





 , Xiaolei Zhang 1,†, Tian Tian 1, Yang Li 1, Shukai Qiao 1, Feng He 1, Lina Xing 1,*
, Xiaolei Zhang 1,†, Tian Tian 1, Yang Li 1, Shukai Qiao 1, Feng He 1, Lina Xing 1,*
1 Department of Hematology, The Second Hospital of Hebei Medical University, 050000 Shijiazhuang, Hebei, China
†These authors contributed equally.
Abstract
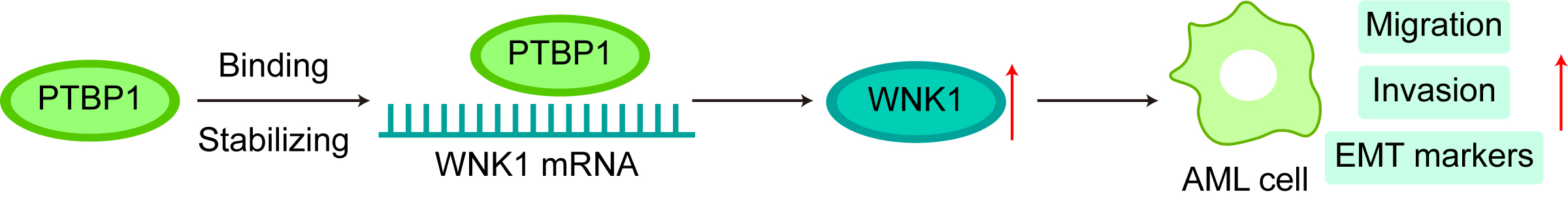
Acute myeloid leukemia (AML) is an aggressive and molecularly diverse hematologic malignancy with unfavorable clinical outcomes and limited options for targeted therapy. This study investigated whether polypyrimidine tract-binding protein 1 (PTBP1), an RNA-binding protein (RBP), affects AML progression by binding to WNK lysine-deficient protein kinase 1 (WNK1).
We first determined the level of WNK1 in AML using the Gene Expression Profiling Interactive Analysis (GEPIA) database and verified it by quantitative reverse transcription polymerase chain reaction (qRT-PCR) and Western blotting (WB) assay. AML cell migration and invasion were analyzed using Transwell assays following WNK1 modulation. Epithelial-to-mesenchymal transition (EMT) marker level was confirmed by WB assay. The influence of WNK1 on the in vivo metastasis of AML was verified via tail vein injection of WNK1-knockdown AML cells into Non-Obese Diabetic/Severe Combined Immunodeficiency (NOD/SCID) mice. Mechanistically, RNA pull-down and RNA immunoprecipitation (RIP) assays were utilized to interpret the relationship between PTBP1 and WNK1 and to determine whether PTBP1 affects AML cell migration and invasion by regulating WNK1, using rescue experiments.
WNK1 was highly expressed in AML. WNK1 inhibition hindered AML cell migration, invasion, and the expression of EMT markers. WNK1 depletion markedly suppressed the metastasis of AML cells in vivo. Mechanistically, PTBP1 directly bound to WNK1 and increased its mRNA stability. Furthermore, PTBP1 facilitated AML cells migration, invasion, and the expression of EMT markers via WNK1.
We demonstrate that PTBP1 promotes AML progression by modulating WNK1. PTBP1 may therefore represent a potential therapeutic target in AML.
Graphical Abstract
Keywords
- polypyrimidine tract-binding protein 1
- WNK lysine deficient protein kinase 1
- acute myeloid leukemia
- cell migration
- cell invasion
- RNA-binding proteins
In Acute myeloid leukemia (AML), immature myeloid blasts undergo clonal outgrowth, leading to their accumulation in the bone marrow (BM) and, in many cases, their appearance in peripheral blood [1, 2, 3]. The disease is characterized by rapid progression, leading to impaired hematopoiesis and multiorgan failure if left untreated [3]. Aberrant cell migration, invasion, and epithelial-to-mesenchymal transition (EMT) marker expression in AML contribute to tumor progression and metastasis [4, 5]. These processes promote disease aggressiveness, leading to poor prognosis and therapeutic resistance [6]. Despite advances in induction chemotherapy and hematopoietic stem cell transplantation, AML is still marked by frequent relapse and poor long-term survival, especially in older patients or those with refractory disease [7]. AML heterogeneity, shaped by intricate mutational landscapes and aberrant signaling, calls for mechanism-based interventions to improve clinical outcomes and therapeutic response [8]. Hence, finding original therapeutic targets is urgent and important.
Recently, polypyrimidine tract-binding protein 1 (PTBP1) has been linked to multiple diseases, especially cancer [9]. For instance, Ni et al. [10] reported that PTBP1 expedites gastric cancer development by modulating c-Myc expression. Wang et al. [11] reported that PTBP1 drives breast cancer progression by regulating PTEN/Akt signaling and autophagy. Luo et al. [12] reported that PTBP1, which is regulated by the lncRNA SFTA1P, plays a driving role in cervical cancer. In addition, several previous studies have highlighted that PTBP1 facilitates the invasion and metastasis of various cancer cells, thus promoting cancer progression [13, 14, 15]. Notably, our previous studies indicated that PTBP1 expression is elevated in AML and tracked with the expression of the proliferative marker Ki67, which could be used as a potential indicator of AML progression [16]. In addition, the binding of deSUMOylated PTBP1 and BECN1 facilitates the proliferation and survival of AML cells by enhancing autophagy, leading to disease progression [17]. However, the influence of PTBP1 on the migration and invasion of AML cells has not been reported.
