


 , Yukiko Yasuoka 2, Masayoshi Nanami 3, Katsumasa Kawahara 2, Hiroshi Nonoguchi 4,*
, Yukiko Yasuoka 2, Masayoshi Nanami 3, Katsumasa Kawahara 2, Hiroshi Nonoguchi 4,*
1 Department of Nephrology, Kumamoto University Graduate School of Medical Sciences, 860-8556 Kumamoto, Kumamoto, Japan
2 Department of Physiology, Kitasato University School of Medicine, 252-0374 Sagamihara, Kanagawa, Japan
3 Department of Cardiovascular and Renal Medicine, Hyogo Medical University, 663-8501 Nishinomiya, Hyogo, Japan
4 Nephrology, Internal Medicine, Sagamihara Red-Cross Hospital, 252-0157 Sagamihara, Kanagawa, Japan
Abstract
Hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitors have recently been approved for clinical use in the treatment of renal anemia. HIF-PH inhibitors were developed following studies investigating the role of HIFs and their molecular mechanisms. Unlike erythropoiesis-stimulating agents, these orally administered agents improve renal anemia in patients with chronic kidney disease (CKD) and chronic inflammation. HIF-PH inhibitors improve renal anemia by stabilizing HIF-2α, which increases erythropoietin production. They also improve iron metabolism by suppressing hepcidin and inducing the expression of iron transport-related genes. The renoprotective effects of HIF-PH inhibitors are controversial, owing to the opposing effects of hypoxia-inducible factor 1-alpha (HIF-1α) and hypoxia-inducible factor 2-alpha (HIF-2α) on kidney injury. They induce the expression of both HIF-1α and HIF-2α. HIF-2α plays a key role in erythropoietin production and mitigates CKD. Although HIF-1α contributes to cell survival under hypoxic conditions, its overexpression is suspected to exacerbate kidney injury in CKD. This review summarizes the development of HIF and HIF-PH, from their discovery to the elucidation of their regulatory mechanisms and the development of HIF-PH inhibitors. We reviewed basic research and recently published clinical studies investigating the effects of HIF-PH inhibitors on renal injury, focusing on their clinical use in treating renal injury rather than renal anemia. We discuss the possible benefits of HIF-PH inhibitors used clinically for acute and chronic kidney injury, especially in the advanced stages of CKD.
Keywords
- HIF-PH inhibitors
- chronic kidney disease
- acute kidney injury
- diabetic kidney disease
- renal anemia
It has been 6 years since hypoxia-inducible factor prolyl hydroxylase (HIF-PH)
inhibitors were approved for the treatment of renal anemia in Japan.
Erythropoiesis-stimulating agents (ESAs) is used for the treatment of renal
anemia in patients with chronic kidney disease (CKD). Given that the production
of erythropoietin decreases as CKD progresses, patients with CKD exhibit anemia.
Endogenous erythropoietin is produced by HIF-2
HIF-1
Several research groups contributed to the discovery of HIF degeneration through
the von Hippel-Lindau tumor suppressor protein (pVHL). Kaelin Jr.’s group discovered that various hypoxia-inducible mRNAs were insensitive to oxygen and
were overexpressed in renal cancer cell lines lacking functional pVHL [14].
Concurrently, Salceda and Caro [15] reported that HIF-1
William G. Kaelin Jr., Peter J. Ratcliffe, and Greg L. Semenza were awarded the Albert Lasker Basic Medical Research Award in 2016 and were subsequently awarded the Nobel Prize in 2019 for their contributions to the discovery of HIF and pVHL-mediated oxygen-sensing regulation of HIF [21, 22]. The first orally active HIF-PH inhibitor, roxadustat, was developed by FibroGen [23, 24] and was approved in China in 2018 for the treatment of renal anemia in patients with dialysis-dependent CKD [25]. There are five HIF-PH inhibitors currently approved for the treatment of renal anemia in patients with CKD: roxadustat, daprodustat, vadadustat, enarodustat, and molidustat [26, 27, 28, 29].
