




 , Xinyi Chen 1,†,§, Xiaoying Pan 1,2,3, Kai Luo 2,3, Zixiong Zhang 1,3, Changyu Zhou 1,3, Haibo Wang 2,3,*
, Xinyi Chen 1,†,§, Xiaoying Pan 1,2,3, Kai Luo 2,3, Zixiong Zhang 1,3, Changyu Zhou 1,3, Haibo Wang 2,3,* , Chuying Huang 1,3,4,*
, Chuying Huang 1,3,4,*1 Hubei Key Laboratory for Translational Research in Traditional Chinese Medicine, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Hubei Minzu University, 445000 Enshi, Hubei, China
2 College of Biological and Food Engineering, Hubei Minzu University, 445000 Enshi, Hubei, China
3 Hubei Provincial Key Lab of Selenium Resources and Bioapplications, 445000 Enshi, Hubei, China
4 Department of Oncology, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, 445000 Enshi, Hubei, China
†These authors contributed equally.
§Current address: School of Public Health, Xiamen University, 361100 Xiamen, Fujian, China.
Abstract
Cadmium (Cd), a widespread environmental pollutant, poses significant risks to human health due to its high bioaccumulation potential and prolonged biological half-life. Selenium (Se) has been reported to exert protective effects against Cd-induced organ toxicity; however, the underlying molecular mechanisms, particularly those associated with lipid metabolism and inflammatory regulation, remain insufficiently elucidated.
The hepatoprotective effects of Se, administered as selenomethionine (SeMet) and Se-enriched Cardamine enshiensis extract (CE), were investigated against Cd-induced hepatic injury using both in vitro (L-02 hepatocytes) and in vivo (C57BL/6J mice) models.
SeMet significantly attenuated Cd-induced cytotoxicity, lipid accumulation, and metabolic dysregulation in L-02 cells. In Cd-exposed mice, treatment with SeMet or CE significantly mitigated hepatic injury, steatosis, and inflammation, as evidenced by normalized serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), and total cholesterol (TC) levels, improved hepatic histoarchitecture, and reduced lipid droplet deposition. Integrated lipidomic and transcriptomic analyses demonstrated that Se supplementation restored Cd-perturbed polyunsaturated fatty acid metabolism, downregulated lipogenic genes (SCD1, Pparγ, Fasn), and suppressed pro-inflammatory mediators (Cxcl2, Ccl2).
Se confers hepatoprotection against Cd toxicity not only through its classical antioxidant activity but also through coordinated modulation of lipid metabolic pathways and inflammatory signaling. This study provides mechanistic insights into Se-mediated defense against Cd-induced hepatotoxicity and highlights the therapeutic potential of Se-enriched phytochemicals for mitigating the adverse effects of environmental Cd exposure.
Keywords
- cadmium
- selenium
- chemical and drug induced liver injury
- lipid metabolism
- inflammation
Cadmium (Cd), a widespread and bioaccumulative environmental toxicant, poses severe threats to human health owing to its prolonged biological half-life and inefficient metabolic clearance [1]. The liver and kidneys, as primary sites of xenobiotic metabolism, are particularly vulnerable to both acute and chronic Cd exposure, making them major targets of Cd-induced toxicity [2, 3, 4]. Extensive evidence indicates that Cd exerts multi-organ toxicity through diverse mechanisms, including oxidative stress, DNA damage, endoplasmic reticulum stress (ERS), apoptosis, autophagy, and reproductive system disorders [5, 6]. Among these, oxidative stress and inflammation are considered key pathogenic events in Cd-mediated tissue injury, despite Cd lacking redox activity as a non-Fenton metal [7, 8]. Although significant advances have been made in delineating the molecular basis of Cd toxicity, effective therapeutic strategies to counteract its deleterious effects remain scarce. Therefore, elucidation of the molecular mechanisms underlying Cd-induced hepatic and renal injury and the development of targeted interventions represent critical research imperatives.
