





 , Karen L. Lankford 2,3, Takahiro Yokoyama 1, Ryo Ukai 1, Ryosuke Hirota 2,4, Shinichi Oka 5, Masanori Sasaki 1,2,6, Jeffery D. Kocsis 2,3, Osamu Honmou 1,2,5
, Karen L. Lankford 2,3, Takahiro Yokoyama 1, Ryo Ukai 1, Ryosuke Hirota 2,4, Shinichi Oka 5, Masanori Sasaki 1,2,6, Jeffery D. Kocsis 2,3, Osamu Honmou 1,2,51 Department of Neural Regenerative Medicine, Institute of Regenerative Medicine, School of Medicine, Sapporo Medical University, 060-8556 Sapporo, Hokkaido, Japan
2 Department of Neurology, Yale University School of Medicine, New Haven, CT 06510, USA
3 Center for Neuroscience and Regeneration Research, VA Connecticut Healthcare System, West Haven, CT 06516, USA
4 Department of Orthopaedic Surgery, School of Medicine, Sapporo Medical University, 060-8556 Sapporo, Hokkaido, Japan
5 Department of Advanced Regenerative Therapeutics, Sapporo Medical University School of Medicine, 060-8556 Sapporo, Hokkaido, Japan
6 Division of Neuroscience, Department of Physiology, Sapporo Medical University School of Medicine, 060-8556 Sapporo, Hokkaido, Japan
Abstract
Spinal cord injury (SCI) initiates a complex secondary cascade characterized by disruption of the blood–spinal cord barrier (BSCB), infiltration of peripheral immune cells, and chronic neuroinflammation. Within this response, macrophages and microglia act as key effectors that critically influence both the progression of injury and the subsequent repair processes. Mesenchymal stromal/stem cell-derived small extracellular vesicles (MSC-sEVs) have recently gained recognition as a promising cell-free therapeutic approach that acts through multiple paracrine mechanisms, including but not limited to immunomodulation. This review summarizes current evidence elucidating how macrophages and microglia contribute to the multifaceted therapeutic actions of MSC-sEVs in the context of SCI. Following intravenous administration, MSC-sEVs preferentially localize to the lesion site, where they are internalized by CD206+ macrophages. This interaction initiates a multifaceted therapeutic program. First, MSC-sEVs not only reprogram myeloid cells toward an anti-inflammatory, M2-like phenotype but also sustain this reparative state, thereby stabilizing a pro-resolving immune environment, attenuating the production of proinflammatory cytokines such as TNF-α and IL-6, and enhancing the expression of anti-inflammatory mediators, including Interleukin-10 (IL-10) and Transforming growth factor-beta (TGF-β). This polarization is partly driven by transferred microRNAs that suppress central inflammatory signaling hubs, notably the Toll-like receptor 4 (TLR4)/Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and NOD-like receptor protein 3 (NLRP3) inflammasome pathways. Second, MSC-sEVs augment macrophage phagocytic capacity, facilitating the removal of myelin debris and apoptotic cells and thereby creating a permissive microenvironment for regeneration. Third, soluble factors released from reprogrammed myeloid cells confer neuroprotective and trophic support to neurons and oligodendrocytes, mitigating secondary degeneration. Finally, these cells contribute to the re-establishment of BSCB integrity by promoting tight junction protein re-expression, reconstituting pericyte–endothelial interactions, and enhancing microvascular remodeling. Collectively, these coordinated mechanisms suppress neuroinflammation, preserve neural tissue, and support functional recovery. The therapeutic benefits of MSC-sEVs in SCI, therefore, depend substantially on the reprogramming of macrophages and microglia. Elucidating this bidirectional communication between MSC-sEVs and myeloid cells provides critical insight into SCI pathophysiology and identifies macrophage and microglial modulation as a strategic target for next-generation sEV-based neuroregenerative interventions.
Keywords
- mesenchymal stromal/stem cell-derived small extracellular vesicles
- spinal cord injury
- macrophages
- microglia
- blood-spinal cord barrier
Spinal cord injury (SCI) is a catastrophic neurological condition that
frequently leads to lifelong sensorimotor disability and reduced quality of life
[1]. Beyond the primary mechanical insult, a secondary injury cascade—including
microvascular failure, blood–spinal cord barrier (BSCB) disruption, and
neuroinflammation—expands tissue damage over hours to weeks and limits
spontaneous recovery [2, 3]. Early BSCB breakdown exposes the injured cord to
blood-borne mediators and promotes infiltration of circulating leukocytes,
amplifying pro‑inflammatory signaling (e.g., Nuclear factor
kappa-light-chain-enhancer of activated B cells (NF‑
After SCI, both microglia and monocyte-derived macrophages are rapidly activated and play indispensable roles in subsequent pathophysiological processes [5, 6]. Despite their distinct developmental origins—microglia arising from yolk sac progenitors as resident immune cells of the central nervous system (CNS) [7], and macrophages being derived from circulating monocytes—both cell types belong to the myeloid lineage and emerge as major contributors to the inflammatory milieu following SCI [8, 9]. While microglia are specialized for CNS surveillance and rapid injury responses [10] and infiltrating macrophages are recruited from the periphery, both populations undergo dynamic activation and polarization after injury, adopting phenotypes along the classical M1 (pro-inflammatory) and alternative M2 (anti-inflammatory) axes [8, 9]. Importantly, the context-dependent activation of these two myeloid populations critically dictate the magnitude of inflammation, scar formation, and ultimately neurological outcome after SCI [6, 10, 11]. Thus, therapeutic strategies aimed at appropriately regulating the functions of macrophages and microglia around the lesion site may represent a critical avenue for the treatment of SCI.
Mesenchymal stromal/stem cell-derived small extracellular vesicles (MSC-sEVs)
are membrane-bound particles with a diameter of approximately 30–200 nm,
released from MSCs [12]. MSC-sEVs are a subclass of extracellular vesicles that
include exosomes which are commonly enriched in markers such as CD63, CD81, and
CD9 [12]. These nano-sized vesicles carry a complex cargo of proteins, lipids,
and nucleic acids (including microRNAs), which is influenced by their parent
cell’s profile and can mediate intercellular communication [13]. Importantly,
MSC-sEVs are widely considered a major effector of the paracrine therapeutic
actions of MSCs [6] and may offer advantages in terms of biological stability,
immunological safety, and ease of standardization compared to whole-cell
therapies [14]. In SCI research, preclinical studies using rodent models report
that intravenously delivered MSC-sEVs can reach the injured spinal cord and
preferentially associate with M2-like macrophages, accompanied by enhanced
TGF‑
SCI is a chronic, life-altering neurological condition with traumatic and non-traumatic causes [17]. Across most registries, road-traffic collisions and falls account for the largest share of traumatic SCI, with a young male predominance. In the aging populaton low-energy falls and non-traumatic etiologies—ischemia/reperfusion, vascular malformations, neoplasms, infection, and inflammatory myelopathies—are increasingly represented [18]. Regardless of cause, a stereotyped sequence—primary mechanical insult followed by secondary biochemical and cellular cascades—governs tissue loss and functional outcome [19].