Both ESAs and HIF-PH inhibitors are currently used for the treatment of renal anemia. ESAs are synthesized from human endogenous erythropoietin and possess additional glycosylation or polyethylene glycol residue to extend their half-life, allowing them to remain in the body and exert a hematopoietic effect for several days [30, 31, 32]. Consequently, ESAs are administered to patients every 2 to 4 weeks. Although their half-life is extended, they are degraded in approximately 1 week, which may cause a wide variation in hemoglobin levels between pre- and post-injections. HIF expression is induced under hypoxic conditions and further induces the transcription of hypoxia-inducible genes, including erythropoietin and iron metabolism-related genes. Once hypoxic conditions are corrected to normoxic conditions, HIFs are hydroxylated by HIF-PHs and promptly degraded by the ubiquitin-proteasome pathway. HIF-PH inhibitors stabilize HIFs regardless of the oxygen level by inhibiting the hydroxylation of HIF-PHs, which maintains the transcription of hypoxia-inducible genes. In contrast to ESAs, they are administered to patients every day or thrice a week, which induces stable erythropoietin production at physiological levels. Given that HIF-PH inhibitors promote iron use by regulating iron metabolism-related genes such as hepcidin, they improve renal anemia even in patients with chronic inflammation [32]. They are better for patients because they are oral medications.
Several studies have demonstrated the protection of renal tubules under AKI by
HIF-PH inhibitors. An overview of the studies introduced in this section is shown
in Table 1 (Ref. [33, 34, 35, 36, 37, 38]). Ito et al. [33] demonstrated
that enarodustat offers a protective effect against AKI using a rat model of
renal ischemia–reperfusion injury (IRI). Pretreatment with enarodustat reduced
plasma creatinine 48 h after ischemia reperfusion, dependent on glycogenesis.
Performing an in vitro experiment using proximal tubular-derived cell
lines, they demonstrated that enarodustat induced the expression of
HIF-1
| Treatment or genetic modification | Experimental model | Results/Mechanisms | Role of HIF-1 |
Role of HIF-2 |
Ref. No. |
| Enarodustat | IRI | Enarodustat improved IRI. Enarodustat increased glycogenesis and suppressed ROS via HIF-1 |
Protective | Not mentioned | [33] |
| Enarodustat | IRI | Roxadustat enhanced the expression of HIF-1 |
Protective | Not mentioned | [34] |
| Roxadustat | IRI | Roxadustat induced erythropoietin in renal tubular cells and maintained the expression of nephron-specific genes after IRI, suggesting the prolonged renoprotective effects by inducing HIF-2 |
Not mentioned | Protective | [35] |
| Endothelial cell-specific HIF-PH2 KO mice | IRI | The mice exerted protective effects against AKI by inhibiting the infiltration of macrophages and inflammation in the kidney. The renoprotective effect was attributed to the activation of HIF-1 |
Protective | Irrelevant | [36] |
| Endothelial cell-specific HIF-2 |
IRI or ureteral obstruction | Renal injury was exacerbated in the mice, which was reversed by blockade of VCAM1 and VLA4. | Irrelevant | Protective | [37] |
| GSK1002083A, an HIF-PH inhibitor | IRI | Renoprotective effect of an HIF-PH inhibitor was observed only when the kidney was pretreated with it. Treatment after IRI did not exert the renoprotective effect on the kidneys. | Generally protective, not distinguished | [38] | |
HIF-PH, hypoxia-inducible factor prolyl hydroxylase; AKI, acute kidney injury; KO, knockout; AIM2, absent in melanoma 2; IRI, ischemia–reperfusion injury; ROS, reactive oxygen species; VCAM1, vascular cell adhesion molecule-1; VLA4, very late antigen-4.
