





 , Peixin Guo 1,2,*
, Peixin Guo 1,2,*
1 Yunnan Key Laboratory of Dai and Yi Medicine, Yunnan University of Chinese Medicine, 650500 Kunming, Yunnan, China
2 College of Ethnic Medicine, Yunnan University of Chinese Medicine, 650500 Kunming, Yunnan, China
3 College of Chinese Medicine, Yunnan University of Chinese Medicine, 650500 Kunming, Yunnan, China
4 College of Basic Medical Sciences, Yunnan University of Chinese Medicine, 650500 Kunming, Yunnan, China
†These authors contributed equally.
Abstract
Neurodegenerative diseases (NDs) are incurable, progressively disabling disorders marked by sustained neuronal degeneration and loss. Their molecular basis involves intricate regulatory networks, while current therapeutic strategies remain inadequate. Oxidative stress (OS) constitutes a major driver in the initiation and progression of age-related pathologies. Kelch-like enoyl-CoA hydratase-associated protein-1 (Keap1)-Nuclear factor Erythroid 2-related factor 2 (Nrf2) signaling pathway, an essential antioxidant system, exerts protective effects by limiting OS-mediated cellular injury. Extensive evidence demonstrates a close association between Nrf2 signaling and the pathological processes of NDs, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS). Traditional Chinese medicine, characterized by multi-target and multi-pathway regulatory actions of its bioactive constituents, offers distinctive therapeutic potential for NDs. This review provides an integrated analysis of current advances of Nrf2 involvement in NDs and evaluates therapeutic strategies based on traditional Chinese medicine and its active components, with the aim of guiding future clinical translation.
Keywords
- NF-E2-related factor 2
- oxidative stress
- neurodegenerative diseases
- molecular mechanisms of pharmacological actions
- traditional Chinese medicine
With the rapid demographic transition toward an aging population, neurodegenerative diseases (NDs) have become a major healthcare challenge worldwide [1]. Disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are characterized by progressive functional impairments, including cognitive, memory, and motor function [2]. The global burden of NDs is considerable; the prevalence of AD alone has already exceeded 50 million and is projected to reach 152 million by 2050 [3, 4]. Likewise, the number of PD cases surpassed 6 million in 2015 and is expected to exceed 17 million by 2040 [5].
The development of NDs arises from a complex interplay of mechanisms, including inflammatory signaling, oxidative stress (OS), pyroptosis, mitochondrial dysfunction, amyloid accumulation, and infection-related factors [6, 7, 8, 9, 10]. Exposure to harmful stimuli provokes excessive generation of reactive species such as reactive oxygen species (ROS), disrupting redox equilibrium and initiating OS [11]. Elevated ROS levels induce structural and functional injury to lipids, proteins, and DNA, thereby destabilizing cellular homeostasis. This oxidative imbalance is closely associated with both neurodegenerative progression and biological aging [12, 13, 14, 15]. In brain tissues of patients with NDs, high levels of oxidative products are consistently observed, accompanied by extensive neuronal oxidative damage [12, 16].
Nuclear factor E2-related factor 2 (Nrf2), an endogenous antioxidant transcription factor, serves as a principal regulator of cellular defense against OS-induced injury [17]. By controlling the transcription of antioxidant genes, cytoprotective enzymes, and efflux transporters, Nrf2 establishes an integrated protective network that preserves intracellular redox equilibrium. Increasing evidence demonstrates that pharmacological modulation of Nrf2 attenuates NDs [9]. In AD models, enhanced Nrf2 activity alleviates ROS-driven damage and mitochondrial impairment [18]. In MPTP-induced PD mice, Nrf2 upregulation improves motor coordination, diminishes dopaminergic neuronal loss, suppresses neuroinflammation, and prevents ferroptotic degeneration, collectively yielding neuroprotective outcomes [19, 20]. In 3-NPA-induced HD rats, protopanaxtriol enhances Nrf2 nuclear translocation and elevates HO-1 and NQO1 expression, thereby reducing OS and restoring neuronal structure [21]. Targeting Nrf2 thus represents a promising therapeutic strategy for NDs by modulating oxidative stress, neuroinflammation, and mitochondrial homeostasis. Given the multifactorial pathogenesis of NDs, interventions designed to influence multiple molecular targets and signaling pathways present substantial potential. Notably, bioactive compounds derived from traditional Chinese medicine provide a compelling direction for ND treatment [22, 23].
This review summarizes recent progress in elucidating the structural and functional roles of Nrf2 in NDs, highlights the contribution of Nrf2 dysregulation to disease progression, and examines emerging therapeutic strategies with potential clinical relevance.
Relevant literature was collected using PubMed database search using (“NF-E2-Related Factor 2” OR “Nrf2”) AND (neurodeg* OR “Alzheimer Disease” OR “Parkinson Disease” OR “Huntington Disease” OR “amyotrophic lateral sclerosis” OR “Medicine, Chinese Traditional”) retrieved 2493 articles published between Jan. 2015 and May. 2025. Inclusion criteria were: ① Articles addressing neurodegenerative disease regulation via the Nrf2 signaling pathway; ② Article titles or abstracts containing either ‘NF-E2-Related Factor 2’ or ‘Nrf2’ alongside any other search term. Exclusion criteria: ① Articles lacking full text were excluded; ② Articles unrelated to neurodegenerative diseases were excluded. This yielded 936 articles. Following final screening based on manuscript subject and full-text content, 70 articles met the criteria: 33 review articles and 37 experimental or other types of articles.
The Keap1–Nrf2 signaling axis plays a central role in maintaining oxidative
equilibrium. Keap1 consists of five distinct domains: NTR, BTB, IVR, DGR (Kelch
repeat), and CTR (Fig. 1A) [24]. Nrf2, a basic leucine zipper (bZip)
transcription factor, is organized into seven conserved domains (Neh1–Neh7)
(Fig. 1B) [25, 26]. Neh1 associates with sMaf proteins to recognize and bind the
ARE, thereby driving the transcription of antioxidant and anti-inflammatory genes
[27]. Neh2 functions as the principal regulatory module, containing the conserved
DLG and ETGE motifs that engage the Keap1 Kelch domain to govern Nrf2 degradation
[28]. The ETGE motif binds with high affinity, while the DLG motif exhibits
weaker interaction [29, 30]. Neh3–Neh5 act as transcriptional activation regions
through co-activator recruitment, although their precise roles remain unclear
[31, 32]. Neh6 carries DSGIS and DSAPG degrons; phosphorylation of DSGIS by
GSK-3
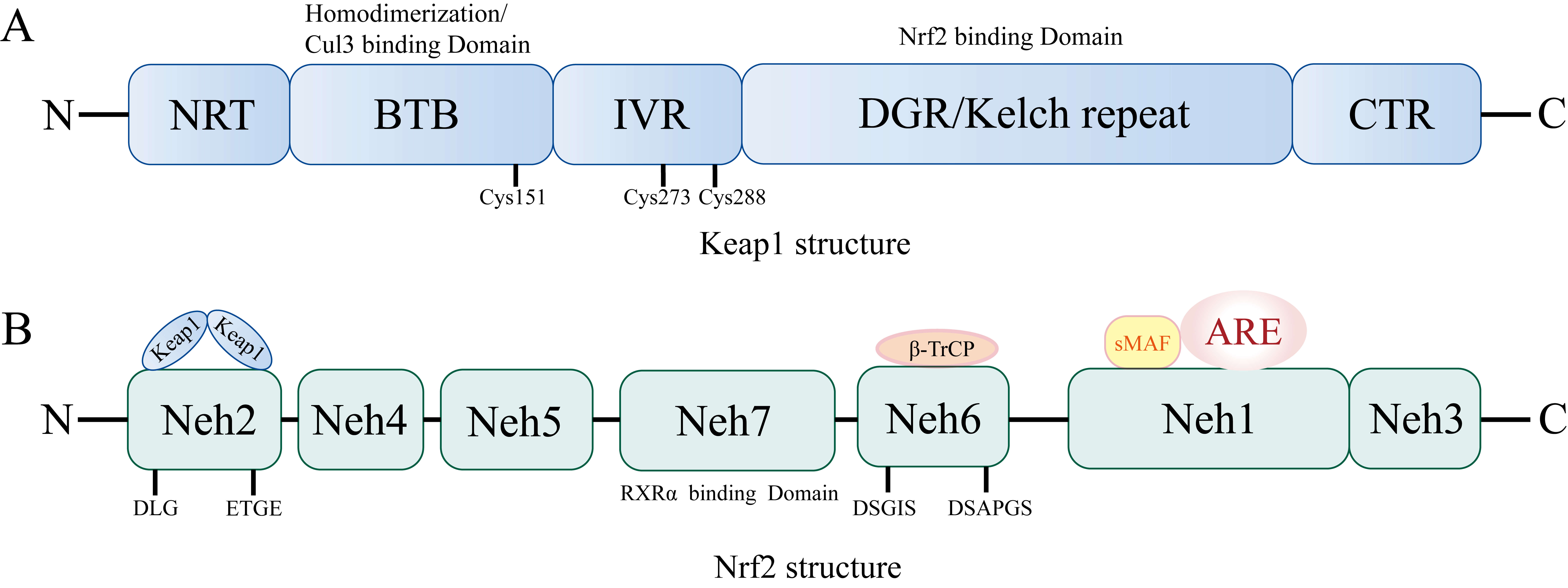 Fig. 1.
