











 , Feihuang Han 1,2,5,†, Zheng Qiao 3,4,6, Sheng Yuan 3,4,6, Xiaohui Bian 3,4,6, Bin Zhang 1,2,5, Kefei Dou 3,4,6,*
, Feihuang Han 1,2,5,†, Zheng Qiao 3,4,6, Sheng Yuan 3,4,6, Xiaohui Bian 3,4,6, Bin Zhang 1,2,5, Kefei Dou 3,4,6,* , Dunliang Ma 1,2,5,*
, Dunliang Ma 1,2,5,*1 Department of Cardiology, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Southern Medical University, 510080 Guangzhou, Guangdong, China
2 Guangdong Provincial Key Laboratory of Clinical Pharmacology, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Southern Medical University, 510080 Guangzhou, Guangdong, China
3 Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, 100037 Beijing, China
4 Coronary Heart Disease Center, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, 100037 Beijing, China
5 Medical Research Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Southern Medical University, 510080 Guangzhou, Guangdong, China
6 Cardiometabolic Medicine Center, National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, 100037 Beijing, China
†These authors contributed equally.
Abstract
Atherosclerosis is a chronic inflammatory disease characterized by lipid-driven immune dysregulation. Argininosuccinate synthase 1 (ASS1) has been implicated in macrophage inflammation, yet its precise mechanistic role in foam cell-mediated vascular injury during atherosclerosis remains unclear. This study investigates whether ASS1 promotes disease progression via the NLRP3/IL-33/ST2 axis.
An in vitro foam cell model was established using phorbol 12-myristate 13-acetate (PMA)-differentiated U937 macrophages treated with oxidized low-density lipoprotein (ox-LDL). The role of ASS1 was assessed via knockdown (si-ASS1) and overexpression (ASS1 overexpression) plasmids. Co-culture systems with human umbilical vein endothelial cells (HUVECs) and human aortic vascular smooth muscle cells (HAVSMCs) were used to evaluate endothelial apoptosis and VSMC proliferation/migration. In vivo, atherosclerosis was induced in apolipoprotein E‑deficient (ApoE)-deficient mice via a 12-week high-fat diet, and ASS1 expression was modulated using AAV9 vectors. Molecular analyses included ROS detection, enzyme-linked immunosorbent assay (ELISA), qPCR, western blot, and immunofluorescence. Plaque burden was assessed via Oil Red O staining.
Ox-LDL treatment significantly upregulated ASS1 expression in U937-derived foam cells. ASS1 overexpression enhanced intracellular ROS production, NLRP3 inflammasome activation, STAT3 phosphorylation, and IL-33 secretion. These effects were reversed by ASS1 knockdown. Rescue experiments demonstrated that STAT3 is required for ASS1-mediated NLRP3 activation and IL-33 upregulation. ASS1 altered IL-33 receptor ST2 signaling by increasing the soluble decoy isoform (sST2) and decreasing the membrane-bound signaling isoform (ST2L). In co-culture, ASS1-overexpressing foam cells promoted HUVEC apoptosis (via mitochondrial pathway) and HAVSMC proliferation, migration, and dedifferentiation. NLRP3 overexpression alone mimicked the pro-inflammatory effects of ASS1 and reversed the anti-inflammatory effects of ASS1 knockdown. In vivo, ASS1 knockdown in ApoE-/- mice reduced plaque lipid deposition, serum levels of IL-33 and IL-1β, and vascular expression of NLRP3 and p-STAT3, while ASS1 overexpression exacerbated these parameters.
ASS1 drives atherosclerosis by activating the STAT3/NLRP3 inflammasome axis, shifting the IL-33/ST2 balance toward a pro-inflammatory state, and amplifying foam cell–mediated endothelial injury and smooth muscle cell dysfunction. Targeting ASS1 may offer a novel therapeutic strategy for inflammatory vascular disease.
Keywords
- atherosclerosis
- ASS1
- NLRP3 inflammasome
- IL-33
- ST2
- foam cells
- endothelial cells
- vascular smooth muscle cells
Atherosclerosis (AS) is a chronic inflammatory disease characterized by the gradual accumulation of cholesterol within arterial walls, which triggers a persistent and unresolved immune response [1]. The onset of AS involves endothelial dysfunction, initiating a cascade of events that includes the release of low‑density lipoproteins (LDLs), infiltration of macrophages, and activation of vascular smooth muscle cells (VSMCs). These processes lead to foam cell formation and extracellular matrix remodeling, thereby driving disease progression [2]. Foam cells—a hallmark of all AS stages—originate from monocytes that infiltrate developing atheromas and subsequently internalize modified LDL [3]. Reactive oxygen species (ROS) produced by foam cells initiate inflammatory responses that recruit additional macrophages into the vascular intima, further promoting the development of fatty streaks and exacerbating AS [4, 5]. A deeper understanding of the mechanisms regulating foam cell‑mediated inflammation may therefore reveal novel therapeutic targets for AS.
The pyrin domain‑containing protein 3 (NLRP3) inflammasome—composed of NLRP3,
caspase‑1, and apoptosis‑associated speck‑like protein containing a CARD
(ASC)—represents a form of inflammatory cell death implicated in the
pathogenesis of AS. Activation of the NLRP3 inflammasome can be triggered by
specific molecules, including lipopolysaccharide as a pathogen‑associated
molecular pattern (PAMP), as well as oxidized LDL (ox‑LDL) and cholesterol
crystals acting as damage‑associated molecular patterns (DAMPs) [6, 7]. At the
molecular level, dysregulated NLRP3 inflammasome activation elevates the levels
of pro‑inflammatory cytokines, namely interleukin (IL)‑1
In recent years, argininosuccinate synthase 1 (ASS1) has gained attention for its role in catalyzing the penultimate step of arginine biosynthesis. Together with argininosuccinate lyase (ASL), ASS1 facilitates arginine synthesis from aspartate, citrulline, and ATP in various tissues and cells [15, 16]. ASS1 is known to regulate inflammatory macrophage activation and antibacterial defense by depleting cellular citrulline [17]. Elevated arginase levels and activity have been observed in several cardiovascular disorders, including AS [18, 19, 20]. Moreover, a recent study linked ASS1 expression to the anti‑inflammatory effects of canagliflozin, which subsequently inhibited AS progression [19]. During early AS, ox‑LDL accumulation induces macrophage dysfunction and foam cell formation [21]. Emerging evidence indicates that ox‑LDL upregulates ASS1 expression in macrophages, promoting ROS generation and pro‑inflammatory cytokine secretion [17, 22]. ASS1 has also been shown to regulate NLRP3 inflammasome activation and influence IL‑33 expression. Nevertheless, the precise mechanistic relationship between ASS1 and the NLRP3/IL‑33/ST2 axis in foam cell‑driven endothelial injury and VSMC dysfunction remains poorly understood.
This study investigates the hypothesis that ASS1 exacerbates AS by activating
the STAT3/NLRP3 inflammasome axis, disrupting the IL‑33/ST2 balance, and
amplifying foam cell‑mediated inflammation. We used PMA‑differentiated U937
macrophages exposed to ox‑LDL to generate foam cells and examined the role of
ASS1 in regulating ROS, NLRP3 activation, and IL‑33 secretion. Co‑culture systems
with human umbilical vein endothelial cells (HUVECs) and human aortic vascular
smooth muscle cells (HAVSMCs) were employed to assess the effects of ASS1 on
endothelial apoptosis and smooth muscle cell proliferation/migration.
Furthermore, an in vivo atherosclerosis model was established using
apolipoprotein E‑deficient (ApoE-/-) mice fed a high‑fat diet for 12 weeks,
with ASS1 expression modulated via AAV9 vectors. Lipid accumulation was evaluated
by Oil Red O staining, and key inflammatory markers—including NLRP3,
phosphorylated STAT3 (p‑STAT3), IL‑33, and IL‑1
The U937 human monocytic cell line (PCS-100-011) was procured from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS), and incubated at 37 °C under a humidified atmosphere containing 5% CO2. To differentiate U937 cells into foam cells, they were first stimulated with 100 nM phorbol 12-myristate 13-acetate (PMA) for 48 h, followed by treatment with 50 µg/mL oxidized low-density lipoprotein (ox-LDL; Sigma-Aldrich, St. Louis, MO, USA) for an additional 48 h.
Small interfering RNA targeting ASS1 (si-ASS1: 5′-UCAAUGAACACCUUUUUGGCC-3′), ASS1 overexpression plasmid (pCDNA3.0-ASS1), NLRP3 overexpression plasmid (pCDNA3.0-NLRP3), and their corresponding negative controls (scrambled siRNA [si-NC] for knockdown and empty vectors for overexpression) were commercially obtained from GenePharma (Shanghai, China). Foam cells were transfected with the indicated plasmids or siRNAs, either alone or in combination with their respective controls, using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA). To further investigate the functional link between ASS1 and STAT3 signaling, we performed rescue experiments using STAT3-specific small interfering RNA (si-STAT3) and a constitutively active STAT3 mutant plasmid (STAT3-CA). The si-STAT3 (target sequence: 5′-GAACCAUGUGGAUAUCAUA-3′) and STAT3-CA overexpression plasmid (pCDNA3.1-STAT3-CA) were purchased from GenePharma (Shanghai, China). U937 foam cells were co-transfected with either si-STAT3 or STAT3-CA along with ASS1 overexpression or knockdown (si-ASS1) plasmids using Lipofectamine 2000 according to the manufacturer’s instructions. Cells were harvested 48 hours post-transfection for subsequent analyses.
Intracellular reactive oxygen species (ROS) levels were measured using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA; Sigma-Aldrich). Cells were incubated with 10 µM DCFH-DA for 30 min at 37 °C, washed thoroughly, and analyzed immediately on a FACScan flow cytometer (Becton-Dickinson, Mountain View, CA, USA). Data were processed with CellQuest software (version 4.1; BD Biosciences, Franklin Lakes, NJ, USA) and presented as fluorescence intensity histograms.
Total RNA was isolated from foam cells using Trizol reagent from Invitrogen
(Carlsbad, CA, USA). Subsequently, cDNA synthesis was performed using the M-MLV
reverse transcriptase kit from TaKaRa Co. (Dalian, Shandong, China). Quantitative
real-time PCR amplification was conducted on an ABI 7500 Real-Time PCR System by
Applied Biosystems. The amplification reaction was performed using the
1
ASS1 forward: 5′-TGAAATTTGCTGAGCTGGTG-3′ and ASS1 reverse: 5′-ATGTACACCTGGCCCTTGAG-3′; ST2L (membrane-bound isoform) forward: 5′-CCTGGTGAAGTTCAACAGCA-3′ and reverse: 5′-TGTAGTAGGCGATGCCACCT-3′;
sST2 (soluble isoform) forward: 5′-GCTCCAGGAACTCCAACCTC-3′ and reverse: 5′-AGCTCCAGGTACATCGGTGT-3′; GAPDH forward: 5′-CAGCCTCAAGATCATCAGCA-3′ and GAPDH reverse: 5′-TGTGGTCATGAGTCCTTCCA-3′.
Human umbilical vein endothelial cells (HUVECs) were obtained from the ATCC
(Rockville, MD, USA), while the Human aortic vascular smooth muscle cells
(HAVSMCs) were purchased from the Institute of Biochemistry and Cell Biology
(Shanghai, China). Both HUVECs and HAVSMCs were cultured in RPMI-1640 medium
supplemented with 10% heat-inactivated FBS (HyClone, Logan, UT, USA), 1%
penicillin and 1% streptomycin. To establish an indirect co-culture system, a
Transwell chamber comprising a culture insert with a 10-µm thick
porous membrane featuring 0.4-µm pores was used. In this system,
foam cells (1
Apoptosis was quantified by flow cytometry using an Annexin V-FITC/PI Apoptosis Detection Kit (Cat# KGA108, KeyGen Biotech, Nanjing, Jiangsu, China) according to the manufacturer’s protocol. Briefly, cells were harvested with trypsin-EDTA, washed twice with PBS, and stained with FITC-labeled Annexin V (1 µg/mL) and propidium iodide (PI, 10 µg/mL) for 20 min in the dark. Samples were then analyzed on a FACScan flow cytometer (Beckman Coulter, Brea, CA, USA).
