




 , Emiko Takeuchi 2, Masashi Satoh 2, Takayuki Imanishi 2, Makoto Otsu 3, Tadahiro Suenaga 2, Yasuo Takeuchi 1
, Emiko Takeuchi 2, Masashi Satoh 2, Takayuki Imanishi 2, Makoto Otsu 3, Tadahiro Suenaga 2, Yasuo Takeuchi 11 Department of Nephrology, Kitasato University School of Medicine, 252-0329 Sagamihara, Kanagawa, Japan
2 Department of Immunology, Kitasato University School of Medicine, 252-0329 Sagamihara, Kanagawa, Japan
3 Division of Hematology, Kitasato University School of Allied Health Sciences, 252-0329 Sagamihara, Kanagawa, Japan
Abstract
Chronic granulomatous disease (CGD) is an inherited immunodeficiency characterized by impaired phagocytic cells due to mutations in genes encoding the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex, leading to recurrent severe infections. In the innate immune system, NETosis, which is a program for the formation of neutrophil extracellular traps (NETs) has been highlighted. Over the past years, NETs have been recognized as being involved in the pathogenesis of immune-mediated inflammatory renal diseases. On the mechanism underlying usual NETosis, reactive oxygen species (ROS) activation by NADPH oxidase (NOX) has been considered to be essential. CGD patients are theoretically unable to form such usual NETosis because of a deficiency of NOX-dependent ROS activation. However, according to previous reports, stimulated neutrophils derived from CGD patients showed NOX-independent unusual NETosis enriched in mitochondrial DNA. The mitochondrial ROS activation on NETosis in CGD was reported to induce an elevated inflammatory response, instead of the usual ROS generation, which might cause autoimmune diseases. We are interested in the renal autoimmune disorder in CGD, especially the inflamed renal disorder caused by unusual mitochondria-rich NETosis in CGD. Indeed, in vivo analysis has revealed severe renal disorder in MRL/lpr lupus-prone mice lacking NOX activity compared to wild-type of MRL/lpr. A few clinical reports showed aggravation of inflamed glomerulonephritis in patients with CGD. Taken together, we presumed that a defect of usual NOX-dependent NETosis might be a clue for aggravated renal disorder in CGD. In the present review, we summarize the features of NETosis in CGD and discuss the aggravation mechanism for renal disorder in CGD by referring to previous reports.
Keywords
- CGD
- NETosis
- NADPH oxidase
- mitochondrial ROS
- renal disorder
Chronic granulomatous disease (CGD) is a rare inherited immunodeficiency that was first reported in 1959 [1]. CGD is usually diagnosed in the first 1 to 3 years of life. The average worldwide birth prevalence has been estimated to be around 1/200,000 [2, 3]. Essentially, CGD is caused by the impaired phagocytic function of the innate immune cells, which is attributable to mutations in genes encoding nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex (OEC) [3, 4]. X-linked CGD secondary to mutation of the CYBB gene for gp91phox is the most prevalent and severe form worldwide [5].
The NADPH oxidase, so-called NOX, consists of the following subunits: gp91phox encoded by the CYBB, p22phox encoded by the CYBA, p47phox encoded by the NCF1, p67phox encoded by the NCF2, and p40phox encoded by the NCF4. Although RAC encoded by the p21gene is not a component of NADPH oxidase, it activates p67phox in its GTP-bound form during cell activation induced by inflammatory or phagocytic stimuli [4, 6, 7]. In patients with CGD, such components for NOX production in phagocytes are defective. In more detail, the NADPH OEC comprises five subunits: two are localized in the cell membrane, and three are localized in the cytosol (Fig. 1) [3]. The two membrane-bound subunits are gp91phox and p22phox. These proteins form a heterodimeric complex (flavocytochrome b558). When neutrophils become activated in response to inflammatory or phagocytic stimuli, cytochrome b558 is activated; thereafter, three cytoplasmic subunits (p47phox, p67phox, and p40phox) form a hetero-trimer which is translocated to cytochrome b558 to induce NOX formation. RAC is also activated and recruited [3, 4]. The p47, p67, and p40 complex activates electron transfer from cytosolic NADPH via cytochrome b558, generating reactive oxygen species. The enzyme responsible for ROS production is called the phagocyte NADPH oxidase. Generally, gp91phox related phagocyte NADPH oxidase for ROS production is so-called NOX2 [6, 7]. NOX2-derived ROS in neutrophils is critical for killing catalase-positive microorganisms. However, uncontrolled and excessive ROS production in neutrophils could induce tissue injury and prolonged inflammatory reactions, which cause inflammatory diseases [5, 7]. Thus, activation of NOX must be tightly regulated.
