



1 Department of Psychiatry, First Faculty of Medicine, Charles University and General University Hospital, 116 36 Prague, Czech Republic
2 Mind-Cell LLC, Baltimore, MD 21230, USA
Abstract
Alzheimer’s disease (AD) is increasingly associated with mitochondrial dysfunction and disrupted metabolism. Thus, the maintenance of nicotinamide adenine dinucleotide (NAD+) homeostasis is proposed as a potential therapeutic strategy. Toward this end, we suggest that AD-related mitochondrial dysfunction might be viewed as a regulatable, redox-dependent vulnerability rather than an inherently degenerative and irreversible process. This perspective advances an evolutionary model in which NAD+-mediated redox systems represent a conserved regulatory axis, and that destabilization of this axis during aging may increase susceptibility to degeneration. Here, we assess the potential of a therapeutic approach that combines this understanding of mitochondrial energy metabolism with results from preclinical studies demonstrating the impact of pharmacologic correction of NAD+ homeostasis (e.g., P7C3-A20) as contextual motivation. We explicitly elevate redox balance, rather than absolute NAD+ abundance, as the mechanistically dominant variable that shapes mitochondrial resilience, inflammatory tone, and neurovascular stability. Accordingly, the key unresolved issue is whether specific physiologic benefits might accrue from increased NAD+ availability per se or rather, the restoration of the NAD+/NADH redox ratio, with important implications for the interpretation of the results of directed metabolic interventions. Within this framework, metabolic failure in