Selenium (Se), an essential micronutrient, is vital for maintaining human health due to its key roles in antioxidative defense, anti-inflammatory regulation, and immune modulation [9, 10, 11, 12]. Accumulating evidence supports the protective effects of Se against Cd toxicity, with exogenous Se supplementation reported to attenuate Cd-induced cellular and tissue damage through multiple mechanisms [13]. One of the primary protective pathways involves the formation of Se–Cd complexes, which promote Cd detoxification and excretion from the body [14, 15, 16]. Furthermore, Se mitigates Cd-induced oxidative stress and ERS in renal tissues, as well as oxidative damage and programmed necrosis in the liver [17]. Se exerts its antioxidant and anti-inflammatory functions through incorporation into selenoproteins, including glutathione peroxidase (GPx) and thioredoxin reductase (TrxR), which collectively maintain redox homeostasis and suppress inflammatory signaling cascades [18, 19]. Moreover, Se has been reported to attenuate Cd toxicity through activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway [20]. Recent studies have demonstrated that various forms of Se, including sodium selenite, selenomethionine (SeMet), nano-Se, and Se derived from Cardamine hupingshanensis, effectively alleviate Cd-induced toxicity in multiple organs such as the liver, kidneys, spleen, brain, and heart in both in vivo and in vitro models [20, 21]. Despite these advances, the precise molecular mechanisms underlying the protective actions of Se, particularly its modulation of lipid metabolic pathways and suppression of inflammatory processes in Cd-induced hepatotoxicity, remain insufficiently elucidated.
The significance of this study is derived from the considerable public health burden associated with environmental cadmium exposure, for which no pharmacological intervention with an established safety and efficacy profile is currently available. Clinical management remains largely dependent on chelation therapy, which is frequently limited by off-target toxicity and organ-related adverse effects. This situation emphasizes the need to identify safe and effective alternative strategies and to advance beyond the conventional antioxidant framework toward a precise characterization of the molecular pathways responsible for selenium-mediated protection. Based on previous observations [21, 22], the aim of this study is to elucidate the molecular mechanisms by which selenium, particularly in the form of a Cardamine enshiensis–derived extract enriched in SeCys2 (CE), protects against cadmium-induced hepatotoxicity, using integrated lipidomic and transcriptomic analyses to define its regulatory effects on polyunsaturated fatty acid metabolism, lipogenic gene networks, and inflammatory signaling pathways.
The L-02 hepatocyte line (kindly provided by Prof. Jiangfeng Wu, China Three Gorges University) was cultured in DMEM/F-12 (Gibco, 11320082, Grand Island, CA, USA) supplemented with 10% fetal bovine serum (FBS; Vivalcell, C04001-050X10, Shanghai, China) and 1% penicillin/streptomycin (Gibco, 15140-122, Grand Island, CA, USA). Cells were maintained at 37 °C in a humidified incubator containing 5% CO2. The L-02 cells used in our laboratory have undergone recent STR profiling authentication, and the validation report has been uploaded as an attachment. Additionally, PCR testing confirmed the absence of mycoplasma contamination. As detailed in Section 2.1, the forward and reverse primer sequences employed for PCR-based mycoplasma identification in this study were 5′-GGGAGCAAACAGGATTAGATACCCT-3′ and 5′-TGCACCATCTGTCACTCTGTGTTAACCTC-3′, respectively.
L-02 cells were seeded in 96-well plates at a density of 1
L-02 cells were plated in 6-well plates at 2
C57BL/6J mice (sourced from Hua Lian Ke, Wuhan, China) were housed under standardized conditions at the Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, with temperature maintained at 20–26 °C, relative humidity at 40–70%, chamber pressure at 20–50 Pa, and a 12 h/12 h light-dark cycle. Animals had ad libitum access to food and water. All experimental procedures were approved by the Institutional Animal Ethics Committee (Approval No.: 202101001). A chronic Cd exposure model was established by intraperitoneal injections of CdCl2 (2 mg/kg) [21, 23] every 48 h for 4 weeks. Se supplementation was administered weekly via oral gavage at 7.5 µg per 20 g body weight, a dose extrapolated from human pharmacokinetics based on a maximum tolerable intake of 400 µg/day [24]. Mice were randomly assigned to six groups (n = 10 per group) [21, 25]: Control (vehicle), Se (SeMet only), CE (Se-rich CE extract only), Cd (CdCl2 only), Se+Cd (SeMet plus CdCl2), and CE+Cd (CE extract plus CdCl2). One week before Cd exposure, Se and Se+Cd groups received a single SeMet pretreatment, while CE and CE+Cd groups were pretreated with CE extract to prime Se-dependent biochemical pathways. Dosing volumes were adjusted every 4 days according to body weight.
Throughout the study, mice were closely monitored for behavioral indicators (locomotor activity, fur condition), physiological parameters (food and water intake, body weight), and general clinical signs.