The expression of ASS1 and NLRP3 in foam cells, and of the endothelial marker CD34 in HUVECs, was examined by immunofluorescence. Cells were seeded in sterile Millicell® EZ-Slide eight-well glass plates (PEZGS0816; Millipore, MA, USA). After reaching appropriate confluency, they were rinsed with PBS, fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.1% Triton X-100 for 30 min. Non-specific binding was blocked with 3% bovine serum albumin (BSA) for 1 h at room temperature. Cells were then incubated overnight at 4 °C with primary antibodies diluted as follows: anti-ASS1 (ab172730, 1:100; Abcam, Cambridge, UK) and anti-NLRP3 (ab270449, 1:100; Abcam) for foam cells, or anti-CD34 (ab81289, 1:200; Abcam) for HUVECs. After washing, samples were incubated for 2 h with a FITC-conjugated secondary antibody (BA1105, Boster, Wuhan, Hubei, China). Nuclei were counterstained with DAPI (10 µg/mL) for 5 min in the dark. Following final PBS washes, fluorescence images were acquired using a fluorescence microscope (Leica, Wetzlar, Germany).
Cell viability of HAVSMCs was assessed using the Cell Counting Kit-8 (HY-K0301, CCK-8; MedChem Express, Monmouth Junction, NJ, USA). Cells from different experimental groups were seeded in 96-well plates. After the respective treatments, 10 µL of CCK-8 solution was added to each well, followed by incubation at 37 °C for 2 h. Absorbance was then measured at 450 nm using a microplate reader.
Cell proliferation was evaluated using the Cell-Light EdU
Apollo®567 In Vitro Imaging Kit (C10371-1, RiboBio,
Guangzhou, Guangdong, China) according to the manufacturer’s instructions.
HAVSMCs in logarithmic growth phase were seeded into 96-well plates at a density
of 4
Cell migration and invasion assays were performed using Transwell® chambers (Corning Life Sciences, Corning, NY, USA). For migration, uncoated inserts were used; for invasion, inserts pre-coated with Matrigel® matrix were employed. HAVSMCs were suspended in 100 µL of serum-free RPMI-1640 medium and seeded into the upper chamber. The lower compartment was filled with 500 µL of complete medium containing 10% FBS to serve as a chemoattractant. After incubation for 24 h at 37 °C under 5% CO2, cells that had migrated or invaded to the lower surface of the membrane were fixed with methanol, stained with 0.1% crystal violet for 35 min, and counted in five randomly selected fields per insert under a light microscope.
Total cellular protein was extracted using RIPA lysis buffer (Pierce, Thermo
Fisher Scientific), and protein concentration was determined by Bradford assay
(Bio-Rad, Hercules, CA, USA). Protein samples (30 µg per lane) were
separated on 12% SDS-PAGE gels and transferred onto PVDF membranes. After
blocking with 5% non-fat milk for 1 h at room temperature, membranes were
incubated overnight at 4 °C with the following primary antibodies (all
from Abcam, unless otherwise noted): anti-ASS1 (ab172730), anti-ICAM-1
(ab282575), anti-VCAM-1 (ab134047), anti-STAT3 (ab68153), anti-phospho-STAT3
(ab267373), anti-ST2 (total; ab194113), anti-sST2 (soluble isoform; ab272372),
anti-SMHC (ab133567), anti-CNN1 (ab46794), anti-
IL-33 levels in foam cell supernatants and IL-1
Apoptotic endothelial cells were detected by TUNEL staining using a fluorescence
FITC kit (12156792910, Roche, Indianapolis, IN, USA) according to the
manufacturer’s instructions. HUVECs grown on coverslips were fixed with 4%
paraformaldehyde and permeabilized with 0.1% Triton X-100. After washing, cells
were incubated with the TUNEL reaction mixture for 1 h at 37 °C in the
dark. Following three washes with PBS, nuclei were counterstained with DAPI
(10 µg/mL; Solarbio, Beijing, China). Images were captured using a
fluorescence microscope (Olympus BX51, Tokyo, Japan) at 200
Male apolipoprotein E-deficient (ApoE-/-) mice were obtained from the Laboratory Animal Center of Fuwai Hospital, National Center for Cardiovascular Diseases. All animal procedures were approved by the Institutional Animal Care and Use Committee of Peking Union Medical College (Approval No. 0105-1-6-ZX(X)-4). Mice were housed under specific pathogen-free conditions with a 12 h light/dark cycle and free access to food and water. Atherosclerosis was induced by feeding 8-week-old mice a high-fat diet (21% fat, 0.15% cholesterol, 3.2% cholate) for 12 weeks. Body weight and food intake were recorded weekly.
After the 12-week dietary intervention (high-fat diet #D12079B, Research Diets Inc., New Brunswick, NJ, USA), mice were euthanized in strict accordance with institutional animal care guidelines. Euthanasia was performed via intraperitoneal injection of sodium pentobarbital (Euthasol®) at a dose of 200 mg kg-1 body weight. The stock solution (39% w/v) was diluted 1:10 with 0.9% saline to obtain a 3.9% w/v working solution. Prior to injection, mice were gently restrained to minimize stress. Depth of anesthesia was confirmed by loss of pedal reflex and cessation of spontaneous respiration. Following this confirmation, euthanasia was carried out as a result of the administered pentobarbital overdose, leading to cardiac arrest. Cardiac arrest was verified by thoracotomy within 5 min of respiratory cessation. Cervical dislocation was performed immediately after cardiac arrest as a secondary physical method to ensure death. All procedures were conducted by trained personnel between 08:00 and 11:00 to minimize circadian influences. Tissues were subsequently harvested for analysis.
To modulate mouse ASS1 (NCBI reference: NM_007494.3) expression in vivo, adeno-associated virus serotype 9 (AAV9) vectors were constructed for overexpression and knockdown. For overexpression, the full-length ASS1 coding sequence was subcloned into the pcDNA3.1 plasmid (Invitrogen, Carlsbad, CA, USA) and then inserted into the AAV9 backbone plasmid pAV-F4/80 (Weizhen Biotechnology Co., Jinan, Shandong, China). For knockdown, potential shRNA target sites were screened by co-transfecting the pcDNA3.1-ASS1 plasmid and the pAV-CAG-GFP-mir30 shRNA library plasmid (Boshang Biotechnology Co., Jinan, Shandong, China) into HEK-293 cells. The most efficient shRNA sequence was subsequently cloned into the pAV-F4/80-GFP-mir30 shRNA vector (Wuyue Biotechnology Co., Jinan, Shandong, China) and packaged into AAV9 particles. Control vectors included AAV9 expressing a scrambled shRNA (for knockdown experiments) and empty pAV-F4/80 (for overexpression experiments).
Mice were randomly assigned to five groups: (1) wild-type sham, (2)
atherosclerosis model, (3) negative control (empty vector), (4) ASS1 knockdown,
and (5) ASS1 overexpression. After 12 weeks of a high-fat diet, mice received an
intravenous injection of the respective AAV9 vector (1
Plaque lipid content was evaluated by Oil Red O staining. Tissue samples from
the transverse cardiac section (TCS) and aortic arch (AAC) were fixed in 10%
neutral-buffered formalin for 24 h at room temperature, embedded in paraffin, and
sectioned at 5 µm. Sections were deparaffinized in xylene, rehydrated
through a graded ethanol series (100%
Sections were stained with Oil Red O working solution (prepared by dissolving Oil Red O powder [Sigma-Aldrich, Cat. No. O0625] in isopropanol and diluting with distilled water at a 3:2 ratio) for 15 min at room temperature. After rinsing with distilled water, nuclei were counterstained with hematoxylin (Merck, Cat. No. 109297) for 1 min, differentiated in 70% ethanol, and mounted with neutral mounting medium. Lipid droplets were visualized as red areas under a light microscope.
The Oil Red O-positive area was quantified as a percentage of total plaque area using image-analysis software. For each mouse, three non-consecutive sections were analyzed, and the mean value was used for statistical comparisons (n = 6 mice per group).
Data are presented as mean
To examine the role of ASS1 in atherosclerosis, foam cells were generated by treating U937 human monocytes with phorbol 12-myristate 13-acetate (PMA) and oxidized low-density lipoprotein (ox-LDL). ELISA showed that PMA + ox-LDL treatment significantly increased IL-33 secretion (Fig. 1a), and flow cytometry confirmed elevated intracellular reactive oxygen species (ROS) levels (Fig. 1b), confirming successful foam cell differentiation.
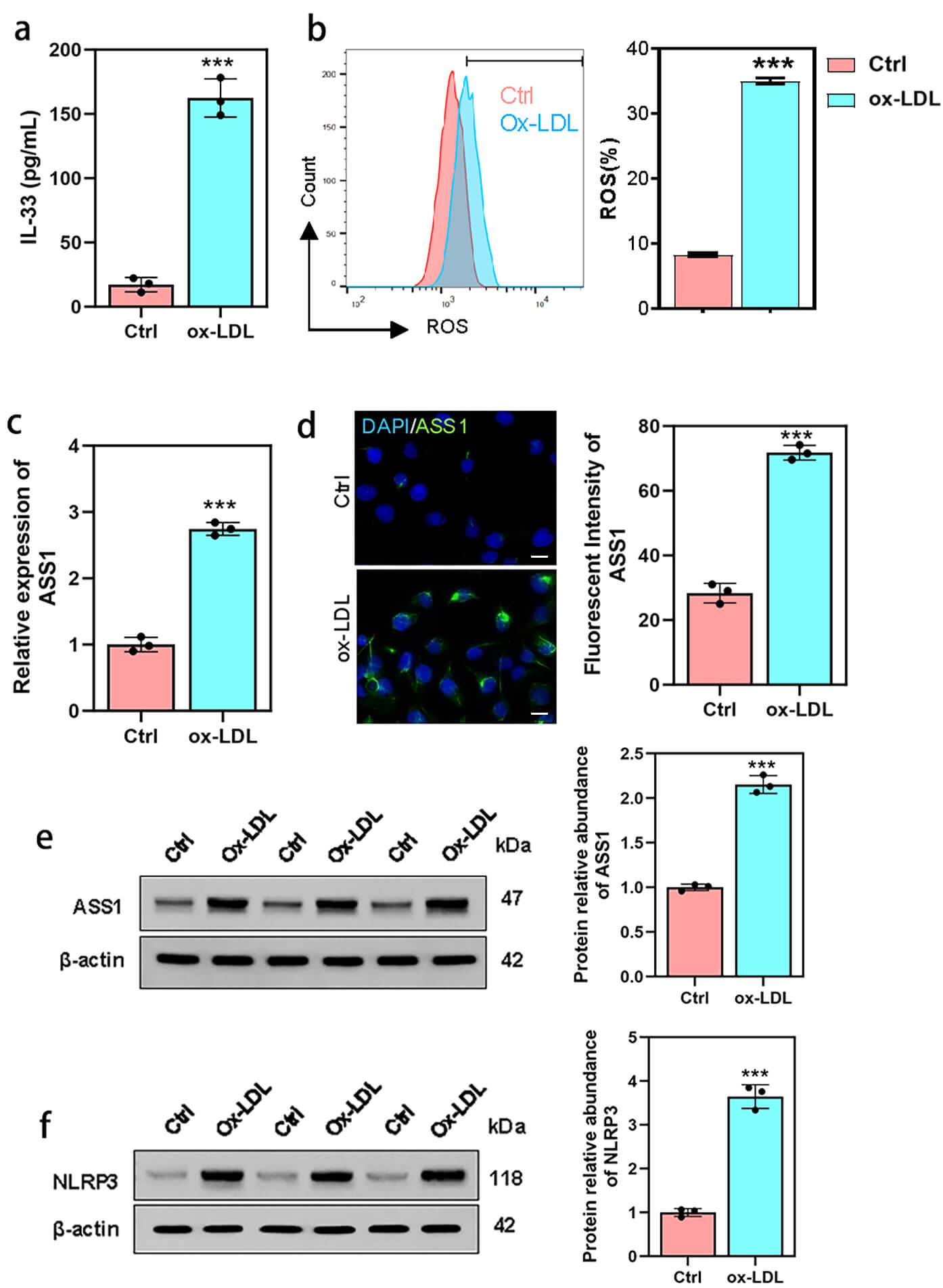 Fig. 1.