Fig. 1.
Structures of Kelch-like enoyl-CoA hydratase-associated
protein-1 (Keap1) and Nuclear factor Erythroid 2-related factor 2 (Nrf2). (A)
Keap1 comprises five distinct regions: the N-terminal region (NTR); the BTB
(Broad-Complex, Tramtrack, and Bric-à-brac) domain, which mediates
homodimerization and interaction with Cul3; the intervening region (IVR)
containing Cys273 and Cys288; the double glycine repeat (DGR)/Kelch repeat domain
responsible for Nrf2 binding; and the C-terminal region (CTR). (B) Nrf2 consists
of seven functional domains (Neh1–Neh7). Neh1 facilitates heterodimerization
with sMaf proteins and binds to antioxidant response elements (ARE). Neh2
includes the DLG and ETGE motifs essential for Keap1 interaction and subsequent
Nrf2 degradation. Neh3–Neh5 serve as transcriptional activation regions. Neh6
contains DSGIS and DSAPG degron motifs, regulating Nrf2 stability. Neh7 interacts
with retinoic acid receptor alpha (RXR
Under redox equilibrium, Nrf2 and Keap1 form homodimers in the cytoplasm. Keap1 dimers interact with the Cul3-Rbx1 E3 ubiquitin ligase complex, directing Nrf2 toward ubiquitination and subsequent proteasomal degradation via the 26S system [24]. When intracellular oxidative or electrophilic stress increases, Nrf2 activation follows the “hinge-lock” paradigm. Covalent modifications of key cysteine residues on Keap1, particularly Cys151 within the BTB domain and Cys273 and Cys288 in the IVR domain, trigger zinc release and structural rearrangements. In this altered state, the DLG motif of Nrf2, functioning as a lock, disengages, whereas the ETGE motif remains bound as a hinge. This partial dissociation prevents ubiquitination and degradation of Nrf2 [29, 36]. Newly synthesized Nrf2 then evades Keap1-mediated repression, translocates into the nucleus, and binds ARE sequences to initiate transcription of antioxidant genes (Fig. 2) [37].
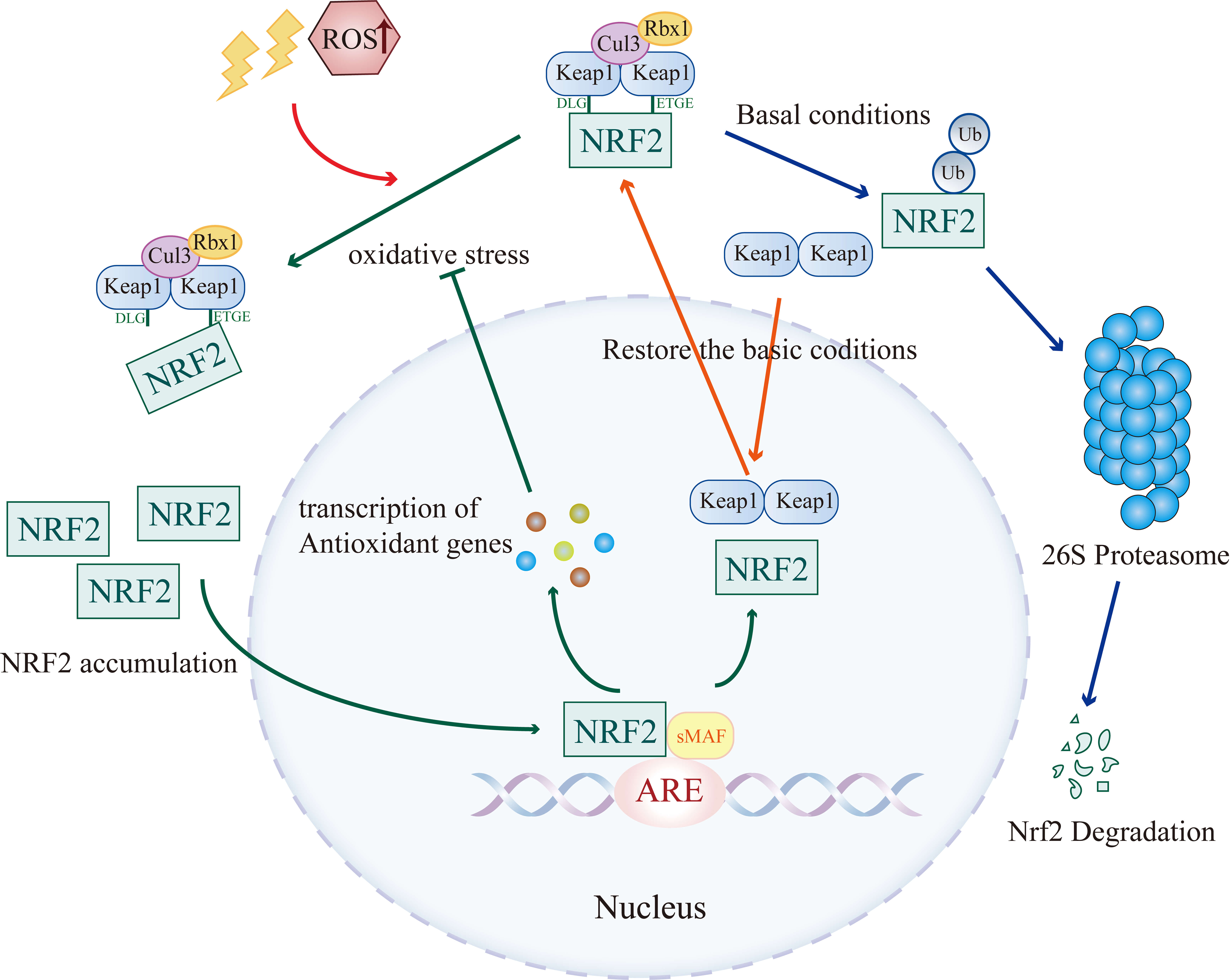 Fig. 2.
Fig. 2.