Mice were anesthetized via intraperitoneal injection of 1.25% tribromoethanol
(0.4 mL/10 g body weight). Adequate anesthesia was confirmed by the absence of a
withdrawal reflex upon hind paw pinching. The animals were then euthanized by
cervical dislocation, and death was verified by complete cessation of respiration
and heartbeat, along with pupil dilation. Following euthanasia, blood was
collected by cardiac puncture into heparinized tubes. Serum was obtained by
centrifugation at 2000
The selenium content of the Cardamine extract was quantified by inductively coupled plasma–mass spectrometry (ICP–MS; Thermo Fisher Scientific, iCAP TQe, Serial No. ICAPTQe00081, Waltham, MA, USA). Calibration curves were generated using a series of working standard solutions prepared by serial dilution of a multi-element standard stock solution with ultrapure water. An internal standard solution was continuously introduced online into all calibration standards and sample solutions to correct for instrumental drift and matrix effects. Before analysis, Cardamine extract samples were sequentially diluted with ultrapure water (100-fold and 1000-fold) and analyzed directly under the same instrumental conditions.
Total RNA was extracted from liver tissues using the EASYspin Tissue & Cell RNA
Kit (Aidlab, RN07, Beijing, China) following the manufacturer’s protocol. RNA
concentration and purity were evaluated spectrophotometrically. One microgram of
total RNA was reverse-transcribed into first-strand cDNA using ToloScript
All-in-One RT EasyMix for qPCR (TOLOBIO, 22107, Shanghai, China). qPCR was
performed in triplicate on a CFX96 Touch Real-Time PCR Detection System using
2
All data are expressed as mean
To investigate the protective role of SeMet against Cd-induced cytotoxicity in L-02 cells, the IC50 of CdCl2 was first determined to be 139.1 µM (Fig. 1a), while 0.1 µM was identified as the non-toxic concentration of SeMet (Fig. 1b). SeMet treatment significantly reduced Cd-induced cytotoxicity in a dose-dependent manner, with stronger protection observed at higher Cd concentrations (Fig. 1c,d). Furthermore, SeMet ameliorated Cd-induced metabolic dysregulation, as reflected by decreased levels of hepatic injury markers (AST and ALT) and lipid metabolites (TG and TC) (Fig. 1e–h). Treatment with 0.1 µM SeMet also significantly reduced lipid accumulation in L-02 cells exposed to 140 µM CdCl2 (Fig. 1i,j).