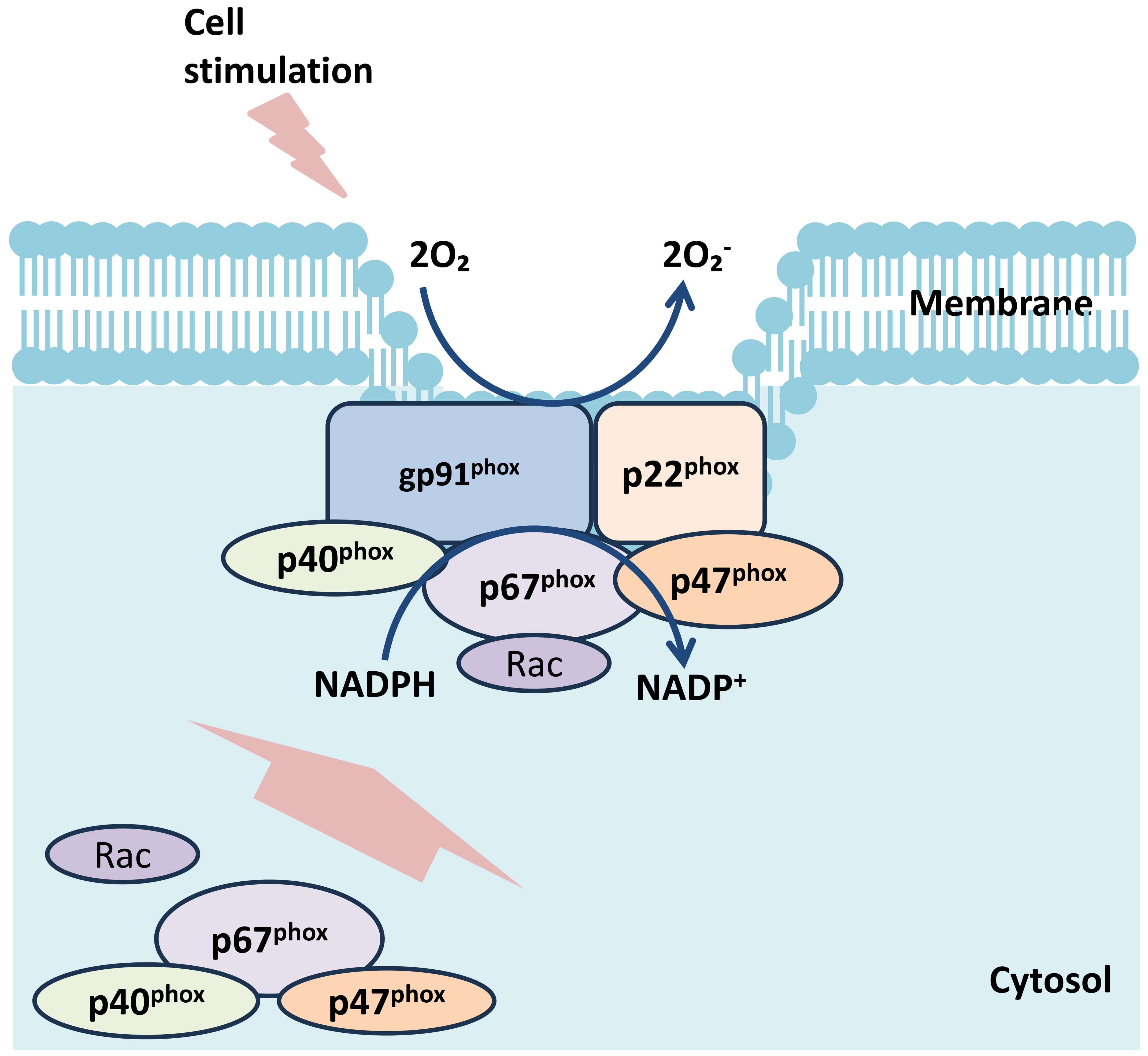 Fig. 1.
Fig. 1.
Scheme of the NADPH oxidase complex subunits. The NADPH oxidase consists of the following subunits: gp91phox encoded by the CYBB, p22phox encoded by the CYBA, p47phox encoded by the NCF1, p67phox encoded by the NCF2, and p40phox encoded by the NCF4. The two membrane-bound subunits are gp91phox and p22phox. Three cytoplasmic subunits are p47phox, p67phox, and p40phox. The membrane subunits gp22phox and gp91phox also include the molecule for the reactive oxygen species (ROS), which interacts with other proteins in the membrane. NADPH, nicotinamide adenine dinucleotide phosphate.
Neutrophil plays pivotal role in the clearance of extracellular bacteria by phagocytosis [8]. In addition, neutrophils have a particular biological defense reaction that produces neutrophil extracellular traps (NETs). The process of NETs formation is referred to as NETosis [9, 10]. On the mechanism underlying NETosis, ROS generation via NOX is essential for the formation of bactericidal lytic NET [8, 10]. Meanwhile, NETs are involved in the pathogenesis of immune-mediated inflammatory diseases, including renal diseases [11, 12, 13, 14]. The relation of NETs to the development of glomerulonephritis (GN) or tubulointerstitial nephritis has been gradually elucidated [12, 13, 15, 16].
In patients with CGD, the inability to induce NOX-derived ROS activation, meaning the deficiency of NOX-dependent usual NETosis, is considered to lead to systemic granuloma formation and recurrent severe infection [3, 4, 5]. Additionally, of note, it has been demonstrated that CGD patients showed a significant elevation of inflammatory responses, and they are at a high risk of autoimmune disease [17, 18, 19]. Thus, we have an interest in features of unusual NETosis in CGD, and elucidating the mechanism for elevated inflammation in CGD is curious. Moreover, it appeared to be intriguing to assess the degree of renal disorder in CGD, although evidence regarding the association of renal disorder with CGD is limited. Herein, we focused on the features of NETosis and renal disorder in CGD based on the previous reports.
