissues from the indicated
groups. Top row: Whole-heart gross morphology. Second row: H&E staining of
cardiomyocytes. Third row: Immunofluorescence staining of pulmonary vessels for
Immunofluorescence staining revealed robust co-localization of NDRG2 with the
smooth muscle marker
We next elucidated the functional role of NDRG2 in HPASMCs. Hypoxia treatment led to a time-dependent increase in NDRG2 expression, which peaked at 24 h (Fig. 2A); this time point was thus used for subsequent experiments.
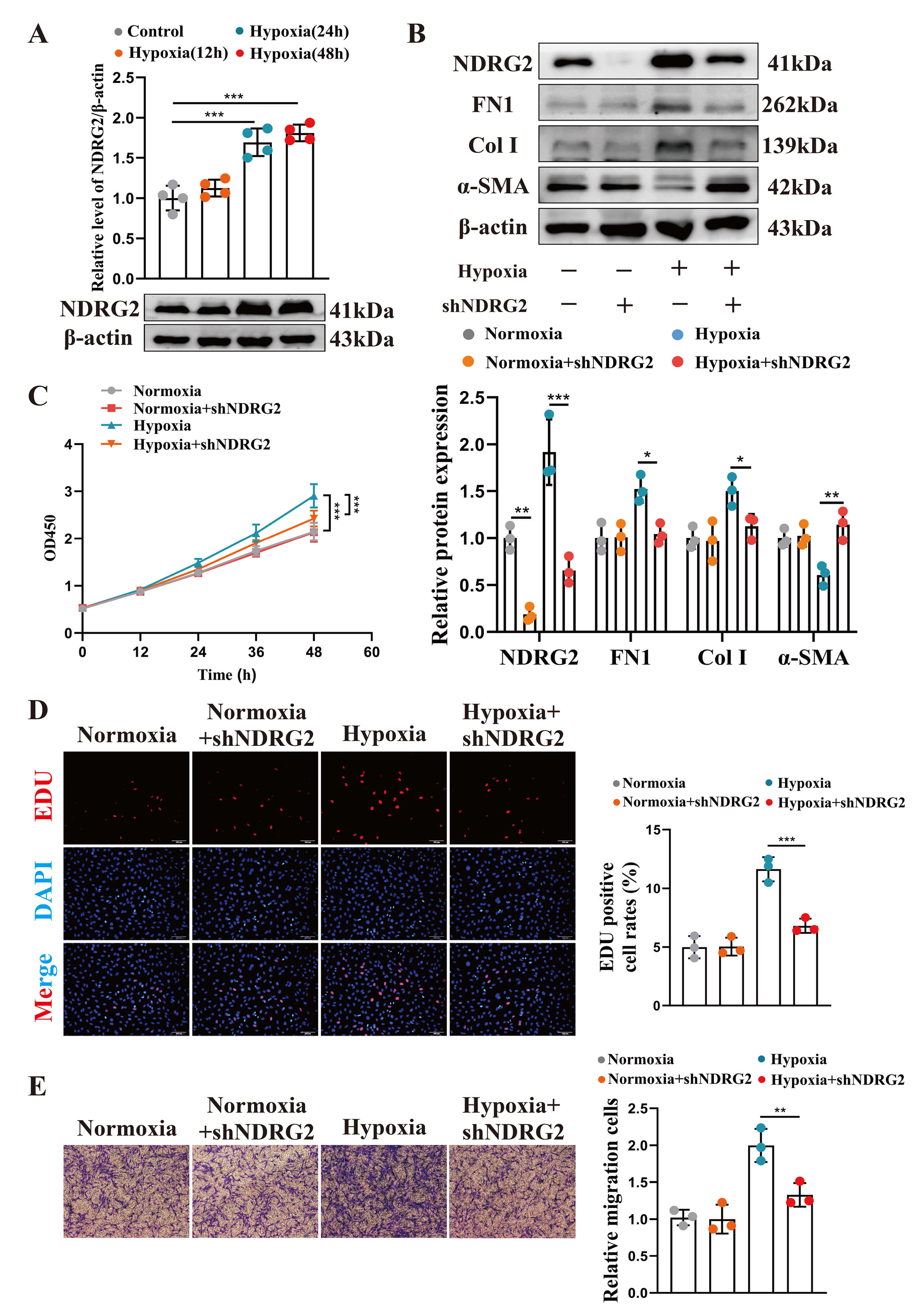 Fig. 2.
