



 , Qi Zhang 1,†, Yuting Chang 1, Gaohua Han 1,*
, Qi Zhang 1,†, Yuting Chang 1, Gaohua Han 1,*
1 Department of Oncology, The Affiliated Taizhou People’s Hospital of Nanjing Medical University, 225300 Taizhou, Jiangsu, China
†These authors contributed equally.
Abstract
Lung cancer is associated with high global incidence and mortality, with non–small cell lung cancer (NSCLC) comprising the majority of cases. Although targeted therapies including epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) inhibitors and immunotherapies, such as programmed cell death 1 (PD-1) and programmed death ligand 1 (PD-L1) inhibitors, have advanced NSCLC treatment, approximately 70% of patients, particularly those with lung squamous cell carcinoma (LUSC), lack actionable driver mutations and therefore depend on chemotherapy, which offers limited efficacy and considerable toxicity. Previous studies indicate that aberrant expression of aldo-keto reductase family 1 member C1 (AKR1C1) is strongly associated with tumor progression, invasiveness, and prognosis in several cancers. As AKR1C1 is implicated in ferroptosis, its targeting may modulate ferroptosis pathways and influence proliferation of lung squamous cell carcinoma cells, suggesting a potential therapeutic strategy for LUSC.
Keywords
- carcinoma
- non-small-cell lung
- ferroptosis
- metabolism
- molecular targeted therapy
Lung cancer is among the most common malignancies worldwide, accounting for approximately 11–12% of all cancer cases in both men and women [1]. It remains the leading cause of cancer-related mortality globally. In China, the disease is particularly aggressive, exhibiting exceptionally high incidence and fatality rates. Non-small cell lung cancer (NSCLC) constitutes the predominant histological subtype, representing roughly 85% of all lung tumors; its two major forms are lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) [1].
Advances in tumor molecular biology and immunology have established targeted therapy and immunotherapy as cornerstone treatment strategies. For LUAD patients with specific mutations, targeted agents against epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), ROS proto-oncogene 1 (ROS1), or B-Raf proto-oncogene, serine/threonine kinase (BRAF) have markedly improved clinical outcomes and prolonged overall survival [2, 3, 4, 5]. However, approximately 30% of NSCLC cases are classified as LUSC, a subtype characterized by a low frequency of targetable driver mutations that limits the utility of targeted therapy [6, 7]. Despite considerable progress in precision medicine, therapeutic efficacy in NSCLC remains heterogeneous. Patients without actionable mutations often depend on chemotherapy, which carries risks of myelosuppression and organ toxicity. Therefore, elucidating key molecular drivers and regulatory mechanisms in NSCLC continues to be of critical importance.
AKR1C1 (aldo-keto reductase family 1 member C1), an enzyme within the aldo-keto reductase superfamily, participates in diverse biochemical reactions such as metabolism, hormone synthesis, and hormone degradation [8, 9]. Its dysregulated expression has been increasingly documented in multiple malignancies and is closely linked to tumor development, invasiveness, and prognosis [10, 11]. Among AKR1 family members, AKR1C1 is frequently reported as upregulated in non-small cell lung cancer (NSCLC) and contributes to tumor proliferation, metastasis, therapy resistance, and ferroptosis regulation [12, 13]. Given this strong association, the present review concentrates on AKR1C1 while briefly acknowledging the potential roles of other family members, including AKR1C2 and AKR1C3, in NSCLC pathogenesis [14, 15, 16].
Fig. 1 presents a schematic overview of the pathways mediated by AKR1C1 in NSCLC progression.