Fig. 1.
ASS1 expression is upregulated in ox-LDL-induced U937 foam
cells. U937 foam cells were generated through co-treatment with 100 nM phorbol
12-myristate 13-acetate (PMA) for 48 h and 50 µg/mL oxidized
low-density lipoprotein (ox-LDL) for an additional 48 h. (a) IL-33 concentration
measured by ELISA assay. (b) ROS generation analyzed by flow cytometry. (c) ASS1
expression assessed via quantitative real-time PCR. (d) Immunofluorescence assay
and (e) Western blot showing ASS1 expression in foam cells. (f) Western blot
analysis demonstrating NLRP3 expression in foam cells. The scale bar is 50
µm in (d). For all panels, n = 3 biologically independent experiments, each
performed on separate days with 1
ASS1 expression was then evaluated in these cells. qPCR analysis revealed a marked upregulation of ASS1 mRNA in foam cells compared with untreated controls (Fig. 1c). Consistent with this, both immunofluorescence and Western blotting demonstrated increased ASS1 protein expression in foam cells (Fig. 1d,e). Interestingly, Western blot also indicated an upward trend in NLRP3 protein levels in foam cells (Fig. 1f).
To determine the functional role of ASS1 in foam cells, we modulated its expression by siRNA-mediated knockdown and plasmid-mediated overexpression. qPCR confirmed efficient reduction of ASS1 mRNA after si-ASS1 transfection and increased expression after ASS1 overexpression (Fig. 2a).
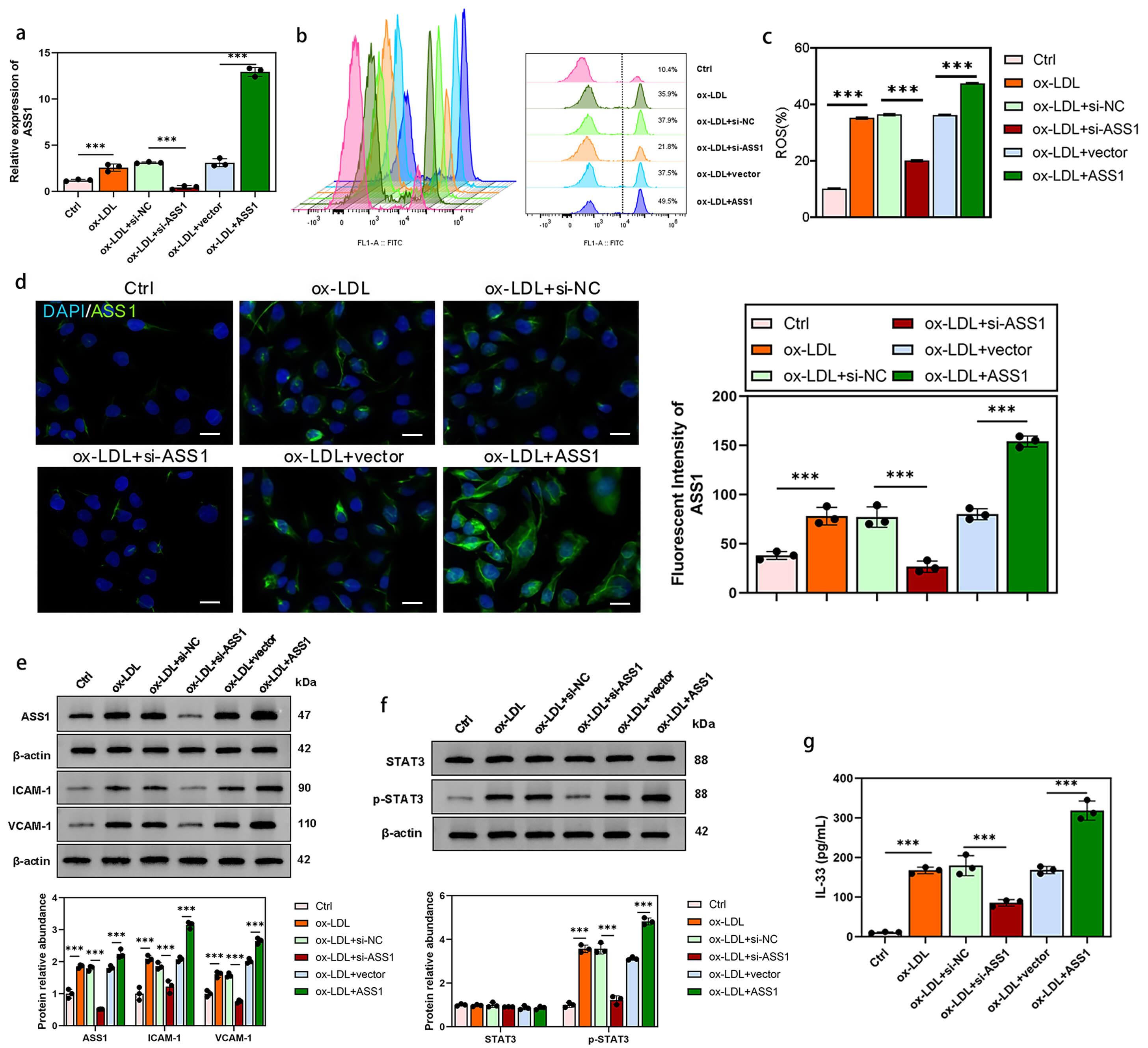 Fig. 2.
Fig. 2.
ASS1 modulates ROS production, inflammatory marker expression,
and IL-33 secretion in foam cells. U937 foam cells were transfected with si-ASS1
or ASS1 overexpression plasmid for 48 h. (a) Quantitative real-time PCR analysis
of ASS1 mRNA expression. (b,c) Flow cytometry analysis of ROS generation. (d)
Immunofluorescence assay showing ASS1 protein levels. (e) Western blot analysis
of ASS1, ICAM-1, VCAM-1, (f) STAT3, and p-STAT3 protein levels. (g) ELISA assay
for IL-33 concentration. The scale bar is 50 µm in (d). For all panels, n =
3 biologically independent experiments (performed on different days with
different cell passages, 1
Intracellular ROS levels, measured by DCFH-DA fluorescence, were elevated in ox-LDL-treated foam cells. ASS1 knockdown significantly attenuated this increase, whereas ASS1 overexpression further enhanced ROS production (Fig. 2b,c).
Western blotting verified the corresponding changes in ASS1 protein (Fig. 2d,e). Notably, ASS1 knockdown reduced the protein levels of the adhesion molecules ICAM-1 and VCAM-1, while ASS1 overexpression increased them (Fig. 2e). Phosphorylation of STAT3 (p-STAT3) was also diminished by ASS1 knockdown and augmented by ASS1 overexpression (Fig. 2f).
Consistent with these pro-inflammatory changes, IL-33 secretion—quantified by ELISA—was decreased upon ASS1 knockdown and elevated upon ASS1 overexpression (Fig. 2g). Together, these data indicate that ASS1 drives oxidative stress and inflammatory activation in ox-LDL-stimulated foam cells.
To determine whether ASS1 acts through STAT3 signaling, rescue experiments were conducted using STAT3-specific siRNA (si-STAT3) and a constitutively active STAT3 mutant (STAT3-CA) in U937 foam cells. Western blot analysis revealed that ASS1 overexpression upregulated NLRP3, ICAM-1, and VCAM-1, an effect that was largely abolished by co-transfection with si-STAT3 (Fig. 3a). Conversely, expression of STAT3-CA reversed the downregulation of these proteins caused by ASS1 knockdown (Fig. 3a).
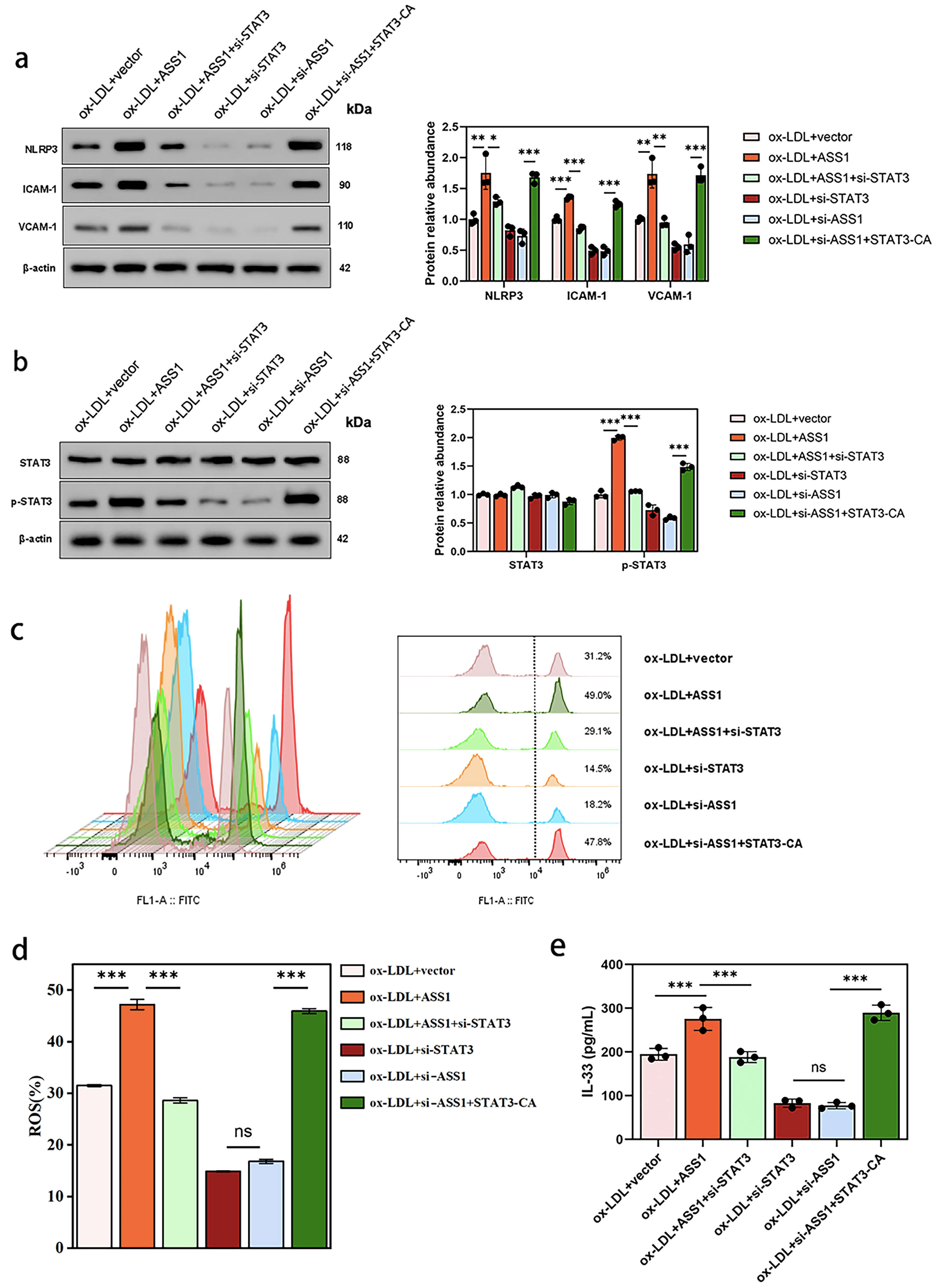 Fig. 3.