Fig. 2.
NDRG2 knockdown attenuated key pathological phenotypes in
hypoxic HPASMCs. (A) Western blot analysis of NDRG2 protein expression in
HPASMCs exposed to hypoxia for the indicated durations. (B) Western blot analysis
of NDRG2, FN1, Col I, and
The knockdown of NDRG2 with specific shRNA (Fig. 2B) attenuated hypoxia-induced phenotypic switching from a contractile to a synthetic secretory phenotype (Fig. 2B), and suppressed cell proliferation (Fig. 2C,D) and migration (Fig. 2E). On the contrary, NDRG2 overexpression under hypoxia (Fig. 3A) exacerbated phenotypic switching (Fig. 3A), potentiated proliferation (Fig. 3B,C), and enhanced migration (Fig. 3D). These data establish NDRG2 as a key driver of pathological HPASMC behavior under hypoxic conditions.
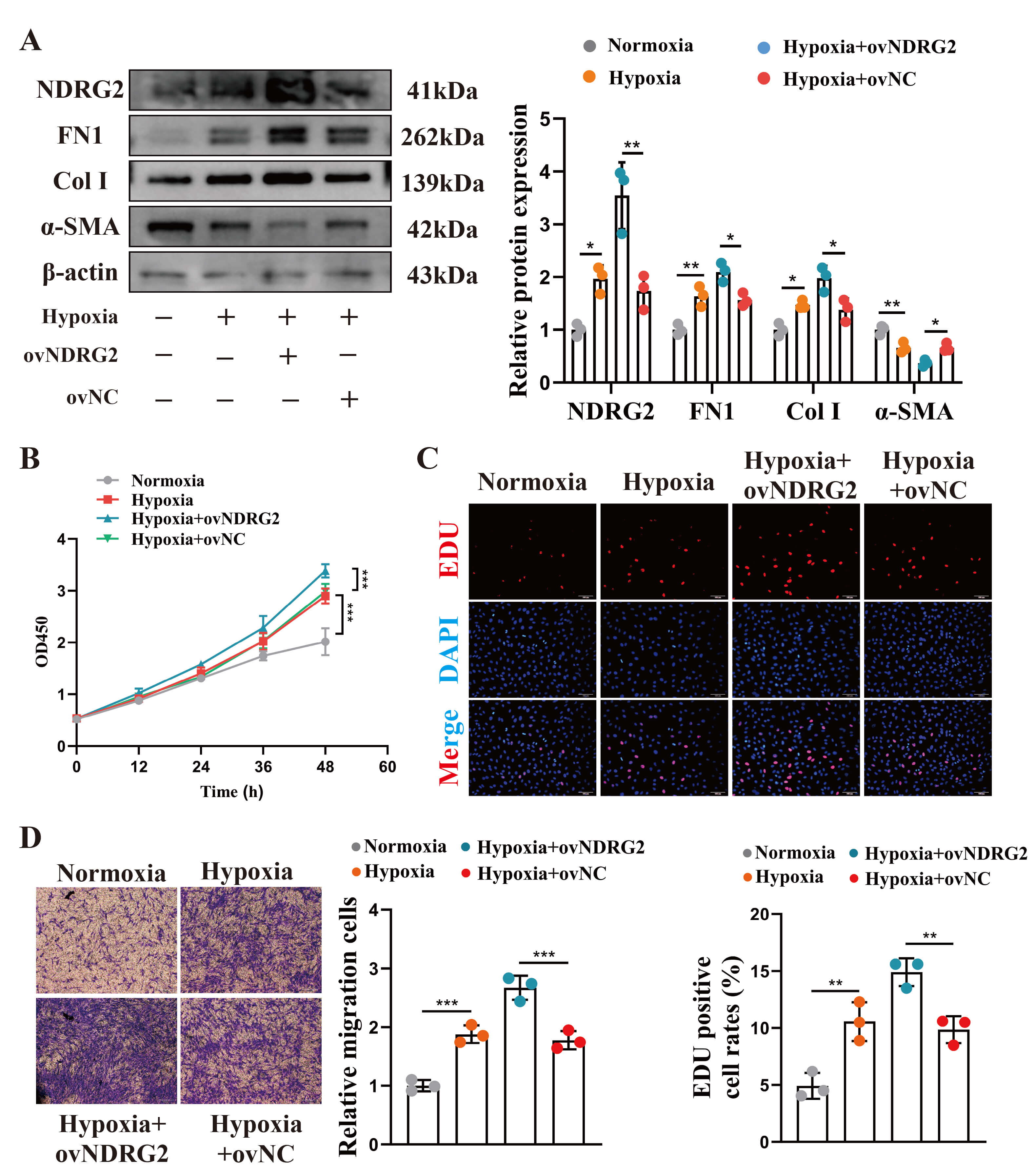 Fig. 3.