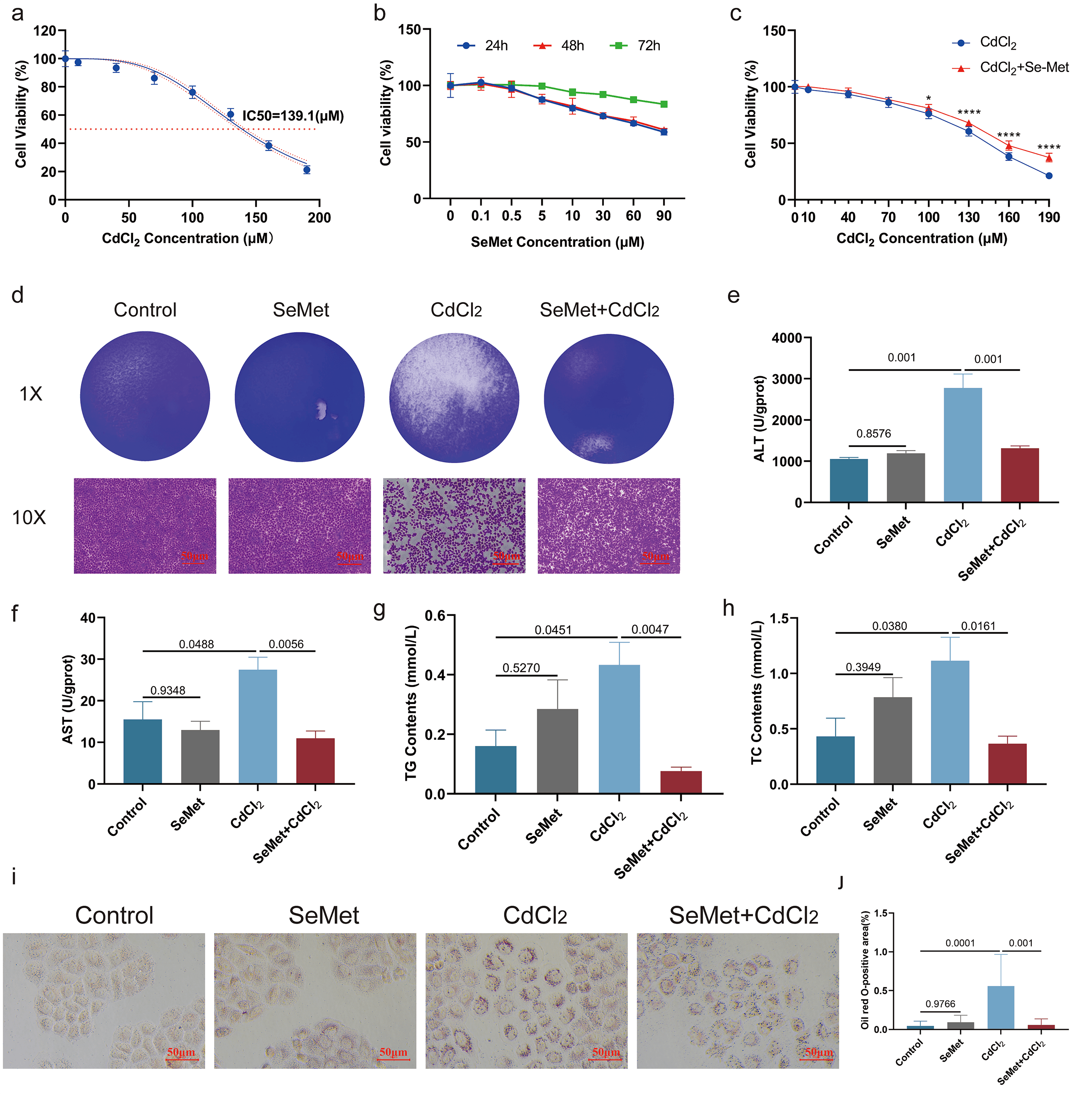 Fig. 1.
Fig. 1.
Protective effects of SeMet against Cd-induced toxicity in L-02
cells. (a) Cytotoxicity of CdCl2 in L-02 cells following 24-hour exposure.
The half-maximal inhibitory concentration (IC50) was determined to be
139.1 µM. (b) Assessment of L-02 cell viability after 24-hour
treatment with increasing concentrations of SeMet (0–100 µM). A
concentration of 0.1 µM was identified as non-cytotoxic. (c,d)
Concentration-dependent cytoprotective effect of SeMet (0.1 µM)
against varying CdCl2 doses (0–190 µM). Cell viability was
measured after 24-hour co-treatment. SeMet pre-treatment significantly mitigated
Cd-induced cytotoxicity, with stronger protection observed at higher CdCl2
concentrations (*p
To investigate the hepatoprotective effects of Se against Cd-induced toxicity, selenomethionine (SeMet) and Se-enriched Cardamine extract (CE) were administered in a mouse model. Cd exposure caused significant hepatic inury, characterized by a rough liver surface and firm texture, which were significantly alleviated by Se supplementation (Fig. 2a,b). Histopathological assessment using H&E staining revealed that Se supplementation significantly reduced Cd-induced hepatocellular ballooning and inflam matory cell infiltration, while Oil Red O staining demonstrated a significant attenuation of hepatic lipid accumulation (Fig. 2c,d). Serum biochemical analyses showed that Cd exposure significantly elevated the activities of liver injury markers (ALT, AST, and the AST/ALT ratio) [27, 28] whereas Se supplementation restored these parameters to near-normal levels (Fig. 2e–g). In comparison, markers of hepatic synthetic function (ALB, GLO, and TP) showed no significant changes across the experimental groups (Fig. 2h–j). Moreover, Cd exposure disrupted hepatic lipid metabolism, as indicated by increased TC and TG levels, both of which were significantly reduced following Se supplementation (Fig. 2k,l). These findings indicate that Se supplementation mitigates Cd-induced hepatotoxicity by preserving liver architecture and alleviating lipid accumulation.