NETosis is one of the cell-death forms, such as necrosis or apoptosis. During the process of underdoing NETosis, nuclear antigens might be modified and released to extracellular space as specialized form called NETs [20]. Basically, NETs are three-dimensional meshworks consisting of chromatin, antimicrobial components such as myeloperoxidase (MPO) and neutrophil elastase (NE) [8]. These fibrous networks can effectively catch and eliminate bacteria, fungi, and viral particles [21, 22, 23]. Additionally, increasing evidence has demonstrated that NETs itself deeply involved in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE) or antineutrophil cytoplasmic antibody (ANCA) associated vasculitis [12, 13, 15, 24]. According to previous articles, persistently activated NETs without proper processing could be an inducing factor for ANCA-related glomerulonephritis or lupus nephritis (LN) [13, 24].
Regarding detail mechanism of NETosis, the essence of usual lytic NETs formation is summarized in Fig. 2a. NETs are formed via various signaling pathways in response to different triggers. Stimulation with a wide range of substances, including bacteria, phorbol-12-myristate-13-acetate (PMA), and viruses are known to be a trigger for the occurrence of NETosis [25]. Sterile inflammatory signals (activated platelets, endothelial cells, crystals, autoantibodies, immune complexes (ICs), and various proinflammatory cytokines) are also reported to induce NETosis and consequently extrude NETs [9, 13, 26, 27, 28, 29]. After recognizing PMA stimulation, NADPH oxidase is activated, and then NADPH oxidase catalyzes O2 to generate ROS in neutrophils. The generated ROS stimulates MPO to trigger the activation and translocation of NE from the azurophilic granules to the nucleus, where NEs digest nucleosomal histones and promote chromatin relaxation, which favors the citrullination of histone (CitH3) by peptidylarginine deiminase 4 (PAD4) [8, 30]. Physiologically, PAD4 catalyzes the conversion of histone arginine to citrullination, resulting in chromatin decondensation in the nuclei of neutrophils [31, 32, 33]. In addition, it was demonstrated that PAD4 expressed in neutrophils directly initiates the translocation of NE and MPO from granule to nuclei [8]. The citrullination of histone by PAD4 activation leads to chromatin decondensation, which causes entropic chromatin swelling [21]. Subsequently, MPO associates with chromatin and, synergistically with NE and PAD4, promotes massive chromatin decondensation [34]. Eventually, decondensed chromatin is released into the extracellular space as NETs formation in association with diverse granular and cytoplasmic proteins [35]. NOX-dependent ROS activation and histone citrullination mediated by PAD4 are essential steps for the development of usual lytic NETs. Fundamentally, citrullination is a post-translational modification that plays an important role in many physiological processes such as cell differentiation and renewal. However, abnormal or aberrant citrullination can lead to the development of diseases, including autoimmune diseases, cancers, and cardiovascular diseases [8, 36].
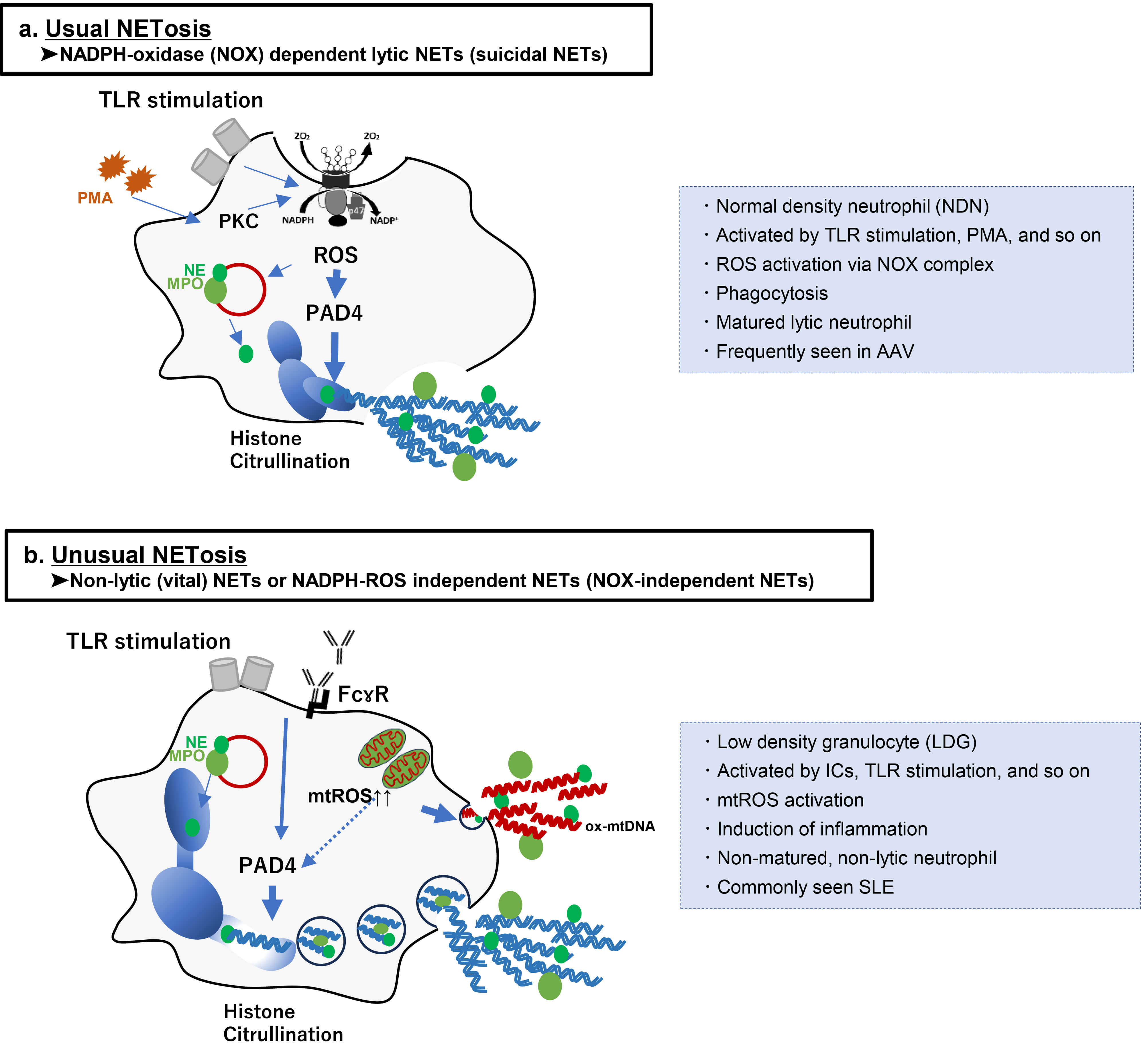 Fig. 2.