Fig. 3.
NDRG2 overexpression exacerbated key pathological phenotypes in
hypoxic HPASMCs. (A) Western blot analysis under indicated conditions. (B)
Proliferation detected using CCK-8 assay (n = 6). (C) Proliferation by
EdU staining (red). Scale bar, 100 µm. (D) Migration detected using the
Transwell assay. Scale bar, 50 µm. (one-way ANOVA with Tukey’s
test). *p
As hypoxia disrupts cellular metabolism, we assessed the impact of NDRG2 on bioenergetics. Hypoxia reduced adenosine triphosphate (ATP) generation and increased reactive oxygen species (ROS) production in HPASMCs, both of which were rescued by NDRG2 knockdown (Fig. 4A,B).
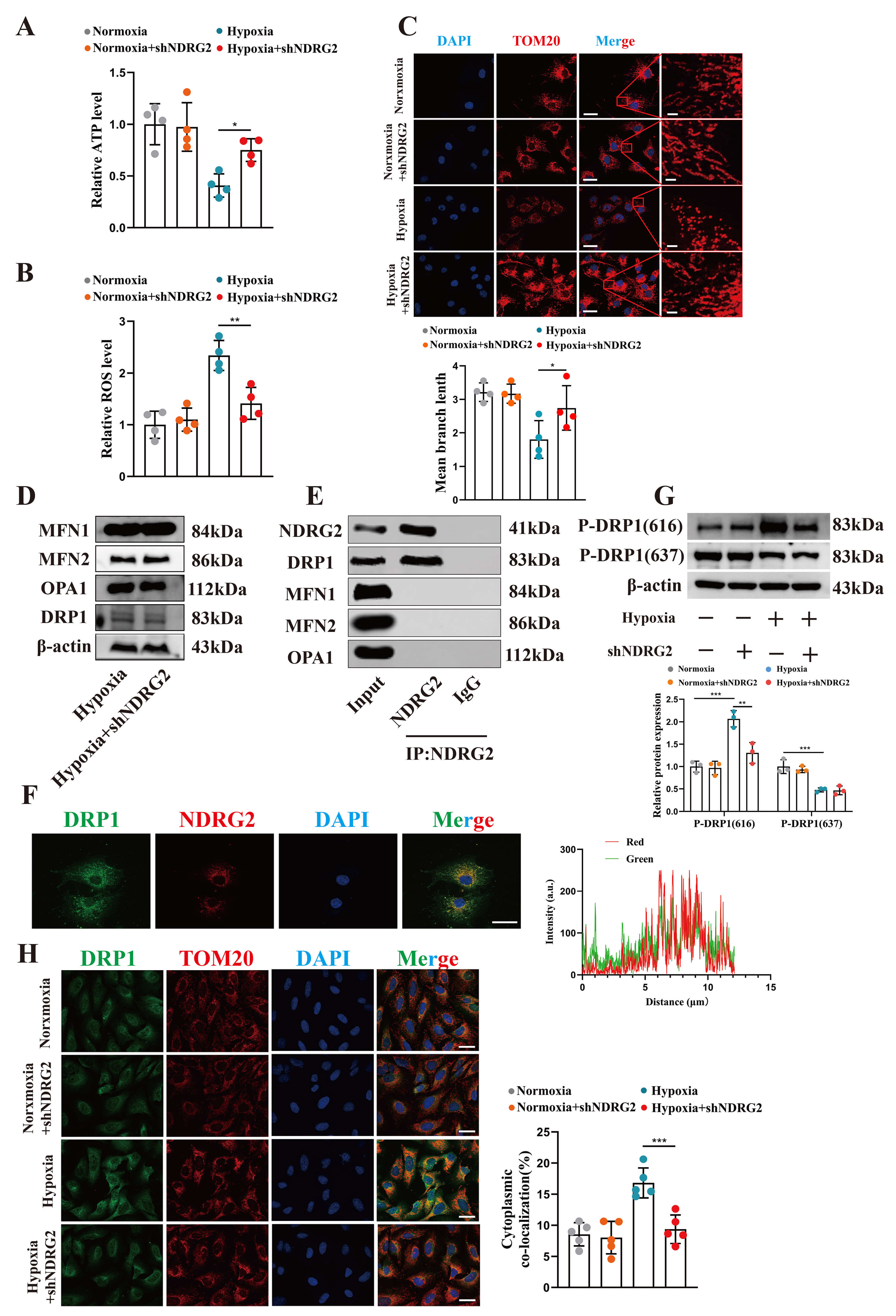 Fig. 4.