WNK lysine deficient protein kinase 1 (WNK1) is a serine/threonine protein kinase that plays a critical role in modulating multiple cellular processes [18]. In breast cancer, WNK1 has been linked to tumor growth and progression [19]. In colon cancer, WNK1 plays a cancer-promoting role by promoting angiogenesis [20]. In addition, Jaykumar et al. [19] reported that WNK1 inhibitors can inhibit the migration of breast cancer cells. However, the role of WNK1 in AML is uncertain. Through GEPIA database analysis, we found that WNK1 expression differed across diverse tumors and that its expression was abnormally elevated in AML patients. However, the function of WNK1 in AML metastasis is not understood. In addition, the ENCORI database predicts that PTBP1 may target WNK1.
The objective of this work was to elucidate how the PTBP1/WNK1 axis contributes to AML development. Through a series of experiments, we identified that PTBP1, as an RNA-binding protein (RBP), regulates WNK1 to influence cell invasion, migration, and the expression of EMT markers. Our findings provide a new direction for AML treatment.
In this study, four human AML cell lines—HL-60, THP-1, KG-1, and AML193—were used to cover distinct differentiation stages and molecular subtypes. HL-60 is a type of peripheral blood leukocyte derived from a patient with acute promyelocytic leukemia [21]. Both THP-1 and AML-193 cells exhibit monocytic lineage characteristics and serve as complementary models for monocytic leukemia [22]. KG-1 represents an early myeloblastic leukemia cell line with immature hematopoietic characteristics [23]. Normal human bone marrow stromal cell line HS-5 and AML cell lines (HL-60, AML-193, KG-1, and THP-1) were purchased from ATCC and cultured in RPMI-1640 medium (12633020, Gibco, Grand Island, New York, USA) supplemented with 10% FBS (A5256701, Gibco, Grand Island, New York, USA) and 100 U/mL penicillin/streptomycin (15140122, Gibco, CA, USA). All the cell lines were incubated at 37 °C in a 5% CO2 humidified atmosphere. All the cell lines were authenticated by STR analysis and regularly tested for mycoplasma contamination using PCR-based methods, with all the results being negative. Detailed methods and results are provided in the Supplementary Material.
Gene overexpression vectors (oe-WNK1 and oe-NC) and short hairpin RNA plasmids targeting WNK1 or PTBP1 (sh-WNK1-1/2/3, sh-PTBP1-1/2/3) were obtained from TranSheepBio (Shanghai, China). These sequences are provided in the Supplementary Material. For transfection, AML cells were seeded into 6-well plates and cultured to approximately 70–80% confluence. Cells were transfected with 2 µg of plasmid DNA per well using Lipofectamine 3000 reagent (L3000015, Invitrogen, Carlsbad, CA, USA). Briefly, plasmid DNA was diluted in Opti-MEM medium and mixed with Lipofectamine 3000 at the ratio recommended by the manufacturer. The transfection mixtures were added to the cells in a dropwise manner, followed by incubation at 37 °C under 5% CO2. After 6 h, the medium was replaced with fresh complete medium. Cells were collected at 48 h for subsequent analyses and at 24–48 h for functional assays, including migration and invasion.
Proteins from AML cells and liver tumor nodules were extracted using RIPA buffer (P0013B, Beyotime, Shanghai, China) containing protease inhibitor cocktails. Protein concentrations were measured using a BCA kit (P0012, Beyotime, Shanghai, China). Proteins (30 µg) were resolved on SDS-PAGE and electrotransferred to PVDF membranes (IPVH00010, Merck Millipore, Darmstadt, Germany). Following a 2-h block with 5% nonfat milk, blots were incubated overnight at 4 °C with antibodies against WNK1 (ab316279, 1/1000; Abcam, Cambridge, UK), PTBP1 (ab133734, 1/10,000; Abcam, Cambridge, UK), E-cadherin (ab314063, 1/1000; Abcam, Cambridge, UK), N-cadherin (ET1607-37, 1/1000; HUABIO, Hangzhou, Zhejiang, China), Vimentin (ET1610-39, 1/20,000; HUABIO, Hangzhou, China), Snail (ER1706-22, 1/1000; HUABIO, Hangzhou, China), and GAPDH (R1210-1, 1:5000; HUABIO, Hangzhou, China). After being washed with TBST, the PVDF membranes were incubated with secondary antibodies (ab6721, 1/2000; Abcam, Cambridge, UK) for 1 hour. Protein detection was performed using an ECL kit (ab133406, Abcam, Cambridge, UK).
RNA was prepared from cells using TRIzol (Invitrogen), followed by cDNA synthesis with the High-Capacity reverse transcription kit (4374966, Applied Biosystems, Waltham, MA, USA). qPCR amplification was conducted on the LightCycler 480 II system in the presence of ChamQ Universal SYBR Master Mix (Vazyme). Primers are provided in Table 1. Expression values were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reported as fold changes derived from 2-ΔΔCt method.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
| WNK1 | GCCGTCAGATCCTTAAAGGTC | CCAGTAGGGCCGGTGATAA |
| PTBP1 | CTCCAAGTTCGGCACAGTGTTG | CAGGCGTTGTAGATGTTCTGCC |
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
WNK1, WNK lysine-deficient protein kinase 1; PTBP1, polypyrimidine tract-binding protein 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Transwell assays were utilized to assess the migration and invasion abilities of
AML cells, as previously described with minor modifications [24]. Briefly, cells
were harvested 48 h after transfection and resuspended in serum-free RPMI-1640
medium at a density of 1
The T7 RiboMAXTM Express Large-Scale RNA Production System (P1320, Promega, Madison, WI, USA) was used to generate abundant WNK1. The Pierce™ RNA 3′ End Desthiobiotinylation Kit (20163, Thermo Fisher Scientific, Waltham, MA, USA) was used to biotinylate the 3′ ends of WNK1 RNAs, both sense and antisense, with RNA probes. The mixture was incubated with AML cell lysates, and RNA‒protein interactions were analyzed using the RNA-Protein Pull-Down Kit (20164, Thermo Fisher Scientific, USA).