Keap1-dependent regulation of Nrf2. Under homeostatic conditions, Keap1 interacts with the ETGE and DLG motifs of Nrf2, promoting its ubiquitination via the Keap1/Cul3/Rbx1 complex and subsequent proteasomal degradation by the 26S proteasome (blue arrow). In response to electrophilic agents or elevated ROS levels, the Keap1–Nrf2 association is attenuated, permitting Nrf2 to evade ubiquitination and degradation. Stabilized Nrf2 accumulates in the cytoplasm, translocates to the nucleus, forms heterodimers with sMaf proteins, and binds to AREs to initiate transcription of antioxidant genes, thereby mitigating oxidative stress (green arrow). Upon reestablishment of redox balance, Keap1 shuttles into the nucleus, promotes the nuclear export of Nrf2, and re-engages the cytoplasmic ubiquitination machinery, restoring its degradation (orange arrow).
Nrf2 is also subject to regulation beyond Keap1 control through several mechanisms, including modulation by p62/SQSTM1, the cyclin-dependent kinase inhibitor p21, and epigenetic processes [38].
p62/SQSTM1 acts as a selective autophagy receptor containing distinct structural elements such as the N-terminal Phox-BEM1 (PB1) domain, ZZ-type zinc finger domain, TRAF6-binding sequence (TBS), LC3-interacting region (LIR), Keap1-interacting region (KIR), and the C-terminal ubiquitin-associated (UBA) domain (Fig. 3A). It additionally functions as an activator of the non-canonical Keap1–Nrf2 pathway [39, 40]. Within the KIR domain, the 349-DPSTGE-354 motif mimics the ETGE motif located in the Neh2 domain of Nrf2, thereby enabling strong Keap1 binding [41]. Phosphorylation of S351 in the KIR domain markedly strengthens the interaction of p62 with Keap1, displacing Nrf2 from Keap1. The resulting p62–Keap1 complex is targeted to autophagosomes, which leads to Nrf2 stabilization, its nuclear import, and transcriptional induction of cytoprotective genes (Fig. 3B) [42]. In addition, p62/SQSTM1 enhances the association of AMPK with ULK1, promoting ULK1 phosphorylation and initiating bulk autophagy. This process accelerates Keap1 degradation, further reinforcing Nrf2 activation [43].
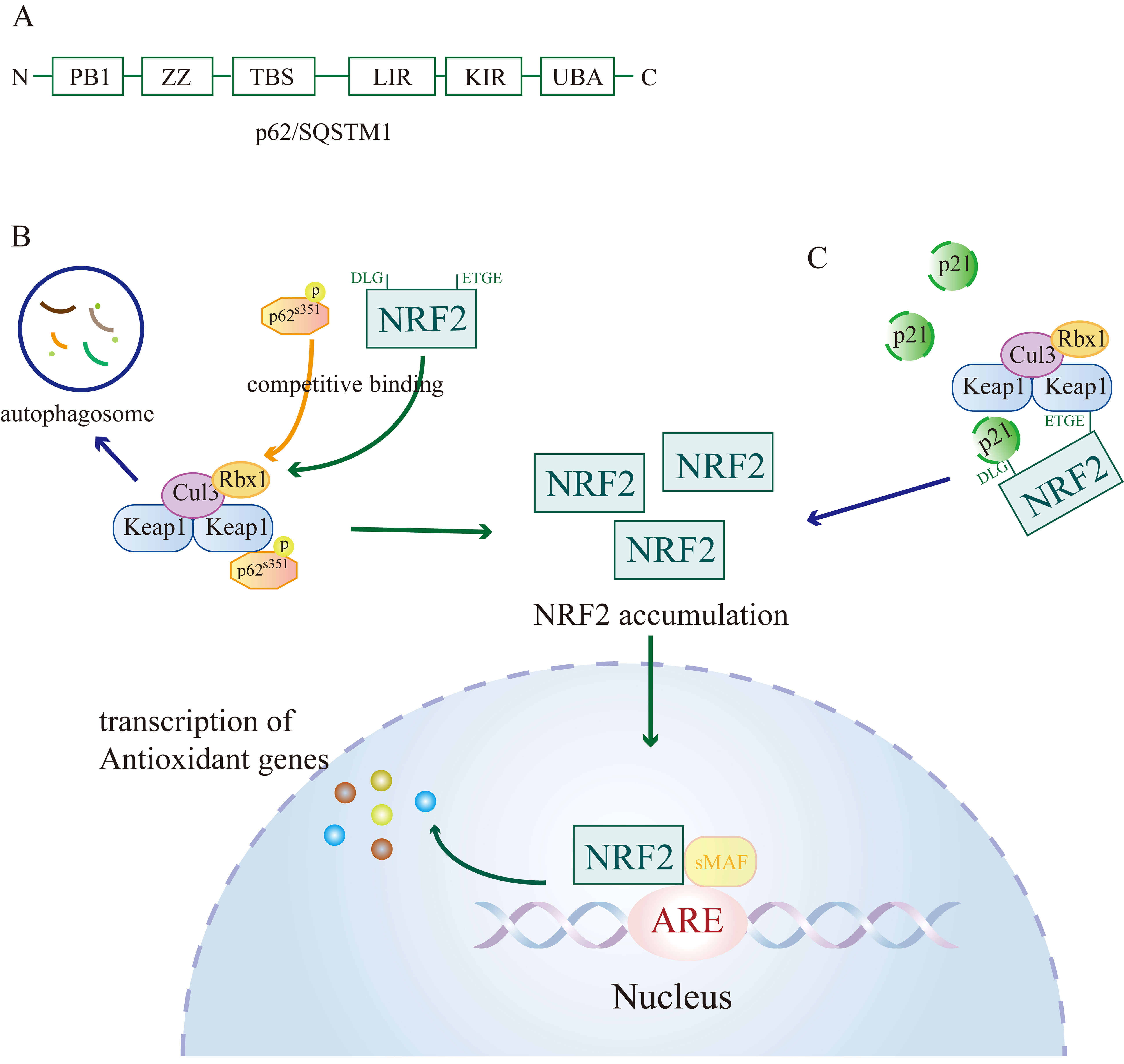 Fig. 3.
Fig. 3.
Non-dependent regulation of Nrf2 by p62 and p21. (A) Domain structure of p62/SQSTM1, comprising the N-terminal Phox-BEM1 (PB1) domain, ZZ-type zinc finger domain, TRAF6-binding sequence (TBS), LC3-interacting region (LIR), Keap1-interacting region (KIR), and the C-terminal ubiquitin-associated (UBA) domain. (B) Phosphorylation of p62 at S351 enables competitive binding with Keap1, displacing Nrf2. The p62–Keap1 complex is sequestered into autophagosomes, resulting in Nrf2 stabilization, nuclear translocation, and activation of antioxidant gene expression. (C) p21 binds to the Nrf2 motif, while Keap1 interacts with ETGE, and the combined effect blocks Nrf2 ubiquitination and degradation, leading to Nrf2 accumulation in the cytoplasm and subsequent nuclear translocation, thereby promoting antioxidant gene transcription.
p21 engages Keap1 while simultaneously binding to Nrf2 via its DLG motif, whereas Keap1 remains attached to the ETGE motif of Nrf2 [44]. This ternary complex disrupts Nrf2 ubiquitination and subsequent proteasomal degradation (Fig. 3C), thereby stabilizing Nrf2 and enhancing cellular antioxidant defense capacity [45].