Fig. 4.
NDRG2 drove mitochondrial fission by binding to and promoting
the phosphorylation of DRP1. (A,B) Quantitative detection of intracellular ATP
levels (A) and ROS levels (B) in HPASMCs under different treatment conditions.
(C) Upper panel: Representative images of mitochondrial morphology in HPASMCs
under different conditions, observed by immunofluorescence staining of TOM20.
Scale bar, 10 µm. Lower panel: Quantitative analysis of mitochondrial
branch length in the corresponding groups. (D) Western blot analysis of
mitochondrial dynamics–related proteins (MFN1, MFN2, OPA1, and DRP1) in HPASMCs
after NDRG2 knockdown. (E) Interaction between NDRG2 and DRP1 detected by
co-immunoprecipitation (Co-IP) of NDRG2 from HPASMC lysates. (F) Representative
immunofluorescence images and quantitative co-localization analysis of NDRG2
(red) and DRP1 (green) in hypoxic HPASMCs. Scale bar, 10 µm. (G)
Representative Western blot bands and quantitative analysis of DRP1
phosphorylation levels at Ser616 and Ser637 in HPASMCs under different
conditions. (H) Representative images of DRP1 mitochondrial translocation
observed by co-staining of DRP1 (green) and TOM20 (red) under different
conditions. Scale bar, 10 µm. *p
Given the central role of mitochondria in energy production, we examined mitochondrial morphology. Staining for the mitochondrial marker TOM20 revealed that hypoxia induced extensive mitochondrial fragmentation, which was prevented by NDRG2 depletion (Fig. 4C). Although NDRG2 knockdown did not alter the total protein levels of key mitochondrial dynamics regulators (MFN1, MFN2, OPA1, and DRP1) (Fig. 4D), Co-IP using an anti-NDRG2 antibody confirmed an interaction between NDRG2 and DRP1 (Fig. 4E), which was further supported by their strong subcellular co-localization (Fig. 4F).
We then investigated DRP1 activation. Hypoxia promoted the phosphorylation of DRP1 at the Ser616 activation site and suppressed phosphorylation at the inhibitory Ser637 site. NDRG2 knockdown specifically abrogated hypoxia-induced DRP1 Ser616 phosphorylation without impacting Ser637 (Fig. 4G). Since Ser616 phosphorylation facilitates DRP1 translocation to mitochondria, we assessed this event and found that hypoxia-enhanced mitochondrial localization of DRP1 was markedly reduced upon NDRG2 knockdown (Fig. 4H). These results demonstrated that NDRG2 interacted with DRP1 and specifically promoted its phosphorylation at the Ser616 activation site under hypoxia. The specific upstream kinase responsible for this effect remains to be identified. However, the aforementioned findings establish NDRG2 as a key regulator of DRP1 activation in this setting.
We performed rescue experiments to definitively establish DRP1 as the key downstream effector of NDRG2. The knockdown of DRP1 (Fig. 5A) reversed the exacerbating effects of NDRG2 overexpression on phenotypic switching (Fig. 5A), proliferation (Fig. 5B,C), and migration (Fig. 5D) under hypoxia. These results demonstrated that NDRG2 drove HPASMC dysfunction primarily in a DRP1-dependent manner.