Fig. 2.
Schematic diagram of the mechanisms for undergoing NETosis and
dissimilarity between usual NOX-dependent Lytic (suicidal) NETs and Non-lytic
(vital) NETs or NOX-independent NETs. (a) Suicidal NETosis is a usual NADPH
oxidase-dependent death process. It can be triggered by PMA and TLR stimulation.
The ROS production mediated by NADPH oxidase 2 initiates the activation of PAD4
to mediate chromatin decondensation. Then, decondensed chromatin is released into
the cytoplasm as the parts of genomic DNA, mixing with granule proteins such as
NE and MPO. Finally, the plasma membrane permeabilizes, allowing for lytic
suicidal NETs expulsion while the neutrophil dies. The suicidal NETs exert
phagocytic function, and it is commonly seen in patients with ANCA-associated
vasculitis. Moreover, suicidal NETs are frequently detected in normal density
neutrophil (NDN) in vitro experiments. (b) Vital NETs, like unusual
NETs, appear without altering the membrane. Several stimuli, such as ICs, TLR
stimulation, and so on, induce NETosis without cell lysis. Although it is unable
to imply that NOX-independent NETosis is equivalent to vital NETosis, activated
pathways for the formation of vital NETs are assumed to be NOX-independent, and
stimulated neutrophils for leading to vital NETs release of mitochondrial DNA
(mtDNA). It is known that the induction of vital NETosis significantly activates
the inflammatory response. Vital NETs are commonly seen in patients with systemic
lupus erythematosus (SLE), and it is frequently detected in low density
granulocytes (LDGs) in vitro experiments. Abbreviation: AAV,
anti-neutrophil cytoplasmic antibody associated vasculitis; ANCA, anti-neutrophil
cytoplasmic antibody; Fc
In the field of basic research, another type of NETs other than the above-described usual lytic NETs has recently been highlighted. Such unusual NETs without cell lysis are termed vital NETs in contrast to usual lytic NETs called suicidal NETs [24]. According to recent reviews [24, 37, 38, 39], two mechanisms of NETosis have been identified: NETosis leading to suicidal NETs, in which neutrophils die after expelling the filaments; and NETosis leading to vital NETs, in which expulsion appears without altering the membrane. Fig. 2a,b summarizes the features and differences between suicidal NETs and vital NETs. Among the two types of NETs, there are several differences in features, including not only pathological mechanism or morphology but also occurrence rate in each autoimmune disease or neutrophil population, although the molecular mechanism of each NET’s formation has not been completely elucidated. On the other hand, an interesting opinion was described in a recent review article in which vital NETs may be a complementary mode of suicidal NETs at the early stage in response to stimuli [39]. They implied that suicidal or vital formation may be simply categorized as a reaction at a different phase within one large framework of the self-defense system. Further evidence regarding the similarity between the two types of NETosis is desirable.
Suicidal NET formation is based on the NADPH oxidase-dependent mechanism. The production of ROS mediated by NADPH oxidase 2 (NOX2) has critical roles in suicidal NETosis as it triggers the downstream signaling cascade for chromatin decondensation [8, 39]. Fundamentally, the NADPH oxidase-dependent suicidal NETs formation has been reported to occur primarily at 3 to 8 hours following the activation of neutrophils by various stimuli, such as PMA, and TLR stimulations, and so on [24, 30]. The induction of NADPH oxidase following the activation of protein kinase C (PKC) via the ERK-MEK signaling route [40, 41] leads to an increase in ROS, initiating the activation of PAD4 to mediate chromatin decondensation. Then, decondensed chromatin is released into the cytoplasm as the parts of genomic DNA, mixing with granule proteins such as NE and MPO. Finally, the plasma membrane permeabilizes, allowing for lytic suicidal NETs expulsion while the neutrophil dies [24, 37] (Fig. 2). The NOX-dependent suicidal NETs fundamentally exert the function as phagocytosis, and a recent review article reported that these suicidal NETs are frequently seen in patients with ANCA-associated with vasculitis (AAV) [24]. Moreover, intriguingly, neutrophils have a heterogeneous population categorized from mature type to immature type in vitro experiments [42], and it has been demonstrated that suicidal NETs are frequently detected in normal density granulocytes (NDGs) [42, 43].