The RNA-Binding Protein Immunoprecipitation Kit Magna RIPTM (17-700, Merck Millipore, Darmstadt, Germany) was used to perform the RIP assay. Briefly, AML cells were harvested using an anti-PTBP1 antibody. Total RNA was collected using Dynabeads® Protein G (10003D, Thermo Fisher Scientific, USA) and TRIzol. WNK1 level was also determined.
The cells were treated with 10 µg/mL actinomycin D (Act D) once they reached 60–80% confluency. RNA was extracted at 0, 3, and 6 hours post-treatment for quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis.
Male NOD/SCID mice (8 weeks) were procured from Gempharmatech Co. Ltd. Each
mouse was injected with 1
For histology, liver, kidney, and spleen tissues were fixed in 4%
paraformaldehyde (
Spleen, liver, and kidney tissues were fixed in 4% paraformaldehyde, dehydrated in 30% sucrose, embedded in OCT, and cryosectioned at 8 µm. The sections were permeabilized with 0.3% Triton X-100, blocked with 5% BSA, and incubated with anti-CD45 antibody (1:100, CL594-60287; Proteintech, Wuhan, Hubei, China) overnight at 4 °C. After being washed, the sections were incubated with Goat anti-Mouse IgG (H+L) Superclonal™ Secondary Antibody (1:500, A28180; Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Nuclei were counterstained with DAPI, and images were acquired using a fluorescence microscope.
The results are expressed as the mean
According to the GEPIA database, WNK1 is abnormally expressed in multifarious tumor tissues (Fig. 1A), and its level is abnormally increased in AML patients (Fig. 1B). Moreover, we verified its level in AML cell lines compared with that in HS-5 cells, and WNK1 levels were enhanced in AML cell lines (HL-60, AML-193, KG-1, and THP-1 cells) (Fig. 1C,D). These findings suggest that WNK1 may influence AML progression. HL-60 and THP-1 cell lines were chosen for further experiments.
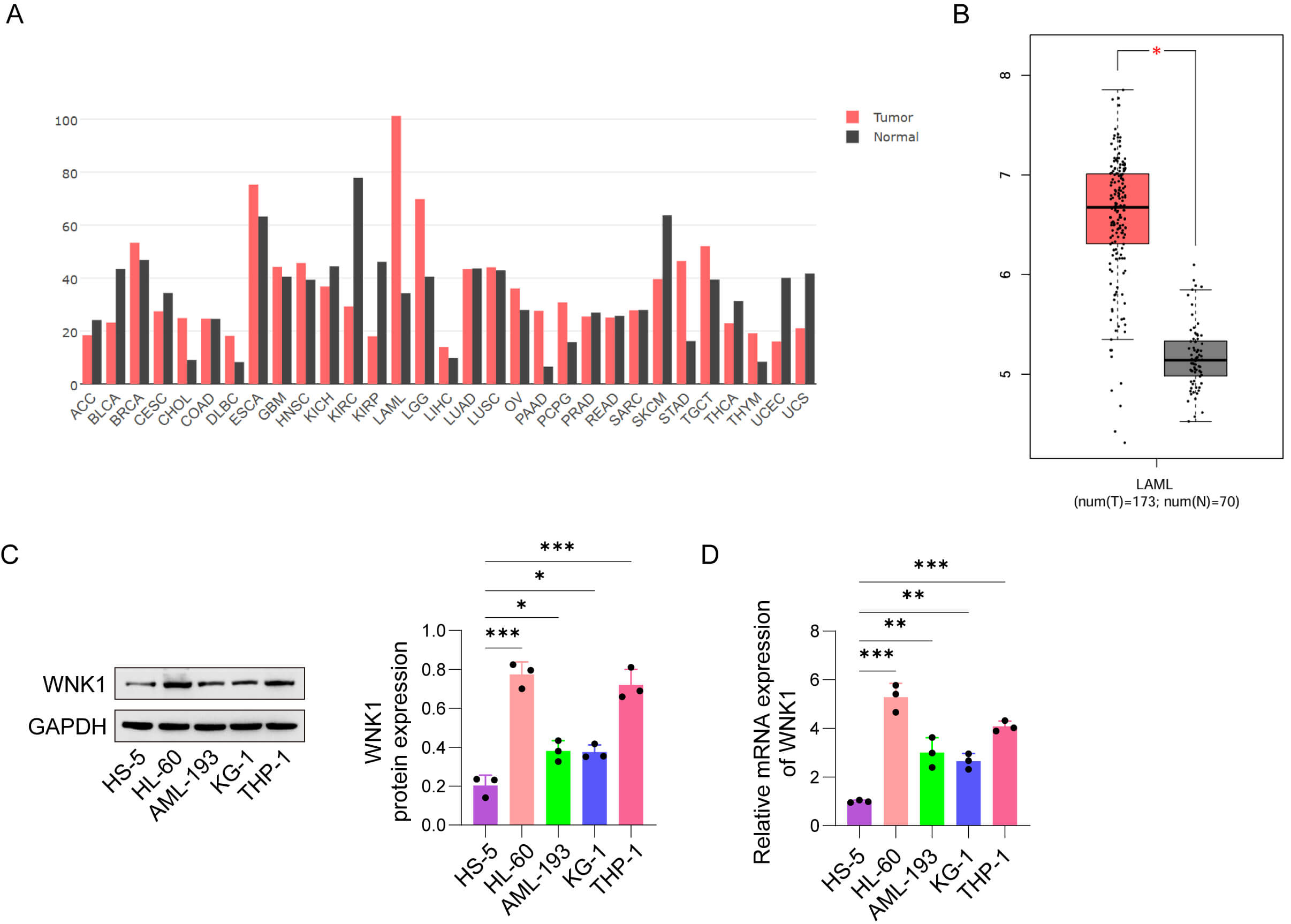 Fig. 1.