Fig. 3.
ASS1 regulates NLRP3 inflammasome activation and IL-33
secretion through STAT3 phosphorylation in U937 foam cells. Foam cells were
co-transfected with indicated plasmids/siRNAs and treated with ox-LDL (50
µg/mL). The experimental groups were as follows: ox-LDL + vector,
ox-LDL + ASS1, ox-LDL + ASS1 + si-STAT3, ox-LDL + si-STAT3, ox-LDL + si-ASS1, and
ox-LDL + si-ASS1 + STAT3-CA. (a) Western blot analysis of NLRP3, ICAM-1, and
VCAM-1 expression. (b) STAT3 and p-STAT3 protein levels. (c,d) ROS production
measured by DCFH-DA flow cytometry. (e) IL-33 concentration determined by ELISA.
For all panels, n = 3 biologically independent experiments (performed on
different days, 1
ASS1 overexpression increased STAT3 phosphorylation (p-STAT3), while ASS1 knockdown reduced it; these changes were respectively blocked by si-STAT3 or restored by STAT3-CA (Fig. 3b). Similarly, ASS1-induced elevation of ROS production and IL-33 secretion was suppressed by si-STAT3, whereas the decreases resulting from ASS1 knockdown were rescued by STAT3-CA (Fig. 3c–e).
Collectively, these data demonstrate that STAT3 activation is an essential downstream event through which ASS1 promotes NLRP3 inflammasome activity and IL-33 release in foam cells.
To further examine the role of ASS1 in ox-LDL-induced endothelial injury, we co-cultured HUVECs with U937-derived foam cells. Apoptosis was significantly increased in HUVECs exposed to ox-LDL-induced foam cells, as shown by flow cytometry (Fig. 4a). This effect was attenuated by ASS1 knockdown in foam cells and further exacerbated by ASS1 overexpression, which was confirmed by TUNEL staining (Fig. 4b).
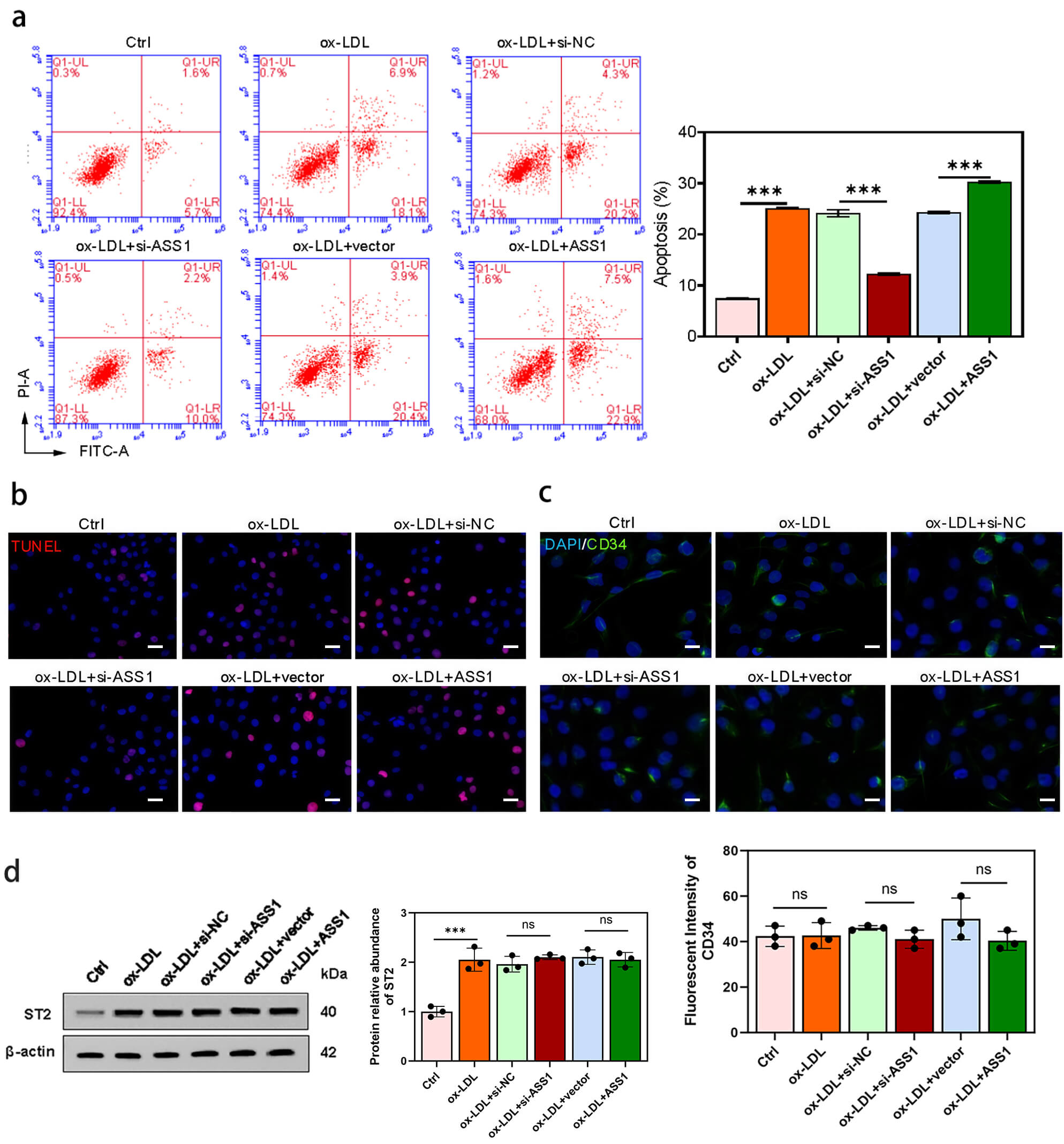 Fig. 4.
Fig. 4.
ASS1 in foam cells promotes HUVEC apoptosis via the
mitochondrial pathway without affecting CD34 or total ST2 expression. HUVECs
were co-cultured with U937 foam cells from control, si-NC, si-ASS1, vector, and
ASS1 groups. (a) Flow cytometry analysis of the apoptotic rate of endothelial
cells. (b) TUNEL fluorescence FITC assay for apoptotic endothelial cells. (c)
Immunofluorescence staining of the vascular marker CD34. (d) Western blot
analysis of ST2 protein levels. The scale bar is 50 µm in (b,c). For all
panels, n = 3 biologically independent experiments (performed on different days
with different HUVEC donors and different U937 passages); each experiment used 1
Western blot analysis of HUVECs revealed that ASS1 knockdown reduced the levels of cleaved PARP and Bax while increasing Bcl-2, whereas ASS1 overexpression had the opposite effect. The Bax/Bcl-2 ratio was approximately 3-fold higher in the ASS1-overexpression group than in the vector control, indicating activation of the mitochondrial apoptosis pathway (Supplementary Fig. 1).
Immunofluorescence staining showed that the endothelial marker CD34 was not altered by ASS1 manipulation in foam cells (Fig. 4c). Interestingly, while co-culture with ox-LDL-induced U937 foam cells significantly enhanced total ST2 expression in HUVECs, these levels were maintained independently of ASS1 manipulation (knockdown or overexpression) (Fig. 4d).
These data suggest that ASS1 in foam cells enhances ox-LDL-driven endothelial apoptosis through the mitochondrial pathway, without affecting endothelial marker expression or total ST2 levels.
Given the opposing roles of the soluble decoy receptor sST2 and the membrane-bound signaling receptor ST2L, we examined how ASS1 regulates their expression. In U937 foam cells, ASS1 overexpression significantly increased sST2 mRNA and decreased ST2L mRNA, raising the sST2/ST2L ratio. Conversely, ASS1 knockdown reduced sST2 and increased ST2L mRNA, lowering the sST2/ST2L ratio (Supplementary Fig. 2a,b).
A similar regulatory pattern was observed in HUVECs and HAVSMCs co-cultured with ASS1-modulated foam cells. Co-culture with ASS1-overexpressing foam cells elevated sST2 mRNA and reduced ST2L mRNA in both HUVECs (Supplementary Fig. 2c,d) and HAVSMCs (Supplementary Fig. 2e,f), again increasing the sST2/ST2L ratio. Western blot analysis confirmed these changes at the protein level: ASS1 overexpression in foam cells increased sST2 protein in co-cultured HUVECs (Supplementary Fig. 2g) and HAVSMCs (Supplementary Fig. 2h), whereas ASS1 knockdown produced the opposite effect.
Together, these findings indicate that ASS1 drives a shift in the ST2 isoform balance toward a pro-inflammatory state—characterized by elevated sST2 and reduced ST2L—thereby potentiating IL-33-mediated signaling activity.
Given the contribution of vascular smooth muscle cells (VSMCs) to atherosclerotic plaque progression, we examined whether ASS1 in foam cells influences HAVSMC behavior. HAVSMCs were co-cultured with ox-LDL-induced U937 foam cells in which ASS1 was either knocked down or overexpressed.
CCK-8 and EdU assays showed that ox-LDL-treated foam cells stimulated HAVSMC proliferation, an effect that was further enhanced by ASS1 overexpression and reduced by ASS1 knockdown (Fig. 5a,b).