Contusion, compression, distraction, or laceration shear axons and microvessels,
producing intraparenchymal hemorrhage and an immediate dysfunction of the BSCB
that initiates the secondary injury cascade [20]. Within this secondary phase,
gray-matter neurons and oligodendrocytes are disrupted; ionic disequilibrium (Na+
influx/K+ efflux) and conduction block develop within minutes [21]. Edema and
local ischemia are compounded by ongoing spinal cord compression and vasospasm
[22]. Excess extracellular glutamate activates N-methyl-D-aspartate (NMDA)/
Macrophages and microglia are the dominant innate immune effectors in the injured spinal cord, and their phenotypic states decisively shape the pathological trajectory by influencing both recovery and inflammation mediated secondary injury (Fig. 1A). In the acute phase (typically 0–3 days post injury), activated microglia and infiltrating macrophages rapidly respond to mechanical and ischemic damage, contributing to debris clearance and containment of cellular damage, while also releasing pro-inflammatory mediators that can exacerbate neuronal and glial loss [9]. These dual roles—both protective and deleterious—underscore their importance as central modulators of the injury microenvironment [8]. Because these myeloid programs are highly plastic and responsive to local cues, they represent tractable targets for therapeutic intervention aimed at modulating both primary and secondary injury cascades [27].
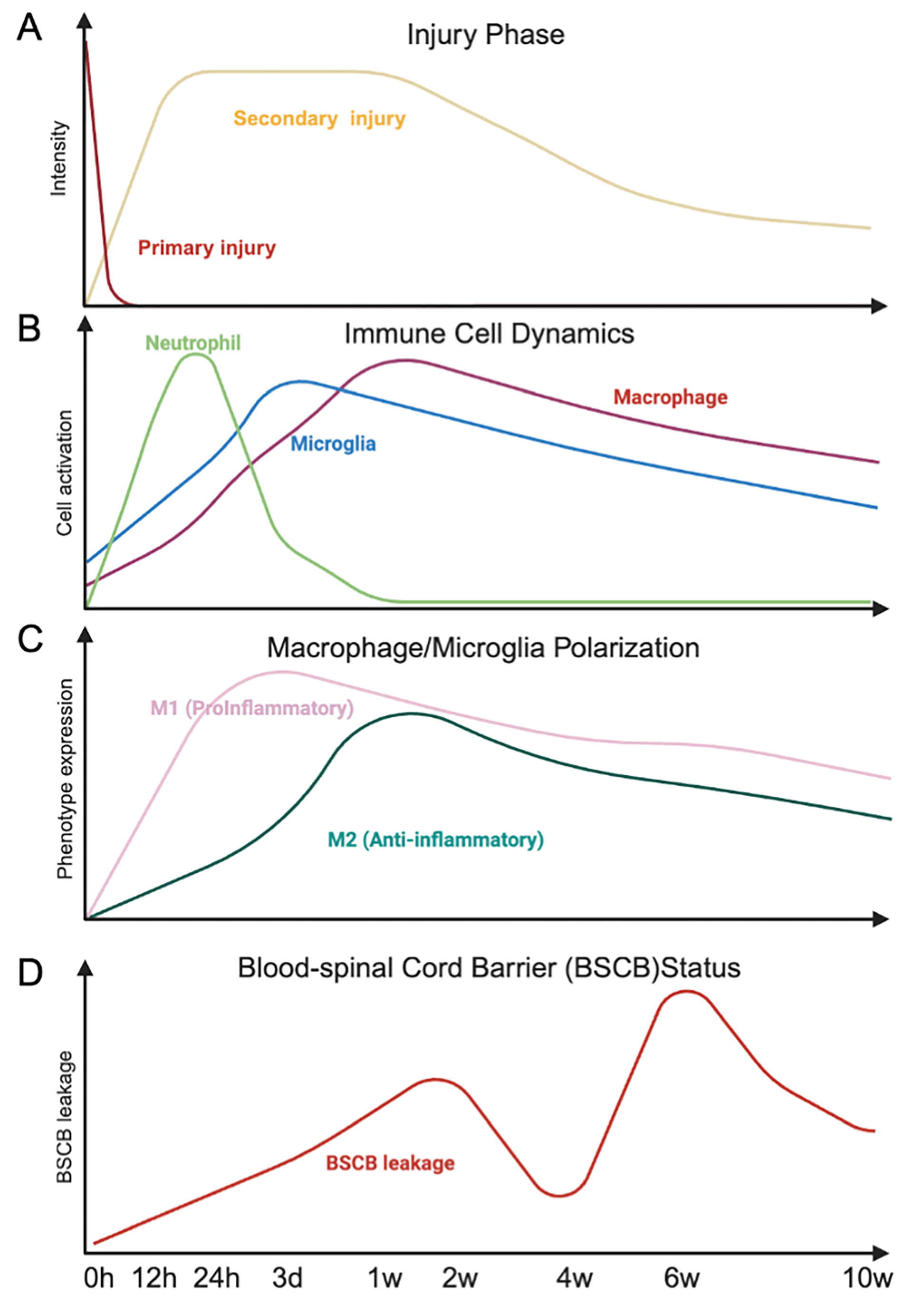 Fig. 1.
Fig. 1.
Temporal dynamics of injury progression, immune responses, and blood–spinal cord barrier disruption after spinal cord injury. Schematic representation of the temporal profile of spinal cord injury (SCI). The upper panel (A) illustrates the rapid primary injury followed by a prolonged secondary injury phase. Subsequent panels depict immune cell dynamics (B), including neutrophil infiltration, microglial activation, and macrophage accumulation, as well as macrophage/microglia polarization from proinflammatory (M1) to anti-inflammatory (M2) phenotypes (C). The lower panel (D) shows the time-dependent increase and partial recovery of blood–spinal cord barrier (BSCB) leakage over 10 weeks after injury. Created in BioRender. Nakazaki, M. (2026) https://BioRender.com/z0lx89y.