Fig. 1.
WNK1 abundance was increased in AML. (A) WNK1 expression in a
variety of tumor tissues was analyzed through the GEPIA database. (B) Comparison
of WNK1 expression levels between AML patients and normal controls on the basis
of GEPIA database analysis (AML, n = 173; normal, n = 70). (C) Representative
western blot images showing WNK1 protein expression in normal human bone marrow
stromal cells (HS-5) and AML cell lines (HL-60, AML-193, KG-1, and THP-1). (D)
Relative mRNA expression levels of WNK1 in HS-5 cells and AML cell lines
determined by qRT‒PCR. The data are presented as the mean
The effects of WNK1 on cell proliferation, migration and EMT markers were investigated by constructing a WNK1 knockdown AML cell model. The knockdown efficiency of WNK1-specific short hairpin RNAs (sh-RNAs) (sh-WNK1-1/2/3) in AML cells was validated, as shown in Fig. 2A,B. Follow-up experiments were performed using sh-WNK1-1, which resulted in the highest knockdown efficiency. As depicted in Fig. 2C,D, knockdown of WNK1 hindered the migration and invasion of HL-60 and THP-1 cells. Besides, E-cadherin level was enhanced and N-cadherin, Vimentin, and Snail expressions were decreased in WNK1-knockdown AML cells (Fig. 2E). In summary, WNK1 inhibition curtailed AML cell invasiveness and limited EMT-associated features.
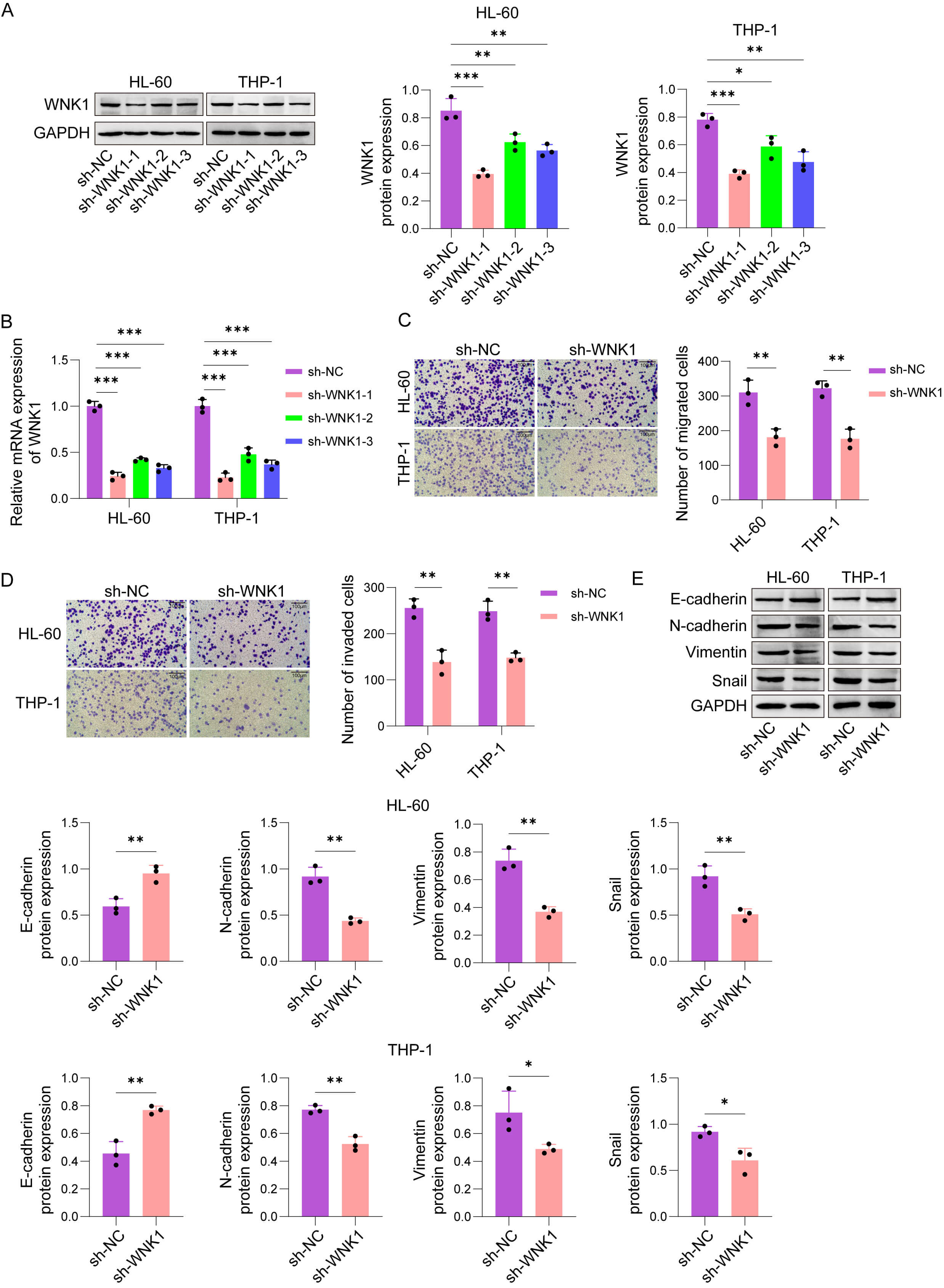 Fig. 2.