Epigenetic regulation, defined as heritable alterations in gene expression without modification of the DNA sequence, represents a central mechanism in controlling cellular adaptation to OS. Principal processes include DNA methylation, histone modification, and non-coding RNA-mediated regulation [31]. Among them, epigenetic modifications modulate Nrf2 activity through Keap1-independent mechanisms. For example, miR-139 directly suppresses cJUN and the nuclear transport regulator KPNA2, leading to reduced Nrf2 activation and heightened sensitivity of NSCLC cells to radiation-induced oxidative stress, thereby restraining tumor progression [46]. Likewise, miR-144-3p targets the 3′-UTR of Nrf2 mRNA in renal tubular epithelial cells, diminishing Nrf2 signaling and accelerating chronic kidney disease (CKD) progression [47]. Under hypoxic conditions, upregulation of miR-140-5p directly suppresses Nrf2 activity, limiting the proliferative capacity of breast cancer (BC) cells [48]. In addition, sulforaphane (SFN) promotes histone H3 acetylation within the Nrf2 promoter region, activating the cardiac Nrf2 pathway and conferring cardioprotective effects [31]. Conversely, CpG methylation within the Nrf2 promoter region reduces Nrf2 and NQO1 protein expression, strongly correlating with prostate cancer development [49] (Fig. 4).
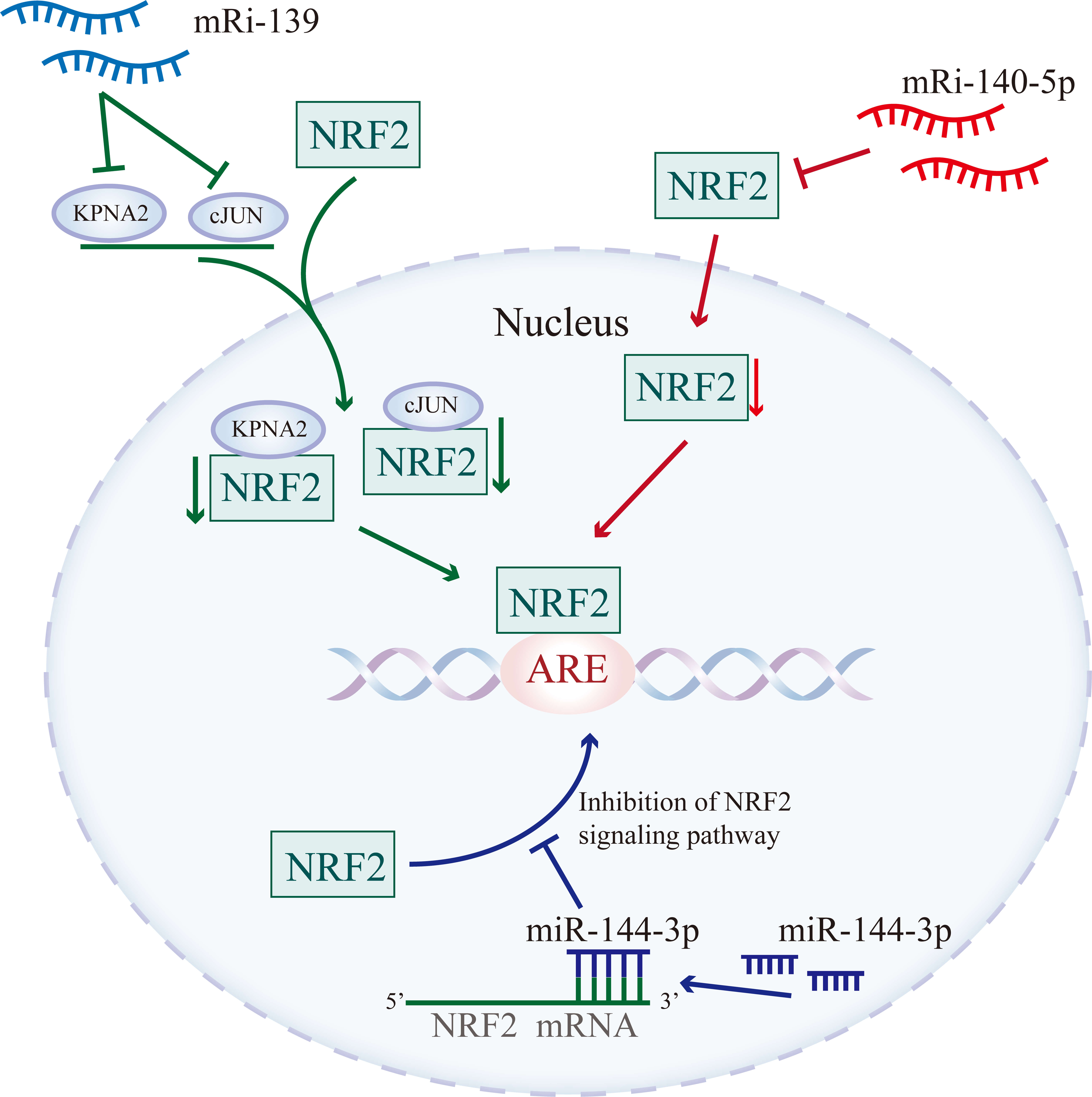 Fig. 4.
Fig. 4.
Epigenetic mechanisms of Nrf2 regulation. miR-139 represses Nrf2 activation by targeting cJUN and KPNA2, thereby limiting nuclear translocation of Nrf2; miR-144-3p binds to the 3′-UTR of Nrf2, suppressing downstream signaling and attenuating activation; miR-140-5p diminishes Nrf2 activity through direct interaction.
AD is a progressive neurodegenerative disorder characterized by cognitive
decline, with its pathological hallmarks being senile plaques derived from
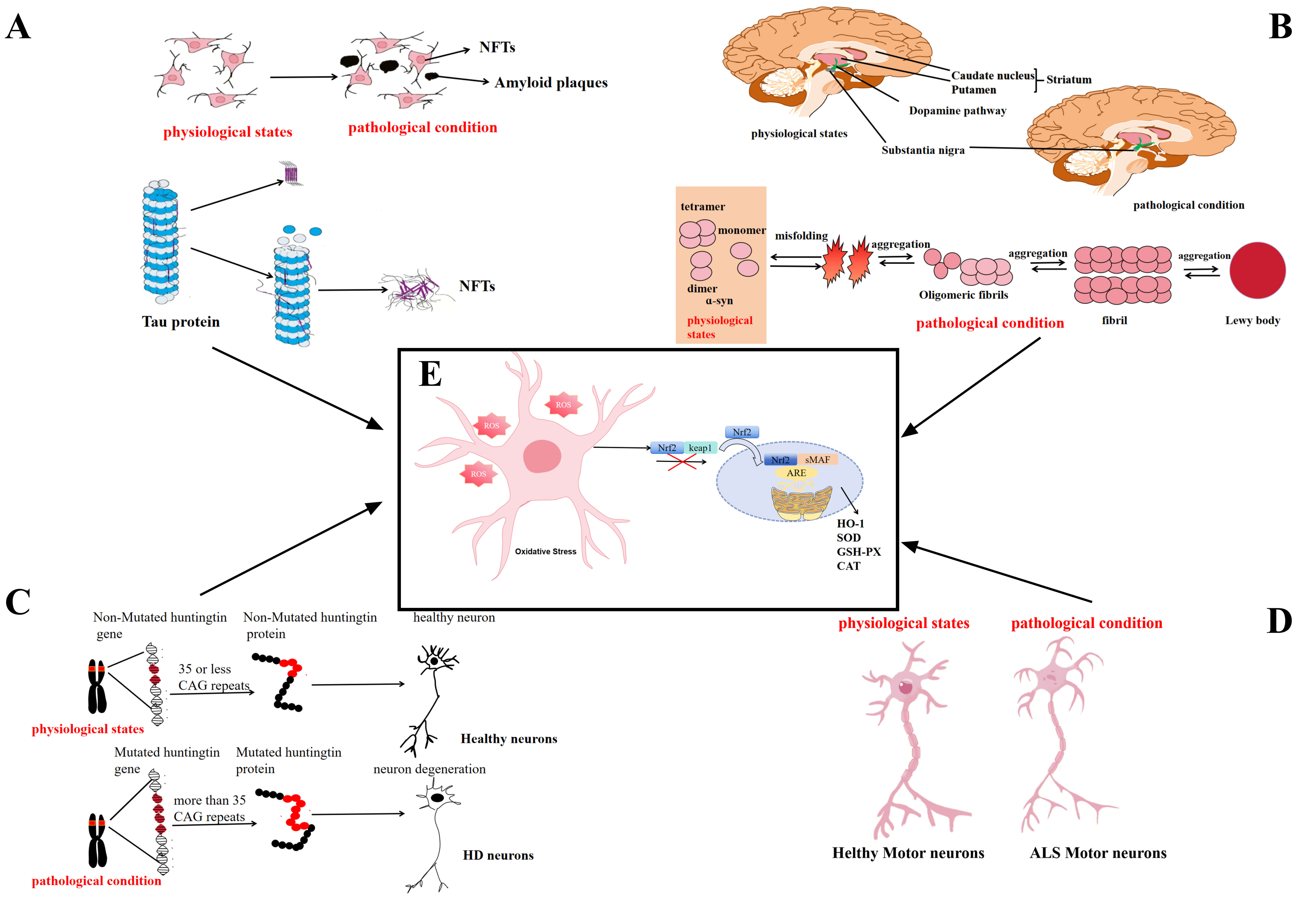 Fig. 5.
