














 , Liting Li 2,†, Xiaohui Li 2, Qiulong Zhang 2,3, Yan Liu 2,4,*
, Liting Li 2,†, Xiaohui Li 2, Qiulong Zhang 2,3, Yan Liu 2,4,*
1 Central Laboratory, The Second People’s Hospital of Foshan (Affiliated Foshan Hospital of Guangdong Pharmaceutical University), 528000 Foshan, Guangdong, China
2 MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, 510275 Guangzhou, Guangdong, China
3 Key Laboratory of Pharmaceutical Analysis and Laboratory Medicine of Fujian Province, School of Pharmacy and Medical Technology, Putian University, 351100 Putian, Fujian, China
4 Department of Biochemistry and Molecular Biology, School of Basic Medicine, Guangdong Medical University, 524023 Zhanjiang, Guangdong, China
†These authors contributed equally.
Abstract
The clustered regularly interspaced short palindromic repeats CRISPR-associated protein 9 (CRISPR/Cas9) system has emerged as a versatile platform for genome editing, transcriptional regulation, and chromosomal imaging. Recent advances in synthetic biology have enabled the engineering of single guide RNA (sgRNA) to confer conditional responsiveness on the CRISPR/Cas9 system. By integrating functional nucleic acid elements, such as aptamers, ribozymes, and aptazymes, into specific structural regions of the sgRNA, researchers have developed systems that respond to a variety of molecular signals, including small molecules, proteins, and endogenous metabolites. These engineered sgRNAs enable spatiotemporal control of gene editing, activation, repression, and imaging in both prokaryotic and eukaryotic cells. This review summarizes the structural principles, design strategies, and applications of condition-responsive CRISPR/Cas9 systems, highlighting their potential in synthetic biology, disease modeling, and therapeutic development. Current challenges and future directions for improving the specificity, efficiency, and applicability of these systems are also discussed.
Keywords
- CRISPR-Cas systems
- guide RNA
- RNA aptamers
- ribozymes
- genetic engineering
- gene expression regulation
The Clustered Regularly Interspaced Short Palindromic Repeats/Cas CRISPR-associated protein 9 (CRISPR/Cas9) system is an adaptive immune mechanism found in bacteria and archaea that protects them from viral or phage invasions. Due to its simple composition, high specificity, and efficient cleavage, researchers have repurposed the CRISPR/Cas9 system into a new generation of gene-editing tools, rapidly expanding its applications [1, 2, 3]. It has now been validated for precise genome editing in a wide range of organisms [4, 5, 6, 7, 8]. Simultaneously, the system has shown great potential in the field of molecular diagnostics, sparking widespread interest in the development of condition responsive technologies based on the CRISPR/Cas9 platform. Engineering of the Cas9 protein has not only endowed it with gene editing functions but also enabled transcriptional regulation (CRISPR Interference [CRISPRi] for gene repression and CRISPR Activation [CRISPRa] for gene activation) [9, 10, 11]. Furthermore, structural design of the guide RNA (gRNA) has achieved conditional responsiveness at the cellular level [12, 13, 14].
Researchers have leveraged the CRISPR/Cas9 platform alongside molecular tools like dynamic nucleic acid structures to regulate cellular functions. This includes strategies such as aptamer-mediated regulation and strand-exchange regulation, which have been further applied in both prokaryotic and eukaryotic cells to control gene expression levels, protein production, and cellular functions [15, 16, 17, 18]. These approaches have significantly broadened the application dimensions of the CRISPR/Cas9 system. By integrating environment-responsive elements, the system can now perceive and respond to specific stimuli, such as small molecules or metabolites, enhancing its spatiotemporal precision within complex biological environments [19, 20, 21, 22]. This strategy also provides key technical support for building intelligent gene circuits and cell-based therapies, demonstrating broad prospects in disease diagnosis and precision medicine.