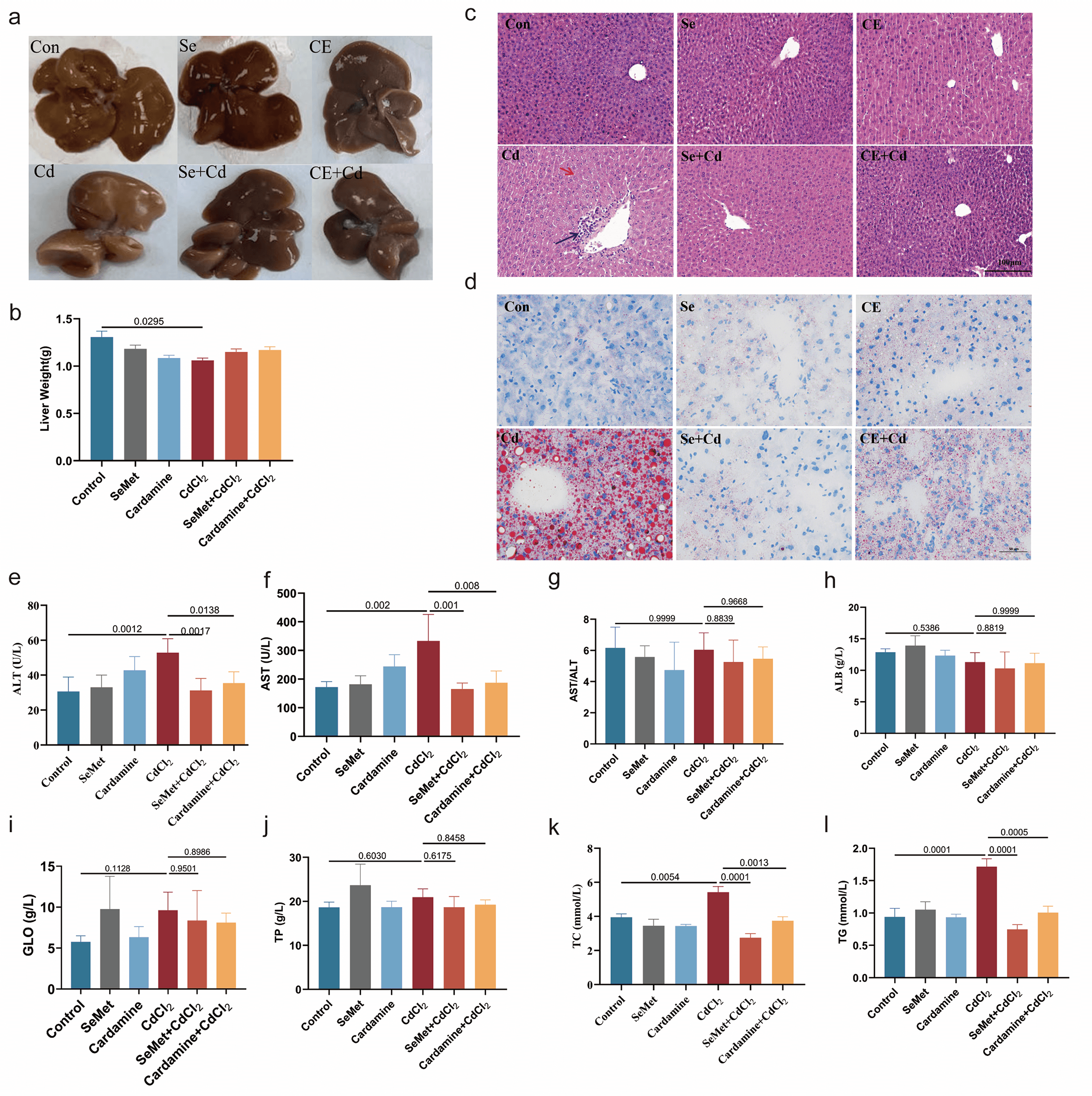 Fig. 2.
Fig. 2.