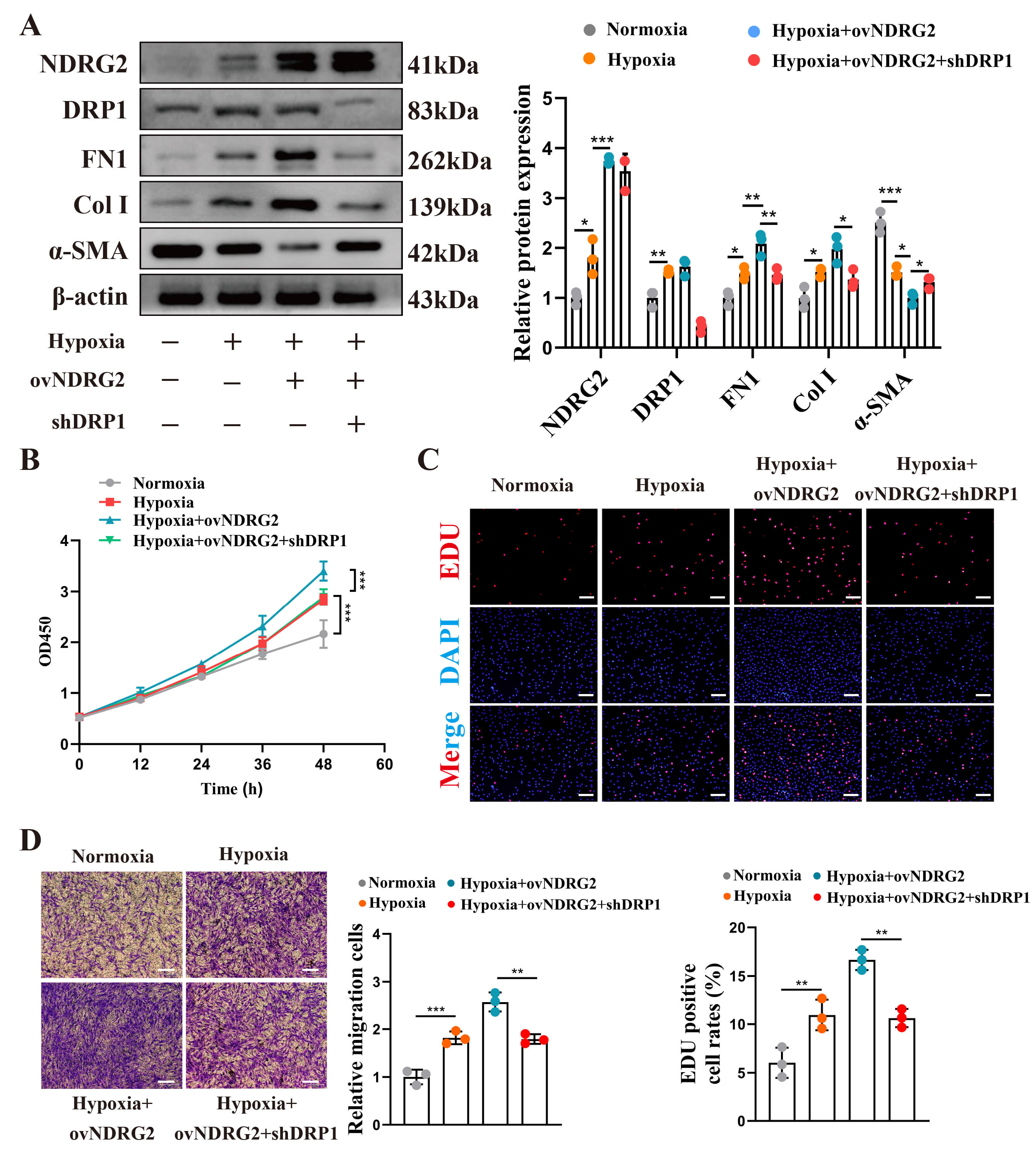 Fig. 5.
Fig. 5.
NDRG2 mediated hypoxia-induced pathological behaviors in
pulmonary arterial smooth muscle cells through a DRP1-dependent mechanism. (A)
Western blot analysis under the indicated conditions. (B) Proliferation detected
using CCK-8 assay (n = 6). (C) Proliferation detected using EdU staining
(red). Scale bar, 200 µm. (D) Migration detected using the Transwell assay.