Aptamers are artificially synthesized single-stranded deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) sequences first reported in 1990 [23, 24, 25]. They fold into secondary and tertiary structures, enabling extremely high specificity for binding their target molecules [26]. Due to their high specific affinity for targets—ranging from small molecules and proteins to entire cells—RNA aptamers are particularly suitable for integration with single guide RNA (sgRNA) to construct CRISPR/Cas9 systems regulated by specific molecules [27, 28]. Similar to protein enzymes, certain ribonucleic acid (RNA) molecules, known as “ribozymes”, can catalyze various biochemical reactions, such as the cleavage and formation of phosphodiester bonds, peptide bonds, and other chemical bonds [29, 30, 31]. In 1988, Thomas R. Cech was awarded the 1989 Nobel Prize in Chemistry for discovering the catalytic properties of RNA. Specific cleavage can immediately render a target RNA molecule non-functional. Incorporating self-cleaving ribozymes with sgRNA allows the construction of ribozyme-regulated CRISPR/Cas9 systems [32, 33].
Utilizing dynamic nucleic acid structures to regulate gene expression offers the advantages of simple engineering and broad applicability. Since the modified sgRNA in such systems can be produced intracellularly via plasmid expression rather than requiring synthetic synthesis, it holds strong potential for in vivo applications. This review primarily summarizes research from the past decade on constructing condition responsive CRISPR/Cas9 systems using aptamers and ribozymes. By engineering the structure of sgRNA—through individual RNA aptamers, ribozymes, or combined aptamer-ribozyme constructs (also called aptazyme)—these systems can respond to specific compounds or proteins in prokaryotic or eukaryotic cells, thereby achieving gene editing, expression regulation, and imaging via the CRISPR/Cas9 system.
Cas9 has been widely adopted for gene editing in a variety of organisms. Similar to other nucleases, Cas9-mediated gene editing is achieved through a two-step process: double-strand DNA breakage followed by DNA repair. The sgRNA guides Cas9 to a specific genomic site, inducing a double-strand break (DSB) [9, 34]. This break subsequently triggers intrinsic cellular DNA repair mechanisms, primarily non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Fig. 1) [35, 36, 37, 38, 39].
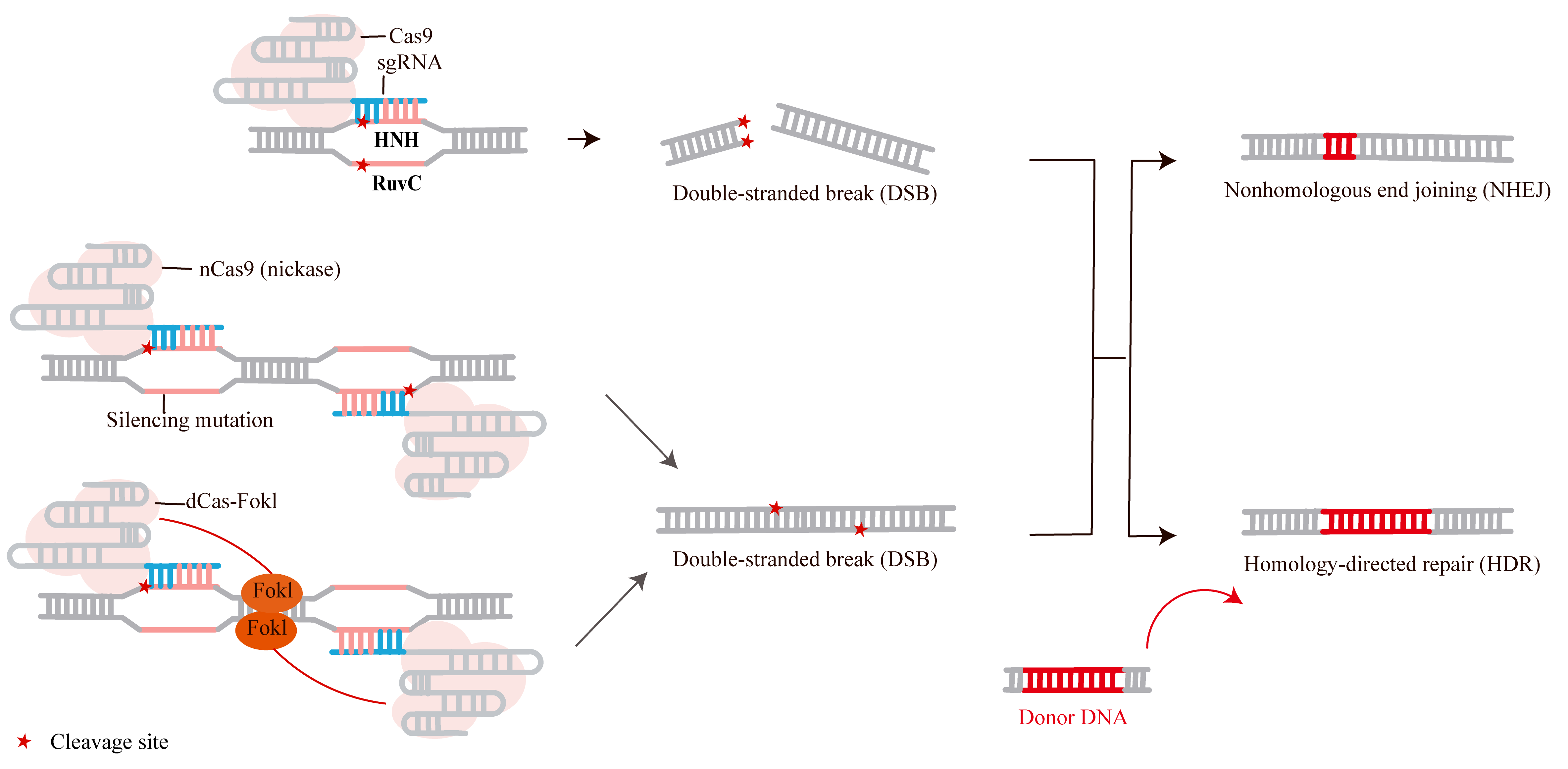 Fig. 1.
Fig. 1.