Protective effects of SeMet and CE against Cd-induced
hepatotoxicity in mice. Macroscopic images of livers from six experimental
groups, Control (1
To elucidate the molecular mechanisms underlying the protective effects of Se
against Cd-induced hepatotoxicity, a comprehensive analysis of hepatic lipid
metabolism was performed on liver tissues from control, Cd-exposed, and
CE+Cd-treated mice (Table 1). Cd exposure resulted in significant perturbations
of polyunsaturated fatty acids (PUFA) metabolic pathways compared with controls,
reflecting a disruption of hepatic lipid homeostasis. The CE supplementation
effectively restored these Cd-induced metabolic alterations. Furthermore, hepatic
lipidomic profiling revealed that CE supplementation significantly modulated
liver lipid metabolite composition in Cd-exposed mice (Fig. 3a–c,
Supplementary Fig. 1a–d). Quantitative analysis showed significant
reductions in specific phospholipid and TG species in the CE+Cd group relative to
Cd-exposed animals, including phosphatidylethanolamine (PE) species (16:1_22:6,
18:0p_22:5, and 36:7e), phosphatidylcholine (PC) derivatives (18:1_14:0, 36:1e,
44:4e, and 45:2), and triacylglycerol (TG) molecules (6:0_14:3_18:2,
11:0_12:2_18:2, and 12:0e_6:0_22:6) (Fig. 3d–k). Transcriptomic analysis
revealed significant enrichment of the cytochrome P450 pathway in CE-treated mice
compared with Cd-exposed animals (Fig. 3l, Supplementary Fig. 1e–g),
suggesting a key role of Se in regulating PUFA metabolism. Gene expression
profiling demonstrated that Cd exposure significantly upregulated major lipogenic
regulators (SCD1 and Ppar
| Accession | LCd_Mean | LCE_Cd_Mean | FC | FDR |
| PE(18:0p_22:5) | 17450872.6 | 15183624.29 | 1.15 | 0.969985079 |
| PC(18:1_14:0) | 39435557.59 | 32130622.06 | 1.23 | 0.969985079 |
| PC(36:1e) | 302313163 | 179843370.8 | 1.68 | 0.887144456 |
| PC(44:4e) | 4882880.47 | 3840599.59 | 1.27 | 0.887144456 |
| PC(45:2) | 307264.63 | 430622.29 | 0.71 | 0.969985079 |
| TG(6:0_14:3_18:2) | 490932.45 | 363589.98 | 1.35 | 0.969985079 |
| TG(11:0_12:2_18:2) | 13452059.04 | 9598922.68 | 1.4 | 0.887144456 |
| TG(12:0e_6:0_22:6) | 365343327.4 | 280980887 | 1.3 | 0.969985079 |
| Cer(d18:0_24:1) | 8623679.74 | 7392526.2 | 1.17 | 0.969985079 |
| BisMePA(40:7e) | 17450872.6 | 15183624.29 | 1.15 | 0.969985079 |
| WE(22:1_16:0) | 1681799.25 | 1438236.09 | 1.17 | 0.887144456 |
| ChE(38:5) | 11793593.01 | 9583919 | 1.23 | 0.887144456 |
| PG(18:2_20:4) | 6301020.77 | 4319869.14 | 1.46 | 0.887144456 |
| PE(36:7e) | 527141.14 | 565008.1 | 0.93 | 0.969985079 |
| PE(16:1_22:6) | 62629348.27 | 73153444.88 | 0.86 | 0.969985079 |
| dMePE(36:7) | 62629348.27 | 73153444.88 | 0.86 | 0.969985079 |
| Hex1Cer(d20:1_20:4) | 4829663.26 | 5519227 | 0.88 | 0.969985079 |
| PS(20:5_18:2) | 2049286.61 | 2911064.25 | 0.7 | 0.969985079 |
| CL(82:11) | 21702584.65 | 25589340.39 | 0.85 | 0.969985079 |
| SM(d18:1_24:3) | 198290475.5 | 221630231 | 0.89 | 0.969985079 |
| PC(45:2) | 307264.63 | 430622.29 | 0.71 | 0.969985079 |
| MGDG(34:1e) | 1533407.06 | 1823557.65 | 0.84 | 0.887144456 |
| MGMG(34:1) | 1533407.06 | 1823557.65 | 0.84 | 0.887144456 |
PE, Phosphatidylethanolamine; PC, phosphatidylcholine; TG, triacylglycerol; Cer, Ceramide; BisMePA, Bis(methylthio)phenylacetic acid; WE, Wax Ester; ChE, Cholesteryl Ester; PG, Phosphatidylglycerol; dMePE, Dimethylphosphatidylethanolamine; hex1cer, Hexosylceramide; PS, Phosphatidylserine; CL, Cardiolipin; SM, Sphingomyelin; MGDG, Monogalactosyldiacylglycerol; MGMG, Monogalactosylmonoacylglycerol.