Yang et al. [34] reported the involvement of absent in melanoma 2
(AIM2) activation and 5’-Nucreotidase Ecto (CD73) expression in the
renoprotective effect of roxadustat using an IRI mouse model. They confirmed that
the expressions of the AIM2 inflammasome complex and CD73 were induced in the
proximal tubular cells of the kidneys of patients with AKI and post-transplant
kidneys. In IRI mice, treatment with roxadustat enhanced the expression of
HIF-1
Both the renoprotective and cardioprotective effects of HIF-PH inhibitors have
been suggested. Deguchi et al. [39] reported the cardioprotective
effects of a HIF-PH inhibitor. Pretreatment with roxadustat markedly reduced
infarct size and suppressed plasma creatinine kinase activity in the mouse
cardio-IRI model. The protective effect was accompanied by an increase in
HIF-1
Both HIF-1
The involvement of hypoxia-inducible factor prolyl hydroxylase 2 (HIF-PH2; also
called PHD2) in the renoprotective effects of endothelial cells has been
suggested. Tamoxifen-induced endothelial cell-specific deficits in HIF-PH2 in
adult mice exerted protective effects against AKI caused by bilateral renal IRI,
in which the infiltration of macrophages and expression of inflammation-related
genes were significantly inhibited in HIF-PH2 knockout (KO) mice [36]. The
renoprotective effect was attributed to the activation of HIF-1
Collectively, the renoprotective effects of HIF-PH inhibitor in AKI are likely
exerted in proximal tubular cells via activation of HIF-1
Kapitsinou et al. [38] reported that the renoprotective effect of an HIF-PH inhibitor was observed only when the kidney was pretreated with an HIF-PH inhibitor. Treatment after ischemic reperfusion did not exert the renoprotective effect on the kidneys. Treatment with GSK1002083A, a structural analog of 2-oxoglutarate, at 48 and 6 h before 20 min of bilateral renal pedicle clamping significantly suppressed the increase in blood urea nitrogen levels. IRI-induced fibrosis and inflammation were significantly improved following treatment with an HIF-PH inhibitor 21 days after IRI [38]. Therefore, HIF-PH inhibitors may be useful for the prevention of AKI due to ischemia, sepsis, and post-transplant kidney injury. In particular, in case of kidney transplantation, the time when renal ischemia occurs is clear. We believe that the use of HIF-PH inhibitors for pretreatment of transplanted kidney before the transplantation is practical. A search of ClinicalTrials.gov (https://clinicaltrials.gov) revealed that there appear to be no clinical studies examining the protective effect of HIF-PH inhibitors on AKI. The clinical studies are warranted to clarify the benefit of HIF-PH inhibitors for AKI.
Treatment with HIF-PH inhibitors induces the expression of both HIF-1
| Treatment or genetic modification | Experimental model | Results/Mechanisms | Role of HIF-1 |
Role of HIF-2 |
Ref. No. |
| Proximal tubular cell-specific HIF-1 |
UUO | Renal fibrosis is caused by EMT, which is induced by the activation of HIF-1 |
Harmful | Not mentioned | [42] |
| Systemic HIF-1 |
UUO | Systemic KO and activation of HIFs exacerbated and attenuated renal fibrosis, respectively. The effects were partially due to activation of HIFs in myeloid cells, which regulates the infiltration of macrophages in the kidney. | Generally protective, not distinguished | [43] | |