Fig. 5.
Core pathological features of NDs in normal and pathological
states. (A) Core pathological features of AD. Abnormal A
A
Marked disruption of redox balance in the brains of AD patients initiates lipid peroxidation, which reduces membrane fluidity and impairs ion channel activity, thereby diminishing neuronal excitability and weakening synaptic transmission, ultimately progressing to neuronal dysfunction and apoptosis [63, 64]. The apoE4 isoform has been implicated in lowering neuronal tolerance to oxidative stress and in aggravating mitochondrial impairment, thereby heightening vulnerability to cell death [65]. Progressive neuronal loss disrupts local microcircuitry and diminishes integrative network function, driving further cognitive decline and reinforcing a pathological cycle that accelerates AD progression. Within this setting, Nrf2 acts as a central regulator of oxidative defense by inducing endogenous antioxidant systems. Upon oxidative challenge, Nrf2 migrates to the nucleus, dimerizes with sMAF, and binds to ARE sequences in promoter regions of target genes, initiating the transcription of antioxidant enzymes such as HO-1, GSH-Px, and CAT (Fig. 5E) [66, 67]. The concerted activity of these enzymes neutralizes excess ROS and supports the repair of oxidative damage, thereby restoring redox balance and attenuating neurodegenerative processes in AD [68]. Emerging evidence further indicates that ApoE expression may intersect with Nrf2 activity, providing new insight into the relationship between genetic susceptibility and antioxidant regulation in AD [65].
PD, the second most common NDs, is defined by the progressive loss of
dopaminergic neurons in the substantia nigra pars compacta and by abnormal
HD is an autosomal dominant neurodegenerative disorder caused by expansion of CAG repeats in the HTT gene, producing mutant Huntington protein (mHTT). Pathological characteristics include striatal neuronal loss and abnormal aggregation of mHTT (Fig. 5C) [87]. Transcriptional dysregulation, mitochondrial dysfunction, and excessive ROS have been identified as major drivers of disease progression [88], with disrupted mitochondrial Ca2+ regulation occupying a central position. Impaired buffering by mHTT leads to matrix Ca2+ overload, which in turn activates NADPH oxidase and promotes ROS generation [89, 90]. The resulting oxidative stress induces lipid peroxidation, protein oxidation, and mitochondrial DNA (mtDNA) damage, ultimately triggering neuronal apoptosis through caspase-dependent mechanisms [91]. This self-amplifying cycle is especially pronounced in the striatum, providing a plausible explanation for its heightened vulnerability in HD [92].
Activation of the Nrf2/ARE signaling cascade in the presence of antioxidant compounds produces a synergistic enhancement of antioxidant enzyme expression, thereby reducing oxidative stress and restricting glutamate release, which in turn delays the manifestation of HD symptoms [93, 94]. Experimental studies have further shown that stimulation of the Keap1/Nrf2 axis sustains intracellular ATP levels and preserves mitochondrial membrane potential, thus protecting astrocytes from oxidative injury [95]. During oxidative stress, astrocytes activate Nrf2–ARE signaling, inducing upregulation of antioxidant enzymes such as HO-1 and SOD, thereby strengthening redox homeostasis within neural tissues (Fig. 5E) [96, 97]. In HD mouse models, astrocyte-specific Nrf2 overexpression substantially decreases striatal oxidative damage and alleviates motor deficits [98], indicating that glial regulatory pathways contribute significantly to neuroprotection. Impairment of Nrf2 signaling has been associated with disease progression in HD and may drive the gradual neurodegeneration characteristic of the disorder. Future research should assess Nrf2-responsive genes as potential therapeutic targets for HD.
ALS is an aggressive and ultimately fatal form of motor neuron disease (MND) characterized by the progressive degeneration of motor neurons in the cerebral cortex, brainstem, and spinal cord (Fig. 5D) [99]. While most cases occur sporadically, approximately 5–10% are inherited, commonly associated with mutations in C9orf72, SOD1, TARDBP, and FUS [100, 101]. Among these, SOD1 mutations hold particular significance, as the Cu2+/Zn2+-binding SOD protein directly influences OS and inflammatory signaling cascades [102, 103]. Both sporadic and familial ALS consistently display a pronounced OS profile, widely regarded as a central factor in neuronal degeneration and disease progression. Clinical studies have identified characteristic changes in oxidative stress biomarkers in ALS patients, including diminished total antioxidant capacity (TAC) and reduced enzymatic activities of glutathione peroxidase (GPX), SOD, and glutathione reductase (GR), along with elevated levels of malondialdehyde (MDA), a marker of lipid peroxidation, and 8-OHdG, indicative of oxidative DNA damage [104, 105, 106]. Reports of increased total serum antioxidant levels in certain cohorts may instead represent an endogenous adaptive response to persistent free radical burden [107]. Regulation of intracellular ROS and reinforcement of antioxidant defense systems are therefore considered essential therapeutic avenues, with antioxidant capacity itself serving as a promising target for intervention.
The Nrf2/Keap1-ARE signaling cascade constitutes a major protective system against ROS generated under oxidative stress, and its disruption contributes to the pathogenesis of diverse disorders, including NDs (Fig. 5E) [108]. In ALS models carrying the hSOD1 G93A mutation, ferroptotic activation is accompanied by impaired nuclear retention of Nrf2 and reduced expression of the ferroptosis regulators SLC7A11 and GPX4 [19]. Consistently, diminished Nrf2 expression has been documented in the motor cortex and spinal motor neurons of ALS patients [109]. Astrocyte-targeted Nrf2 overexpression alleviates the neurotoxic influence of mutant astrocytes on motor neurons, delaying disease onset and extending lifespan in ALS mice, thereby highlighting the therapeutic potential of reinforcing Nrf2 signaling to counteract progressive neurodegeneration [110].