Within minutes to hours after SCI, microglia shift from a surveillant to an
activated state; neutrophils surge over the first 24–48 h; and monocyte-derived
macrophages infiltrate and, by ~day 3–7, dominate the lesion
core [28], limiting injury spread through elimination of damaged cells (Fig. 1B)
[29]. Within a week of SCI, macrophages predominate in the lesion core, with both
deleterious (M1, inducible nitric oxide synthase (iNOS)/Tumor necrosis
factor-alpha (TNF-
The rapid activation of microglia and infiltrating macrophages following the primary mechanical insult constitutes a double-edged response; essential for debris clearance and neuroprotection yet liable to exacerbate tissue damage through dysregulated inflammatory signaling. This is in contrast to peripheral wounds that resolve rapidly [29]. As these myeloid populations persist and adapt to the evolving microenvironment, their phenotypic transitions increasingly influence vascular integrity, glial reactivity, and extracellular matrix dynamics. These shifts mark the transition from the acute innate immune response to a chronic, myeloid-driven phase of secondary injury, characterized by sustained BSCB dysfunction and scar formation, which will be discussed in the next section.
Activated myeloid cells (microglia and infiltrating monocyte-derived
macrophages) function as key drivers of the non-permissive microenvironment that
defines secondary injury, primarily by coordinating a pathological crosstalk
between sustained blood-spinal cord barrier (BSCB) dysfunction and maladaptive
matrix remodeling [33, 34]. Release of pro-inflammatory cytokines (e.g.,
TNF-
MSC therapies for SCI has been investigated for more than two decades in animal models and have advanced into multiple early-phase clinical studies [43]. Although multiple studies have shown improved functional outcomes after intravenous or intrathecal delivery of MSCs in animal models of SCI, little to no engraftment of MSCs has been found within the lesioned areas [2], arguing that MSCs do not directly participate in tissue repair, but rather secrete factors which travel to the injury sites. One of the early effects of MSC treatments on the injured spinal cord was a rapid stabilization of the BSCB. MSC administration reduces macromolecular leakage and edema, restores tight-junction organization (e.g., ZO-1, occludin, claudins) [6], and enhances pericyte–endothelial coupling [34], thereby curbing leukocyte influx and limiting the number of inflammatory cells in the paranchyma [34]. These changes also correlate with a suppression of key proteases (calpains, MMP-2/9), rebalancing toward increased production of the metalloprotease inhibitor TIMPs [44].
Mesenchymal stromal/stem cell-derived small extracellular vesicles (MSC-sEVs) are largely responsible for the therapeutic effects observed following intravenous MSC administration. The term “sEVs” encompasses heterogeneous vesicle populations (30–200 nm in diameter) released by MSCs, including exosomes and other subtypes; definitive assignment to a specific biogenesis pathway requires demonstration of endosomal origin markers [45]. In our studies western blot analysis showed that hMSC-sEVs samples were highly enriched in three key exosomal surface marker proteins (CD63, CD9, and Alix), when compared to equivalent protein amounts from the cultured MSCs from which the sEVs were isolated [6, 11]. Thus, while we feel that the sEVs we use are highly enriched in exosomes, we use the conservative term sEVs. MSC-sEVs are increasingly recognized as key mediators of paracrine activity, capable of delivering microRNAs, proteins, and lipids to target cells [6]. Although intravenously infused MSCs do not reach the lesion site, they become trapped in the lung microvasculature, where they survive for 2–3 days and continuously release MSC-sEVs into the systemic circulation (Fig. 2) [2, 6]. These circulating MSC-sEVs subsequently accumulate at the site of SCI and are preferentially internalized by activated CD206+-macrophages/microglia within the lesion microenvironment [6]. This mechanism explains how MSCs exert therapeutic effects at distant injury sites without direct cellular engraftment.
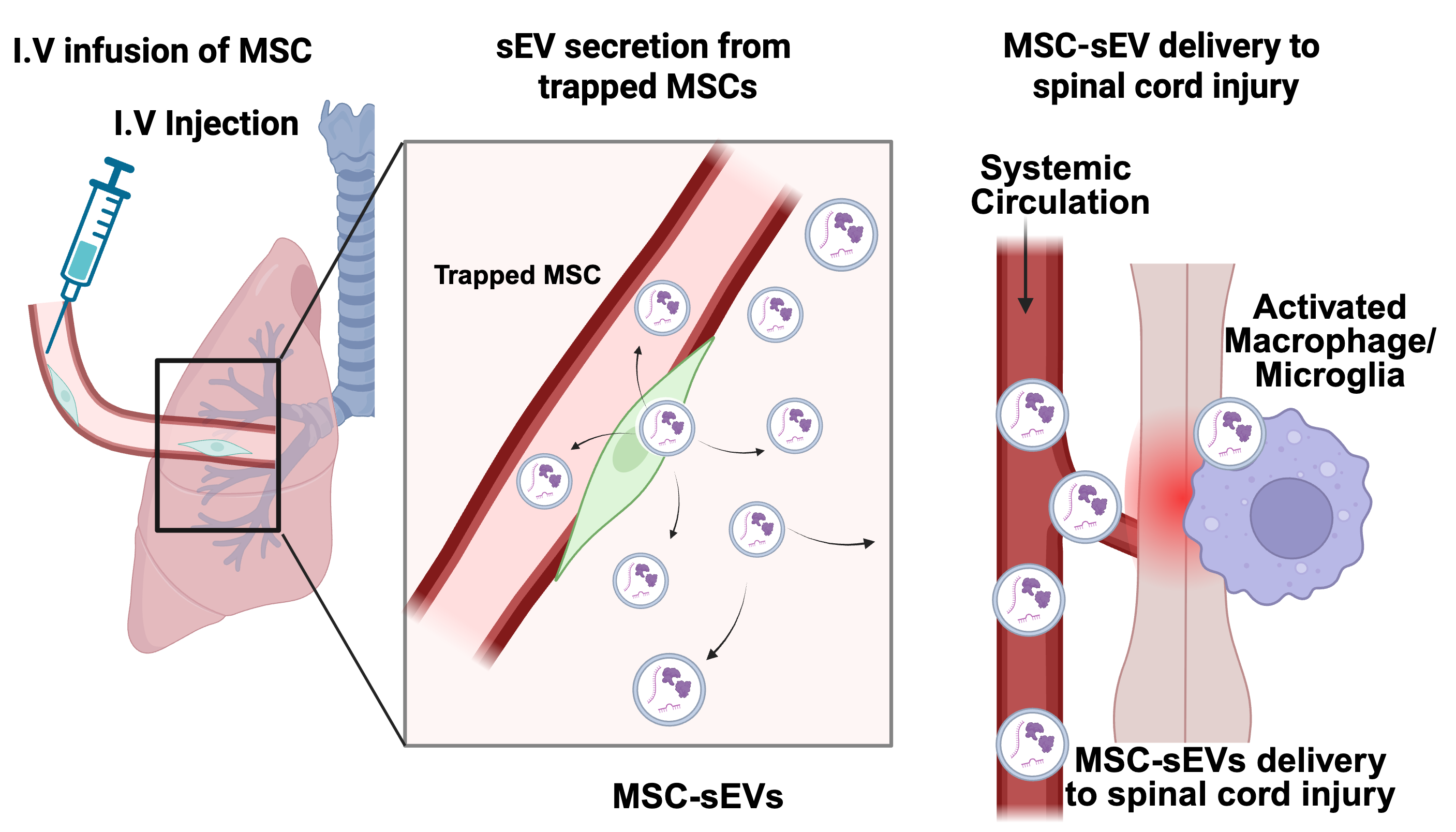 Fig. 2.