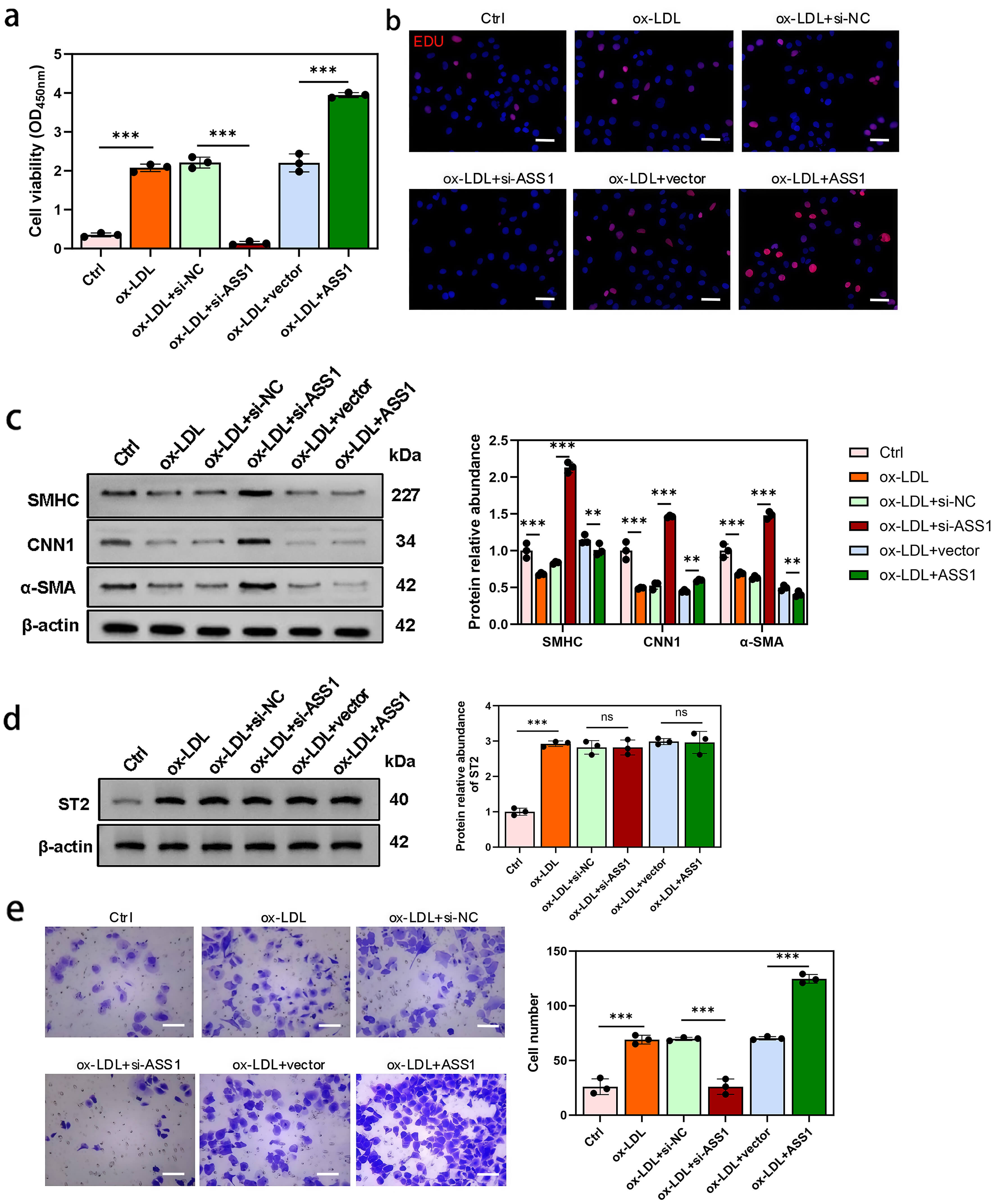 Fig. 5.
Fig. 5.
ASS1 in foam cells promotes HAVSMC proliferation, migration, and
dedifferentiation. HAVSMCs were co-cultured with U937 foam cells from control,
si-NC, si-ASS1, vector, and ASS1 groups. (a) Cell viability was assessed by the
CCK-8 assay. (b) Cell proliferation of HAVSMCs was evaluated using EdU staining.
(c) Western blot analysis of SMHC, CNN1, and
Western blot analysis revealed that co-culture with ox-LDL-induced foam cells
downregulated the VSMC differentiation markers SMHC, CNN1, and
Notably, total ST2 protein levels in HAVSMCs were elevated following co-culture with ox-LDL-treated foam cells, but were not significantly altered by modulating ASS1 expression in foam cells (Fig. 5d).
Finally, Transwell migration assays demonstrated that ASS1 knockdown in foam cells attenuated the enhanced migration of HAVSMCs induced by co-culture, whereas ASS1 overexpression further increased migratory capacity (Fig. 5e).
Together, these results indicate that ASS1 in foam cells drives HAVSMC proliferation, migration, and dedifferentiation—key processes in atheroma progression—without affecting total ST2 expression in co-cultured HAVSMCs.
To determine whether NLRP3 mediates the pro-inflammatory effects of ASS1, rescue experiments were performed in U937 foam cells. ELISA showed that ASS1 knockdown reduced IL-33 secretion, and this reduction was largely reversed by NLRP3 overexpression (Fig. 6a). Similarly, the decrease in ROS production caused by ASS1 knockdown was restored upon NLRP3 overexpression (Fig. 6b,c).
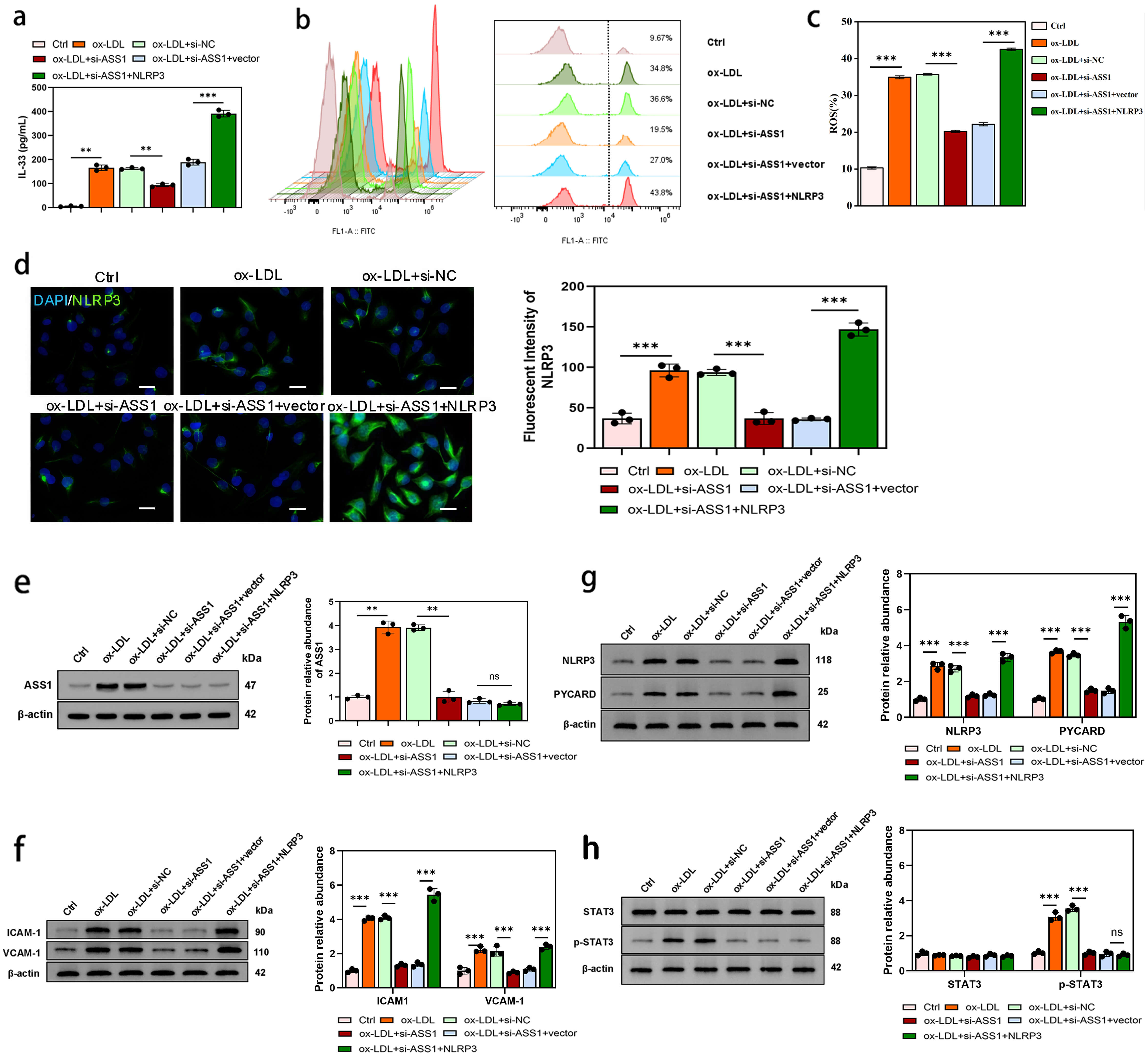 Fig. 6.
Fig. 6.
NLRP3 overexpression rescues the anti-inflammatory effects of
ASS1 knockdown in foam cells. U937 foam cells were co-transfected with si-ASS1
and NLRP3 overexpression plasmid or transfected with si-ASS1 alone. (a) IL-33
concentration determined by ELISA assay. (b,c) Flow cytometry analysis of ROS
generation. (d) Immunofluorescence assay showing NLRP3 protein expression in U937
foam cells. (e) Western blot analysis of ASS1 protein levels. (f) Western blot
analysis of adhesion molecules ICAM-1 and VCAM-1, (g) inflammasome-associated
proteins NLRP3 and PYCARD, and (h) STAT3 and p-STAT3 associated with the STAT3
signaling pathway. The scale bar is 50 µm in (d). For all panels, n = 3
biologically independent experiments (performed on different days with different
cell passages); each experiment contained 3 technical replicates (wells) per
condition, with 2
Immunofluorescence confirmed successful NLRP3 overexpression (Fig. 6d). ASS1 protein downregulation by siRNA was not altered by NLRP3 overexpression (Fig. 6e), indicating that NLRP3 acts downstream of ASS1.
Western blot analysis revealed that the downregulation of adhesion molecules (ICAM-1 and VCAM-1) induced by ASS1 knockdown was abolished by NLRP3 overexpression (Fig. 6f). Likewise, the reduced expression of NLRP3 itself and its adaptor protein PYCARD after ASS1 knockdown was restored by NLRP3 overexpression (Fig. 6g). In contrast, NLRP3 overexpression did not reverse the decrease in STAT3 phosphorylation (p-STAT3) caused by ASS1 knockdown (Fig. 6h).
These results demonstrate that NLRP3 activation is a key downstream event responsible for ASS1-driven inflammatory signaling and oxidative stress in foam cells, independent of changes in ASS1 expression or STAT3 phosphorylation.
To determine whether NLRP3 activation itself is sufficient to induce ASS1-like inflammatory responses, we overexpressed NLRP3 alone in ox-LDL-treated U937 foam cells without altering ASS1 expression. Western blot analysis showed that NLRP3 overexpression significantly increased the protein levels of adhesion molecules ICAM-1 and VCAM-1 (Fig. 7a), as well as NLRP3 and its adaptor protein PYCARD (Fig. 7b), to a degree comparable to ASS1 overexpression.
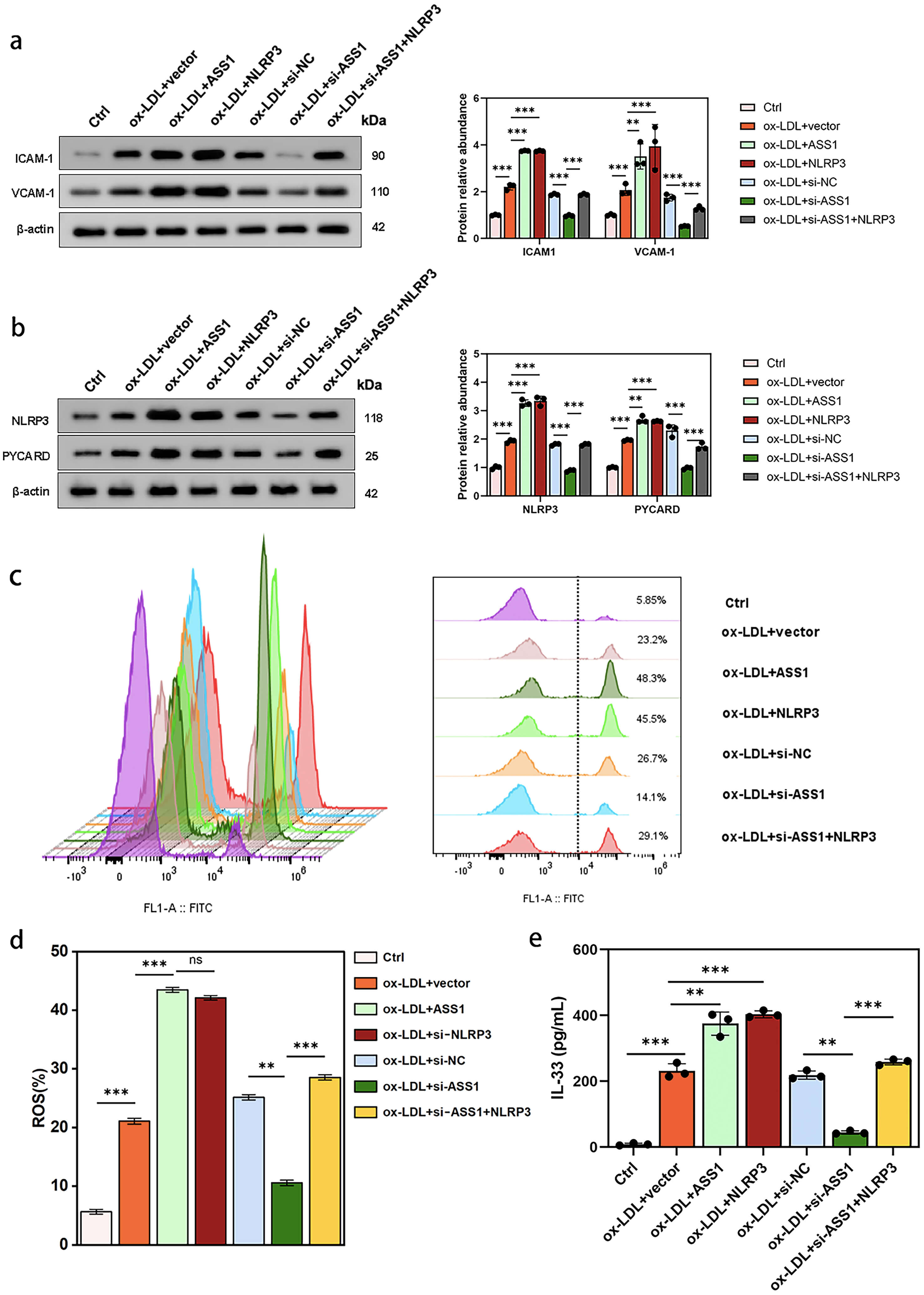 Fig. 7.