AD can be understood as an upstream permissive condition that explains, rather than replaces, canonical amyloid-β and tau-associated pathologies. While extended human lifespan may expose late-life vulnerabilities in otherwise conserved metabolic systems, claims of causal primacy, disease reversibility, and cross-neurodegenerative generalization remain premature, underscoring the need for redox-resolved, genetic, and clinical validation.
Graphical Abstract
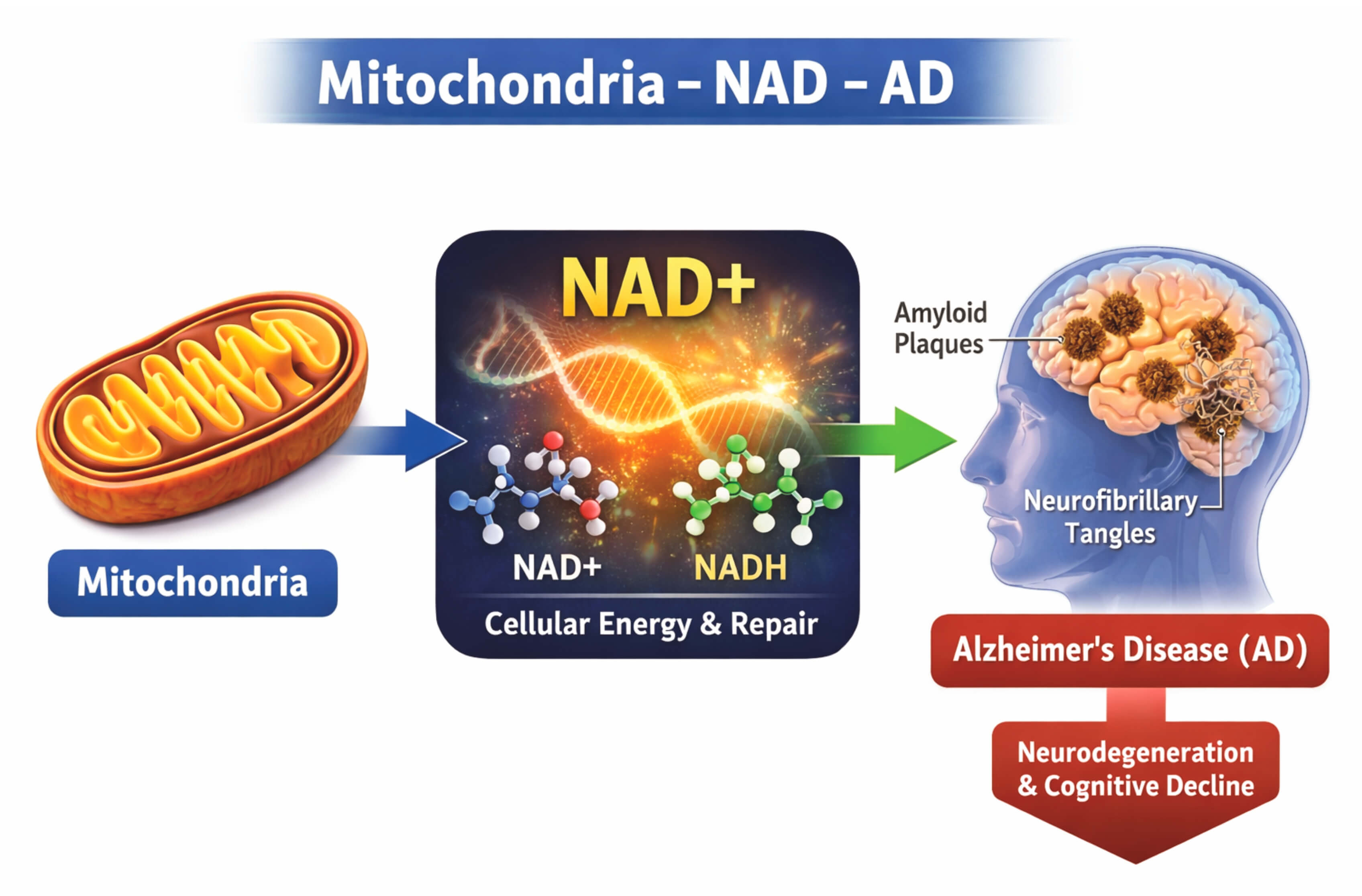
Keywords
- nicotinamide adenine dinucleotide
- mitochondrial dysfunction
- Alzheimer’s disease
- neurodegenerative diseases
- evolution
- neuroinflammatory diseases
- cognition
In this manuscript, we propose a hypothesis based on an evolutionary rationale
that connects Alzheimer’s disease (AD)-associated pathology with the biology of
mitochondria and nicotinamide adenine dinucleotide (NAD+) homeostasis. The
interpretive paradigm presented does not suggest that a decline in NAD+ is
the sole cause of Alzheimer’s disease pathology, but rather proposes that the
decline in NAD+-mediated redox regulation reveals a fundamental metabolic
weakness that may manifest as late-life neurodegenerative disease pathology.
According to this paradigm, mitochondria, which originated from
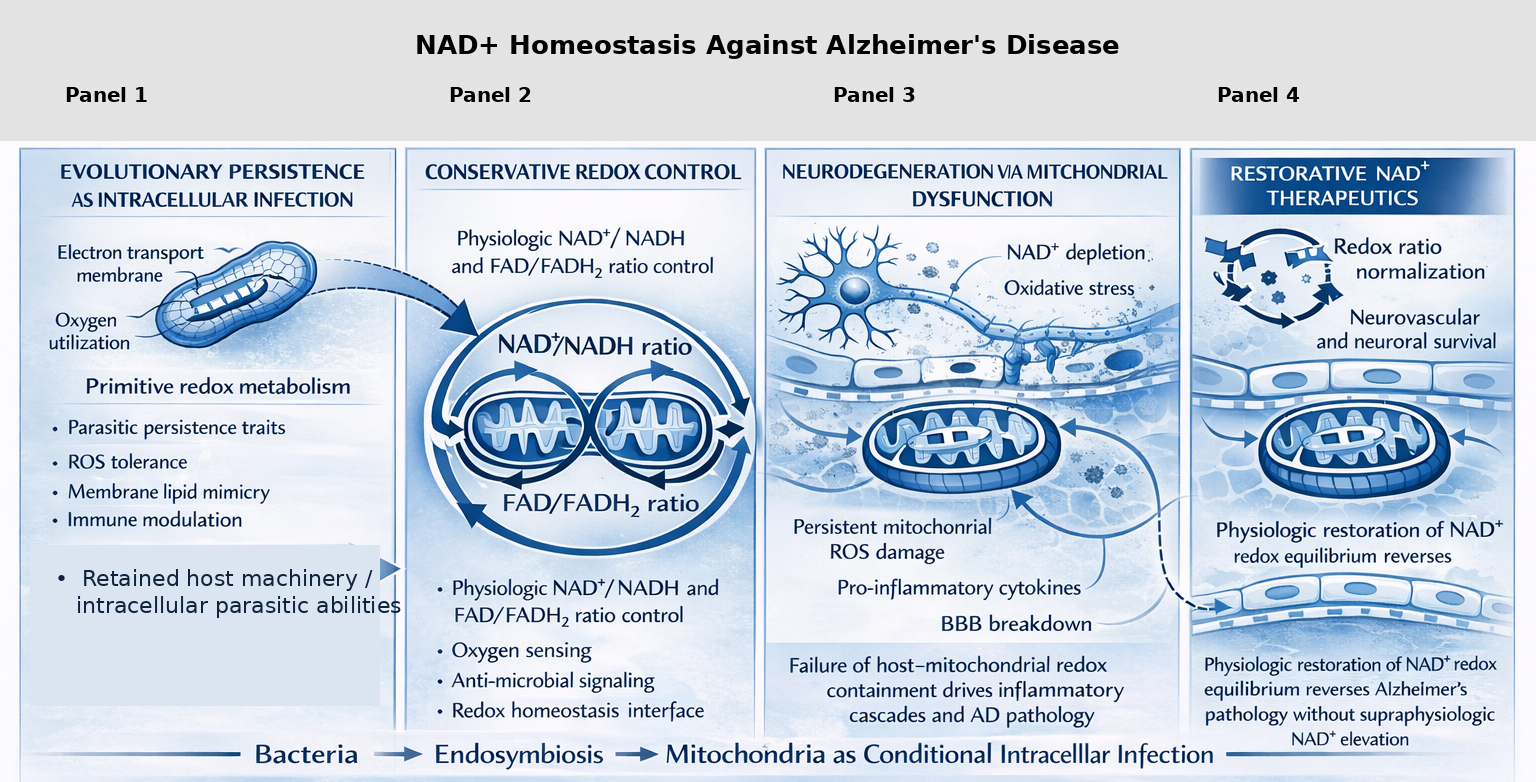 Fig. 1.
Fig. 1.
Nicotinamide adenine dinucleotide (NAD+) homeostasis is an evolutionarily conserved determinant of mitochondrial function, neurovascular integrity, and Alzheimer’s disease (AD) reversibility. The schematic diagram integrates evolutionary, bioenergetic, and pathological approaches that might be used to understand why physiological