Nrf2 activation enhances its expression in the brain, triggers transcription of cytoprotective genes, reduces OS, and alleviates neurodegenerative pathology, thereby positioning Nrf2-directed agents as therapeutic candidates for NDs. Evidence from both in vitro and in vivo studies demonstrates that multiple traditional Chinese medicines and their active constituents modulate Nrf2 signaling and confer neuroprotection through antioxidant mechanisms (Table 1, Ref. [111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126]) [127]. Such agents are increasingly regarded as potential interventions in the therapeutic landscape of NDs.
| Category | Active ingredient | Source | Models | Treatment | Major Finding | Ref. |
| Flavonoids | Baicalein | Scutellaria baicalensis Georgi | 1 mM 6-OHDA treated PC12 cells 8 h | 50 |
[111] | |
| Puerarin | Pueraria lobata (Willd.) Ohwi | 250 µM MPP+ treated PC12 cells for 24 h | 10 µM Pueraria Mirifica treated PC cells for 1 h | [112] | ||
| APP/PS1 transgenic mice | Puerarin (30 mg/kg) dissolved in 1,2-propanediol and administered by gavage for 28 consecutive days | [113] | ||||
| Apigenin | Apium graveolens L. | 1 mg/L doxycycline (Dox) treatment of SNCAWTSH-SY5Y cells for 24 hours | SNCAWTSH-SY5Y cells were treated with 50 µM and 100 µM apigenin for 24 h, respectively | [114] | ||
| Male C57BL/6J mice were taken and rotenone (3 mg/kg) was injected subcutaneously continuously for 5 weeks | Male C57BL/6J mice were administered orally, low dose (25 mg/kg) and high dose (50 mg/kg) apigenin for 5 weeks prior to modeling | |||||
| Tg(SOD1*G93A)1Gur/J transgenic mice | Oral apigenin (80 mg/kg) once daily, sh-ALDH1A2 treatment initiated at symptom onset (day 90) | [115] | ||||
| Quercetin | Flos Sophorae Immaturus; platycladus orientalis (L.) Franco | PC12 cells were treated with 20 µmol/L |
PC12 cells were pretreated with 10 µmol/L, 20 µmol/L, 40 µmol/L and 80 µmol/L quercetin for 24 h, 48 h and 72 h, respectively | [116] | ||
| APP/PS1 transgenic mice | Quercetin (100 mg/kg) administered by gavage for 6 consecutive months | [117] | ||||
| Luteolin | Lonicera japonica Thunb; Dendranthema morifolium Ramat. | Mouse neuroblastoma Neuro2A (ATCC) cells were transfected with a plasmid encoding an htt N-terminal fragment containing 20Q (htt20Q) and 160Q (htt160Q) to obtain cell models expressing normal (20Q htt) and mutant (160Q htt) cells | Mutant (160Q htt) cells were treated with lignocaine (5 ng/mL) for 48 h | [118] | ||
| Icariin | Epimedium brevicornu Maxim. | 40 µM 6-OHDA treatment of PC12 cells for 24 h | 0.005 µM and 0.05 µM Icariin pretreated PC12 cells for 24 h | [119] | ||
| Terpenoids | Andrographolide | Andrographis paniculata (Burm. f.) Wall. ex Nees | 1.5 mM MPP+ treated SH-SY5Y cells 24 h | 1.5 µM Andrographolide Pretreatment of SH-SY5Y cells 24 h | [120] | |
| Tanshinone IIA | Salviae Miltiorrhizae Radix et Rhizoma | 100 µM 6-OHDA treated SH-SY5Y cells for 24 h | 20 µg/mL tanshinone IIA treated SH-SY5Y cells 24 h | [121] | ||
| Celastrol | Tripterygium wilfordii Hook. F. | Continuous 5 days intraperitoneal injection of 20 mg/kg MPTP induced PD in mice | 10 µg/kg tretinoin intraperitoneally for 7 days | [122] | ||
| AAV-mediated human wild-type |
10 µg/kg tretinoin intraperitoneal injection for 42 days | |||||
| Phenols | Curcumin | Curcuma longa L. | Subcutaneous injection of 2.5 mg/kg of rotenone induced PD in mice for 35 days | Pregavage administration of 80 mg/kg Curcumin for 7 days, then co-treatment with rotenone for 35 days for a total of 42 days | [123] | |
| Resveratrol | grapes; Reynoutria japonica Houtt | 50 µM MPP+ treated SH-SY5Y cells for 24 h | Cells were replaced from medium containing MPP+ to human neural stem cell-derived exosomes (hNSCs-Exos) medium treated with 10 µg/mL resveratrol for another 48 h | [124] | ||
| Others | Dimethyl fumarate | Synthetic | App-KI Mouse | 300 mg/kg dimethyl fumarate administered by gavage 3 times weekly for 1 month (short-term dosing) and 5 months (long-term dosing) | [125] | |
| Primary astrocytes | Primary astrocytes treated with 35 µM dimethyl fumarate for 1 h | |||||
| Sulforaphane | Brassica oleracea L. var. Italica Plenck; Brassica oleracea var. capitata Linnaeus | GFP, GFP - T4, and GFP-T4C3 were injected into the right hippocampal CA1 region of pure C57BL/6J tau knockout (tau (-/-)) male mice (1.5 µL of each AAV at a titer of 1013), respectively. Two months later, the expression of AAVs in the hippocampus was observed and quantified | 50 mg/kg radicicol intraperitoneally three times per week for two weeks | [126] |
Abbreviations: 4-HNE, 4-hydroxynonenal; 6-OHDA, 6-hydroxydopamine;
Flavonoids, characterized by a 2-phenylchromenone (C6-C3-C6) backbone, display
extensive structural variability that determines their bioactivity. Specific
subclasses contain
Baicalein, a principal constituent of Scutellaria baicalensis Georgi, exhibits anti-inflammatory, antioxidative, antineoplastic, and neuroprotective properties relevant to NDs [105]. Experimental studies further indicate that baicalin protects PC12 cells from 6-OHDA-induced oxidative stress through activation of the Keap1/Nrf2/HO-1 pathway and marked suppression of Keap1 protein expression [111].
Puerarin, a bioactive isoflavone primarily derived from Pueraria lobata(Willd.) Ohwi, demonstrates cardioprotective, neuroprotective, antioxidant,
anticancer, and anti-inflammatory properties [130]. Experimental studies indicate
that puerarin mitigates MPP+-induced oxidative stress through activation of the
Nrf2 signaling cascade. In PC12 cells, puerarin enhances GSH synthesis, promotes
nuclear accumulation of Nrf2, and elevates GCLC expression via ARE-dependent
transcriptional and translational regulation. These molecular adaptations improve
motor performance in wild-type mice exposed to MPP+ and reduce oxidative injury
within the ventral midbrain [112]. In addition, puerarin regulates the
Akt/GSK-3
Apigenin, widely distributed in dietary sources such as Apium
graveolens L., spinach, chamomile, grapes, and apples, exerts diverse
pharmacological actions including antioxidant, antibacterial, and antidiabetic
effects [131]. Experimental studies demonstrate that apigenin suppresses neuronal
apoptosis by accelerating
Quercetin, mainly derived from traditional Chinese medicinal plants such as
Flos Sophorae Immaturus and Platycladus orientalis (L.) Franco,
exhibits broad pharmacological activities including anti-cancer,
anti-inflammatory, antiviral, and antioxidant effects [132]. Experimental results
reveal that quercetin enhances PC12 cell survival under A
Luteolin, a flavonoid abundant in traditional Chinese medicinal plants such as Lonicera japonica Thunb. and Dendranthema morifolium Ramat., exhibits antioxidant, anti-inflammatory, antitumor, and neuroprotective properties [133]. Experimental studies demonstrate that luteolin improves cell survival and suppresses apoptosis, thereby reducing the cytotoxicity associated with mutant huntingtin protein. In neuroblastoma cells expressing 160Qhtt, luteolin diminishes both soluble and insoluble mHTt aggregates, indicating its potential to modulate protein misfolding and aggregation pathways [118].