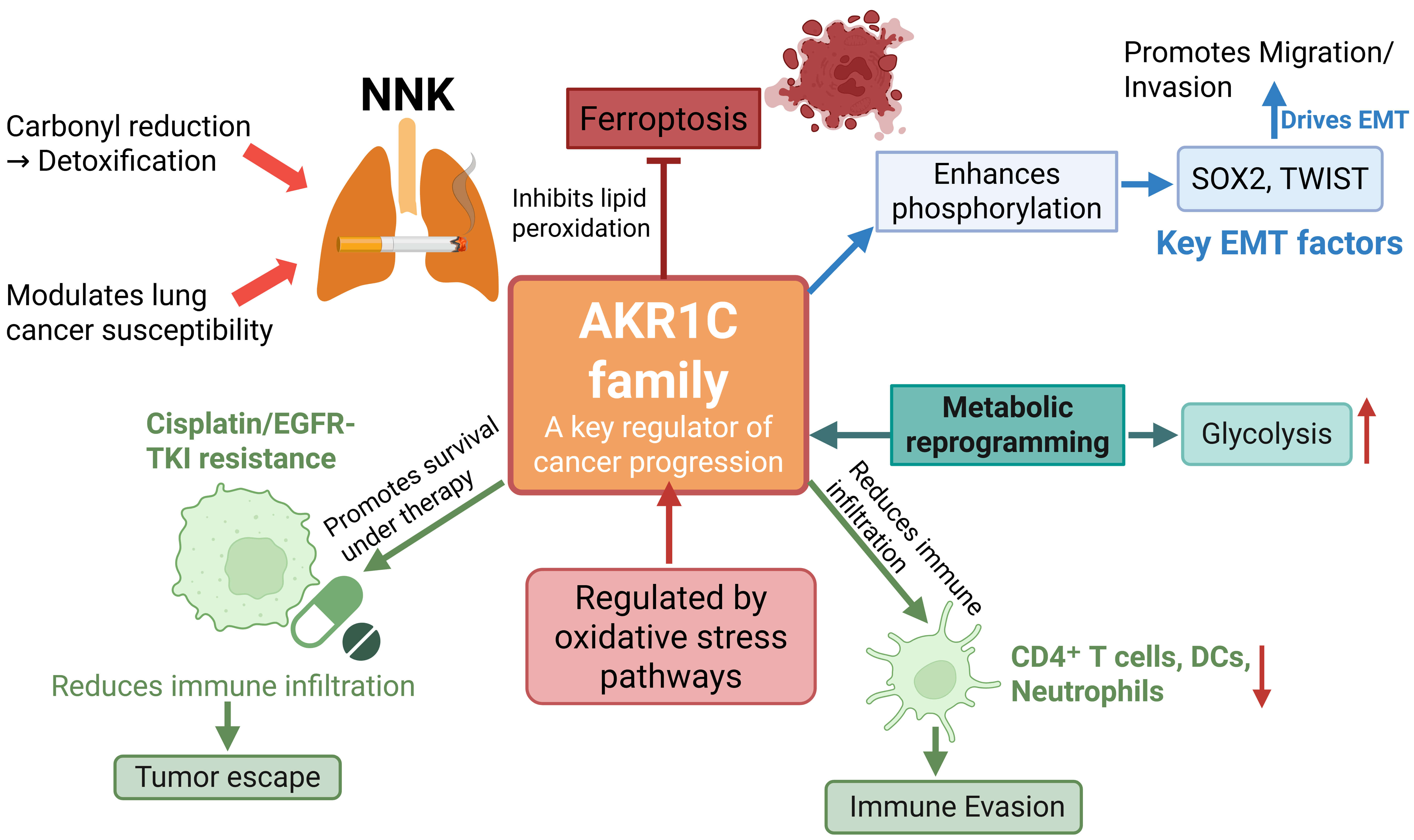 Fig. 1.
Fig. 1.
Integrated schematic of AKR1C1-mediated mechanisms in NSCLC.
AKR1C1 influences NNK detoxification and lung cancer susceptibility, activates
STAT3 signaling to drive EMT and metastasis, and stabilizes HIF-1
The aldo-keto reductase (AKR1) family includes four enzymes, AKR1C1 through AKR1C4, that catalyze NADP+-dependent reductions and participate in metabolizing steroid hormones, prostaglandins, fatty acids, and polycyclic aromatic hydrocarbons [17, 18, 19]. Table 1 summarizes the known biological functions of the AKR1C family.
| Classification | Biological function |
| AKR1C1 | AKR1C1 catalyzes the reduction of progesterone to its inactive metabolite, 20 |
| AKR1C2 | AKR1C2 exhibits high affinity for bile acids and primarily acts as a 3 |
| AKR1C3 | AKR1C3 displays both overlapping and unique substrate specificities. It functions as a prostaglandin D2 (PGD2) synthase in prostaglandin metabolism, catalyzing the conversion of PGD2 to 9 |
| AKR1C4 | AKR1C4 mediates the bioconversion of the organochlorine pesticide chlordecone to chlordecone alcohol in the liver. This predominantly hepatic enzyme efficiently forms 5 |
The AKR1 family contributes to nitroaromatic metabolism in human lung cells, potentially leading to DNA adduct formation, mutagenesis, and carcinogenesis [18]. AKR1C3, a key member of this family, regulates androgen receptor (AR) signaling. Its overexpression activates the AR cascade, which can drive tumor recurrence and resistance to anti-tumor therapies [20, 21, 22]. Elevated AKR1C3 expression promotes cell growth, invasion, and metastasis, and is strongly associated with poor prognosis in cancer patients [23, 24, 25].