NAD+ redox balance is important for maintaining mitochondrial and host cell homeostasis and brain resilience. The evolutionary origin of mitochondria from bacteria through endosymbiosis is shown in Panel 1, with special reference to maintaining redox machinery and conditional intracellular signaling capabilities that persist in modern eukaryotic cells. Panel 2 illustrates conservative redox control through tight regulation of the NAD+/NADH and flavin adenine dinucleotide (FAD+)/FADH2 ratios, which are critical for oxygen sensing, antimicrobial signaling, and mitochondrial–host redox homeostasis. Panel 3 shows how failure of host–mitochondrial redox containment leads to NAD+ depletion, persistent mitochondrial reactive oxygen species (ROS) production, proinflammatory cytokine signaling, blood–brain barrier (BBB) breakdown, and progressive neurodegeneration characteristic of AD. Panel 4 highlights physiologic restoration of NAD+ redox equilibrium—without supraphysiologic NAD+ elevation—as a means to normalize redox ratios, preserve neurovascular and neuronal survival, and reverse AD pathology, supporting a model in which mitochondrial dysfunction reflects a reversible breakdown of an ancient host–symbiont redox relationship rather than irreversible cellular failure. Subheading: Ancient bacterial chemistry preserved within mitochondria governs cognitive survival- Mitochondria retain ancestral bacterial redox and bioenergetic systems—particularly NAD+-dependent electron transfer—that continue to regulate neuronal resilience and cognitive function. This figure was generated using ChatGPT (version 5.2) under the direction of the author.