Fig. 2.
Knockdown of WNK1 hindered AML cells migration,
invasion, and EMT markers expression. (A) Representative western blot images and
quantitative analysis of WNK1 protein levels in HL-60 and THP-1 cells transfected
with sh-NC or WNK1-specific sh-RNAs (sh-WNK1-1/2/3). (B) Relative mRNA expression
levels of WNK1 in HL-60 and THP-1 cells transfected with sh-NC or sh-WNK1-1/2/3
determined by qRT‒PCR. (C) Representative images and quantification of migrated
HL-60 and THP-1 cells in transwell migration assays after WNK1 knockdown (Scale
bar, 100 µm). (D) Representative images and quantification of
invaded HL-60 and THP-1 cells in Matrigel-coated transwell invasion assays after
WNK1 knockdown (Scale bar, 100 µm). (E) Representative western blot
images and quantitative analysis of EMT-associated marker proteins (E-cadherin,
N-cadherin, Vimentin, and Snail) in HL-60 and THP-1 cells transfected with sh-NC
or sh-WNK1-1. The data are presented as the mean
By tail vein injection of HL-60 cells transfected with sh-WNK1 or sh-NC, we established a xenogeneic AML model in NOD/SCID mice to explore the function of WNK1 in AML metastasis. After 4 weeks, mice transplanted with sh-WNK1-carrying HL-60 cells had significantly reduced liver and spleen weights (Fig. 3A). Besides, the mice transplanted with WNK1-knockdown HL-60 cells displayed lower proportions of human CD45+ cells in the BM (Fig. 3B). The positive signal of human CD45 in the spleen, liver, and kidney was also significantly reduced by WNK1 knockdown. In particular, the spleen, liver, and kidney tissues of the sh-NC group were obviously damaged, which was alleviated by WNK1 knockdown (Fig. 3C). Furthermore, E-cadherin level was elevated and N-cadherin, Vimentin, and Snail expressions were diminished in the liver tumor nodules of the sh-WNK1 group (Fig. 3D). In summary, WNK1 knockdown markedly slowed AML cell metastasis in vivo.
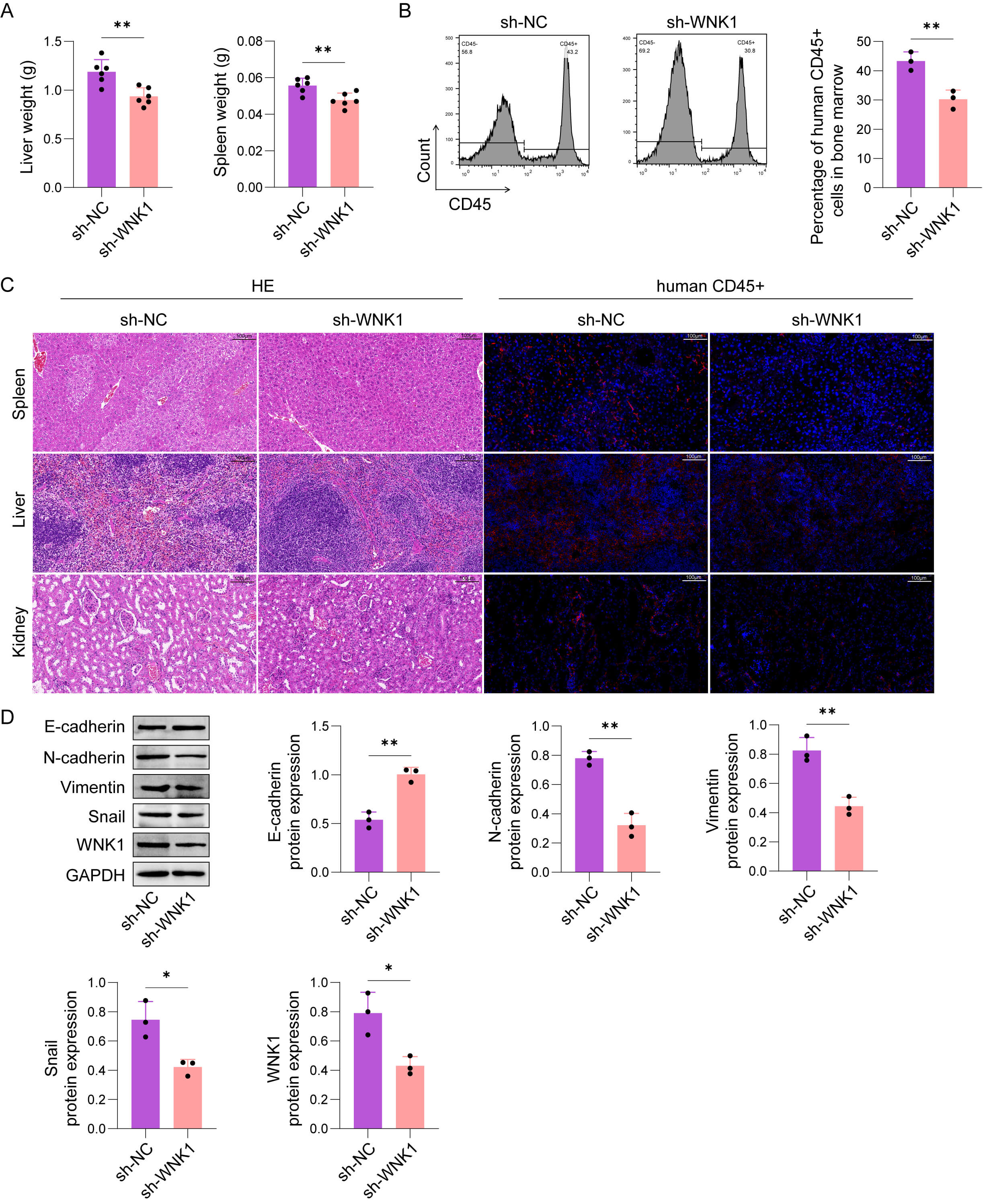 Fig. 3.