Beyond their role in AR signaling, AKR1C1, AKR1C2, and AKR1C3 also participate in estrogen and androgen metabolism. These enzymes influence cellular responses to tamoxifen (TAM) in breast cancer, thereby impacting treatment efficacy [26, 27]. AKR1 family members further regulate key oncogenic pathways such as rat sarcoma virus/mitogen-activated protein kinase (RAS/MAPK), which are critical for cell survival, proliferation, and metastasis [26, 27, 28]. Consequently, the AKR1 family affects cancer progression not only through hormonal regulation but also by modulating these signaling pathways.
Members of the AKR1 family also show significant expression in gastric cancer (GC), where their levels correlate with tumor progression, prognosis, and immune infiltration, indicating their potential as diagnostic and therapeutic biomarkers [29, 30]. For example, AKR1C1 and AKR1C3 are overexpressed in signet ring cell gastric cancer (SRCGC) and contribute to cisplatin resistance. Suppressing these enzymes augments cisplatin-induced autophagy, thereby sensitizing resistant cells to drug-mediated death [29, 31].
Similarly, in hepatocellular carcinoma (HCC), AKR1C1 and AKR1C3 expression serves as a potential diagnostic and prognostic indicator [32, 33, 34]. Elevated levels of AKR1C1–3, especially of AKR1C3, are associated with poor overall survival [24, 32, 35].
These findings indicate that AKR1C3 represents a promising target for novel anti-tumor therapies [16, 36, 37, 38]. Therapeutic strategies directed at AKR1C3 fall into two main categories: inhibitors of the enzyme and prodrugs selectively activated by it. The pan-AKR1C inhibitor S07-2010, for instance, overcomes platinum resistance mediated by the AKR1C family and acts synergistically with chemotherapeutic agents [29, 37, 39]. In the prodrug category, AST-3424 exhibits potent anti-tumor activity that strongly depends on AKR1C3 expression levels, suggesting its potential as a broad-spectrum targeted therapy [14, 40, 41, 42]. Such prodrugs could offer significant advantages by overcoming resistance to chemotherapy, immunotherapy, and radiotherapy, while simultaneously reducing systemic toxicity and adverse effects [40, 43, 44].
Lung cancer development is driven by distinct risk factors and molecular mechanisms. Smoking represents the predominant risk, as tobacco smoke contains more than 4000 chemicals, including at least 69 known carcinogens [45, 46, 47, 48]. Chronic respiratory diseases such as COPD and tuberculosis, along with occupational exposures to asbestos or diesel exhaust, contribute to approximately 5–10% of cases [49, 50, 51]. At the molecular level, malignant transformation is driven by oncogene activation (e.g., EGFR, KRAS) and tumor suppressor inactivation (e.g., TP53, RB1) [52, 53]. Aberrant signaling through EGFR, BRAF, or KRAS promotes uncontrolled proliferation, while impaired apoptosis due to Bax/Bcl-2 imbalance, cell-cycle dysregulation, and immune evasion via PD-1/PD-L1 or CTLA-4 pathways further support tumor progression [54].
Given these processes, metabolic enzymes like AKR1C1 are particularly interesting due to their dual roles in carcinogen detoxification and tumor biology. Among AKR1 isoforms, AKR1C1 is frequently overexpressed in NSCLC and exhibits multifunctional oncogenic activities, including the regulation of redox balance, ferroptosis susceptibility, and metastatic signaling [16, 23]. Although AKR1C1 is emphasized here given the breadth of supporting evidence, AKR1C2 and AKR1C3 have also been implicated in NSCLC progression, particularly in redox homeostasis and chemoresistance [23, 36]. This review therefore focuses on AKR1C1 while acknowledging the potential cooperative roles of other AKR1 family members.
The tobacco-derived carcinogen NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone) undergoes metabolic activation, while its carbonyl reduction to NNAL constitutes a detoxification pathway [55, 56]. AKR1C1 functions as the principal enzyme responsible for this carbonyl reduction [57]. In genetic association studies examining 53 tag single nucleotide polymorphisms (SNPs), AKR1C1 polymorphisms demonstrated a significant association with lung cancer risk, suggesting that genetic variation in AKR1C1 influences NNK metabolism and modifies individual susceptibility to NSCLC [57].
In NSCLC, AKR1C1 expression strongly correlates with tumor metastatic potential [12]. AKR1C1 physically interacts with STAT3 to enhance its phosphorylation, thereby activating downstream target genes such as SOX2 and TWIST that drive epithelial–mesenchymal transition (EMT), migration, and invasion [9, 12]. The acetylation status of AKR1C1 is critical for this pro-metastatic activity; although acetylation does not alter its enzymatic function, it remains indispensable for STAT3 binding and subsequent signaling activation [10]. Elevated AKR1C1 expression stimulates proliferation and motility in NSCLC cells and is associated with poor clinical outcomes [9, 12, 13]. Additionally, AKR1C1 interacts with sequestosome 1(SQSTM1/p62), linking it to autophagy and oxidative stress pathways that further promote cancer progression [58, 59]. Fig. 2 summarizes these interactions and illustrates how AKR1C1 overexpression enhances STAT3 signaling and metastatic potential.