Regarding the vital NETs, several stimuli such as LPS, activated platelets, ICs, TLR stimulation, granulocyte/macrophage colony-stimulating factor (GM-CSF), C5a, calcium ionophores, and nicotine induce NETosis without cell lysis [24, 27, 39, 44, 45]. Vital NETosis occurs so faster than suicidal NETosis, and its pathways is assumed to be NOX-independent [24, 46], and neutrophils primed by above-described stimuli are reported to form vital NETs with release of mitochondrial DNA (mtDNA) [47] (Fig. 2b). In accordance with previous reports, the release process of vital NETs composed of mtDNA requires the production of glycolytic ATP to rearrange the microtubule network and F-actin [48]. Douda et al. [27] described a rapid NOX-independent NETs formation process that is mediated by mitochondrial ROS and a calcium-activated small conductance potassium channel. However, the source of ROS generation and the degree of PAD4 activation in the process for the formation of vital NETosis remain completely elucidated. In addition, intriguingly, induction of vital NETosis could significantly activate an inflammatory response, which leads to autoimmune diseases [39, 44]. It has been reported that vital NETs in autoimmune diseases are enriched in oxidized mtDNA instead of nuclear chromatin, which is known to act as a potent inflammatory inducer [44]. Vital NETs with oxidized mtDNA were more detected in patients with LN than AAV [49]. Moreover, in contrast to suicidal NETs, non-lytic NETs are frequently formed in low-density granulocytes (LDGs) in vitro experiments [11, 24]. It has been demonstrated that LDGs synthesize increased levels of proinflammatory cytokines and are prone to spontaneously form NETs compared with NDGs [43]. However, most studies on NETs induction have been conducted in vitro or ex vivo experiments. Further studies are needed to evaluate vital NETs using experimental animal disease models or human samples.
In patients with CGD, induction of NETosis via NOX-dependent ROS activation is
theoretically impossible because of the lack of phagocyte NADPH oxidase activity.
Indeed, a previous report indicated that CGD neutrophils were resistant to
NETosis by the analysis using NDG [50]. However, although not sufficient, there
has been evidence that identified the presence of NETosis in CGD. Several
previous reports indicated the possibility of NOX-independent NETosis in CGD,
which is different from usual lytic NETosis [12, 27, 44, 51, 52] (Table 1, Ref.
[27, 44, 51, 52, 53, 54]). A recent review article by Nishi and Mayadas [12]mentioned that NETs can be generated even if the NADPH oxidase is absent. In
accordance with their previous data, neutrophils still can make NETs in response
to IC engagement of FC
| Ref. No | Author (year) | Model/Sample | Inducing stimuli | NOX dependence | Feature of NETosis/Notes for mtROS, mtDNA, and ox-mtDNA | Key Findings/Notes for type I IFN |
| [27] | Douda et al. (2015) | In vitro/ex vivo | Calcium ionophore | NOX-independent pathway was activated. | ➢Activated NOX-independent NET-osis was fast and mediated by mitROS. | NA |
| [44] | Lood et al. (2016) | In vitro/ex vivo | RNP ICs | NADPH-oxidase activity (-); NOX2-independent | ➢CGD-LDGs showed enhanced mtDNA-rich NETosis driven by increased mitROS, and the generated NETs had a higher proportion of damaged, ox-mtDNA. | ➢In CGD, increased NOX-independent mtDNA-rich NETs were associated with elevation of type I IFN activity. |
| [44] | Lood et al. (2016) | Human blood | N/A | NADPH-oxidase activity (-); NOX2-independent | NA | ➢CGD patients showed elevated serum type I IFN activity, including 92.3% of those with autoantibodies. |
| [51] | Fuchs et al. (2007) | In vitro/ex vivo | Gox | NADPH-oxidase activity (-); NOX2-independent | ➢Stimulation with S. aureus and PMA did not make usual NETs, but neutrophils released lytic NETs after Gox stimulation. | NA |
| [52] | Chen et al. (2012) | In vivo | BSA/anti-BSA ICs | NADPH-oxidase activity (-); NOX2-independent | ➢Neutrophils still can make NETs in response to ICs engagement of FC |
NA |
| [53] | Kelkka et al. (2014) | Human blood | N/A | NADPH-oxidase activity (-); NOX-independent | NA | ➢CGD patients developed a type I IFN signature. |
| [54] | Campbell et al. (2012) | In vivo | N/A | Lack of functional NADPH oxidase; NOX-independent | NA | ➢Cybb-deficient, as in CGD, showed a worsened lupus phenotype compared with wild-type MRL/lpr mice. |
Abbreviation: CGD, chronic granulomatous disease; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; mtROS, mitochondria reactive oxygen species; mtDNA, mitochondrial DNA; ox-mtDNA, oxidized mitochondrial DNA; Ref. No, reference number; LDGs, low-density granulocytes; RNP-ICs, ribonucleoprotein immune complexes; IFN, interferon; Gox, glucose oxidase; PMA, phorbol myristate acetate; ICs, immunocomplexes; N/A, not applicable; NA, not available.