Scale bar, 50 µm. *p
Finally, we assessed the preventive potential of targeting NDRG2 in vivo. Rats received intratracheal AAV9-shNDRG2 to knock down lung NDRG2 expression 2 weeks before initiation of SuHx treatment, allowing evaluation of whether NDRG2 inhibition could attenuate disease development. The intratracheal delivery of AAV9-shNDRG2 efficiently knocked down NDRG2 expression in the lungs of SuHx rats (Fig. 6A,H). This intervention markedly attenuated disease progression, as evidenced by reduced right ventricular hypertrophy (Fig. 6B), inhibition of pulmonary vascular remodeling (Fig. 6D,E,F), and lower RVSP (Fig. 6C). Consequently, the exercise capacity of treated rats was significantly improved (Fig. 6G). Furthermore, NDRG2 knockdown reversed the pathological phenotypic switching of smooth muscle cells in isolated pulmonary arteries (Fig. 6H).
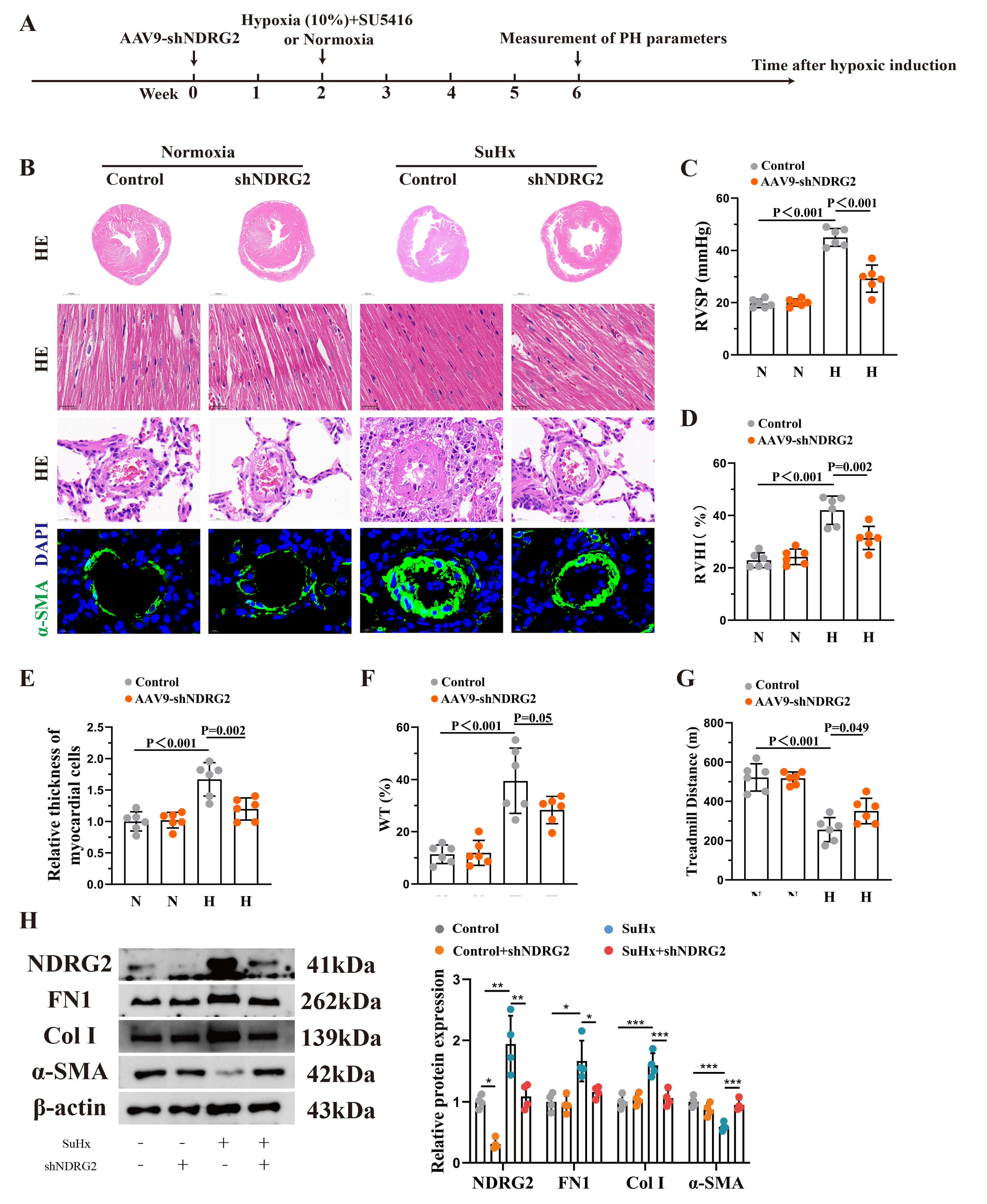 Fig. 6.