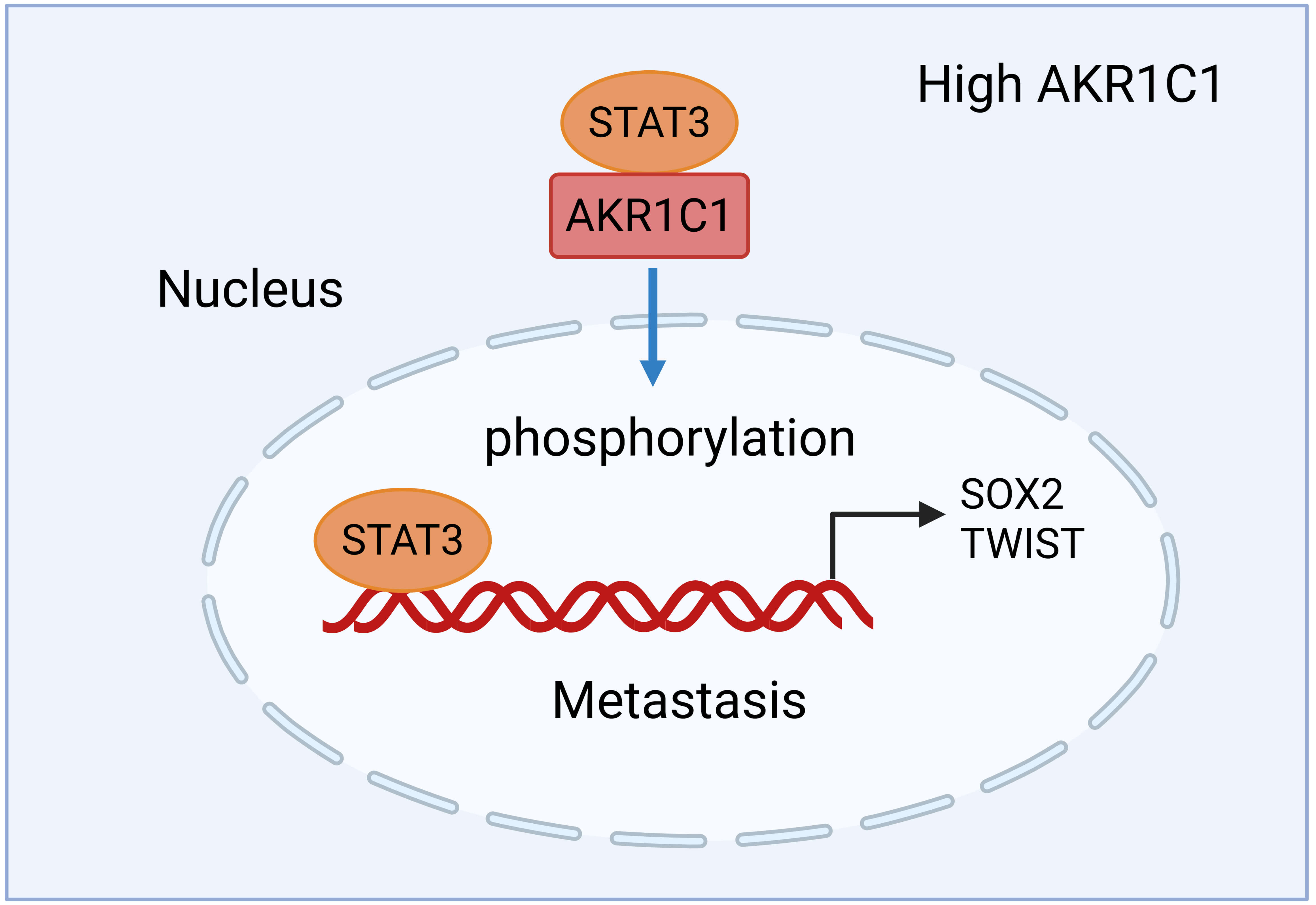 Fig. 2.
Fig. 2.
AKR1C1-driven STAT3 activation promotes NSCLC metastasis. High AKR1C1 expression enhances STAT3 phosphorylation, thereby activating the downstream transcription factors SOX2 and TWIST that regulate genes controlling epithelial–mesenchymal transition, migration, and invasion. Although AKR1C1 acetylation is essential for STAT3 binding and signaling activation, it does not influence the canonical enzymatic activity of AKR1C1. Together, these mechanisms drive metastatic behavior and correlate with aggressive tumor phenotypes in NSCLC. Created in BioRender.com. Dongping, L. (2026) https://BioRender.com/1g3nnak. SOX2, SRY-Box transcription factor 2; TWIST, twist family bHLH transcription factor.