| Renal epithelial cell-specific HIF-2 |
Adenine-induced kidney injury | Activation of renal tubular HIF-2 |
Not mentioned | Harmful at early stage and protective at late stage kidney injury | [44] |
| ICA/Roxiadustat | Adenine-induced chronic tubulointerstitial nephritis | Both ICA and Roxiadustat inhibited the progression of renal tubular damage and interstitial fibrosis by activating a regulatory, anti-inflammatory MNP phenotype. | Generally protective, not distinguished | [45] | |
| Endothelial cell-specific HIF-PH2 KO mice | Angiotensin II-induced renal fibrosis | Deficit of HIF-PH2 in endothelial cells inhibited angiotensin II-induced renal fibrosis owing to decreased expression of angiotensin II receptor type 1 with subsequent suppression of ROS production and TGF- |
Generally protective, not distinguished | [46] | |
| L-mimosine, a HIF-PH inhibitor | Subtotal nephrectomy | Administration of L-mimosine at an advanced stage significantly improved renal function, renal fibrosis, and anemia, exacerbating them at the early stage and not having any effects at the end stage. | Harmful at early stage | Protective at advanced stage | [47] |
CKD, chronic kidney disease; ICA, Isoquinoline derivative
2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido) acetate; EMT,
epithelial-mesenchymal transition; HIFs, HIF-1
Higgins et al. [42] found that progression of renal fibrosis was
HIF-1
Kobayashi et al. [43] examined the effect of global deletion or
activation of HIF-1
The expression of HIF-2
Schley et al. [45] reported the renoprotective effect of an HIF-PH
inhibitor on tubulointerstitial nephritis caused by an adenine-containing diet in
mice. In the study, the effects of isoquinoline derivative
2-(1-chloro-4-hydroxyisoquinoline-3-carboxamido) acetate (ICA), an inhibitor for
PHD enzymes, and roxadustat were examined. Both ICA and roxadustat inhibited the
progression of renal tubular damage and interstitial fibrosis induced by adenine,
accompanied by a reduced infiltration of mononuclear phagocytes. Notably, the
renoprotective effects of ICA were independent of HIF-1
The roles of HIF-PH2 in chronic kidney injury have been examined. Zhao
et al. [46] demonstrated that a deficit of HIF-PH2 in endothelial cells
inhibited angiotensin II-induced renal fibrosis owing to decreased expression of
angiotensin II receptor type 1 with subsequent suppression of ROS production and
TGF-
Previous studies suggest that the timing of HIF-PH inhibitor administration is
important for exerting the renoprotective effects in CKD. Yu et al. [47, 51]
examined the effect of L-mimosine, a HIF-PH inhibitor, on kidney injury in rats
subjected to subtotal nephrectomy. L-mimosine was administered during
early, advanced, or end-stage kidney damage. The study demonstrated that the
administration of L-mimosine at an advanced stage significantly improved renal
function, renal fibrosis, and anemia, exacerbating them at the early stage and
not having any effects at the end stage. The study further demonstrated that the
renoprotective effect of L-mimosine at the advanced stage of kidney injury was
accompanied by significantly increased expression of HIF-2
These findings suggest the possible use of HIF-PH inhibitors in the treatment of
patients with progressive CKD. Treatment with HIF-PH inhibitors probably
maintains the expression of HIF-2
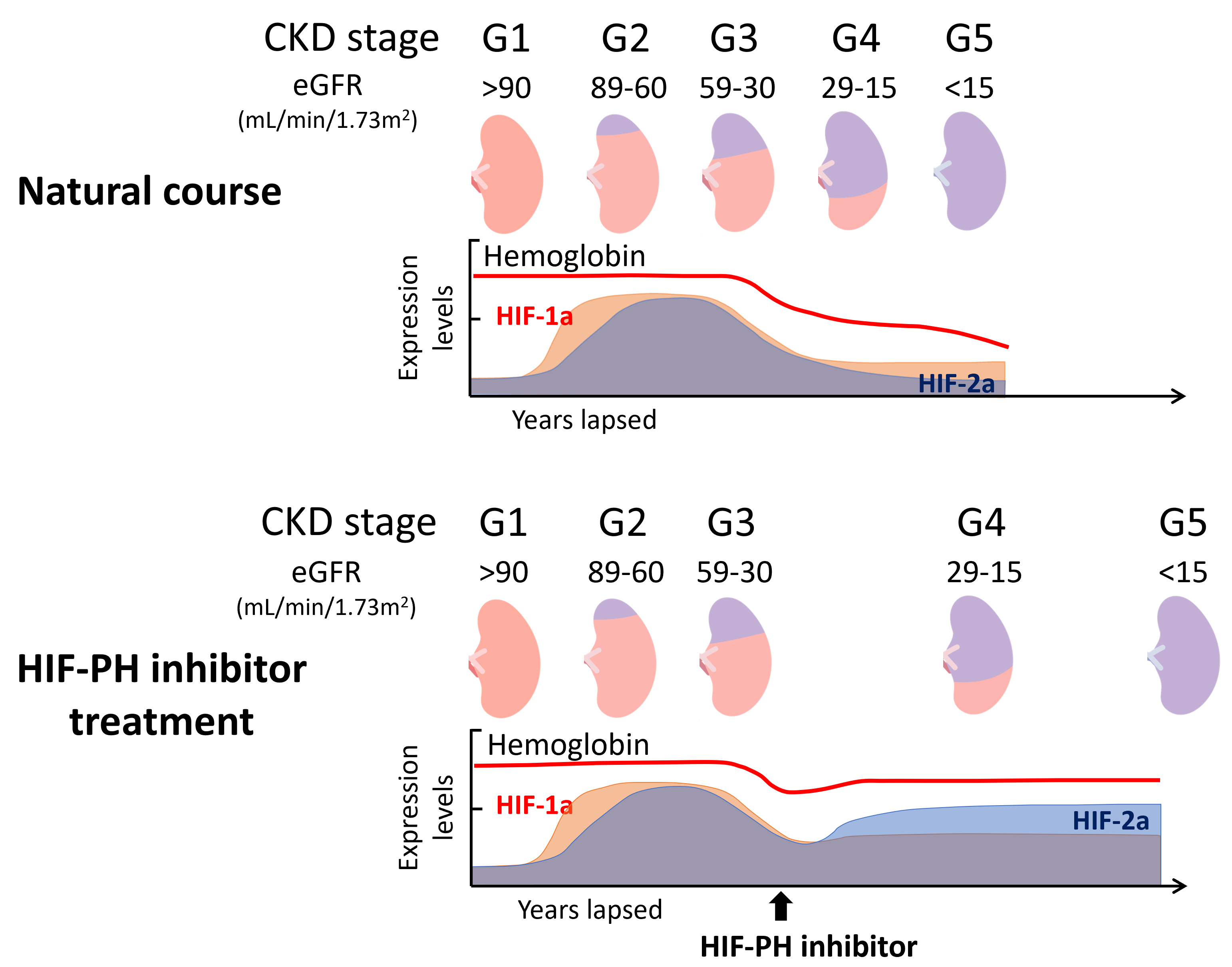 Fig. 1.