Fig. 3.
WNK1 knockdown markedly restrained the metastasis of AML cells
in vivo. The cells were divided into the following two groups: sh-WNK1
or sh-NC. (A) Liver and spleen weights. (B) Statistical analysis of the
percentage of human CD45+ cells in the BM. (C) H&E and human CD45 staining
were used to evaluate the spread of leukemia in the spleen, liver, and kidney
(scale bar, 100 µm). (D) E-cadherin, N-cadherin, Vimentin, and Snail
protein levels in live tissues were measured by western blot assay. The data are
presented as the mean
Previously, we noted that PTBP1 modulates AML disease progression [16, 17], but its effect on the migration and invasion of AML cells was unclear. The ENCORI database predicted that PTBP1 and WNK1 had binding sites (Fig. 4A). Therefore, we probed the relationship between WNK1 and PTBP1. An RNA pull-down assay revealed that PTBP1 was distinctly pulled down by WNK1-sense (Fig. 4B). In addition, an RIP assay confirmed the binding between PTBP1 and WNK1 (Fig. 4C). To determine whether PTBP1 could disturb WNK1 levels in AML cells, we first silenced PTBP1 by using PTBP1-specific sh-RNAs (sh-PTBP1-1/2/3). sh-PTBP1 significantly downregulated PTBP1 levels in AML cells, among which sh-PTBP1-1 had the highest knockdown efficiency and was selected for subsequent studies (Fig. 4D,E). As depicted by the results of the actinomycin D assay, PTBP1 knockdown decreased the stability of WNK1 mRNA (Fig. 4F). Furthermore, WNK1 levels decreased in PTBP1-silenced AML cells (Fig. 4G,H). In sum, PTBP1 directly bound to WNK1 and augmented its mRNA stability.
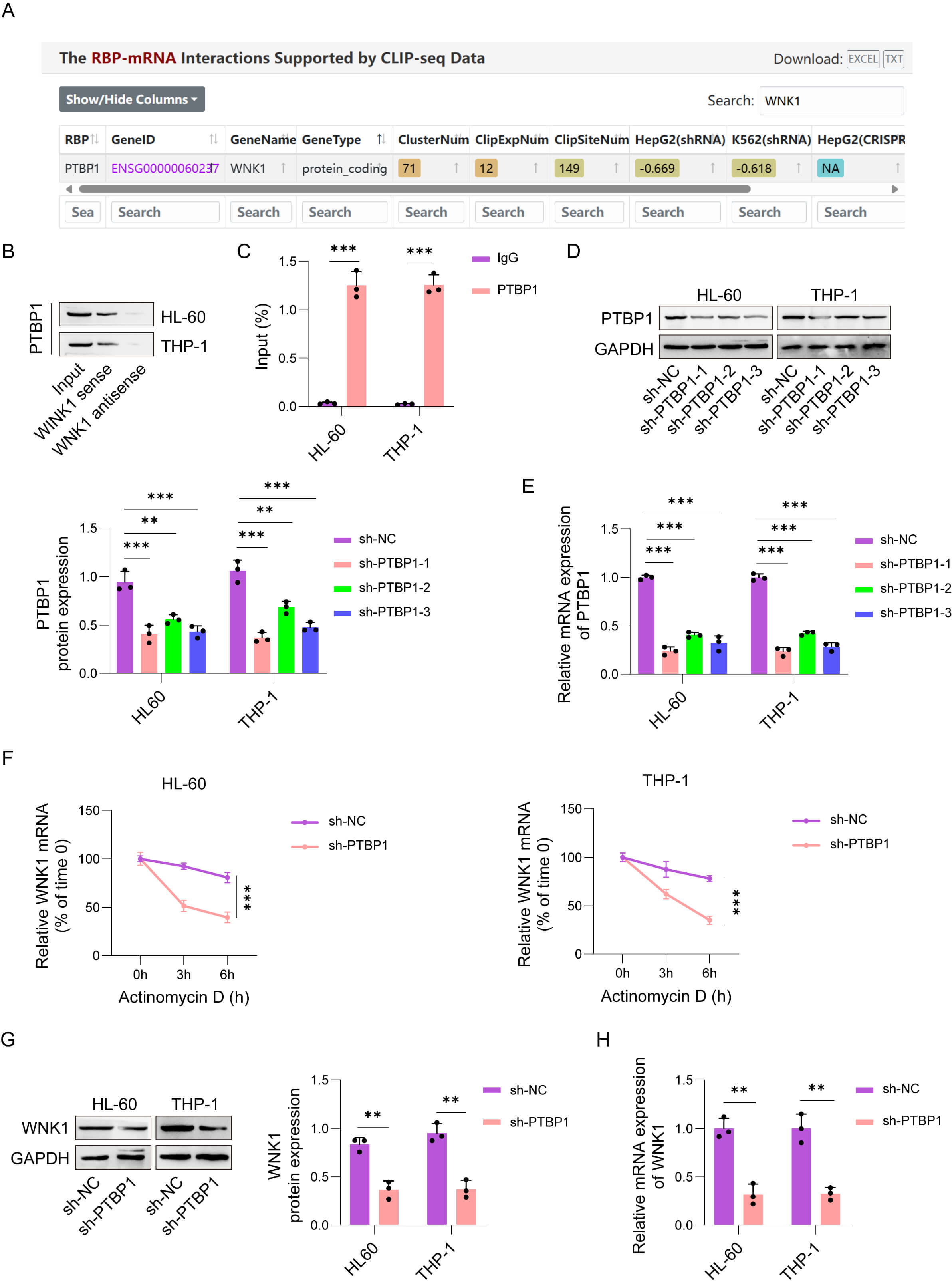 Fig. 4.