AKR1C1 functions as a downstream effector of YTH N6-methyladenosine RNA binding protein 1(YTHDF1). In NSCLC, diminished YTHDF1 expression promotes cisplatin (DDP) resistance via the Keap1–Nrf2–AKR1C1 axis, which is associated with poor prognosis [60, 61]. Elevated AKR1C1 expression is linked to resistance of malignant lung cells to EGFR-TKI (epidermal growth factor receptor-tyrosine kinase inhibitor) drugs. In LUAD, increased AKR1C1 correlates specifically with gefitinib resistance, indicating that AKR1C1 facilitates the evasion of targeted therapies by malignant cells [62, 63].
High AKR1C1 expression independently predicts reduced overall survival in NSCLC and correlates strongly with aggressive tumor phenotypes and recurrence [12]. Patients exhibiting YTHDF1 negativity alongside AKR1C1 positivity demonstrate significantly shorter overall survival [59]. In lung adenocarcinoma, AKR1C1 expression may indicate both the response to EGFR-TKI treatment and overall prognosis [62, 63].
AKR1C1 expression in NSCLC inversely correlates with immune cell infiltration, such as CD4+ T cells, neutrophils, and dendritic cells, reflecting an immunosuppressive tumor microenvironment [13]. AKR1C1 also activates STAT3, a pathway known to suppress T-cell responses and promotes immune evasion, thereby linking AKR1C1 activity to diminished antitumor immunity in NSCLC [9]. These findings collectively indicate that AKR1C1 not only promotes tumor progression but may also indirectly compromise immune surveillance.
AKR1C1 is consistently upregulated in non-small cell lung cancer
(NSCLC), where it contributes to poor prognosis by modulating redox homeostasis,
ferroptosis, and metabolic adaptation [12, 13]. Functional studies demonstrate
that AKR1C1 activates the HIF-1
Clinically, elevated AKR1C1 expression is associated with reduced response to EGFR-TKIs and shorter progression-free survival in certain LUAD cohorts [62]. These findings position AKR1C1 as a potentially important therapeutic target in LUSC, while retaining its prognostic relevance in LUAD. Further subtype-specific investigations and functional validation are needed to confirm these distinctions.
In summary, AKR1C1 contributes broadly to NSCLC pathogenesis by mediating carcinogen metabolism-including tobacco-derived NNK-promoting metastatic progression through STAT3 signaling, conferring resistance to agents such as cisplatin and EGFR-TKIs, and exhibiting strong associations with adverse clinical outcomes and tumor immunosuppression. Thus, AKR1C1 represents both a promising prognostic biomarker and a potential therapeutic target for NSCLC management.
Accumulating evidence indicates that AKR1C1 is upregulated in various cancer types, where it contributes critically to tumor development and malignant progression [11, 66, 67]. In oropharyngeal squamous cell carcinoma, elevated expression of both AKR1C1 and AKR1C3 correlates with reduced patient survival [36, 68]. In acute myeloid leukemia (AML), AKR1C1 overexpression in mesenchymal stem cells (MSCs) promotes AML cell viability and is linked to poorer survival outcomes [68, 69].
AKR1C1 also mediates therapeutic resistance in multiple tumor types. In nasopharyngeal carcinoma, suppressing AKR1C1 expression enhances cisplatin sensitivity and promotes cisplatin-induced apoptosis [67]. Melanoma cells are protected from ferroptosis through AKR1C1’s regulation in lipid peroxide metabolism; inhibiting this enzyme sensitizes them to ferroptosis and improves the efficacy of ferroptosis-inducing agents [70]. AKR1C1 has been implicated in resistance against cisplatin and methotrexate in colon cancer [66, 71]. In bladder cancer, it increases invasiveness through modulation of tyrosine kinase signaling pathways [72, 73]. For breast cancer, elevated AKR1C1 expression correlates with an altered immune microenvironment and drives tumor progression along with multidrug resistance, highlighting its therapeutic relevance [74, 75, 76, 77, 78].
In cervical cancer, AKR1C1 enhances tumor progression by upregulating TWIST1 expression and activating the PI3K/Akt (phosphatidylinositol 3-kinase/protein kinase B) signaling pathway, thereby promoting cell migration [79]. In endometrial cancer, AKR1C1 functions as a pivotal progesterone-metabolizing enzyme within the Nrf2-TET1 (ten-eleven translocation 1) axis, mediating resistance to progesterone therapy (the regulatory mechanisms of Nrf2 will be discussed in detail in subsequent sections). Inhibition of this signaling pathway restores sensitivity to progesterone treatment [45].