Icariin, the principal bioactive component of Epimedium brevicornu Maxim., exerts antioxidant, anti-inflammatory, anti-apoptotic, and antitumor effects [134]. Evidence shows that epimedium glycoside attenuates apoptosis by lowering the Bax/Bcl-2 ratio, suppressing cytochrome c release, and inhibiting caspase-3 activation. Moreover, epimedium glycoside activates the Nrf2 pathway, thereby limiting intracellular ROS accumulation, enhancing SOD activity, and upregulating Nrf2, HO-1, and NQO1 expression, ultimately reducing neuronal damage induced by 6-OHDA [119].
Furthermore, the above-mentioned flavonoids possess certain medicinal potential and are commonly used as dietary supplements, such as puerarin, apigenin, and quercetin [135]. Nevertheless, due to challenges such as low bioavailability and toxicity, the development of many flavonoids into clinical drugs has been unsuccessful. The majority of research on flavonoids is based on in vitro or animal models, lacking large-scale clinical trials to support their efficacy as medications [136].
Terpenoids constitute a diverse group of natural compounds categorized by
structural features into monoterpenes, sesquiterpenes, and diterpenes. Among
them, andrographolide, a diterpenoid lactone mainly derived from
Andrographis paniculata (Burm. f.) Wall. ex Nees., exhibits
anti-inflammatory, antioxidant, anticancer, and antihyperglycemic activities
[137]. The
Tanshinone IIA, a lipophilic diterpene isolated from Salviae Miltiorrhizae Radix et Rhizoma, demonstrates anti-inflammatory, antibacterial, and antioxidant properties [138]. Experimental evidence indicates that it preserves dopaminergic neurons in the substantia nigra-striatal pathway by activating Nrf2/ARE signaling. In SH-SY5Y cells, it alleviates 6-OHDA-induced toxicity, reduces LDH release and ROS production, and induces the expression of ARE-regulated genes such as HO-1 and GCLC [121].
Celastrol, a pentacyclic triterpenoid with cork-like characteristics primarily
obtained from Tripterygium wilfordii Hook. F, exhibits
anti-inflammatory, antioxidant, and anti-obesity activities, with emerging
relevance for NDs [139]. Leucoanthraquinone has been reported to prevent
dopaminergic neuronal loss, preserve nigrostriatal integrity, suppress
neuroinflammation, and improve motor outcomes in both MPTP-induced PD mouse
models and AAV-mediated
Phenolic compounds act as electrophilic agents that covalently interact with
cysteine thiol residues of Keap1, disrupting the Keap1–Nrf2 complex and
releasing Nrf2 into the cytoplasm. Following liberation, Nrf2 translocates to the
nucleus, associates with ARE sequences in promoter regions, and activates
transcription of phase II cytoprotective genes, thereby strengthening cellular
defense against OS-induced injury [38]. These electrophilic agents, collectively
referred to as Nrf2 activators, frequently contain reactive moieties such as
Curcumin, a natural compound isolated from the rhizome of Curcuma longa
L., displays antibacterial, antineoplastic, antioxidant, and anti-inflammatory
activities [141]. Its
Resveratrol, a polyphenolic compound derived from grapes, Reynoutria
japonica Houtt, and various other botanicals, exerts diverse pharmacological
actions, including antioxidant, anti-inflammatory, cardioprotective, antitumor,
antidiabetic, anti-obesity, neuroprotective, and anti-aging effects [143]. In
hNSCs-Exos exposed to resveratrol, PGC1
Currently, dimethyl fumarate (DMF) is the only Nrf2 activator approved for clinical use in NDs, with its indication restricted to multiple sclerosis (MS). Developed by Biogen and marketed as Tecfidera, DMF acts as a prodrug that is metabolized in vivo to generate the active metabolite monomethyl fumarate (MMF), which mediates its therapeutic efficacy [145, 146]. Regulatory authorization was first granted by the US FDA in March 2013 as a first-line therapy for MS, followed by European Medicines Agency approval in January 2014 [147]. Preclinical evidence demonstrates that DMF activates Nrf2 signaling in primary astrocytes, leading to reduced expression of proinflammatory mediators, attenuation of neuroinflammation, and improvement of cognitive deficits. Furthermore, DMF suppresses STAT3/C3 and C3 receptor expression in astrocytes and microglia derived from App gene-knockin (App-KI) mice, indicating its capacity to modulate AD-associated inflammatory pathways [125].
Other Nrf2 agonists under clinical evaluation (Table 2) include sulforaphane (SFN), curcumin, and resveratrol. SFN, a phytochemical abundant in cruciferous vegetables such as Brassica oleracea L. var. Italica Plenck and Brassica oleracea var. capitata Linnaeus, exhibits antioxidant, anti-inflammatory, and anti-apoptotic activities [148]. In tau-knockout mice, SFN mitigates caspase-3–cleaved tau–induced ROS accumulation, mitochondrial depolarization, and ATP depletion, while simultaneously upregulating OPA-1, strengthening mitochondrial antioxidant capacity, and improving recognition memory in truncated tau–expressing mice, thereby enhancing spatial cognitive performance [126].
| Active ingredient | NDs | Clinical Progress | Clinical Trials.gov Identifier |
| Sulforaphane | PD | Phase II | NCT05084365 |
| Curcumin | AD | Phase I/II | NCT00164749 |
| Phase II | NCT00099710 | ||
| Resveratrol | AD | Phase I | NCT02502253 |
| Phase II | NCT01504854 | ||
| Phase III | NCT00743743 | ||
| Phase III | NCT00678431 | ||
| HD | Phase III | NCT02336633 |
Recent investigations highlight single-compound Chinese herbal medicines as potential therapeutic agents for NDs, acting through modulation of key signaling networks such as Nrf2 and regulation of pathological mechanisms including oxidative stress, inflammation, and apoptosis via multi-target, multi-pathway interactions (Table 3, Ref. [149, 150, 151, 152, 153, 154, 155]).
| Traditional Chinese medicine | Models | Treatment | Major Finding | Ref. |
| Uncaria rhynchophylla (Miq.) Miq. ex Havil. | Male C57BL/6 J mice were injected intraperitoneally with 30 mg/kg MPTP once daily for 3 weeks. | 20, 40, 60 mg/kg Uncaria rhynchophylla (Miq.) Miq. ex Havil. extract administered by gavage for 3 weeks | [149] | |
| 100 ng/mL LPS stimulation of BV-2 microglia and SH-SY5Y cells for 24 h | 5, 10, 20 µg/mL Uncaria rhynchophylla (Miq.) Miq. ex Havil. extract treated BV2 microglia and SH-SY5Y cells for 4 h | |||
| Pilose antler | Male Wistar rats stereotactic right brain (A: –4.8 mm, L: +2.0 mm, H: 8.0 mm from substantia nigra (SN), A: –4.8 mm, L: +1.2 mm, H: 8 mm from ventral tegmental area (VTA)) injected with 6-OHDA (8 µg, 2 µL/min) | 60, 180 mg/kg Pilose antler extract administered by continuous gavage for 14 d | [150] | |
| 200, 400 and 800 µg/mL MPP+, Pre-treatment of PC12 cells for 24 h | 200, 400 and 800 µg/mL Pilose antler extract treated PC12 cells for 24 h | [151] | ||
| Male C57BL/6 mice were injected intraperitoneally with 30 mg/kg MPTP daily from d 5 to d 10 | 30 mg/kg Pilose antler extract administered by gavage for 10 d | |||
| Melissa officinalis | 0.1 µg/mL LPS treated BV2 cells for 18 h | 10, 50, and 100 µg/mL ethanol extract of Melissa officinalis treated BV2 cells for 18 h | [152] | |
| Hericium coralloides | APP/PS1 transgenic mice | 5 mL/kg Hericium coralloides administered by gavage for 49 d | [153] | |
| Centella asiatica | 5xFAD mice | 5xFAD were given Centella asiatica extract in water (2 g/L) for a total of 4 months of treatment | [154] | |
| Moschus | 10 µM Erastin treated HT22 cells for 24 h | 30 µg/mL and 60 µg/mL Moschus extract pretreated HT22 cells for 24 h | [155] |
Abbreviations: 5-HT, 5-hydroxytryptamine; DA, dopamine; DOPAC, 3,4-Dihydroxyphenylacetic acid; FTH1, ferritin heavy chain 1; FPN1, ferroportin1; GAP-43, growth associated protein-43; Glu, glutamate; HVA, homovanillic acid; GABA, Gamma-aminobutyric acid; NF-H, neurofilament heavy; SLC7A11, cystine/glutamate antiporter subunit; TFRC, transferrin receptor.