Fig. 2.
Proposed mechanism of MSC-derived small extracellular vesicle (EV) delivery to the injured spinal cord following systemic administration. Following intravenous (I.V.) infusion, mesenchymal stromal/stem cells (MSCs) do not traffic directly to the injury site; instead, due to their relatively large size (15–30 µm), they become entrapped within the lung microvasculature, where they survive transiently for 2–3 days (left panel). During this period, the trapped MSCs continuously secrete small extracellular vesicles (MSC-sEVs, 30–200 nm in diameter) into the systemic circulation (middle panel). These circulating MSC-sEVs subsequently home to sites of tissue injury, including the damaged spinal cord, where they extravasate through the disrupted blood-spinal cord barrier (BSCB) and accumulate within the lesion microenvironment (right panel). At the injury site, MSC-sEVs are preferentially internalized by activated macrophages and microglia. The internalized sEVs deliver bioactive cargo, including immunomodulatory microRNAs, to reprogram macrophage/microglial function and promote a reparative microenvironment conducive to neural repair [6]. Created in BioRender. Nakazaki, M. (2026) https://BioRender.com/6kegcsw.
MSC-sEVs reproduce key MSC effects on functional recovery and restoration of
BSCB function—reducing permeability, reinforcing tight and adherens junctions,
and increasing pericyte coverage—while also reprogramming macrophages/microglia
toward IL-10/TGF-
The cargo of MSC-sEVs is complex, including proteins, mRNAs, microRNAs (miRNAs),
cDNA, and lipids, each with the potential to alter the phenotypical expression of
recipient cells [46]. Experiments showing that RNase treatments of MSC-sEVs
abolished their protective effects on kidney ischemia–reperfusion injury, argued
that RNAs were likely a key active component of MSC-sEVs [47]. In particular,
miRNAs have long been proposed as key mediators: as each miRNA regulates numerous
mRNA targets, reshaping rates of mRNA translation and decay and profoundly
altering gene expression, allowing small quantities of miRNAs to exert outsized
effects on cell functions [34]. miRNAs can regulate key processes such as
inflammation, apoptosis, angiogenesis, and neurogenesis, offering new avenues for
promoting functional recovery after SCI. Intravenous delivery MSC-sEVs have been
shown to modulate gene expression in injured spinal tissue, reducing neuronal
apoptosis, suppressing inflammation, and promoting vascular and neural
regeneration. Specific miRNAs identified in MSC-sEVs—such as miR-381,
miR-21-5p, miR-126, and miR-216a-5p—target signaling pathways (e.g.,
BRD4/WNT5A, FasL, SPRED1/PIK3R2, TLR4/NF-
The potential cell targets for MSC-sEVs in the injured cord include all major cellular compartments within the lesion: tissue-resident microglia and recruited macrophages at the border [6, 11, 34, 50], neurons [51, 52], astrocytes [53], oligodendrocytes [52, 54], endothelial cells [55], and pericytes [56], lining the central canal [49]. Route and timing of delivery shape apparent targeting. After intravenous dosing in the subacute window, labeled sEVs are most consistently detected within lesion phagocytes [6, 11, 50], whereas intralesional or intranasal delivery often yields stronger colocalization with neurons and astrocytes [57]. Caution is warranted about interpretation of MSC-sEVs uptake in some in vivo studies however, as resolution of the micrographs could not always conclusively distinguish between surface-bound sEVs and true sEVs uptake [50].
Although MSC-sEVs influence multiple cellular nodes in SCI, macrophages/microglia emerge as the dominant in vivo conduit translating vesicle cargo into lesion-wide benefits [6, 11, 34, 50]. The direct evidence shows that intravenously delivered MSC-sEVs selectively accumulate within macrophages of the injured spinal cord [50]. Using DiR-labeled sEVs isolated from rat bone marrow MSCs and high-resolution confocal microscopy, it was demonstrated that MSC-sEVs trafficked specifically to contused, but not intact, spinal cord regions, where fluorescence “hotspots” co-localized with CD206+ M2-type macrophages, but not with iNOS+ M1 macrophages, neurons, astrocytes, endothelial cells, or pericytes. Three-dimensional reconstructions confirmed that these sEVs were internalized rather than surface-bound, indicating genuine cellular uptake [50].
The preferential uptake of MSC-sEVs by M2 macrophages can be attributed to the distinctive surface receptor profile and enhanced phagocytic capacity characteristic of the M2 phenotype. M2 macrophages express high levels of scavenger receptors, including CD206 (mannose receptor), CD36, and CD163, which facilitate recognition and internalization of extracellular particles [6, 11, 58, 59]. Phosphatidylserine exposed on the sEV surface serves as an “eat-me” signal recognized by receptors such as TIM4 and MerTK expressed on phagocytic cells [60]. Additionally, MSC-sEVs interact with heparan sulfate proteoglycans on the recipient cell surface, and subsequent internalization proceeds via lipid raft/caveolar-mediated endocytosis [61].