Fig. 4.
PTBP1 bound to WNK1 and augmented its mRNA stability. (A) The
ENCORI database predicted that PTBP1 and WNK1 have binding sites. The
relationship between PTBP1 and WNK1 was confirmed by an RNA pull-down assay (B)
and RNA immunoprecipitation (RIP) (C). (D,E) The transfection efficiency of
PTBP1-specific sh-RNAs (sh-PTBP1#1/2/3) into HL-60 and THP-1 cells was validated
by qRT‒PCR and western blot assays. (F) After treating cells with actinomycin D
for 0, 3, and 6 h, WNK1 mRNA stability was analyzed by qPCR. (G,H) WNK1 mRNA and
protein levels were measured by qRT‒PCR and western blot assay, respectively. The
data are presented as the mean
To further explore whether PTBP1 influences AML progression by regulating WNK1, we designed a salvage experiment. WNK1 overexpression (oe-WNK1) increased WNK1 levels in HL-60 and THP-1 cells (Fig. 5A,B). Besides, knockdown of PTBP1 suppressed AML cells migration and invasion, which were abolished by WNK1 overexpression (Fig. 5C,D). As expected, E-cadherin level was augmented and N-cadherin, Vimentin, and Snail expressions were diminished in PTBP1-deficient AML cells, but these changes were reversed by WNK1 expression (Fig. 5E). These results illustrate that PTBP1 facilitates AML cells migration, invasion, and EMT markers expression through WNK1.
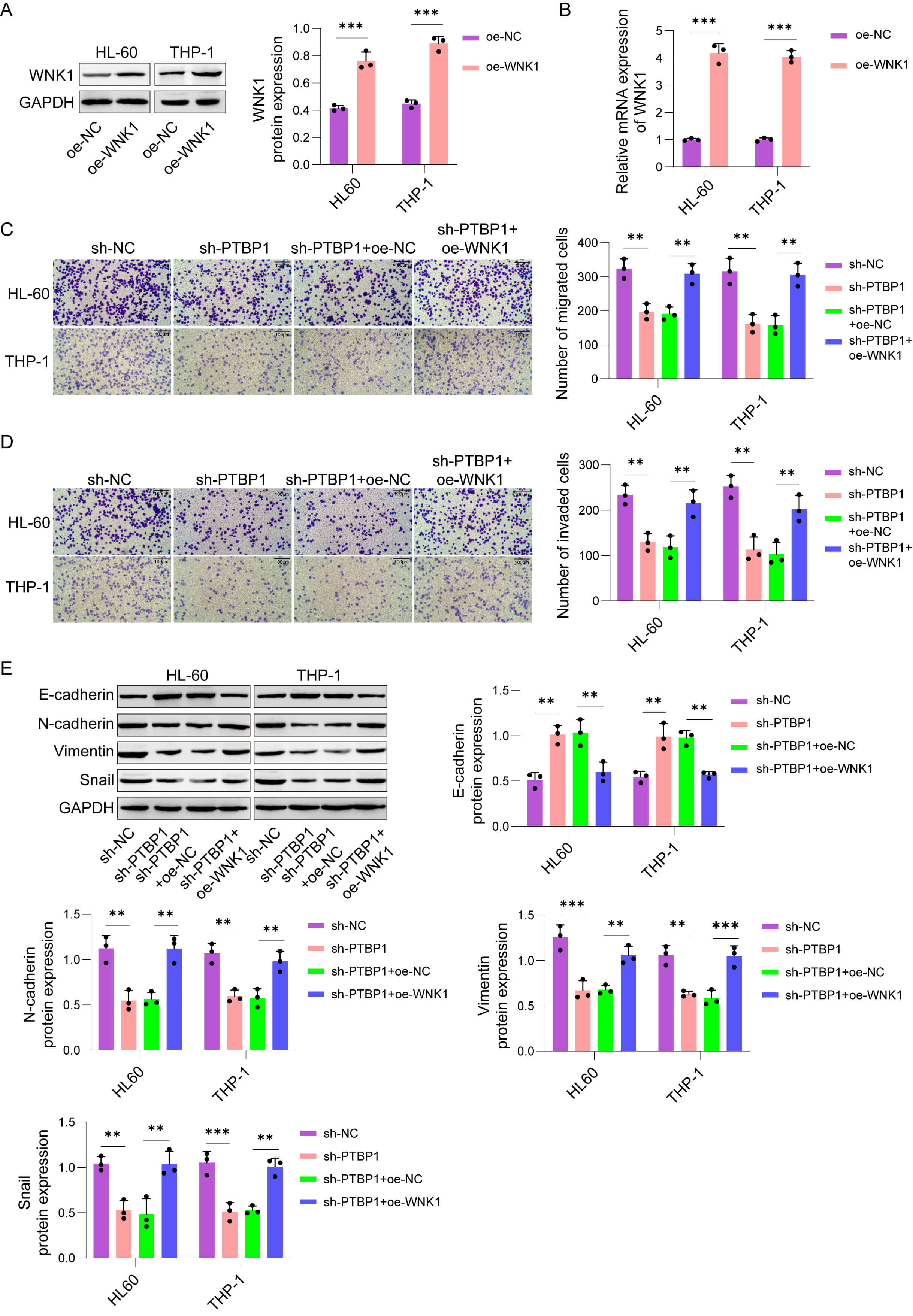 Fig. 5.