Fig. 7.
NLRP3 overexpression increased downstream factors to levels
comparable to ASS1 overexpression, and reversed the effects of ASS1 knockdown.
U937 foam cells were treated as follows: Ctrl (untreated), ox-LDL + vector,
ox-LDL + ASS1, ox-LDL + NLRP3, ox-LDL + si-NC, ox-LDL + si-ASS1, or ox-LDL +
si-ASS1 + NLRP3. (a) Western blot analysis of adhesion molecules ICAM-1 and
VCAM-1. (b) Inflammasome-associated proteins NLRP3 and PYCARD expression. (c,d)
ROS production measured by DCFH-DA flow cytometry. (e) IL-33 concentration
determined by ELISA assay. For all panels, n = 3 biologically independent
experiments (performed on different days, 1
Flow cytometry revealed that NLRP3 overexpression alone markedly elevated intracellular ROS production (Fig. 7c,d), similar to the effect of ASS1 overexpression. ELISA further demonstrated that IL-33 secretion was increased by approximately 70% in NLRP3-overexpressing cells, with no statistically significant difference from the ASS1-overexpression group (Fig. 7e).
Importantly, the anti-inflammatory effects of ASS1 knockdown—reduced ROS, lower adhesion molecule expression, and decreased IL-33 secretion—were largely reversed when NLRP3 was co-overexpressed (Fig. 7a–e).
Together, these results indicate that NLRP3 activation is sufficient to drive a pro-inflammatory phenotype in foam cells, and that the pro-atherogenic effects of ASS1 are mediated primarily through upregulation of NLRP3.
We next examined whether NLRP3 modulates the impact of ASS1 on vascular cells using a foam-cell co-culture model.
HUVECs: ASS1 knockdown in foam cells reduced HUVEC apoptosis, an effect that was reversed by NLRP3 overexpression in foam cells, as shown by flow cytometry and TUNEL staining (Fig. 8a,b). The endothelial marker CD34 was unaffected by either ASS1 or NLRP3 manipulation (Fig. 8c). Total ST2 expression in HUVECs was elevated after co-culture with ox-LDL-induced foam cells, but remained elevated upon ASS1 knockdown or NLRP3 overexpression (Fig. 8d).
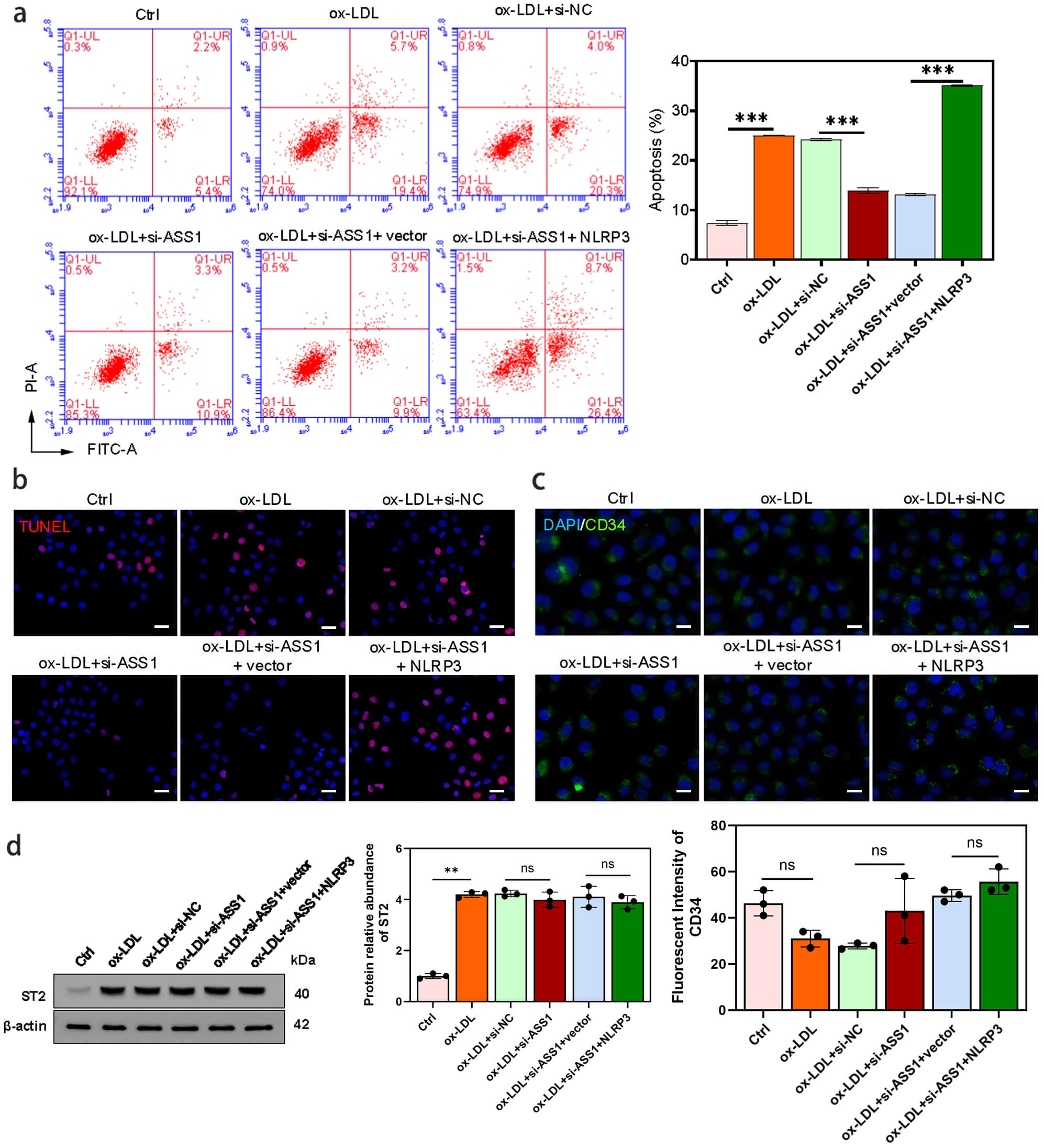 Fig. 8.
Fig. 8.
NLRP3 overexpression in foam cells reverses the protective
effects of ASS1 knockdown on HUVEC apoptosis. HUVECs were co-cultured with U937
foam cells from control, si-NC, si-ASS1, si-ASS1 + vector, and si-ASS1 + NLRP3
groups. (a) Flow cytometry assay to determine cell apoptotic rate. (b) TUNEL
fluorescence FITC kit used to detect apoptotic endothelial cells. (c)
Immunofluorescence staining of the vascular marker CD34. (d) Western blot
analysis of ST2 protein levels. The scale bar is 50 µm in (b,c). For all
panels, n = 3 biologically independent experiments (performed on different days
with different HUVEC donors and different U937 passages); each experiment
contained 3 technical replicates (wells) per group, with 1
HAVSMCs: ASS1 knockdown in foam cells attenuated HAVSMC proliferation, which was
restored by NLRP3 overexpression (CCK-8 and EdU assays; Fig. 9a,b). Western
blotting showed that ASS1 knockdown increased the expression of smooth-muscle
differentiation markers (SMHC, CNN1,
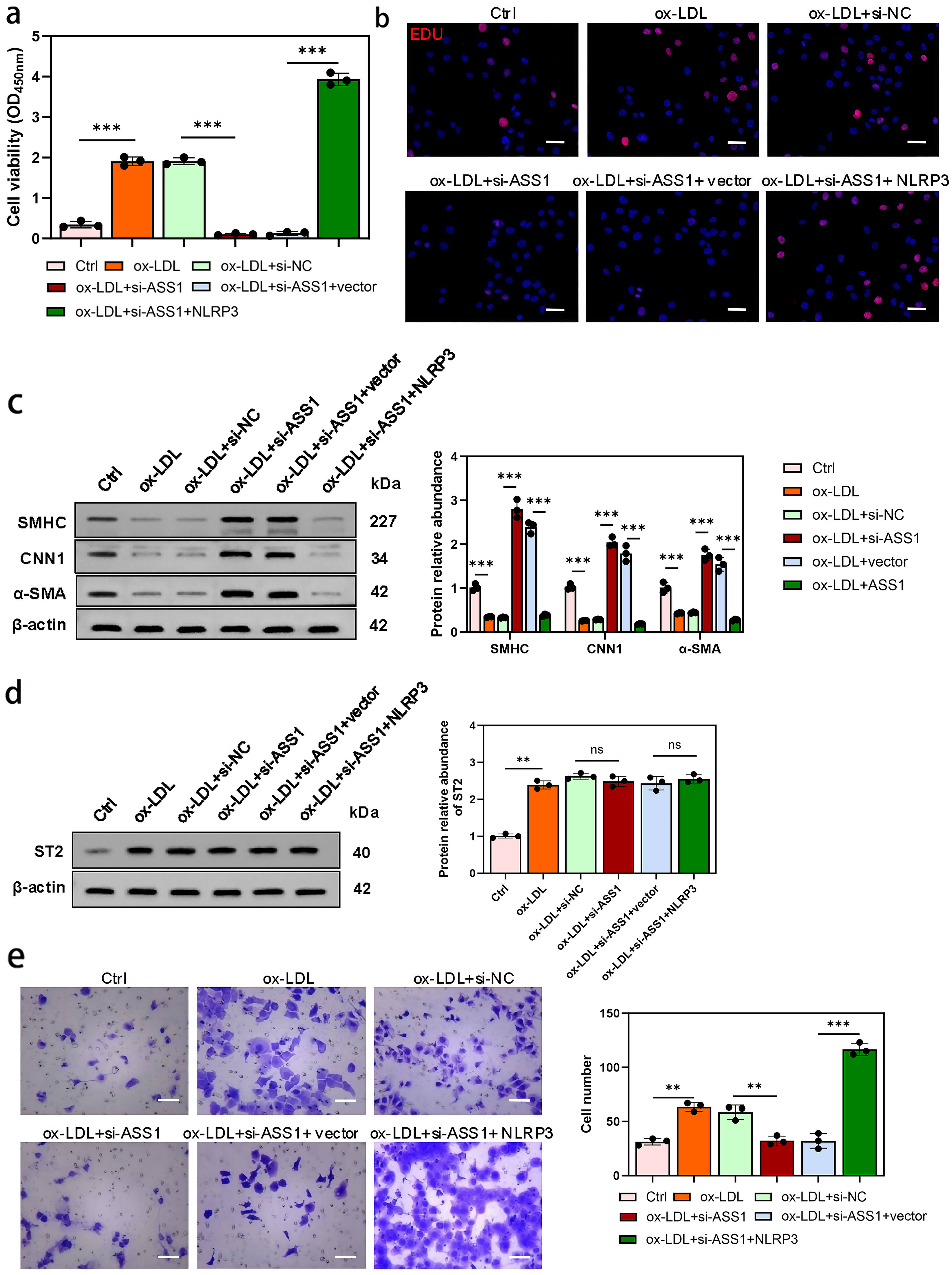 Fig. 9.