The conceptual model presented here is based on a recent study [5] of
pharmacological restoration of NAD+ homeostasis in response to the small
carbazole-type molecule, P7C3-A20. Specifically, Chaubey and colleagues [5]
demonstrated that administration of P7C3-A20 reversed both neuropathology and
cognitive deficits in advanced A
Our overarching thesis is that the restoration of NAD+ homeostasis, as
opposed to non-specific NAD+ supplementation, might be targeted
therapeutically to reverse the AD-like pathologies by re-establishing the
regulation of mitochondrial function and metabolism. In other words, redox
regulation, as exhibited by the NAD+/NADH ratio, constitutes the primary
mechanistic axis through which mitochondrial restoration operates, with absolute
abundance of NAD+ serving in a secondary or permissive role. This
is particularly important as the non-specific supplementation of NAD+
precursors might lead to supraphysiological levels and deleterious effects, e.g.,
oncogenesis and overly-rapid metabolism [7]. This conceptualization is based on
the broader assumption that the maintenance of NAD+-mediated redox balance
is an evolutionary strategy for maintaining brain integrity in response to
various pathologies of neuroinflammation. The redox/evolutionary mechanism is
positioned as an upstream metabolic regulatory pathway. As such, it does not
replace canonical AD markers; rather, it is proposed as a mechanism that may
permit amyloid-
An understanding of the distinction between the absolute concentration of NAD+ vs the NAD+/NADH ratio (Fig. 1) is a core conceptual pillar of our hypothesis. Whereas total NAD+ levels reflect substrate availability, the NAD+/NADH ratio represents the cellular redox state with a differential impact on metabolic flux and redox-sensitive signaling. A return to physiologic NAD+ homeostasis will presumably target and correct both the NAD+/NADH ratio and the NAD+ concentration; this is because the ratio oxidized NAD+ to reduced NADH is critical for establishing the redox potential, which, in turn, regulates the rate of glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation. Changes to this critical ratio have been shown to have an immediate impact on the efficiency of the mitochondrial electron transport chain, the rate of reactive oxygen species (ROS) production, and the signaling pathways affected by ROS [6, 9, 10]. With respect to evolution, the regulatory role of NAD+ and redox control can be considered one of the first and simplest molecular solutions to fluctuating oxygen availability and microbial pressure, an observation that reinforces its plausibility as a modifiable vulnerability rather than a disease-specific trigger [1].
Ratios of NAD+/NADH in treated and untreated AD models would therefore become crucial in determining if therapeutic responses to P7C3-A20 were dependent on its impact on redox balance or NAD+ substrate utilization (Fig. 1). Currently, it is not clear whether the actions of P7C3-A20 favorably affect NAD+ biosynthesis, NADH oxidation, or a combination of both. While P7C3-A20 may have a differential effect on total NAD+ pools rather than redox ratios, this possibility remains unresolved without compartment- and ratio-resolved measurements. These differences would have significant implications for the potential reversibility of its pharmacologic actions. It is also important to note that reversibility data should be viewed with some caution. While the results presented by Chaubey et al. [5] suggest that metabolic intervention can address late AD-associated pathology in mouse models, they do not prove that NAD+ homeostasis is a causal feature. However, these results do suggest that NAD+ homeostasis may be a therapeutically modifiable vulnerability in AD.
Restoration of NAD+ availability is not a unidirectional intervention. By contrast, this intervention increases the flux through multiple NAD+-consuming enzyme systems with potentially divergent effects on neurodegeneration. These include sirtuins, poly (ADP-ribose) polymerases, CD38, and other ADP-ribosyltransferases [6, 7, 9, 11, 12, 13, 14]. Of particular note, aging-associated upregulation of CD38 links NAD+ depletion directly to neuroinflammatory amplification, raising the concern that nonspecific increases in NAD+ availability could exacerbate rather than resolve inflammatory signaling. Accordingly, any framework that assumes uniform benefit across all NAD+-dependent pathways risks oversimplifying a tightly regulated metabolic network. The Chaubey et al. [5] findings do not resolve whether P7C3-A20 selectively channels NAD+ toward neuroprotective pathways or globally increases NAD+ availability. Enzyme-specific activity measurements will be required to elucidate pathway selectivity and guide rational combination strategies, for example, pairing NAD+ stabilization with CD38 inhibition.
A full understanding of NAD+ homeostasis also requires an understanding of the subcellular compartmentalization of NAD+. This is because NAD+ is generated in the nucleus, cytoplasm, and mitochondria by independently regulated processes; one form cannot be converted to another [15]. From a mitochondria-centric standpoint, specific targeting of NAD+ levels in mitochondria is likely the most relevant mechanism. Consistent with this idea, Chaubey et al. [5] have shown that P7C3-A20 corrects NAD+ balance. This correction protects human brain microvascular endothelial cells from oxidative stress, a phenomenon that is directly related to NAD+ balance and blood-brain barrier integrity, with particular emphasis on nitric oxide dynamics [16].