Fig. 1.
Schematic illustration of hemoglobin levels, HIF-1
The pathogenesis of diabetic kidney disease is unique and induced by vascular
damage due to hyperglycemia. Once proteinuria occurs, disease progression is
rapid and unavoidable. The establishment of a treatment method before the
progression to renal fibrosis is necessary. An overview of the studies introduced in this section is shown in Table 3 (Ref. [53, 54, 55, 56, 57, 58]). Nordquist et al. [53]
examined the effects of CoCl2 on kidney injury in rats with
streptozotocin-induced diabetes. In diabetic rats, oxygen consumption was
significantly increased, accompanied by excessive glomerular filtration rate
(GFR), proteinuria, and tubulointerstitial injury. Treatment with CoCl2
reduced oxygen consumption and improved excessive GFR, proteinuria, and
tubulointerstitial injury, suggesting the protective effect of HIFs on diabetic
kidney injury. The beneficial effects of CoCl2 were related to reduced
oxidative stress [53]. In proximal tubule-specific conditional HIF-1
| Treatment or genetic modification | Experimental model | Results/Mechanisms | Role of HIF-1 |
Role of HIF-2 |
Ref. No. |
| CoCl2 | Streptozotocin-induced diabetes | Treatment with CoCl2 reduced oxygen consumption and improved excessive GFR, proteinuria, and tubulointerstitial injury. The beneficial effects of CoCl2 were related to reduced oxidative stress. | Generally protective, not distinguished | [53] | |
| Proximal tubular cell-specific conditional HIF-1 |
Streptozotocin-induced diabetes | In HIF-1 |
Protective | Not mentioned | [54] |
| Systemic HIF-1 |
Streptozotocin-induced diabetes | HIF-1 |
Protective | Not mentioned | [55] |
| Systemic HIF-1 |
Streptozotocin-induced diabetes | HIF-1 |
Protective | Not mentioned | [56] |
| YC-1, an HIF-1 |
OVE26 mice, a type 1 diabetic animal model | The expression of HIF-1 |
Harmful | Not mentioned | [57] |
| Enarodustat | BTBR ob/ob mice, a type 2 diabetic animal model | Enarodustat improved albuminuria and foot process effacement by suppressing palmitate-induced CCL2/MCP-1 production via HIF-1 |
Protective | Not mentioned | [58] |
BTBR, black and tan brachyury; GFR, glomerular filtration rate; CCL2/MCP1, C-C motif chemokine ligand 2/monocyte chemoattractant protein 1; GLUT-1, glucose transporter 1.
The opposite effects of HIF-1
Sugahara et al. [58] demonstrated the renoprotective effect of an
HIF-PH inhibitor in a diabetic kidney disease model. Diabetic black and tan
brachyury (BTBR) ob/ob mice were treated with enarodustat or its vehicle between
4 and 22 weeks of age. BTBR ob/ob mice exhibited albuminuria with foot process
effacement, glomerular hypertrophy, mesangial expansion, and interstitial
fibrosis. Treatment with enarodustat improved albuminuria and foot process
effacement, but did not alter glomerular hypertrophy and interstitial fibrosis.