Thus far, it has become evident that patients with CGD are at a higher risk of autoimmune diseases, including SLE, juvenile idiopathic arthritis, autoimmune thrombocytopenia, and antiphospholipid syndrome [17, 18, 19, 55]. Although the mechanisms underlying the increased risk of CGD-related autoimmune diseases are not completely clarified, defects in phagocytic function are considered to result in repeated antigen stimulation and hypergammaglobulinemia, leading to autoantibody production [56]. In short, a subclinical persistent infection status due to the inability of phagocytic function might amplify the immune response in CGD. Additionally, a previous clinical cohort study in patients with CGD indicated that 92.3% of CGD subjects with clinical autoimmunity and/or positive autoantibodies had elevated type I interferon (IFN) that is a representative marker of inflammatory response [44]. Therefore, the high risk of autoimmune diseases in CGD patients might be attributable not only to the status of persistent infection, but rather to a specific mechanism for elevated inflammatory response.
Regarding increased inflammation in CGD, we propose the involvement of
NOX-independent and mtDNA-rich NETs in CGD. To our knowledge, the functional
effects of NETs have dual aspects. Briefly, NETs induction and clearance may
originally result in a protective antimicrobial effect, but excessive NETs
formation and insufficient removal of NETs could lead to tissue damage and
autoantigen modification and externalization [13, 57]. Previously, it was
demonstrated that NET-derived DNA complexed with danger-associated molecular
patterns converts the DNA to potent immunogenic structures [58, 59]. In fact, NETs
were demonstrated to activate plasmacytoid dendritic cells [58] and autoreactive
B cells in vitro [60], which resulted in the production of
IFN-
In the previous report, LGDs derived from CGD patients showed enhanced spontaneous NETosis, which is comparable to SLE LDG [44]. In addition, those LDGs in CGD patients relied on mitochondrial ROS to form NETs. Inhibition of other ROS source such as MPO, with 3-amino,12,4-triazole or NO with l-NG-monomethyl-l-arginine, did not abrogate NETosis in CGD LDGs [44]. Moreover, NETs from CGD subjects showed not only increased mt-DNA but also a higher amount of MPO and NE. They mentioned that LDG from CGD patients showed an increase in the total amount of NETosis, and it was presented that mitochondrial ROS synthesis might promote NETs enriched in mtDNA in CGD [44]. Moreover, as described in the previous report in which the activation of type I IFN signature was elevated in patients with CGD [53], Lood et al. [44] also demonstrated that mitochondrial ROS production in LDG NETosis was associated with enhanced type I IFN activity in CGD. Collectively, previous evidence led to the following summary: NETosis from CGD patients aggravates inflammation via mitochondrial ROS activation. As a considerable hypothesis, the above-described enhanced mitochondrial ROS production in LDGs from CGD patients may be a compensatory mechanism related to a lack of usual NOX-induced ROS production. Previously, the enhanced levels of circulating neutrophil granular proteins, pro-inflammatory cytokines, and type I IFNs were confirmed in patients with CGD [53, 65, 66]. Also, it was clarified that chronic inflammation in CGD could synergize with neutrophil dysregulation to promote amplification of inflammatory responses [67, 68, 69, 70, 71]. Taken together, CGD patients might need to amplify immune responses as an alternative reaction for biological defense instead of the inability to form usual NOX-dependent NETosis, which might be a key to the high prevalence of autoimmune diseases.
To the best of our knowledge, reports on renal disorders in patients with CGD remain limited. A recent study documented kidney and urinary-tract diseases in 22% of 430 patients with CGD [4]. Other articles have reported an incidence of renal disorders ranging from 2.7% to 23% [72, 73]. Renal insufficiency is considered relatively rare compared with other CGD-associated complications such as pulmonary or gastrointestinal diseases [4]. Moreover, previous studies consistently identified p47phox, encoded by the NCF1, as the predominant genotype among CGD patients presenting with renal disorders [55, 72, 74]. Although the detailed mechanism has not been elucidated, mouse experimental models have shown that NCF1-encoded p47phox mediates autoreactive T-cell responses leading to severe arthritis [75] and mitigates renal fibrosis by suppressing pro-fibrotic factors [76]. Taken together, abnormalities in the p47phox genotype appear to play a key role in modulating the severity of renal involvement in CGD, though current human evidence remains limited and should not be over-interpreted.
Previously, Couser and Johnson [77] and Waldmann [78] proposed that immunological abnormality is deeply involved in the pathogenesis of renal diseases. Abnormal neutrophil activation in the innate immune system could be a trigger or aggravating factor for renal diseases, including AAV or LN [12, 13, 24]. In particular, persistently activated NETs without proper procession has been highlighted as a critical factor for inflammatory renal diseases [24]. In the experimental renal diseases model, Wu et al. [16] revealed that NETs formation in the mouse inflamed kidney tissues after ischemia reperfusion injury (I/R), and infiltration of neutrophils resulting in NETs was increased in acute kidney injury (AKI) due to I/R in a time-dependent manner. Moreover, another study in murine models revealed that NET-derived histones can cause severe crescentic GN [79]. It was demonstrated that NETs themselves had direct cytotoxic effects on glomerular endothelial cells via mediating the activation of histones and MPO in murine models [79, 80, 81, 82]. Taken together, those findings based on the basic research let us presume that renal insufficiency in CGD may be severe and advanced since characteristic NOX-independent NETosis in CGD could amplify the inflammatory response. As evidence in vivo experiment that is compatible with our hypothesis, lupus-prone MRL/lpr mice that lack functional NADPH oxidase because of deficient Cybb, like CGD, develop a worsening lupus phenotype when compared to mice that are not deficient in Cybb [54]. Further experiments that will address our hypothesis are warranted.