Regional heterogeneity in the brain, as well as cellular heterogeneity involving neurons, glia, and endothelial cells, introduces an additional level of complexity. Mitochondria are now recognized as “immuno-metabolic organelles” that are involved in the regulation of innate immune responses through the release of ROS, mitochondrial DNA, inflammasomes, and antiviral pathways [8, 17, 18, 19]. NAD+ balance is a critical regulatory step in these processes. Thus, NAD+ is a plausible target that links these processes to one another, again without implying disease specificity.
The concept of “chronic intracellular infection” as it applies to mitochondria requires detailed scrutiny [2, 3]. Current-day mitochondria are well integrated in their role in cellular biology and do not possess any features suggesting infection, for example, replicative autonomy or transmissibility [20]. The use of what is essentially a metaphor highlights the concept of regulatory asymmetry and the role of immune signaling; it does not suggest a literal infection process. The heuristic use of this metaphor helps us to strike a balance between current evolutionary hypotheses and the biological consensus focused on mitochondria as fully integrated intracellular organelles. Of note, mitochondria can exist independently and function in the extracellular environment; these findings suggest a role for a coordinated immune masking process. While the immunologic role of the mitochondria in signaling processes might be considered to be largely pathogenic in nature, this phenomenon actually represents a regulated host response [8]. Nonetheless, use of the infection metaphor highlights the important concept that mitochondrial function requires active regulation to prevent inflammation and metabolic instability. In most cases, the loss of NAD+ redox regulation “unmasks” (as opposed to initiates) inflammation. The corollary to the chronic intracellular infection metaphor highlights neurodegeneration as a failure of regulatory self-control that might be more accurately described as part of an evolving two-billion-year-old process that has recently gone awry.
The framework must also consider why highly-conserved NAD+ systems become vulnerable to dysfunction late in life, despite presumably having been optimized under strong evolutionary constraints. Possible reasons include fitness pressures experienced in early life, extension of human longevity beyond the evolutionary parameters that defined this optimization, or the plasticity of metabolic regulation rather than evolutionary fragility [21]. From this point of view, homeostatic regulation of NAD+ might be considered a flexible regulatory system that can be therapeutically targeted rather than an evolutionary residue of unresolved host-organelle conflict. However, intentional organization of the mitochondrion to accommodate particular processes also serves to obscure its infectious state [2, 3].
Finally, it is premature to extend these results beyond AD-associated pathology. While the disruption of NAD+ homeostasis has a clear impact on universally relevant cellular processes, neurodegenerative disorders have unique regional vulnerabilities and are susceptible to specific pathogenic mechanisms [22]. It is not appropriate to generalize the efficacy of P7C3-A20 or to claim preeminence of NAD+ in neurodegenerative disorders. First and foremost, no one should perceive these results as an endorsement of over-the-counter NAD+ supplements that lack efficacy data and may be risky.
In summary, our integrated framework positions NAD+ homeostasis as an evolutionarily conserved, metabolically central, and therapeutically modifiable axis of vulnerability in AD. The principal strength of our hypothesis lies in reframing mitochondrial dysfunction as dynamic and regulatable rather than an irreversibly degenerative process. However, claims of causal primacy, disease reversibility, and related broad generalizations currently exceed the available evidence. Near-term priorities for this line of research include identification of redox-resolved NAD+/NADH parameters, compartment-specific targeting strategies, and translational biomarkers capable of distinguishing redox restoration from nonspecific elevations in NAD+. Rigorous mechanistic, genetic, compartment-specific, and longitudinal validation will be required before the evolutionary, mechanistic, and therapeutic implications of NAD+-centered interventions can be fully substantiated.
AD, Alzheimer’s disease; FAD, flavin adenine dinucleotide; NAD, nicotinamide adenine dinucleotide.
The author is accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The author declares no conflict of interest. Despite George B. Stefano being an employee of Mind-Cell LLC, the judgments in data interpretation and writing were not influenced by this relationship.
During the preparation of this work, the author used ChatGpt 5.2. to provide informational organization and figure generation based on his direction. The author reviewed and edited the document as needed and takes full responsibility for its content.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.