Fig. 6.
In vivo knockdown of NDRG2 alleviated the progression
of pulmonary hypertension in rats. (A) Schematic diagram of the animal
experimental design. (B) Representative images of heart and lung tissue sections
from the indicated groups (control, control + AAV9-shNDRG2, SuHx, and SuHx +
AAV9-shNDRG2). Scale bars from top to bottom: 2 mm, 25 µm, 20 µm, and
10 µm. (C–G) Quantification of RVSP (C), RVHI (D), cardiomyocyte diameter
(E), arteriolar wall thickness (%) (F), and running distance (G) (n =
6). (H) Western blot analysis of protein expression in isolated pulmonary
arterial media. *p
Our study identified NDRG2 as an additional member of the NDRG family involved in pulmonary vascular remodeling in PH, demonstrating its crucial role in regulating mitochondrial dynamics. We established that NDRG2 acted as a molecular switch in pathological PASMCs, linking hypoxic stress to mitochondrial fission and metabolic dysregulation through direct interaction with DRP1 and precise modulation of its phosphorylation status. This pathway ultimately drove disease progression. Our findings not only provide novel insights into PH pathogenesis but also reveal a potential therapeutic target.
The biological functions of NDRG2 are highly context dependent, varying by cell type and pathological setting, with reported roles spanning from cytoprotection to disease promotion [24, 25]. This functional diversity is reflected in its variable response to hypoxia across various cell lineages, thereby underscoring the complexity of its regulation [26]. Our data definitively showed that NDRG2 is upregulated and exerts a net pro-pathogenic effect in hypoxic PASMCs, aligning with its characterization as a stress-responsive gene. Notably, our previous work identified a pro-remodeling function for its homolog NDRG1 in PH [12]. The congruent pathogenic role of NDRG2 highlighted in this study points to a conserved importance of the NDRG protein family in pulmonary vascular pathology. A comparative analysis suggests that both NDRG1 and NDRG2 may converge on disrupting mitochondrial homeostasis; however, NDRG1 has also been implicated in modulating additional pathways such as PI3K–Akt signaling. This indicates a shared nodal role in metabolic dysregulation, alongside isoform-specific mechanisms that may collectively drive vascular remodeling. Furthermore, our in vitro studies employed a sustained 1% O2 exposure to robustly elicit cellular responses. In human PH, vascular cells may experience more moderate or intermittent hypoxia. Future studies investigating the threshold and dynamics of NDRG2 induction and DRP1 activation across a range of oxygen tensions may be valuable to better understand how this pathway engages across the spectrum of hypoxic stress relevant to clinical disease.
The central mechanistic advance of our work is the delineation of the NDRG2–DRP1–mitochondrial fission axis. Mitochondrial fragmentation is a well-established driver of PH pathogenesis [27, 28], facilitating vascular remodeling by promoting PASMC proliferation and conferring resistance to apoptosis [17, 29]. DRP1 serves as the master regulator of mitochondrial fission, with its activity predominantly controlled by opposing post-translational modifications: activating phosphorylation at Ser616 and inhibitory phosphorylation at Ser637 [16, 30]. We found that NDRG2 specifically enhanced DRP1 phosphorylation at the Ser616 activation site via a direct molecular interaction, without altering total DRP1 levels or the phosphorylation status at Ser637. This selective effect suggests that NDRG2 interfaces with upstream regulators specific to the Ser616 pathway (e.g., ERK1/2 or CDK1), thereby shifting the phosphorylation balance decisively toward activation to drive pathological fission. This modification is crucial for DRP1 activation and its subsequent translocation to mitochondria. Consequently, we placed NDRG2 at a pivotal regulatory node, directly governing the DRP1 activation switch to transduce extracellular hypoxic cues into intracellular metabolic pathology, evidenced here by bioenergetic failure (reduced ATP levels) and oxidative stress (ROS accumulation). Based on this observed dysfunction, we predicted that NDRG2 overexpression also impaired mitochondrial respiratory function, leading to reduced oxygen consumption rates and compromised membrane potential. Direct measurement of these parameters in future studies can provide a more detailed resolution of the metabolic alterations downstream of the NDRG2-DRP1 axis. These pathological changes collectively propel the proliferation and migration of PASMCs.