Fig. 9.
NLRP3 overexpression in foam cells restores HAVSMC
proliferation, migration, and dedifferentiation suppressed by ASS1 knockdown. HAVSMCs were co-cultured with U937 foam cells from control, si-NC, si-ASS1,
si-ASS1 + vector, and si-ASS1 + NLRP3 groups. (a) Cell viability was
assessed by the CCK-8 assay. (b) Cell proliferation of HAVSMCs was evaluated
using EdU staining. (c) Western blot analysis of SMHC, CNN1, and
These results indicate that NLRP3 activation in foam cells can counteract the regulatory effects of ASS1 on endothelial apoptosis and smooth muscle proliferation/migration, whereas total ST2 expression in vascular cells is influenced by ox-LDL-exposed foam cells but not directly by ASS1 or NLRP3.
To further evaluate the role of ASS1 in atherosclerosis, we performed histological and biochemical analyses in ApoE-deficient mice. Mice were fed a high-fat diet for 12 weeks to induce atherosclerosis, after which ASS1 expression was modulated via AAV9 vectors. Plaques from the transverse cardiac section (TCS) and aortic arch (AAC) were examined.
Oil Red O staining showed that lipid accumulation was markedly increased in atherosclerotic (AS) mice compared with sham-operated controls (Fig. 10a). Quantification confirmed the most severe lipid deposition in the ASS1-overexpression (ASS1OE) group, intermediate levels in the AS and negative control (NC) groups, and significantly reduced lipid in the ASS1-knockdown (ASS1kd) group (Fig. 10b,c). Body weight was also highest in ASS1OE mice, while ASS1kd mice maintained weights similar to the Sham group, suggesting a systemic metabolic influence of ASS1 (Fig. 10d).
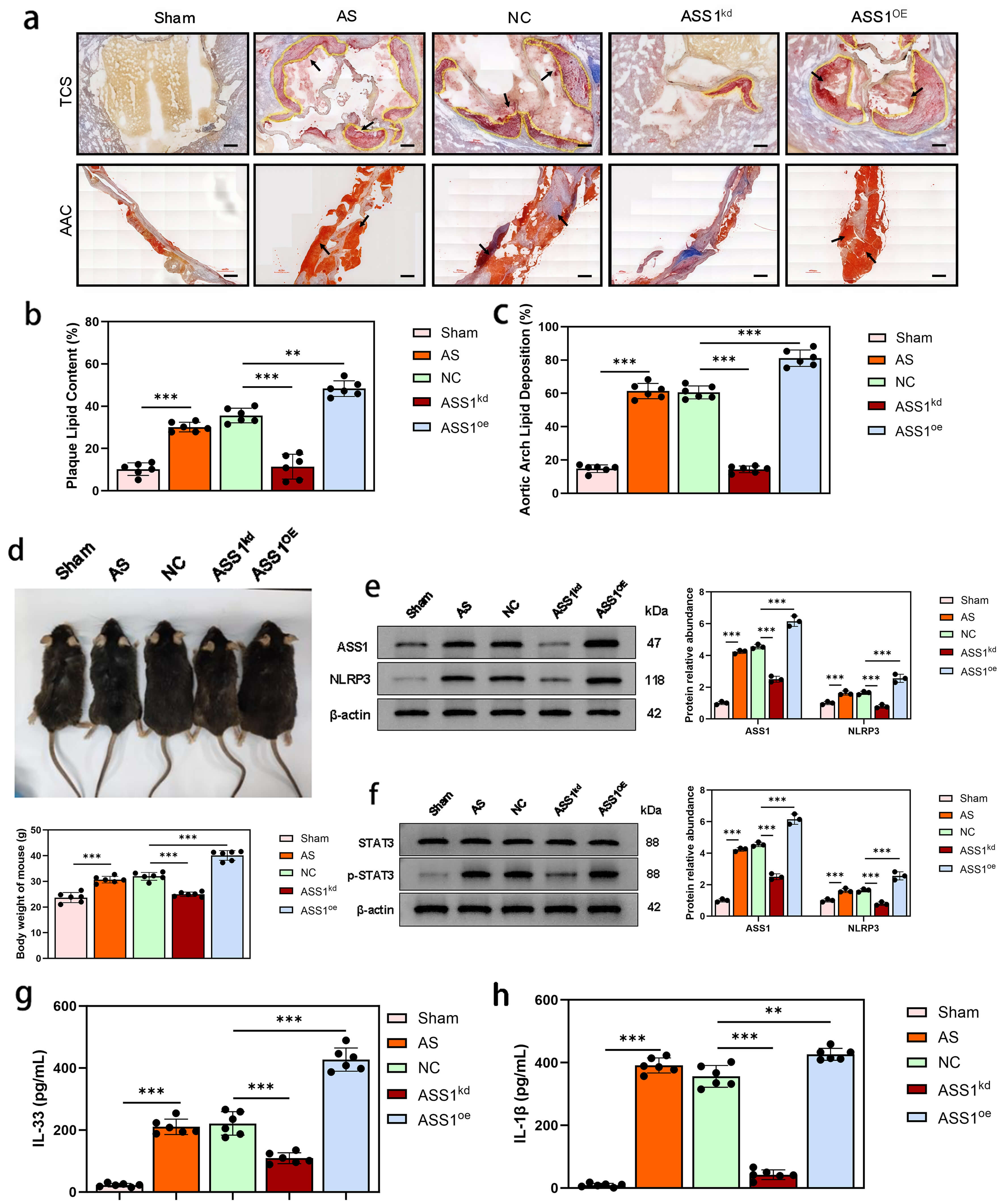 Fig. 10.
Fig. 10.
ASS1 exacerbates atherosclerotic plaque progression,
inflammation, and systemic metabolism in ApoE-deficient mice. Atherosclerosis
was induced by feeding a high-fat diet for 12 weeks, followed by AAV9-mediated
knockdown or overexpression of ASS1. Grouping: wild-type sham-operated group
(Sham), atherosclerotic model group (AS), negative control group (NC, empty
vector), ASS1 knockdown group (ASS1kd), and ASS1 overexpression group
(ASS1OE). (a) Representative images of Oil Red O staining of the
transverse cardiac section (TCS) and aortic arch cut (AAC). The black arrows in
the figure indicate the severe areas of plaque lipid accumulation. (b,c) Quantification of lipid deposition of the TCS and AAC (Oil Red O-positive area
percentage within plaque). (d) Body weight of mice measured at the study
endpoint and comparative analysis between groups. (e) Western blot
analysis (left) and quantitative summary (right) of ASS1 and NLRP3 protein
expression in vascular tissues (n = 3 independent biological replicates). (f)
Western blot analysis (left) and quantitative summary (right) of STAT3 and
phosphorylated STAT3 (p-STAT3) protein levels in vascular tissues. (g,h) Serum
IL-33 and serum IL-1
Western blot analysis of vascular tissues revealed that ASS1 overexpression increased NLRP3 protein levels, whereas ASS1 knockdown decreased them (Fig. 10e). Similarly, phosphorylated STAT3 (p-STAT3) was upregulated by ASS1 overexpression and downregulated by ASS1 knockdown (Fig. 10f), consistent with the in vitro findings.
ELISA of serum cytokines showed that IL-33 was elevated in AS mice, further
increased by ASS1 overexpression, and attenuated by ASS1 knockdown
(Fig. 10g). To determine whether NLRP3 activation also affects other inflammatory
mediators, we measured IL-1
These in vivo results demonstrate that ASS1 exacerbates plaque lipid
deposition, activates the NLRP3 inflammasome and STAT3 signaling, and enhances
the secretion of pro-inflammatory cytokines IL-33 and IL-1
AS is a complex inflammatory disorder affecting medium and large arteries, primarily involving three key cell types: macrophages, endothelial cells, and smooth muscle cells [20, 21]. In the initial stages of AS, the accumulation of oxidized low-density lipoprotein (ox-LDL) leads to macrophage dysfunction, resulting in the formation of foam cells and the production of pro-inflammatory cytokines [22]. Extensive research has established the crucial role of ASS1, a key enzyme in arginine biosynthesis, in regulating inflammatory macrophage activation and providing defense against bacterial infections by depleting cellular citrulline [15]. Zhang et al. [23] demonstrated that arginine can partially regulate the activation of the NLRP3 inflammasome by inhibiting ROS production in vascular endothelial cells. Building on these observations, we hypothesized that ASS1 is implicated in the regulation of pro-inflammatory cytokine production during foam cell formation. Our findings in this present study revealed that ox-LDL treatment significantly increased the production of reactive oxygen species (ROS) and upregulated the expression of argininosuccinate synthase 1 (ASS1) in PMA-induced macrophage-like U937 cells. Importantly, knocking down ASS1 notably reduced ox-LDL-induced ROS production and the secretion of interleukin-33 (IL-33), suggesting a positive regulatory role of ASS1 in IL-33 production in U937 foam cells.
Importantly, our rescue experiments using si-STAT3 and a constitutively active STAT3 mutant (STAT3-CA) provide direct evidence that STAT3 activation is a critical downstream event in ASS1-mediated pro-inflammatory signaling. The fact that STAT3 inhibition completely reversed the effects of ASS1 overexpression on NLRP3, adhesion molecules, ROS, and IL-33, while constitutive STAT3 activation restored the inflammatory phenotype suppressed by ASS1 knockdown, establishes a causal link between ASS1 and STAT3 phosphorylation. This moves beyond correlation and defines a clear signaling axis: ASS1 upregulation enhances STAT3 activation, which in turn drives NLRP3 inflammasome assembly and IL-33 secretion, thereby amplifying foam cell-driven inflammation in atherosclerosis. Furthermore, our study revealed that NLRP3 overexpression reversed the decrease in ROS production and IL-33 secretion induced by ASS1 silencing in U937 foam cells, which aligns with previous research indicating that the accumulation of ox-LDL enhances NLRP3 expression, leading to the assembly of the NLRP3 inflammasome complex in macrophages derived from THP-1 cells [24]. Notably, our rescue experiments with NLRP3 overexpression alone revealed that NLRP3 activation is sufficient to mimic the pro-inflammatory effects of ASS1 overexpression, including enhanced ROS production, upregulation of adhesion molecules, and elevated IL-33 secretion. Importantly, the effects of ASS1 knockdown were largely reversed by NLRP3 overexpression, suggesting that NLRP3 acts downstream of ASS1 and STAT3, and is a primary mediator of its pro-atherogenic functions. Collectively, these findings indicate that the ox-LDL-induced upregulation of ASS1 can activate the STAT3/NLRP3 inflammasome axis in U937 foam cells.