Actually, as clinical evidence for aggravation of renal disorder in CGD, Tanaka et al. [55] summarized the case reports of biopsy-proven GN in patients with CGD. According to their review of literature, including their case, 10 cases of biopsy-proven GN in CGD patients were identified [17, 55, 74, 83, 84, 85, 86, 87]. Table 2 (Ref. [17, 55, 74, 83, 84, 85, 86, 87]) summarizes clinicopathological features of the 10 cases of GN in CGD patients. Of those 10 cases, immunoglobulin A nephropathy (IgAN) and IgA vasculitis nephritis (IgAVN) were the most common glomerular diseases, reported in two patients each. Other cases include membranous nephropathy, focal segmental glomerulosclerosis, tubulointerstitial nephritis, LN, and so on. Remarkably, all four IgAN and IgAV in CGD patients presented with rapidly progressive GN (RPGN) with crescent formation on glomeruli, and one case of them showed poor renal prognosis despite intensive steroid therapy (Table 2) [84], which is an untypical clinical course. Both IgAN and IgAVN tend to exhibit a slowly progressive clinical course [88], and therefore, both diseases are generally categorized as chronic GN [89]. Until now, intensive treatment including steroid therapy has apparently improved the renal outcome of IgAN [90] or IgAVN [91]. Therefore, it is not so hard to presume the involvement of a unique mechanism of CGD in the development of severe crescentic IgAN and IgAVN in those four CGN patients. Clinicopathologically, IgAN and IgAVN are very similar, and both diseases show glomerular capillaritis with infiltration of inflammatory cells [92, 93, 94]. In fact, activation of proinflammatory cytokines was reported to induce the production of galactose-deficient IgA1, which is a critical molecule for the development of IgAN [95]. Also, elevated inflammatory cytokines were significantly higher in patients with IgAVN [94]. Moreover, Heineke et al. [92] emphasized activated neutrophils in the glomerular capillary loop, which causes severe capillaritis with an elevated inflammatory response. On the other hand, the above-mentioned crescentic IgAN exhibiting RPGN is a relatively rare phenotype [92], but it commonly shows a significant elevation of inflammatory response [92]. Collectively, we hypothesized that NOX-independent NETosis enriched in mtDNA may be associated with unusual severe IgAN or IgAVN via aggravating the inflammatory response in patients with CGD. Further investigation with accumulated cases from all over the world is desirable to confirm our hypothesis. In the future, if validated, these accumulated findings may help pave the way for non-invasive diagnostic strategies, such as the detection of urinary mtDNA-NET complexes as a potential early biomarker for renal injury in CGD patients.
| Case | Author (year) [Ref No] | Age/gender | Genotype | Pathological type | RPGN/crescent | Treatment | Outcome |
| 1 | Tanaka et al. (2021) [55] | 22/M | gp91phox | IgAV | (+)/(+) | mPSL 500 mg × 3 days and PSL 35 mg/day | improve |
| 2 | Leiding et al. (2013) [74] | 44/F | gp47phox | MN | NA/NA | NA | NA |
| 3 | Leiding et al. (2013) [74] | 27/M | gp47phox | FSGS | NA/NA | NA | NA |
| 4 | Leiding et al. (2013) [74] | 15/M | gp47phox | FSGS, TIN | NA/NA | NA | ESKD |
| 5 | De Ravin et al. (2008) [17] | 13/M | gp91phox | IgAN | (+)/(+) | PSL30 mg twice daily | improved |
| 6 | Narsipur and Shanley (2002) [83] | 22/M | gp91phox | IgAN | (+)/(+) | NA | NA |
| 7 | Kimpen et al. (1991) [84] | 8/F | gp47phox | IgAV | (+)/(+) | NA | ESKD |
| 8 | Manzi et al. (1991) [85] | 4/M | gp91phox | LN Subendothelial deposits of IgG, IgM, C3, C1q | (-)/(-) | PSL30 mg/every other day | maintain |
| 9 | Frifelt et al. (1985) [86] | 14/M | Autosomal recessive | Mes proliferative GN | NA/NA | Penicillin therapy for 9 months + PSL 2weeks. | ESKD |
| IF study was not done | |||||||
| 10 | van Rhenen et al. (1979) [87] | 18/F | Unknown | Sclerosing GN with crescents | (+)/(+) | Antibiotics therapy and temporary hemodialysis | improved |
| No deposits in glomeruli |
Abbreviations: Ref, reference; RPGN, rapidly progressive glomerulonephritis; M, male; F, female; NA, not available; IgAV, IgA vasculitis; MN, membranous Nephropathy; FSGS, focal segmental glomerular sclerosis; TIN, tubulointerstitial nephritis; IgAN, IgA nephropathy; LN, lupus nephritis; Mes, mesangial; GN, glomerulonephritis; IF, immunofluorescence; mPSL, methylprednisolone; PSL, prednisolone; ESKD, end stage kidney disease.
In the present review, we focused on characteristic immunological features of CGD and then considered the mechanism of aggravated renal disorder in patients with CGD. Fig. 3 provides a graphical summary of our proposed concept. To approach the core of its mechanism, further investigation, including clinical studies and basic researches will be necessary. Currently, the evidence supporting the hypothesis of exacerbated renal dysfunction in CGD patients is primarily derived from in vitro experiments and non-CGD disease models, and thus remains preliminary. In vivo analyses using mouse models have been very limited. To address this limitation, further studies are warranted to advance knowledge in this field, including the use of CGD patient-derived tissues, in vivo CGD animal models, and targeted investigation of NOX-independent NETosis pathways in renal tissue.