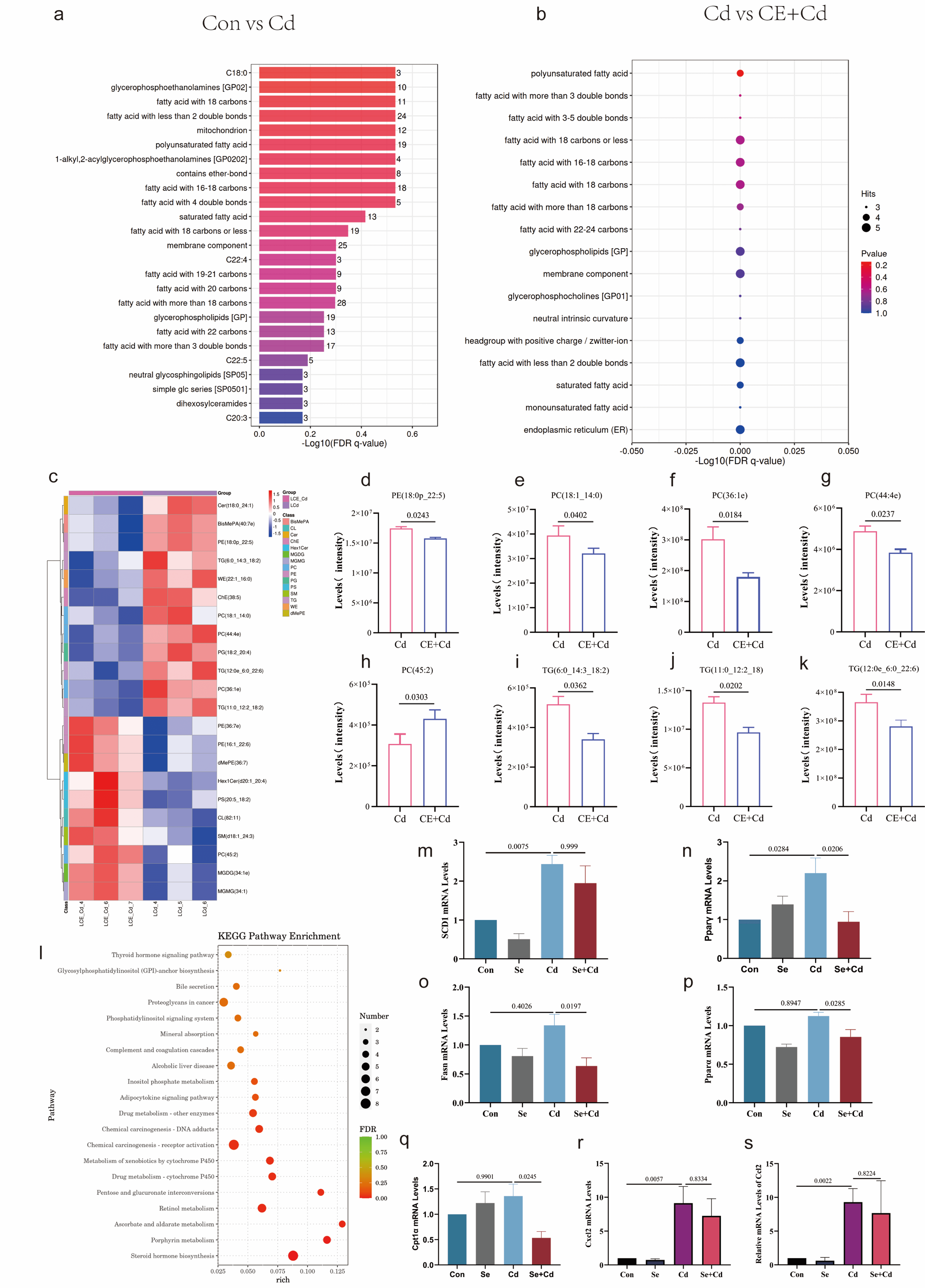 Fig. 3.
Fig. 3.
Se-mediated regulation of lipid metabolism and inflammatory
responses in Cd-exposed liver. Lipid functional enrichment analysis of Control
vs. Cd (a) and Cd vs. CE+Cd (b) groups. A heatmap shows the relative abundance of
significantly altered hepatic lipid metabolites across Control, Cd, and CE+Cd
groups, with each row representing a lipid species and each column representing a
sample. Z-score normalized intensities are indicated by a color scale from blue
(low) to red (high) (c). Quantitative analysis of specific lipid species
identified by lipidomics is presented for PE (d–f), PC (g–i), and TG (j,k) in
the livers of Control, Cd, and CE+Cd groups. KEGG pathway enrichment analysis
based on transcriptomic data from the Cd vs. CE+Cd comparison highlighted
significant enrichment of the cytochrome P450 pathway (l). Hepatic mRNA
expression levels of key genes involved in lipid metabolism, determined by
qRT-PCR, are shown for SCD1 (m), Ppar
This study employed the distinctive biomarker levels reported in the Enshi population, serum selenium (416.977 µg/L) and urinary cadmium (3.848 µg/L), as references for human exposure, allowing reverse calculation of corresponding doses for animal experiments [29, 30]. This approach is consistent with strategies that employ cadmium-based physiologically based pharmacokinetic (PBPK) modeling to establish recommended reference concentrations for diseases such as chronic kidney disease and osteoporosis [31]. Deriving exposure doses from human biomarkers not only demonstrates the feasibility of translating environmental epidemiological data into toxicological studies but also provides a basis for the future development of precise selenium-cadmium combined exposure kinetic models.