Endothelial cell injury is widely acknowledged as a critical and initiating factor in the development of AS [25]. In our co-culture system consisting of U937 foam cells and HUVECs, we observed that the knockdown of ASS1 in foam cells significantly inhibited ox-LDL-induced apoptosis. Moreover, our Western blot analysis revealed that ASS1 overexpression in foam cells significantly upregulated cleaved PARP and Bax expression while downregulating Bcl-2 in co-cultured HUVECs, resulting in an elevated Bax/Bcl-2 ratio. These findings suggest that ASS1 promotes HUVEC apoptosis through the mitochondrial pathway, further supporting the involvement of intrinsic apoptosis in ASS1-mediated endothelial injury during atherosclerosis.
ST2, also known as interleukin-1 receptor-like-1 protein (IL1RL1), is produced
by T cells and endothelial cells [26] and has been shown to have significant
involvement in cardiovascular diseases [27]. The IL-33/ST2 pathway, which is
predominantly found in the endothelium of arterial vessels, is now recognized as
a key player in the inflammatory mechanisms underlying AS [28]. The ST2 receptor
has two isoforms: soluble decoy receptor (sST2) and membrane-bound signaling
receptor (ST2L), which exert opposing biological effects. Their dynamic balance
dictates disease outcome. ST2L mediates protection: IL-33 binding activates the
MyD88/NF-
Critically, our new data reveal that ASS1 does not uniformly increase total ST2,
but specifically shifts the balance between its isoforms. ASS1 upregulation in
foam cells promotes the expression of the soluble decoy receptor (sST2) while
suppressing the membrane-bound signaling receptor (ST2L) in both foam cells
themselves and in co-cultured HUVECs/HAVSMCs. This shift results in an increased
sST2/ST2L ratio. Emerging evidence supports the involvement of the IL-33/ST2
signaling pathway in the inflammatory pathogenesis of various injury-related
diseases [29]. A recent investigation by Ma et al. [30] demonstrated
that co-culturing NLRP3-overexpressing rat renal macrophages with NRK-52E cells
led to NLRP3 activation in resident macrophages, resulting in the upregulation of
IL-33. Subsequently, IL-33 mediated the IL-33/ST2/NF-
In addition to endothelial cells, abnormal proliferation, apoptosis, and
migration of VSMCs have been strongly implicated in the progression of AS [32, 33]. Ox-LDL, a significant contributor to AS progression, has been shown to
impact VSMC apoptosis, proliferation, and migration [34]. In our study, we
observed that ASS1 knockdown in foam cells significantly inhibited ox-LDL-induced
proliferation and migration, as well as the expression of smooth muscle cell
markers (SMHC, SNN1, and
The role of ASS1 in atherosclerosis (AS) was further investigated using an
ApoE-deficient mouse model fed a high-fat diet for 12 weeks. Modulation of ASS1
expression via AAV9 vectors demonstrated significant effects on lipid
accumulation and inflammatory responses in AS mice. Oil Red O staining revealed
that ASS1 knockdown reduced lipid accumulation in the hearts and aortas, while
ASS1 overexpression exacerbated lipid enrichment compared to controls. These
findings highlight ASS1 as a key regulator of lipid deposition in AS. In
addition, ASS1 expression significantly influenced the levels of NLRP3 and
p-STAT3 in vascular tissues, consistent with the regulation of inflammatory
pathways observed in vitro. ASS1 knockdown decreased NLRP3 levels,
whereas overexpression of ASS1 led to elevated NLRP3 expression. Similarly,
p-STAT3 was downregulated with ASS1 knockdown but upregulated with ASS1
overexpression. Furthermore, ELISA assays showed that ASS1 regulated IL-33 levels
in the serum of AS mice, with ASS1 overexpression increasing IL-33 concentration
and knockdown reducing it. In addition to IL-33, our study also demonstrated that
ASS1 overexpression significantly enhanced serum IL-1
Based on our results, a pattern of ASS1-mediated NLRP3/IL-33/ST2 pathway on
inflammatory responses in foam cells during atherosclerosis was proposed (Fig. 11). Upon excessive levels of oxidative stress in in blood vessels, ox-LDL enter
the macrophage through receptor-mediated endocytosis, which results in the
pro-inflammatory polarization of the macrophage and leads to the activation of
JAK2-STAT1 signaling pathway that upregulates intracellular ASS1. This mechanism
of ox-LDL-induced ASS1 elevation has been validated in the study by Mao
et al. [15]. and further verified in our present study. Elevated
intracellular ASS1 induces the phosphorylation of STAT3. Subsequently, p-STAT3
translocates from the cytoplasm to the nucleoplasm and then stimulates gene
expression through binding of p-STAT3 to a conserved DNA element gamma-activated
sequence (GAS), initiating NLRP3-related gene transcription. The activation of
the NLRP3 inflammasome leads to increased expression and secretion of IL-33 (and
IL-1
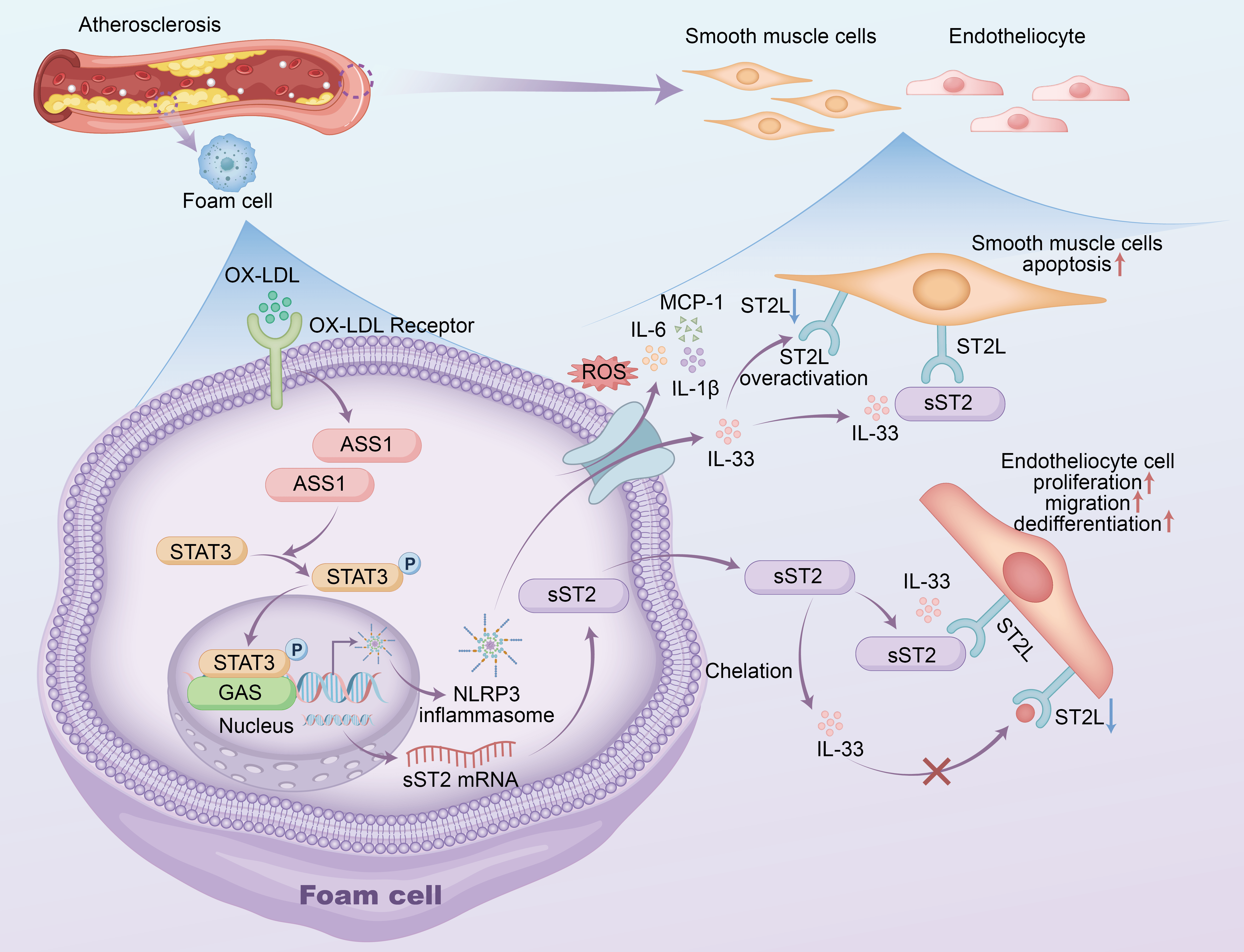 Fig. 11.
Fig. 11.
A pattern of ASS1-mediated NLRP3/IL-33/ST2 pathway on
inflammatory responses in foam cells during atherosclerosis was proposed. A
pattern of ASS1-mediated NLRP3/IL-33/ST2 pathway on inflammatory responses in
foam cells during atherosclerosis was proposed. Our findings delineate a coherent
pro-inflammatory signaling cascade in atherosclerosis: ox-LDL
Several limitations warrant mention. First, while we establish STAT3 as a key
downstream effector of ASS1, emerging data show that JAK2‑STAT1 signaling
upregulates ASS1 [15], raising the possibility of a reciprocal ASS1/STAT1 axis
that may independently modulate atherosclerosis, which was not examined here.
Second, modulating ASS1 could influence foam‑cell formation efficiency under
ox‑LDL stimulation, potentially confounding inflammatory readouts due to
variations in foam‑cell number or lipid‑loading state across groups. Third,
although we propose a self‑amplifying loop where ASS1‑driven IL‑33/sST2 elevation
blocks protective ST2L signaling, the mechanism for concurrent ST2L
downregulation—whether through ligand‑induced internalization or
transcriptional suppression—remains speculative and needs experimental
validation. Finally, our focus on the NLRP3/IL‑33/ST2 axis does not exclude roles
for other NLRP3‑dependent cytokines (e.g., IL‑1
In summary, this study identifies argininosuccinate synthase 1 (ASS1) as a
critical regulator of atherosclerotic inflammation via the STAT3/NLRP3/IL-33/ST2
signaling axis. We show that oxidized low-density lipoprotein (ox-LDL)
upregulates ASS1 expression in macrophage-derived foam cells, which in turn
activates NLRP3 inflammasome assembly in a STAT3-dependent manner and enhances
the secretion of pro-inflammatory cytokines IL-33 and IL-1
The data used to support the findings of this study are available from the corresponding author Dr. Kefei Dou upon reasonable request. The data are not publicly available due to privacy or ethical restrictions.
All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. KD had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. KD and DM contributed to the conception and design of the work. SW and FH performed the cytology experiments and gene expression analysis. ZQ, SY, and XB established the animal model and performed the statistical analysis. BZ participated in data interpretation and the preparation of figures/tables. SW and FH drafted the manuscript. KD, DM, and BZ critically revised the manuscript. KD and DM finalized the manuscript. All authors contributed to editorial changes in the manuscript. All the authors reviewed and approved the final version.
The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Peking Union Medical College (PUMC). All animal procedures were conducted in strict accordance with the guidelines and regulations for the care and use of laboratory animals, and adhered to the principles of the 3Rs (Replacement, Reduction, and Refinement) to minimize animal suffering and distress. The formal ethical approval document was subsequently issued by Fuwai Hospital, an affiliate of the Chinese Academy of Medical Sciences (CAMS) and a constituent of PUMC. A scanned copy of the official ethical approval document is available for submission upon request (Approval No. 0105-1-6-ZX(X)-4).
Not applicable.
The research reported in this publication was supported by the Noncommunicable Chronic Diseases-National Science and Technology Major Project (grant number 2025ZD0548200).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47686.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.