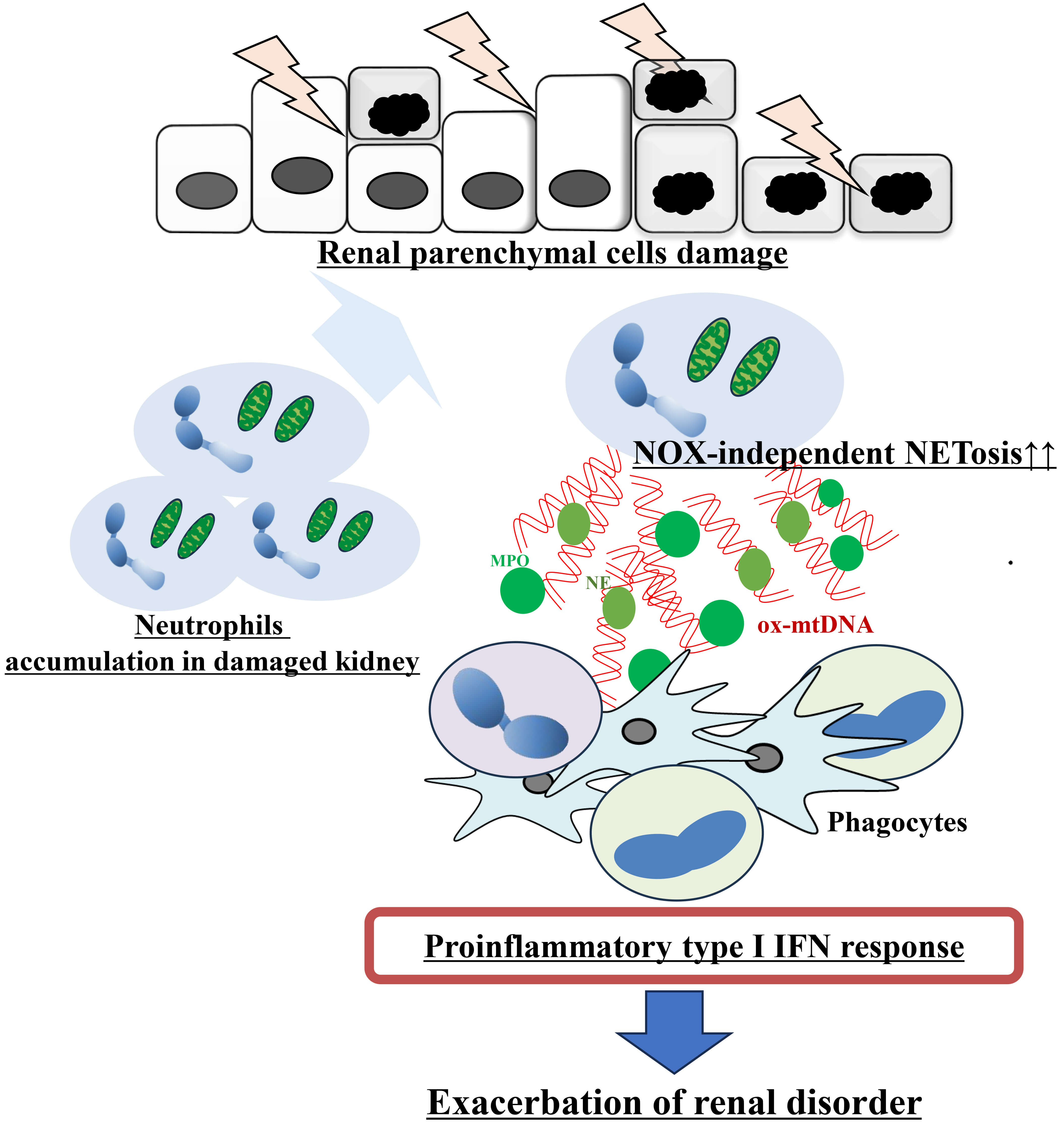 Fig. 3.
Fig. 3.
Schematic representation of the hypothetical mechanism
underlying the exacerbation of renal disease in patients with CGD. Damage to
renal parenchymal cells leads to the recruitment and accumulation of neutrophils
in the injured kidney. In CGD, activated neutrophils undergo NOX2-independent
NETosis and release ox-mtDNA, due to their inability to generate usual
NOX-dependent lytic NETosis. These ox-mtDNA-rich NET components are subsequently
recognized by phagocytes, triggering activation of a proinflammatory type I IFN
response. The amplified type I IFN signaling ultimately contributes to the
exacerbation of renal disorder. Abbreviation: IFN, interferon; MPO,
myeloperoxidase; NE, neutrophil elastase; NOX, nicotinamide adenine dinucleotide
phosphate oxidase; ox-mtDNA, oxidized mitochondrial DNA;
TA, YW, and ET led the conceptualization of the review and performed the primary drafting of the manuscript. TA, YW, ET, MS, TI, MO, TS, and YT contributed significantly to the research design, systematic literature search, and critical evaluation of the included studies. All authors were actively involved in data synthesis, interpreting the findings, and creating the original figures and tables presented in the work. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.