Despite these insights, our study had certain limitations. First, our
mechanistic conclusions were derived primarily from studies on PASMCs. Although
this focused approach allowed detailed dissection of the NDRG2–DRP1 axis, we
acknowledge that PH is a multicellular disease involving endothelial dysfunction,
fibroblast activation, and immune cell infiltration. Our in vivo
AAV9-mediated knockdown was not cell specific, and thus the therapeutic effects
observed might have resulted from NDRG2 inhibition in multiple cell types. The
contribution of NDRG2 within nonsmooth muscle vascular cells to PH pathogenesis
remains an important area for future investigation. Second, the precise upstream
mechanism whereby NDRG2 facilitates DRP1 Ser616 phosphorylation remains to be
fully elucidated. Although our study established the functional consequence of
this modification, identifying the responsible kinase(s) is a key remaining
question. NDRG2 may act as a scaffold to recruit specific kinases known to
phosphorylate DRP1 at Ser616 (e.g., ERK1/2, CDK1, or PKC
In summary, our study identified NDRG2 as a key upstream regulator driving pathological mitochondrial fission and vascular remodeling in PH. We demonstrated that NDRG2 was upregulated in hypoxic PASMCs and experimental PH models, where it promoted a synthetic secretory phenotype, proliferation, and migration. Mechanistically, NDRG2 directly interacted with DRP1 to specifically enhance its phosphorylation at Ser616, leading to excessive mitochondrial fission, bioenergetic dysfunction, and oxidative stress. Crucially, the genetic inhibition of NDRG2 in vivo ameliorated hemodynamic and structural pathologies in rats with SuHx-induced PH. These findings delineate a novel NDRG2–DRP1 axis central to PH pathogenesis in preclinical models, thereby nominating this pathway for further mechanistic and translational research (Graphical Abstract).
The datasets used and analyzed in this study are available from the corresponding author upon reasonable request.
JYL, JYC, YL, YSS, YYZ, YRZ, EFX, YQX, SST, HXL and WMH significantly contributed to the study, including its conception, design, execution, data acquisition, analysis, and interpretation. JYL, JYC, YL, YSS, YYZ, YRZ, EFX, YQX, SST, HXL and WMH were also involved in drafting, revising, or critically reviewing the manuscript; All authors approved the final version for publication; agreed on the journal for manuscript submission; and accepted accountability for all aspects of the study. JYL designed the study, conducted the experiments, interpreted the results, and wrote the manuscript. YSS and JYC conducted the experiments and interpreted the results. YL, YRZ and EFX contributed to the study design, conducted the experiments, and participated in manuscript revision. YYZ, YQX, SST and HXL drew icons for statistical analysis. WMH provided financial support.
The Research Ethics Committee at Ningbo University granted approval for all animal experiments and methodologies (AEWC-NBU20250371). All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. The Laboratory Animal Science Department facilitated the procurement of Sprague–Dawley rats. Strict compliance with the established protocols for the care and use of laboratory animals was maintained throughout all experimental processes.
The authors would like to express their gratitude to all those who helped them in writing this manuscript. They thank all the peer reviewers for their opinions and suggestions.
This work was financially supported by the National Natural Science Foundation of China (Grant No. 82500082). Additional support was provided by the One Health Interdisciplinary Research Project at the Institute of One Health Science, Ningbo University, the project “Establishment of the Coronary Heart Disease Interventional Diagnosis and Treatment Center at the Cixi Integrated Traditional Chinese and Western Medicine Medical Health Group” (Grant No. 812410050), and the project “The Role of Leukocyte-Derived Chemokine 2 in Regulating Macrophage Polarization” (Grant No. 882500370).
The authors declare no conflict of interest.
During the preparation of this work the authors used DeepSeek in order to write and polish manuscripts. After using this tool/service, the authors reviewed and edited the content as needed and takes full responsibility for the content of the published article.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.