The lesion microenvironment appears to prime M2 macrophages for preferential sEV uptake. In vitro studies demonstrated that macrophages polarized to an M2 phenotype with IL-4 stimulation, cultured under low pH conditions (pH 6) mimicking the acidic inflammatory milieu, and exposed to phagocytosis-inducing debris showed markedly enhanced uptake of DiR-labeled MSC-sEVs [6, 11]. Notably, sEV uptake was not observed in M1-polarized or resting M0 macrophages under any condition tested [6, 11, 50]. This suggests that active phagocytic priming, rather than CD206 expression alone, is required for efficient MSC-sEV internalization. The acidic microenvironment at the lesion site, combined with the presence of myelin debris and other damage-associated molecular patterns, likely creates the optimal conditions for M2 macrophage-selective uptake of MSC-sEVs. Collectively, these findings identify macrophages/microglia as a key cellular target through which MSC-sEVs mediate anti-inflammatory, trophic, and vascular-stabilizing effects in SCI. The next section therefore focouses on macrophage/microglia-mediated mechanisms in detail.
Mononuclear phagocytes—tissue-resident microglia together with recruited
monocyte-derived macrophages —are the predominant in vivo recipients
of MSC-sEVs following SCI. Several convergent features account for this primacy
(i) spatiotemporal proximity, with microglia rapidly surveying the parenchyma and
macrophages accumulating at the lesion border from ~24 h onward
[62]; (ii) vesicle-capture capacity driven by phagocytic receptors and
recognition of sEV surface lipids (e.g., phosphatidylserine) [63]; and (iii)
network centrality, as myeloid cells shape the lesion microenvironment by
controlling NF-
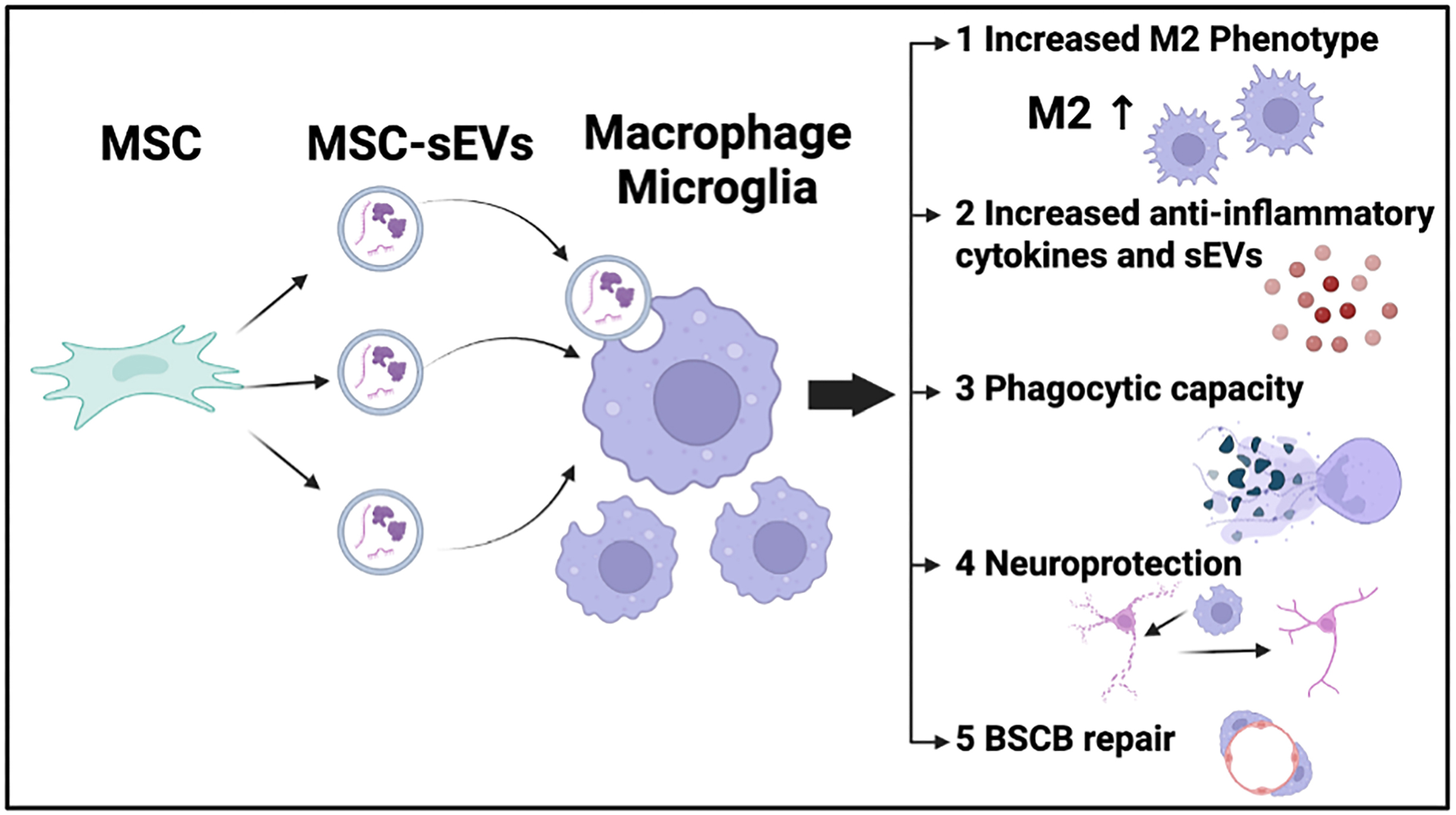 Fig. 3.
Fig. 3.