Fig. 5.
PTBP1 promoted AML cell migration and invasion while
upregulating EMT-associated markers in a WNK1-dependent manner. (A,B) WNK1 mRNA
and protein levels were measured by qRT‒PCR and western blot assay, respectively,
in WNK1-overexpressing HL-60 and THP-1 cells. The cells were divided into the
following groups: sh-NC, sh-PTBP1, sh-PTBP1+oe-NC, and sh-PTBP1+oe-WNK1. (C,D)
Cell migration and invasion in AML cells (Scale bar, 100 µm). (E)
E-cadherin, N-cadherin, Vimentin, and Snail protein levels were measured by
western blot assay in HL-60 and THP-1 cells. The data are presented as the mean
AML features aberrant expansion of immature precursors and continues to have disappointing long-term outcomes. Despite advances in risk stratification and treatment, overall survival in AML remains poor, underscoring the necessity of clarifying the molecular drivers of disease progression and dissemination.
As a serine/threonine kinase, WNK1 participates in the control of multiple cellular processes [18], including ion homeostasis, proliferation, migration, and invasion [25]. Abnormal expression of WNK1 has been observed in diverse cancer types, and its kinase activity is closely linked to tumor aggressiveness, metastasis, and poor prognosis [26]. Specifically, overexpression of WNK1 in cancers such as renal tumors [27], colorectal [20], and hepatocellular carcinoma [28] has been linked to enhanced tumor cell proliferation, survival, and chemoresistance. As the understanding of the biological functions and mechanisms of WNK1 deepens, targeted therapies against WNK1 may offer novel therapeutic strategies for cancer treatment. Here, we demonstrated that WNK1 is markedly augmented in AML tissues and cell lines and that its depletion significantly suppresses AML cell migration, invasion, and EMT-like phenotypes in vitro, as well as leukemic dissemination in vivo. These findings identify WNK1 as an important driver of AML progression.
RBPs play pivotal roles in post-transcriptional gene regulation [29] and have emerged as key contributors to tumorigenesis [30, 31]. Dysregulation of RBPs can profoundly alter mRNA stability and translation, thereby promoting malignant phenotypes [32]. PTBP1, a well-characterized RBP, has been reported to facilitate cancer progression in multiple tumor types by stabilizing oncogenic transcripts [11, 14]. Consistent with these observations, our previous studies revealed elevated PTBP1 expression in AML and its association with disease progression [16]. However, its involvement in AML cell migration and invasion has not been clarified.
In this study, we provide mechanistic evidence that PTBP1 directly binds to WNK1 mRNA and enhances its stability, thereby increasing WNK1 expression in AML cells. Functional rescue experiments further demonstrated that WNK1 mediates the promigratory and pro-invasive effects of PTBP1, establishing PTBP1 as an upstream regulator of WNK1 in AML. This PTBP1-dependent post-transcriptional regulation of WNK1 represents a previously unrecognized mechanism contributing to AML aggressiveness.
AML is a highly heterogeneous malignancy characterized by diverse molecular subtypes and recurrent genetic mutations. While our study demonstrated a critical functional role of the PTBP1/WNK1 axis in promoting AML cell migration, invasion, and EMT-associated marker expression, several limitations should be noted. We did not systematically explore whether WNK1 expression is correlated with specific AML genetic subtypes. Second, our mechanistic findings require validation in primary patient samples. Third, the in vivo analysis assessed leukemic dissemination but not survival. To address these gaps, future work should (i) leverage annotated clinical cohorts (e.g., TCGA and Beat AML) to define the relationship between WNK1 and AML genetic heterogeneity; (ii) validate the expression and clinical relevance of this axis in patient specimens; and (iii) employ extended survival studies in animal models to determine the therapeutic potential of targeting WNK1.
In summary, our findings revealed that the PTBP1/WNK1 axis is a critical regulator of AML cell migration, invasion, and EMT-like features. By stabilizing WNK1 mRNA, PTBP1 promotes leukemic progression, underscoring the importance of post-transcriptional control in AML pathogenesis. These results not only deepen our understanding of AML biology but also suggest that targeting PTBP1 or WNK1 may represent a promising therapeutic strategy for this aggressive disease.
AML, Acute myeloid leukemia; EMT, Epithelial-to-mesenchymal transition; WNK1, WNK lysine-deficient protein kinase 1; RBP, RNA-binding protein; PTBP1, Polypyrimidine tract-binding protein 1; BM, Bone marrow; sh-RNA, Short Hairpin RNA; qRT-PCR, Quantitative Reverse Transcription Polymerase Chain Reaction; WB, Western blot; H&E, Hematoxylin-eosin; RIP, RNA Immunoprecipitation.
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
LX and XQ guaranteed the integrity of the entire study. LX, XQ and SQ designed the study. LX and XQ designed the literature research. LX, YL and SQ defined the intellectual content. XQ, XZ and LX performed the experiment. TT and YL collected the data. XZ and FH analyzed the data. XQ and XZ wrote the main manuscript and prepared figures. All authors reviewed the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal study was approved by the Animal Ethics Committee of the Second Hospital of Hebei Medical University (2025-AE260) and conducted in accordance with the ARRIVE guidelines and the 3R principle (Replacement, Reduction, Refinement).
Not applicable.
This work was supported by the S&T Program of Hebei (No. 246Z7729G, No. H2023206910).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47982.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.