Extracts of Uncaria rhynchophylla (Miq.) Miq. ex Havil. markedly
improve MPTP-induced motor deficits and dopaminergic neurodegeneration in mice
while normalizing serum oxidative and inflammatory markers. In vitro
evidence further indicates that the extract attenuates neuroinflammatory injury
in SH-SY5Y neurons by suppressing the TLR4/NF-
Pilose antler extract demonstrates neuroprotective efficacy by preventing 6-OHDA-induced apoptosis of substantia nigra pars compacta (SNpc) neurons and preserving tyrosine hydroxylase (TH)-positive cell populations [150]. In MPTP-induced PD mouse models, Pilose antler peptides have been shown to limit neuronal apoptosis and oxidative stress through activation of the SIRT1-dependent Akt/Nrf2/HO-1 signaling cascade, thereby protecting dopaminergic neurons and improving neurobehavioral performance [151].
Melissa officinalis exhibits anti-inflammatory, antioxidant, sedative, antibacterial, and antiviral activities. According to Choi JW et al. [152], ethanol extract of this herb exerts neuroprotective effects by attenuating inflammatory responses and oxidative stress through the upregulation of antioxidant mediators such as Nrf2, HO-1, CAT, and SOD2.
Hericium coralloides has demonstrated therapeutic potential in
hyperlipidemia, NDs, and cancer, with evidence indicating modulation of Nrf2
signaling, elevation of antioxidant enzymes including SOD, GSH-Px, and CAT in
brain tissue and serum, and suppression of oxidative markers such as 4-HNE and
MDA in the brain. In addition, inhibition of A
Centella asiatica improves cognitive function in experimental models of aging
and NDs; in 5xFAD mice, four months of extract administration enhanced spatial
memory, episodic recall, and executive performance while markedly decreasing
cortical A
Moschus demonstrates neuroprotective capacity, likely mediated through anti-inflammatory, antioxidant, and anti-apoptotic pathways. By activating the Keap1/Nrf2 axis, Moschus lowers MDA, ROS, and lipid peroxide accumulation, restores intracellular GSH content, upregulates GPX4 and SLC7A11, and normalizes iron metabolism, thereby reducing neuronal damage [155].
Recent scientific and technological progress has enabled innovative strategies to modulate Nrf2 activity, including gene editing, nanotechnology, and applications from cell therapy and regenerative medicine. These approaches improve the therapeutic profile of Nrf2 activators by enhancing efficacy, safety, and disease-specific applicability.
Gene editing enables targeted correction of genetic defects by directly modifying DNA sequences. In one study, Yang J et al. [156] developed AREs-dCAS9-VP64_GFP and Nrf2-sgRNA plasmids to establish oxidative stress–responsive regulatory elements. These constructs were incorporated into nanocarriers, forming a nanozyme-enhanced MOF-CRISPR delivery system. Intravenous administration of this platform markedly improved neuronal integrity and mitigated cognitive deficits in 3xTg-AD mice. The precision of gene editing technology offers critical insights into the regulatory architecture of the Nrf2 signaling axis and supports the advancement of next-generation Nrf2-targeted therapeutics.
Nanotechnology plays a central role in designing delivery systems for Nrf2 agonists, improving bioavailability, targeting specificity, and therapeutic efficiency [157]. Polymer nanoparticles, particularly PLGA-based carriers, represent effective platforms for targeted administration of Nrf2 activators in ALS. Such carriers provide controlled drug release, extending pharmacological activity while increasing stability and systemic availability [158, 159]. In ALS transgenic mouse models, PLGA nanoparticle–mediated delivery of Nrf2 activators significantly delayed motor neuron loss and prolonged survival. Adjustment of PLGA molecular weight and copolymer composition further optimizes release kinetics and biocompatibility, thereby expanding the therapeutic potential of Nrf2 activators in NDs [160].
Cell therapy employs autologous or allogeneic cell transplantation to achieve therapeutic outcomes [161]. Transplantation of Human umbilical cord mesenchymal stem cells (HUC-MSCs) has been shown to reduces MDA levels and increases NO, SOD, and nNOS activity in the hippocampus of Tg2576 transgenic mice, thereby significantly reducing oxidative stress in the hippocampus of AD mice. This intervention markedly improved cognitive impairment and reshaped the brain microenvironment in AD mice, while exhibiting antioxidant and neuroreparative properties in in vitro AD models [162]. Thus, modulation of the Nrf2 pathway through cell-based therapy constitutes a promising direction for ND treatment.
Research on the structural and functional characteristics of Nrf2, along with its disease-associated regulatory network, has progressed substantially, particularly regarding the diversification of its regulatory mechanisms. As a transcription factor governing OS, mitochondrial dysfunction, and inflammatory signaling, Nrf2 exerts a decisive influence on ND progression by modulating downstream antioxidant genes such as HO-1 and SOD. Reduced Nrf2 expression has been closely linked to disease advancement in AD, PD, HD, and ALS, reflecting its essential contribution to cellular homeostasis and therapeutic relevance.
In NDs defined by abnormal protein aggregation and neuronal degeneration, Nrf2 activates antioxidant defenses through both Keap1-dependent and Keap1-independent pathways, with ROS-driven OS acting as a central pathogenic driver. Preclinical studies demonstrate that such activation reduces neuronal injury and improves neurological outcomes. Accordingly, therapeutic strategies targeting Nrf2 hold substantial promise. Natural compounds, including flavonoids, terpenoids, and bioactive constituents of Chinese herbal medicine, act as selective Nrf2 activators and confer neuroprotection through this signaling axis. Beyond its role in antioxidant defense, Nrf2 serves as a master regulator capable of integrating multiple protective pathways, including autophagy and anti-inflammatory processes, thereby overcoming the limitations of single-agent antioxidant therapy. Combining Nrf2 agonists with emerging technologies such as nanocarrier-mediated delivery provides a rational approach for multi-target interventions, offering new opportunities to address the escalating burden of NDs.
3-NPA, 3-nitropropionic acid;
WTC, JYW, and QL jointly conceptualized and structured the manuscript, drafted the original version, and contributed substantially to its subsequent revision. LAZ, XW, and SFL were responsible for figure preparation and literature review. YHX and PXG created the tables and analyzed the data; the draft underwent multiple revisions before the final version was completed. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors express their sincere gratitude to Yan Wan and Feifan Liu for their valuable feedback and suggestions during manuscript preparation.
This research was supported by the National Natural Science Foundation of China (81660671); Yunnan Provincial Science and Technology Department-Applied Basic Research Joint Special Funds of Chinese Medicine (202101AZ070001-172); Yunnan Provincial Key Laboratory of Formula Granules (202105AG070014); High-level Discipline Construction Project of Dai Medicine, National Administration of Traditional Chinese Medicine (zyzdxk-2023192); and Yunnan Key Laboratory for Dai and Yi Medicines (2024SS24045).
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGpt to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.