Illustration of MSC-sEV–mediated modulation of macrophages and
microglia following SCI. Mesenchymal stromal/stem cells (MSCs) secrete small
extracellular vesicles (MSC-sEVs) enriched in bioactive cargo, including
regulatory microRNAs, mRNAs, and proteins. Following systemic administration,
MSC-sEVs are preferentially internalized by macrophages and microglia at the SCI
lesion site, triggering a coordinated immunomodulatory program that shifts these
cells toward an anti-inflammatory, reparative (M2-like) phenotype. The downstream
functional consequences include: (1) Increased M2 phenotype
polarization: MSC-sEV-derived microRNAs suppress TLR4/NF-
MSC-sEVs exhibit strong immunomodulatory capacity, being preferentially incorporated by macrophages and microglia and actively promoting their polarization toward the anti-inflammatory, reparative M2 phenotype, thereby transforming the local microenvironment from a pro-inflammatory to a restorative state [6, 11, 15, 50, 65, 66, 67, 68]. Intravenously delivered hMSC-sEVs predominantly localize to CD206+ M2 macrophages along the edges of the SCI lesion site, showing minimal colocalization with the M1 marker iNOS in vivo [6, 11, 50]. This M1-to-M2 phenotypic shift is primarily mediated by exosomal microRNAs (miRNAs), which target distinct signaling nodes to reprogram myeloid cell function (Table 1, Ref. [15, 66, 67, 69, 70, 71, 72, 73, 74], Fig. 4).
| Micro RNA | Sample source | Expression | Target genes | Target pathways | Cell types | Biological effect | Year/References |
| miR-216a-5p | Hypoxia-preconditioned BM-MSC-sEVs | Down | TLR4 | TLR4/NF-κB | Microglia | M1→M2 shift; Inflammation ↓ | 2020 / Liu et al. [69] |
| miR-181c | hUC-MSC-sEVs | Down | TLR4 | TLR4→NF-κB (p65) | Macrophages | TNF- |
2021 / Zhang et al. [70] |
| miR-23b | BM-MSC-sEVs | Down | TLR4 | TLR4→NF-κB | Microglia (BV2) | IL-6 ↓, IL-1 |
2021 / Nie and Jiang [71] |
| miR-146a-5p | Hypoxia-preconditioned BM-/MSC-sEVs; hUC-MSC-sEVs | Down | TRAF6 | TRAF6→NLRP3 | Macrophage, Microglia | M1→M2 polarization/Pyroptosis ↓ | 2024 / Liang et al. [67] |
| 2022 / Hua et al. [74] | |||||||
| miR-125a | BM-MSC-sEVs | Down | IRF5 | IRF5 signaling | Macrophages | M2 polarization ↑; Neuroprotection | 2021 / Chang et al. [15] |
| miR-124-3p | BM-MSC-sEVs | Down | Ern1 | Ern1 signaling | Macrophage, Microglia | M2 polarization ↑; Inflammation ↓ | 2020 / Li et al. [72] |
| miR-21-5p | IL-4-primed hUC-MSC-sEVs | Down | PDCD4 | PDCD4-mediated apoptosis | Macrophages | M1→M2 shift; Inflammation ↓, Apoptosis ↓ | 2025 / Li et al. [66] |
| miR-21a-5p | BM-MSC-sEVs | Down | PELI1 | NLRP3 mediated Pyroptosis | Macrophage, Microglia | Pyroptosis ↓ | 2024 / Gu et al. [73] |
Abbreviations: BM, bone marrow; hUC, human umbilical cord; MSC-sEVs, mesenchymal
stem/stromal cell-derived small extracellular vesicles; IRF5, interferon
regulatory factor 5; TLR4, Toll-like receptor 4; NF-
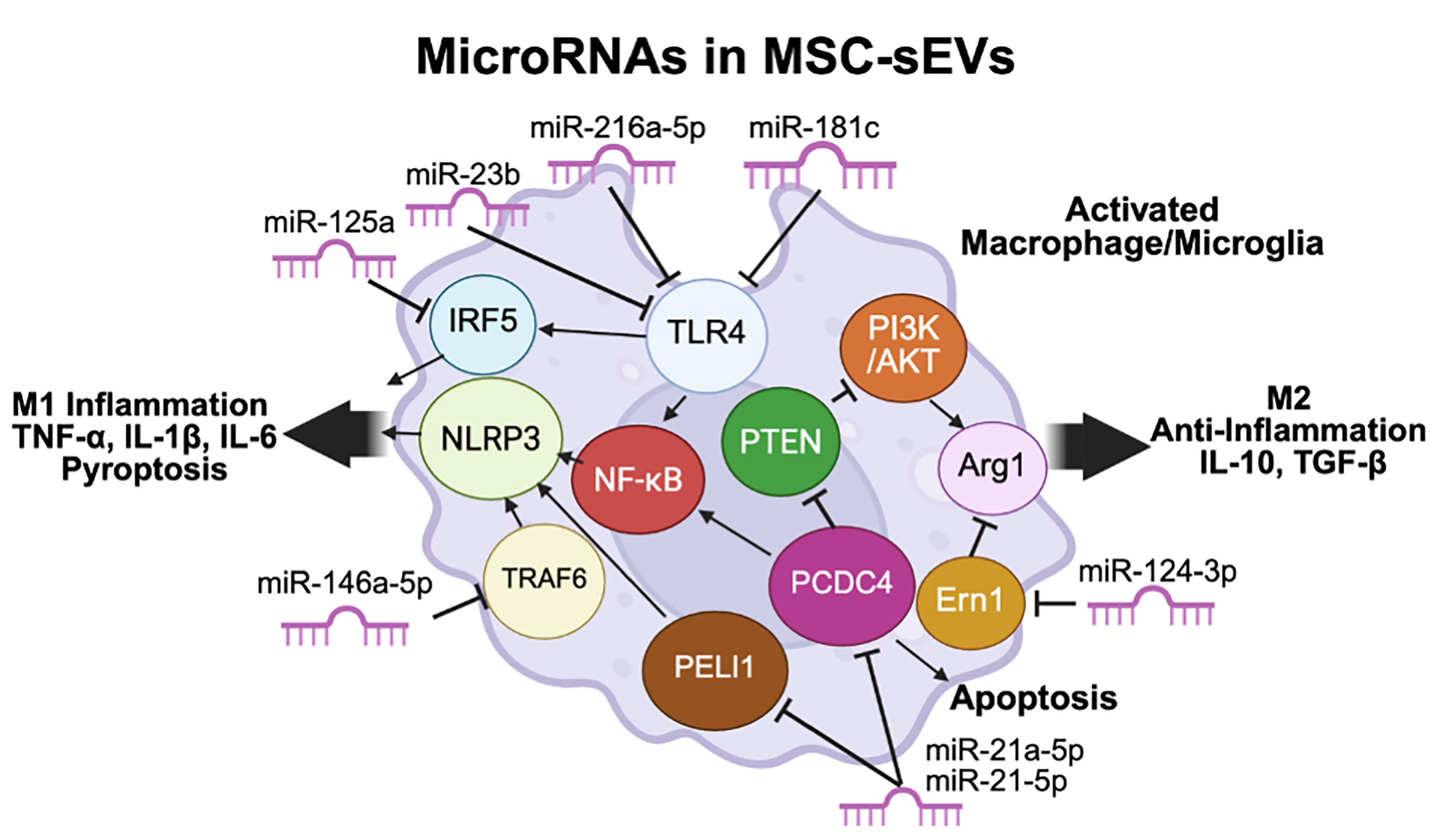 Fig. 4.