AKR1C1 knockout suppresses NSCLC cell proliferation and migration while elevating intracellular iron ion levels and lipid peroxidation, thereby promoting ferroptosis [13]. In recent years, significant advances have been made in elucidating the role of ferroptosis in lung cancer. Substantial evidence now supports the targeting of ferroptosis as a viable therapeutic strategy for NSCLC. This regulated form of cell death is distinguished by iron-dependent, pervasive lipid peroxidation of cellular membranes [80, 81, 82, 83]. The resulting accumulation of lipid-associated reactive oxygen species (ROS) triggers peroxidation outside mitochondria, ultimately leading to ferroptotic cell death [84, 85].
Key regulatory pathways in ferroptosis include the canonical GPX4 pathway, iron metabolism, lipid metabolism, and the mevalonate pathway [85, 86, 87, 88, 89]. The cystine–glutathione (GSH)–GPX4 axis constitutes a central ferroptosis suppression system. Glutathione peroxidase 4 (GPX4) is critical for inhibiting ferroptosis [90, 91, 92, 93]. Lipid peroxides that escape GPX4 reduction (via GSH) are oxidized by ferrous ions, which generates ROS [94, 95, 96]. This progressive accumulation of peroxides finally induces ferroptosis [96, 97, 98].
Recent advances have substantially deepened our comprehension of ferroptosis in tumor biology and cancer therapeutics. Numerous oncogenic signaling pathways have been identified to regulate tumor cell susceptibility to ferroptosis. Consequently, ferroptosis induction has emerged as a highly promising therapeutic strategy for cancer treatment and overcoming resistance to conventional therapies [99, 100, 101, 102].
The RAS (rat sarcoma virus) family oncogenes (HRAS, NRAS, KRAS) represent some of the most commonly mutated genes across diverse human malignancies. Ferroptosis inducers such as erastin and RSL3 exhibit selective cytotoxicity in tumor cells engineered to express RAS mutations. This resistance phenotype can be reversed through either genetic or pharmacological inhibition of RAS or its downstream effector pathways (BRAF, MEK, ERK), demonstrating that ferroptosis induction represents a viable strategy against RAS-driven malignancies [87]. Tumor suppressor P53 mutations may impair its dual capacity to promote both apoptotic and ferroptotic pathways. Under specific conditions, several metabolic genes-including spermidine/spermine N1-acetyltransferase 1 (SAT1), ferredoxin reductase (FDXR), and glutaminase 2 (GLS2) have been identified as direct transcriptional targets of P53 during ferroptotic processes [103, 104].
Hypoxia-inducible factor (HIF), the principal regulator of hypoxia, exerts dual
effects on ferroptosis in cancer cells [105, 106]. Under hypoxic conditions, HIF
suppresses ferroptosis sensitivity by inhibiting ferritinophagy and sustaining
the GSH–GPX4 antioxidant axis; conversely, in specific scenarios characterized
by predominant HIF-2
Morphologically, ferroptotic cells are characterized by the absence of classical apoptotic features, instead displaying shrunken mitochondria with reduced or vanished cristae, disrupted outer membranes, and elevated membrane density [90]. This form of cell death is progressively recognized as a therapeutically exploitable weakness in certain cancer, with its induction being regarded as advantageous for anticancer treatment across diverse malignancies [113].
The molecular regulation of ferroptosis is fundamentally intertwined with core metabolic processes, including iron homeostasis, amino acid catabolism, and lipid turnover [114, 115, 116]. Perturbations in these ferroptosis-related metabolic pathways have been conclusively shown to affect treatment outcomes in lung cancer [117, 118].
In LUAD models, concurrent mutations in STK11 and KEAP1 markedly upregulate expression of the ferroptosis-protective gene AKR1C1, thereby augmenting resistance to pharmacologically induced ferroptosis [119]. These co-mutant cells demonstrate diminished ferroptosis susceptibility through modulation of genes regulating iron metabolism and lipid peroxidation, predominantly via activation of Nrf2-dependent transcriptional pathways [119]. As depicted in Fig. 3, STK11/KEAP1 loss activates Nrf2, facilitating its binding to antioxidant response elements (AREs) and subsequent induction of AKR1C1 expression. System xc–, the cystine/glutamate antiporter, mediates cystine uptake in exchange for glutamate, supplying cysteine for glutathione (GSH) synthesis. GSH maintains glutathione peroxidase 4 (GPX4) activity, consequently inhibiting lipid peroxidation and ferroptotic cell death. While AKR1C1 does not directly catalyze cysteine-to-GSH conversion, it operates downstream of Nrf2 to enhance cellular redox homeostasis and ferroptosis resistance.