Treatment with enarodustat decreased the expression of C-C motif chemokine ligand
2/monocyte chemoattractant protein 1 (CCL2/MCP1) in the glomeruli of BTBR ob/ob
mice, resulting in the suppression of macrophage infiltration. An in
vitro study revealed that enarodustat suppressed palmitate-induced CCL2/MCP-1
production via HIF-1
Generally, few studies have examined the effects of HIF-PH inhibitors in
diabetic kidney disease models. Although they may be effective in diabetic kidney
disease, activation of HIF-1
Given that HIF-PH inhibitors are involved in the regulation of various pathways,
the effects of combining them with other drugs on CKD should be considered. Sung
et al. [59] examined the combination therapy of roxadustat and
dapagliflozin, a sodium glucose co-transporter 2 (SGLT2) inhibitor, for
cardiorenal syndrome-induced damage in rats. SD rats were subjected to remnant
kidney-induced chronic kidney injury, followed by acute myocardial infarction.
The rats were treated with either roxadustat or dapagliflozin, or both, for a
month. Combination therapy significantly improved cardiorenal syndrome—both
cardiac and renal functions improved by suppressing tissue fibrosis. The study
suggested that the combination therapy enhanced HIF-1
The involvement of Zinc (Zn) deficiency in CKD progression and the
renoprotective effects of Zn supplementation have been suggested [61]. Zhang
et al. [62] reported that treatment with Zn inhibited tubular EMT and
attenuated renal tubulointerstitial fibrosis by downregulation of HIF-1
The Rho-kinase inhibitor, fasudil, used to treat cerebral vasospasm after
subarachnoid hemorrhage, is expected to be approved for other diseases such as
pulmonary hypertension and heart failure [63]. Matoba et al. [64]
demonstrated that fasudil improved kidney injury in db/db mice through the
inhibition of HIF-1
HIFs are involved in many signaling pathways, such as mitochondrial and glycolytic metabolism, erythropoiesis, ferroptosis, and inflammation [65]. Some of these pathways are thought to be common to other clinically approved drugs. Although the use of HIF-PH inhibitors in combination therapy is expected to have a renoprotective effect, caution is required as they may also inhibit the effects of combined medication.
Clinical trials have examined the safety and efficacy of HIF-PH inhibitors. The effect of vadadustat on renal anemia and major adverse cardiovascular events (MACE) was examined in non-dialysis patients with CKD who either received or did not receive darbepoetin alfa [66]. Treatment with vadadustat improved renal anemia comparably to darbepoetin alfa, demonstrating the non-inferiority of vadadustat to darbepoetin. However, MACE was more in patients with vadadustat than in patients with darbepoetin alfa (22.0% vs. 19.9%; hazard ratio, 1.17). The increased risk of MACE in patients treated with vadadustat was largely due to an excess of nonfatal myocardial infarctions and a higher incidence of death from non-cardiovascular causes. The effects of daprodustat on renal anemia and MACE were examined in non-dialysis patients with stage 3–5 CKD [67]. The control group comprised non-dialysis patients with stage 3–5 CKD who received an injection of darbepoetin alfa. The effect of daprodustat on Hb elevation was not significantly different from that of darbepoetin, confirming the non-inferiority of daprodustat to darbepoetin alfa. The occurrence of MACE in patients treated with daprodustat was not different from that in patients treated with darbepoetin, suggesting the safety of daprodustat in non-dialysis patients with CKD. These two large-scale clinical studies revealed controversial results regarding the safety of HIF-PH inhibitors. The incidence of MACE in different HIF-PH inhibitors including vadadustat and daprodustat is shown in Table 4 (Ref. [66, 67, 68, 69, 70]) for reference.
| HIF-PH inhibitor | Control | Ref. No. |
| Vadadustat (n = 1739) | Darbepoetin (n = 1732) | [66] |
| 382 (22.0%) | 344 (19.9%) | |
| Daprodustat (n = 1937) | Darbepoetin (n = 1935) | [67] |
| 378 (19.5%) | 371 (19.2%) | |
| Roxadustat (n = 1083) | ESA (n =1059) | [68] |
| 105 (9.7%) | 136 (12.8%) | |
| Molidustat (n = 82) | Darbepoetin (n = 82) | [69] |
| 3 (3.7%) | 1 (1.2%) | |
| Molidustat (n = 82) | Darbepoetin (n = 79) | [70] |
| 6 (7.3%) | 0 (0%) |
The data was referenced from their representative clinical trials. The number of patients who developed MACE and their incidence rates are shown. The incidence of MACE for enarodustat does not appear to be published. MACE, major adverse cardiovascular events; ESA, erythropoiesis-stimulating agents.