Ferroptosis is a novel iron-dependent form of programmed cell death driven by lipid peroxidation [32]. Selenium and its associated selenoproteins are key inhibitors of ferroptosis [33]. Previous study has shown that cadmium can induce ferroptosis through nuclear translocation of the antioxidant transcription factor Nrf2 [34]. In comparison, inhibition of the TXNRD1/Nrf2 pathway, an essential component of mammalian antioxidant defense, via agents such as auranofin enhances Nrf2 activation [35]. Our previous work demonstrated that selenium effectively counteracts ferroptosis induced by RSL3 through targeting TXNRD1 [36]. This study further shows that selenium supplementation (CE) exerts anti-ferroptotic effects by regulating PUFA metabolism. Consistent with previous reports indicating that selenium mitigates doxorubicin-induced ferroptosis by reducing key phospholipids involved in lipid peroxidation, such as PE and PC [24], our data reveal that CE treatment significantly restored cadmium-induced PUFA metabolic disturbances, reversing aberrant changes in specific PEs (e.g., 16:1_22:6, 18:0p_22:5) and PC derivatives.
Further analysis indicates that activation of the PPAR
Different chemical forms of selenium display distinct biological activities
[42]. The major selenium species present in the Se-enriched CE used in this study
were selenocysteine (SeCys2) [43]. Organic selenium generally exhibits
higher bioavailability and tissue retention compared to inorganic forms [44] and
demonstrates greater efficacy in regulating lipid metabolism–related genes, such
as PPAR
Although this study demonstrates selenium’s protective effects against cadmium toxicity via regulation of lipid metabolism and inflammatory responses, several limitations remain in the molecular mechanisms and experimental design. First, the intervention period was four weeks. While significant protective effects were observed, this relatively short duration may have limited the evaluation of long-term outcomes under chronic cadmium exposure. Second, as a water extract, CE represents a complex mixture. Although previous studies indicate that its primary active component is organic selenium, and SeMet was included as a control in this study, other coexisting phytochemicals may exert synergistic effects. To clarify selenium’s specific contribution to CE’s protective effects, future studies should incorporate more refined control groups, including purified SeCys2, selenium-enriched CE, and selenium-depleted CE, to systematically identify the independent and combined actions of each component.
No specific therapeutic agents currently exist for cadmium exposure. Based on the significant antagonistic effects of CE observed here, we plan to implement a human intervention study in cadmium-polluted regions. Participants will receive selenium preparations derived from Cardamine enshiensis, with dynamic monitoring of blood selenium metabolites, cadmium body burden, and hepatic and renal function markers. This study aims to validate the “selenium–cadmium antagonism” mechanism in humans and provide evidence-based strategies for health interventions in populations exposed to environmental heavy metals.
Se-enriched CE effectively mitigates cadmium-induced hepatotoxicity at
environmentally relevant exposure levels. The protective mechanism involves a
dual pathway: CE inhibits overactivation of the PPAR
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
ZW, XC performed the research study. Data analysis was conducted by ZW and XC. The manuscript was written and revised by ZW, XC. XP, KL, CZ and ZZ contributed to data acquisition, participated in experiments, prepared figures and tables, and performed literature searches. They also participated in revising the manuscript. HW, CH designed the research study, contributed to manuscript revision and funding acquisition. All authors participated in editorial changes and read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals of China and relevant institutional guidelines. The experimental protocol was approved by the Animal Ethics Committee of Enshi Tujia and Miao Autonomous Prefecture Central Hospital (Approval No. 202101001).
The authors would like to thank all the reviewers who participated in the review and MJEditor for its linguistic assistance during the preparation of this manuscript.
This work was supported by the Natural Science Foundation of Hubei Province (2025AFD137).
The authors declare no conflict of interest.
During the preparation of this work, the authors used DeepSeek to check spelling and grammar. The scientific content was written entirely independently by the authors, without the use of AI tools. We hope this clarification addresses the issues raised concerning the use of artificial intelligence in our submitted paper. The authors bear full responsibility for the accuracy, completeness, and scientific rigour of the content presented herein.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL50288.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.