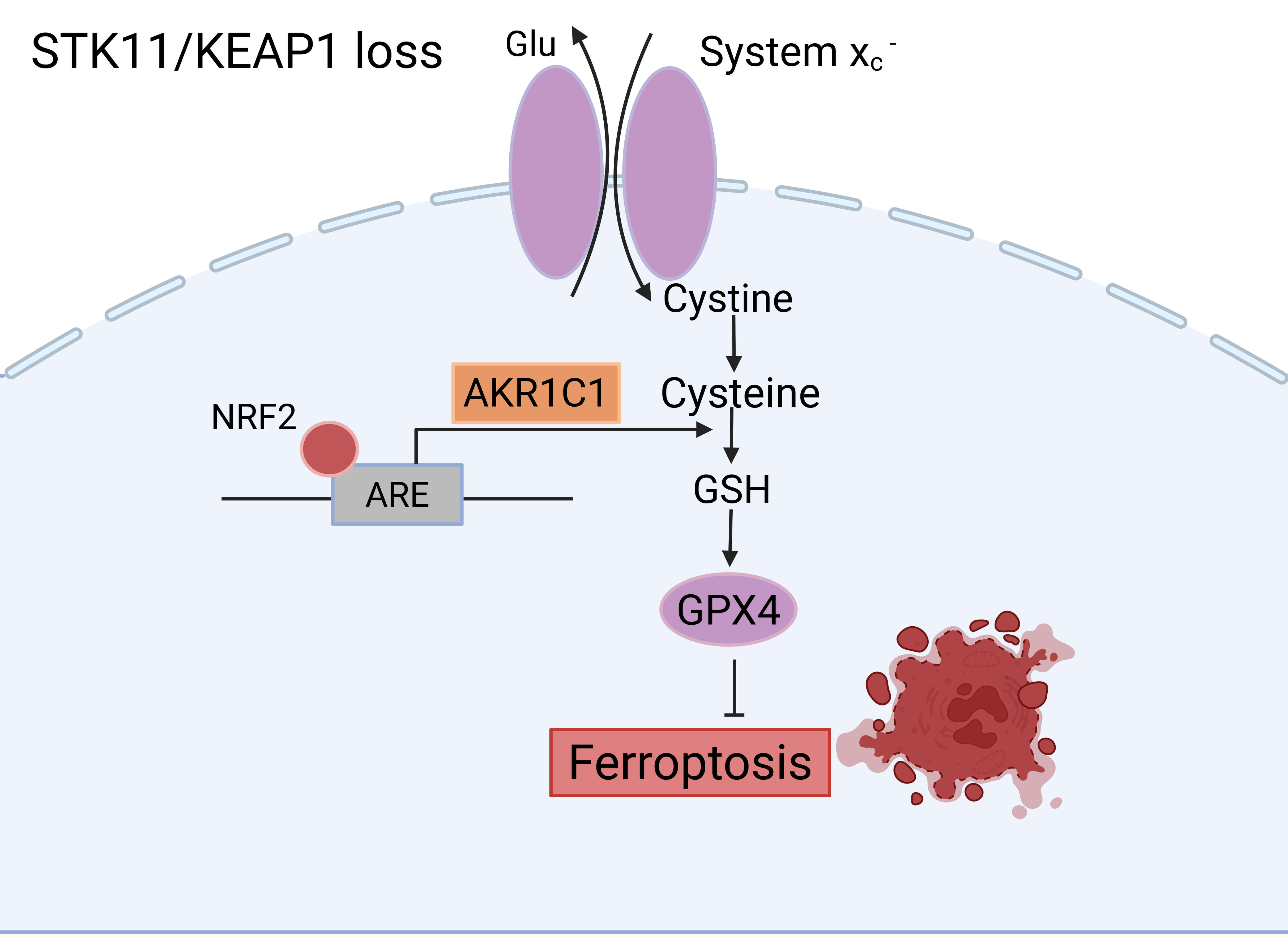 Fig. 3.
Fig. 3.
Mechanistic model linking STK11/KEAP1 co-mutation to ferroptosis resistance in LUAD. Loss of STK11/KEAP1 activates Nrf2, which binds to antioxidant response elements (AREs) and upregulates AKR1C1 expression. System xc– mediates the exchange of extracellular cystine for intracellular glutamate, supplying cysteine for glutathione (GSH) synthesis. This GSH pool supports GPX4 activity, which in turn prevents lipid peroxidation and ferroptosis. AKR1C1 promotes ferroptosis resistance primarily via redox regulation downstream of Nrf2, not by directly engaging in GSH synthesis. Created in BioRender.com. Dongping, L. (2026) https://BioRender.com/l1h1kgk. Glu, glutamate; GSH, glutathione.