Fig. 4.
MicroRNA-mediated regulation of macrophage/microglial
polarization and cell death by MSC-sEVs. MSC-sEVs deliver regulatory microRNAs
to activated macrophages and microglia. miR-216a-5p, miR-23b, miR-181c, and
miR-146a-5p suppress TLR4/NF-
Multiple microRNAs (miRNAs)–target axes converge on TLR4/NF-
In addition to miRNAs, emerging evidence implicates other non-coding RNAs in
MSC-sEV-mediated immunomodulation after SCI. Notably, long non-coding RNA
(lncRNA)-Gm37494-loaded exosomes from adipose tissue-derived MSCs have been shown
to regulate M1/M2 polarization of microglia through the
miR-130b-3p/PPAR
MSC-sEVs coordinate an anti-inflammatory program in injured tissues by dampening
pro-inflammatory signaling while amplifying pro-resolving mediators
[66, 68, 73, 78]. In a rat SCI model, intravenously delivered MSC-sEVs significantly
demonstrated lower level of systemic serum TNF-
In addition to cytokine modulation, MSC-sEVs suppress multiple forms of programmed cell death that amplify tissue damage after SCI. MSC-sEVs potently inhibit inflammasome-mediated pyroptosis: MSC-sEVs potently suppress inflammasome-mediated pyroptosis, a form of inflammatory cell death that amplifies tissue damage after SCI. Exosomal miR-146a-5p from hUC-MSC-sEVs targets TRAF6, inhibiting NOD-like receptor protein 3 (NLRP3) inflammasome activation and reducing pyroptosis in macrophages and microglia (Table 1, Fig. 4) [74]. In parallel, miR-21a-5p from BM-MSC-sEVs targets PELI1, an E3 ubiquitin ligase that promotes NLRP3-mediated pyroptosis; suppression of PELI1 enhances autophagy and inhibits pyroptotic cell death in macrophages and microglia, thereby attenuating neuroinflammation (Table 1, Fig. 4) [73]. In addition to pyroptosis, MSC-sEVs also inhibit apoptosis. Exosomal miR-21-5p from IL-4-primed hUC-MSC-sEVs targets PDCD4 (programmed cell death 4), a pro-apoptotic tumor suppressor that promotes inflammation and cell death. Down-regulation of PDCD4 suppresses apoptosis in macrophages and shifts their phenotype from M1 to M2, with concurrent reduction of inflammatory cytokines (Table 1, Fig. 4) [66]. Thus, MSC-sEVs protect against both pyroptosis and apoptosis, collectively preserving cell viability and promoting a reparative microenvironment.
Taken together, MSC-sEVs exert broad anti-inflammatory and cytoprotective
effects through three interconnected mechanisms: (i) suppression of
pro-inflammatory cytokines (IL-1
After SCI, inefficient clearance of cellular debris—especially myelin fragments laden with axon growth–inhibitory lipids and proteins—perpetuates inflammation and impedes regeneration [80]. Restoring macrophage phagocytic capacity is thus a rational therapeutic target to foster a pro-regenerative milieu in the injured CNS. MSC-sEVs restore macrophage debris clearance and improve outcomes after SCI. In a rodent SCI model, MSC-sEVs rescued the post-injury phagocytic defect of infiltrating monocyte-derived macrophages, increased in vitro and in vivo uptake of myelin debris, and translated these cellular effects into improved axon regrowth and enhanced hindlimb locomotor recovery [16]. Mechanistically, myelin debris is cleared via multiple receptors on phagocytes; among them, MARCO (macrophage receptor with collagenous structure) has been implicated in binding polyanionic ligands and participating in myelin debris recognition/uptake [16]. Pharmacologic antagonism of MARCO with poly-G blunted exosome-driven myelin uptake and attenuated functional recovery [16]. EV-mediated MARCO induction thus provides a mechanistic link between MSC-sEVs and enhanced debris clearance after SCI.
M2-biased macrophages are primed for efferocytosis and high-capacity clearance. Across tissues and disease models, M2 macrophages exhibit strong efferocytic activity (the engulfment of apoptotic cells)—a phenotype associated with resolution of inflammation and tissue repair [81]. Reviews from immunology and drug-discovery perspectives converge on this point and highlight efferocytosis as a tractable therapeutic axis to redirect macrophages toward pro-resolving functions [81, 82]. MSC-sEVs both act on and are internalized by reparative macrophages. In SCI, intravenously administered MSC-sEVs home to the lesion and are preferentially taken up by M2-type macrophages in vivo [6, 11, 50]. Complementary in vitro evidence shows that IL-4–stimulated, M2-like rat macrophages exposed to a central-myelin–enriched fraction (acidic pH conditions) efficiently internalize DiR-labeled human MSC-sEVs, supporting a positive feedback loop in which sEVs both reprogram and are captured by reparative macrophages [6, 11]. MSC-sEVs broadly tune macrophage phenotype and function.
MSC-sEVs enhance macrophage effector function by (i) restoring phagocytosis of inhibitory myelin debris through upregulating debris-recognition machinery such as MARCO, and biasing macrophages toward M2-like, efferocytic states. These coordinated actions accelerate removal of detrimental debris, mitigate the inhibitory microenvironment, and support axonal regrowth and functional recovery after SCI.