Systematic reviews of meta-analyses examining the efficacy and safety of HIF-PH
inhibitors for the treatment of anemia in patients with CKD showed no notable
disparities in safety outcomes between the HIF-PH inhibitor and ESA or placebo
groups [71, 72]. These agents improved anemia as much as ESA by enhancing iron
metabolism, decreasing hepcidin levels, and improving iron transport. Potential
adverse events (AEs) associated with HIF-PH inhibitors have been suggested,
including the development of malignant tumors, diabetic retinopathy, age-related
macular degeneration, and progression of polycystic kidney diseases [73, 74]. The
onset or progression of these diseases has been suggested to be induced by the
activation of HIF-1
Two clinical trials described above reported that CKD progression was not
delayed in patients treated with HIF-PH inhibitors compared to patients treated
with darbepoetin alfa. The effect of the HIF-PH inhibitor on CKD progression has
not been examined in comparison to a placebo. A few clinical studies have
suggested a renoprotective effect of ESA on CKD progression; however, these
studies were small-scale. Recently, we performed a retrospective study to examine
the renoprotective effects of daprodustat [76]. The study included patients with
stage 3–5 CKD (mean eGFR, 26.0 mL/min/1.73 m2
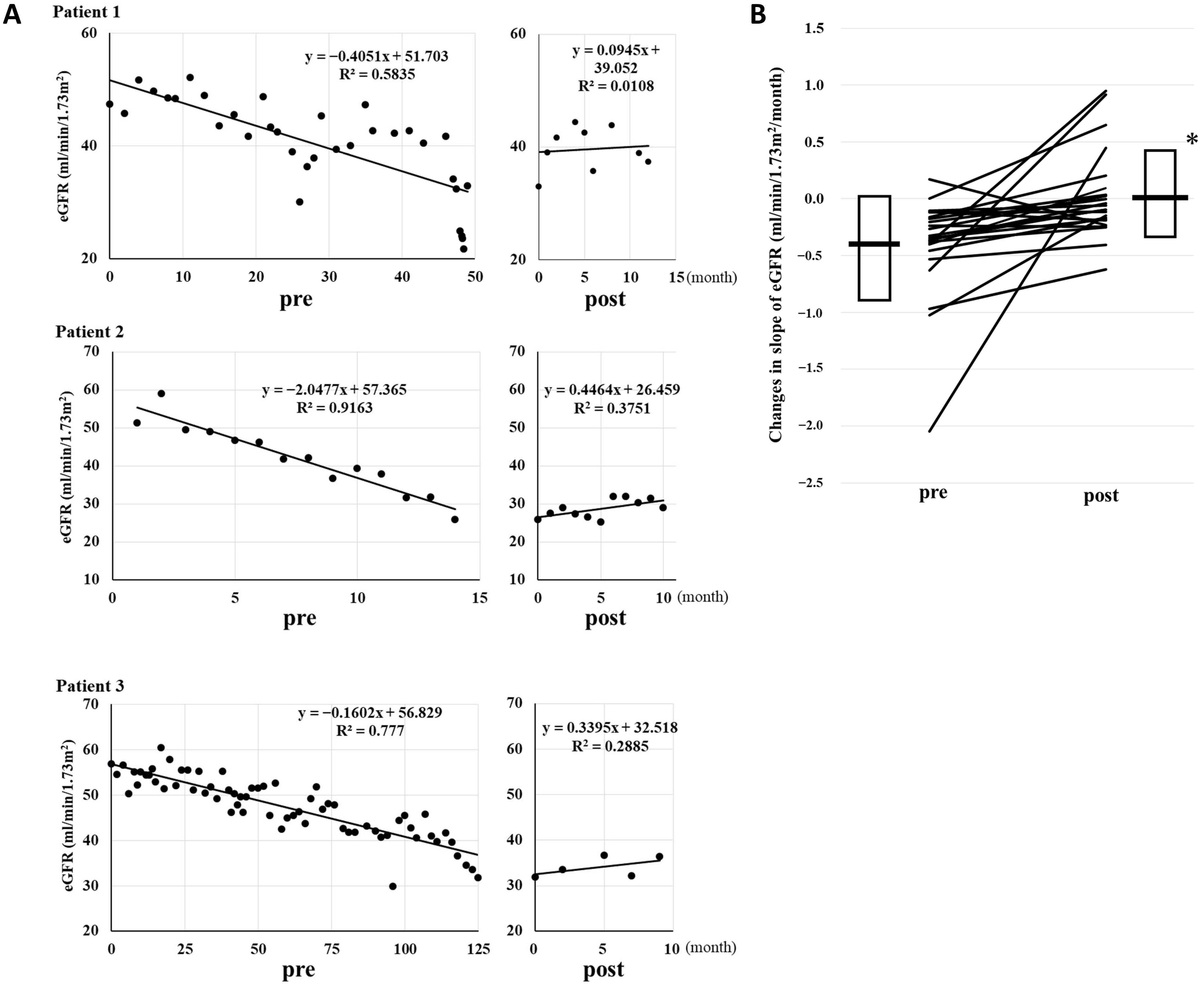 Fig. 2.
Fig. 2.
Renoprotective effect of daprodustat in patients with CKD. (A)
The changes in the eGFR slope in three patients before and after (pre and post,
respectively) the administration of daprodustat. The figures are shown as
representative results from a retrospective study to examine renoprotective
effect of daprodustat. (B) The changes in the slope of the decline of eGFR
(mL/min/1.73 m2/month) by daprodustat. Daprodustat significantly reduced the
decline of eGFR slope. Box and bar show mean
Due to their pharmacological effects, HIF-PH inhibitors have been developed as drugs to improve anemia. Clinical studies have been conducted on renal anemia, leading to their current practical application for renal anemia. It appears that renoprotective effect of HIF-PH inhibitors was not anticipated. A search on ClinicalTrials.gov revealed that currently, very few clinical studies examining the renoprotective effect of HIF-PH inhibitors as a primary endpoint have registered. It is expected that the large-scale clinical trials will be conducted with renoprotective effects as a primary endpoint.