AKR1C1 orchestrates iron homeostasis and lipid peroxidation through multiple molecular pathways. Comprehensive bioinformatic and functional analyses reveal that AKR1C1 expression exhibits positive correlation with ferroptosis-related gene signatures, while its suppression leads to intracellular Fe2+ accumulation and heightened lipid peroxidation in NSCLC cells [13]. Mechanistically, AKR1C1 maintains cellular GSH pools to support GPX4 activity, thereby limiting lipid peroxide accumulation [120]. The flavonoid compound nobiletin, an AKR1C1-targeting agent, facilitates GPX4 ubiquitination and subsequent degradation, ultimately potentiating ferroptosis sensitivity [76]. Furthermore, AKR1C1 also influences cytochrome P450 family 1 subfamily B member 1 (CYP1B1) expression and mediates the cyclic adenosine monophosphate - protein kinase A (cAMP-PKA) signaling pathway in specific cancer contexts, thereby altering cellular vulnerability to iron-dependent lipid peroxidation [11].
Collectively, these findings indicate that AKR1C1 suppresses ferroptosis by sustaining the GSH/GPX4 antioxidant defense system, regulating lipid-metabolizing enzymes, and preserving iron homeostasis. Therefore, targeting AKR1C1 or its associated pathways could increase cellular susceptibility to ferroptosis and may offer a therapeutic strategy for NSCLC, particularly in subtypes without other actionable drivers.
Glutamine serves as a crucial energy source and biosynthetic precursor within the tumor microenvironment [91]. Ferroptosis is characterized by iron-dependent peroxidation of phospholipids containing polyunsaturated fatty acids (PUFAs) in cell membranes, ultimately causing cell death [121, 122]. This process exhibits critical dependence on GPX4, which uses glutathione to detoxify lipid peroxides. Inhibiting glutamine metabolism can suppress ferroptosis, underscoring its essential role of glutamine in this cell death pathway [90]. The tumor microenvironment often exhibits high oxidative stress, where dysregulated lipid metabolism may compromise GPX4 activity and promote ferroptosis in tumor cells. Such metabolic dysregulation increases the pool of peroxidation-susceptible polyunsaturated fatty acids while limiting glutathione availability, thereby indirectly reducing GPX4 catalytic activity and facilitating lipid peroxide accumulation that drives ferroptosis [123, 124]. Glycolysis serves as a primary energy production pathway, especially under hypoxic conditions where it dominates ATP synthesis [114]. During ferroptosis, elevated intracellular ROS may induce mitochondrial dysfunction, impairing cellular energy metabolism and potentially affecting glycolysis flux [125]. Pyruvate, a key glycolytic intermediate, contributes significantly to GSH biosynthesis [126, 127]. The substantial GSH consumption during ferroptosis consequently diminishes cellular antioxidant capacity [128]. Ferroptosis regulation involves multiple signaling pathways; for instance, the tumor suppressor p53 modulates ferroptotic responses through phosphorylation-mediated activation or repression of specific target genes [129, 130].
Cancer is characterized by metabolic reprogramming, which includes enhanced
lactate production from glucose breakdown and glutamine hydrolysis to supply
intermediates and energy for tumor growth and proliferation [131, 132, 133, 134]. Under
hypoxia conditions, hypoxia-inducible factor 1-alpha (HIF-1
Recent study indicates that AKR1C1 modifies tumor metabolism and enhances NSCLC
cell proliferation by activating HIF-1
In summary, AKR1C1, a member of the AKR family, is closely associated with the ferroptosis pathway and merits investigation for its potential role in diagnosing, treating, and predicting the prognosis of various cancers. In NSCLC, elevated AKR1C1 expression promotes cancer cell proliferation, confers resistance to targeted therapies, and facilitates metabolic reprogramming, establishing it as a candidate prognostic biomarker. Consequently, determining whether AKR1C1 inhibition can suppress NSCLC cell proliferation by modulating ferroptosis, and whether targeting AKR1C1 constitutes a viable therapeutic strategy, is of considerable clinical importance. Investigating this ferroptosis-related gene should clarify the mechanisms of NSCLC progression through ferroptosis, thereby informing future targeted and immunotherapeutic interventions.
QZ conceived and designed the study. GHH developed the overall concept. XRZ and YTC conducted the literature search and data collection. XRZ drafted the manuscript. GHH and QZ critically revised the manuscript for important intellectual content. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We gratefully acknowledge the assistance and instruction from professor Wenhao Shen of the the Affiliated Taizhou People’s Hospital of Nanjing Medical University for Xinran Zhang Investigators.
This research was funded by the Nanjing Medical University Science and Technology Development Fund, grant number NMUB20230297.
The authors declare no conflict of interest.
During the preparation of this work the authors used Deepseek in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.