Polarization of macrophages and microglia toward the M2 phenotype driven by upstream modulators such as MSC-sEVs contributes to the establishment of a reparative environment marked by reduced neurotoxicity, diminished glial scarring, and activation of neuronal and axonal regenerative programs [15, 83, 84]. In vitro, conditioned media from IL-4/IL-13–induced M2 macrophages elicit in adult dorsal root ganglion (DRG) neurons a distinctive neurite phenotype characterized by elongation and sparse branching and promote neurite extension across CSPG-rich inhibitory substrates [85]. In contrast, M1 macrophages induce short, highly branched neurites and exert direct neurotoxic effects [86]. Mechanistically, M2 macrophages upregulate arginase-1, thereby diverting L-arginine toward polyamine biosynthesis (putrescine/spermidine/spermine), which functions downstream of cAMP to mitigate myelin- and MAG-mediated growth inhibition [86]. Concurrently, M2-associated proteolytic signatures, defined by restrained MMP-9 activity and matrix-remodeling bias, may enable selective ECM turnover in perineuronal regions without compromising neuronal stability, thus maintaining growth-permissive microenvironments [86]. M2 macrophages also secrete a neurotrophic repertoire including IGF-1, BDNF, and NGF, each capable of directly engaging neuronal receptors to support survival, plasticity, and neurite outgrowth [87]. Notably, macrophage-derived IGF-1 alone induces robust sprouting from DRG explants; inhibition or deletion of neuronal IGF-1R abolishes this effect, confirming a direct neuron-targeted mechanism [88].
Beyond trophic support, M2 macrophages are enriched sources of IL-10 and
TGF-
Microglia similarly confer direct trophic support to neurons. Microglia-derived
IL-10 and TGF-
Disruption of the blood–spinal cord barrier (BSCB) following SCI initiates edema, leukocyte infiltration, and subsequent tissue loss. Among neuroimmune regulators within the neurovascular unit (NVU), macrophages and microglia exhibit dual functions: pro-inflammatory (M1) phenotypes exacerbate oxidative and proteolytic damage to the endothelium, while reparative (M2) states facilitate resolution, vascular restoration, and barrier re-sealing [92]. Convergent evidence from SCI and broader CNS models suggests that promoting M2-like and homeostatic programs is critical for re-establishing microvascular integrity and BSCB functionality, typically accompanied by the reappearance of tight-junction (TJ) and adherens-junction (AJ) proteins (occludin, claudin-5, ZO-1) and functional recovery [41].
A key reparative pathway is characterized by immunomodulatory regulation at the
endothelial interface. Pro-inflammatory cytokines such as TNF-
Collectively, these reports indicate that macrophages and microglia play
coordinated and phase-dependent roles in BSCB restoration. The sustained presence
of anti-inflammatory and homeostatic signals, particularly IL-10 and
TGF-
Despite positive preclinical results in rodent SCI models, several barriers remain for clinical translation of MSC-sEVs.
Current isolation methods produce heterogeneous products with variable potency, and batch-to-batch variability remains a significant concern [95]. Key quality metrics—particle size distribution, surface marker profiles (CD9, CD63, CD81), and cargo composition—differ substantially between protocols, and many clinical trials lack detailed sEV characterization, reducing reproducibility [95, 96]. Potency assays are not yet robust or standardized, and marker-based quantification does not guarantee therapeutic efficacy [96]. Advances in Good Manufacturing Practice (GMP) production have enabled scalable isolation, but defining critical quality attributes and establishing release criteria remain active areas of development.
MSC-sEVs can be stored at –80 °C with preserved bioactivity, offering logistical advantages over live cell therapies [97]. However, optimal storage conditions, freeze-thaw stability, and long-term shelf-life require further standardization for clinical-grade products.
Understanding the biodistribution, clearance kinetics, and target tissue accumulation of systemically administered MSC-sEVs is critical for dose optimization and safety assessment. Current evidence suggests rapid hepatic and splenic uptake following intravenous administration, with limited accumulation at injury sites without targeted delivery strategies [50]. Engineering approaches—including surface modification with targeting peptides, magnetic guidance, and hydrogel-based local delivery—may enhance therapeutic efficacy by improving site-specific retention [98].
MSC-sEVs have low tumorigenic risk compared to cell-based therapies, but do not fit existing drug or cell therapy regulatory categories, complicating approval pathways and GMP compliance [95]. Data on long-term immunogenicity and repeated dosing remain limited [95, 97]. These challenges are important but tractable, and ongoing regulatory dialogue will be essential for establishing appropriate frameworks.
Current preclinical evidence predominantly derives from rodent models, which provide valuable mechanistic insights but may not fully recapitulate human SCI pathophysiology [5, 99]. Large animal models including porcine and canine SCI offer closer anatomical and physiological parallels to humans, particularly regarding spinal cord dimensions, CSF dynamics, and immune responses, potentially providing more predictive efficacy and safety data [99, 100]. Organoid-based in vitro systems and patient-derived induced pluripotent stem cell (iPSC) models represent complementary approaches for mechanistic studies and personalized medicine applications [101].
Integration of MSC-sEV therapy with emerging technologies offers promising avenues for optimization. Personalized genomic approaches may enable patient stratification and prediction of therapeutic response [98]. Additionally, combination strategies incorporating MSC-sEVs with biomaterial scaffolds, electrical stimulation, or rehabilitative training may enhance functional recovery through synergistic mechanisms. Engineering tools that enable targeted drug delivery, such as surface-modified sEVs or stimulus-responsive release systems, represent active areas of development [98, 102, 103, 104]. Future research should prioritize cross-model validation studies that bridge rodent, large animal, and human-derived systems to establish robust therapeutic paradigms and accelerate clinical translation.
Macrophages and microglia are central regulators of the neuroimmune and vascular responses that shape the trajectory of spinal cord repair. Evidence summarized in this review indicates that MSC-sEVs exert their therapeutic efficacy primarily through bidirectional interactions with these myeloid populations. By promoting and maintaining anti-inflammatory, efferocytic and pro-resolving phenotypes, MSC-sEVs attenuate cytokine-driven injury, enhance debris clearance, and restore blood–spinal cord barrier integrity. The resulting stabilization of the neurovascular unit provides a permissive environment for neuronal survival, axonal growth and remyelination.
Future research should focus on defining the precise molecular cargoes responsible for these effects, mapping sEV uptake and signaling in vivo at single-cell resolution, and optimizing delivery strategies that achieve sustained myeloid modulation. Such advances will accelerate translation of MSC-sEV–based therapeutics toward clinically viable interventions for spinal cord injury and other neurological disorders.
MN conceptualized and designed the review. MN, KLL, and TY performed the literature search and data analysis. MN drafted the manuscript. RU, RH, SO, and MS contributed to manuscript revision and figure preparation. JDK and OH provided critical supervision, contributed to the conception and design of the review, and final editorial approval. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported in part by the North Tech Foundation (R7SB-sa-7 to MN), JSPS KAKENHI grant (JP25K12482 to MN) and the RR&D and BLR&D Services of the U.S. Department of Veterans Affairs, Grant (B7335R and B9260L).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.