In the last decade, SGLT2 and nonsteroidal mineralocorticoid receptor (MR) inhibitors have proven their renoprotective effects and have been used as promising new drugs for the treatment of patients with CKD. Although SGLT2 inhibitors are effective in early-stage patients with CKD with proteinuria, their renoprotective effect is not much expected in patients with advanced CKD, in whom proximal tubular function has already been abolished. MR inhibitors have been shown to suppress proteinuria and delay CKD progression; however, an initial dip in eGFR and hyperkalemia limits their use in patients with advanced CKD. Collectively, HIF-PH inhibitors may represent an unmet therapeutic need for delaying the progression of moderately advanced CKD.
HIF-PH inhibitors hold potential clinical applications in kidney injury
treatment. The activation of HIF-1
In this review, we mainly focused on the renoprotective effects of HIF-PH
inhibitors via activation of HIF-1
AEs, adverse events; AIM2, absent in melanoma 2; AKI, acute kidney injury; BTBR, black and tan brachyury; CCL2/MCP1, C-C motif chemokine ligand 2/monocyte chemoattractant protein 1; CKD, chronic kidney disease; CoCl2, cobalt(II)chloride; EMT, epithelial–mesenchymal transition; ESA, erythropoiesis-stimulating agents; GLUT-1, glucose transporter 1; GFR, glomerular filtration rate; HIF, hypoxia-inducible factor; HIFs, HIF-1
YI and HN conceived the study. YI, YY, MN, and KK conducted the literature review from physiological, pathological, and clinical perspectives and interpreted the data. YI and HN contributed to the manuscript writing and editing. YY, MN, and KK reviewed the manuscript and provided critical feedback and suggestions. All authors contributed to editorial changes in the manuscript. All the authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to thank Editage for English language editing.
This research received no external funding.
The authors declare no conflict of interest.
ChatGPT-4o was used for minor grammatical refinement during the drafting of this article. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.