Cas CRISPR-associated protein 9 (Cas9) introduces a double-strand break (DSB) in the deoxyribonucleic acid (DNA), triggering cellular repair via Non-Homologous End Joining (NHEJ) or Homology-Directed Repair (HDR). NHEJ often results in random insertions or deletions (indels), while HDR can be used to introduce precise genetic modifications using a donor DNA template. FokI is a restriction endonuclease whose nuclease domain must dimerize to cleave DNA. When fused to catalytically inactive nuclease-deactivated Cas9 (dCas9), two dCas9-FokI monomers binding at adjacent sites facilitate FokI dimerization, enabling highly specific DNA cleavage near the target site. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
The NHEJ repair pathway randomly introduces insertion or deletion mutations (indels) at the DSB site. By disrupting the reading frame of the target gene or mutating critical regions of the encoded protein, this process can lead to gene knockout (KO) [40]. In contrast, HDR can introduce precise sequence modifications—such as deletions, mutations, insertions, or gene corrections—at the DSB site using a donor DNA template as a guide [41, 42]. Consequently, the CRISPR/Cas9 system provides a powerful platform for sequence-specific genome editing, enabling diverse applications including gene knockout, gene knock-in (KI), and targeted mutation or correction of specific sequences [3, 43, 44].
Beyond its nuclease activity, Cas9 can serve as a unique platform for recruiting functional proteins and RNA factors to specific DNA target sites. Based on this capability, it has been engineered into a sequence-specific gene regulation tool [10, 11, 45, 46]. To achieve this, transcriptional activators and repressors are fused to a nuclease-deactivated Cas9 (dCas9). dCas9 retains the ability to bind sgRNA and target DNA but lacks nuclease activity, thereby functioning as an RNA-guided, sequence-specific DNA-binding platform.
In bacterial cells as shown in Fig. 2a, dCas9 alone can effectively repress transcription of target genes by sterically hindering the transcription machinery [46, 47]. This approach is referred to as CRISPR interference (CRISPRi). While CRISPRi is generally highly efficient in prokaryotes, the dCas9-sgRNA complex alone is less effective at silencing gene expression in mammalian cells [48]. However, CRISPRi in mammalian cells can be enhanced by fusing transcriptional repressor domains—such as the KRAB domain from Kox1—to dCas9, enabling successful repression of reporter or endogenous genes [45, 49].
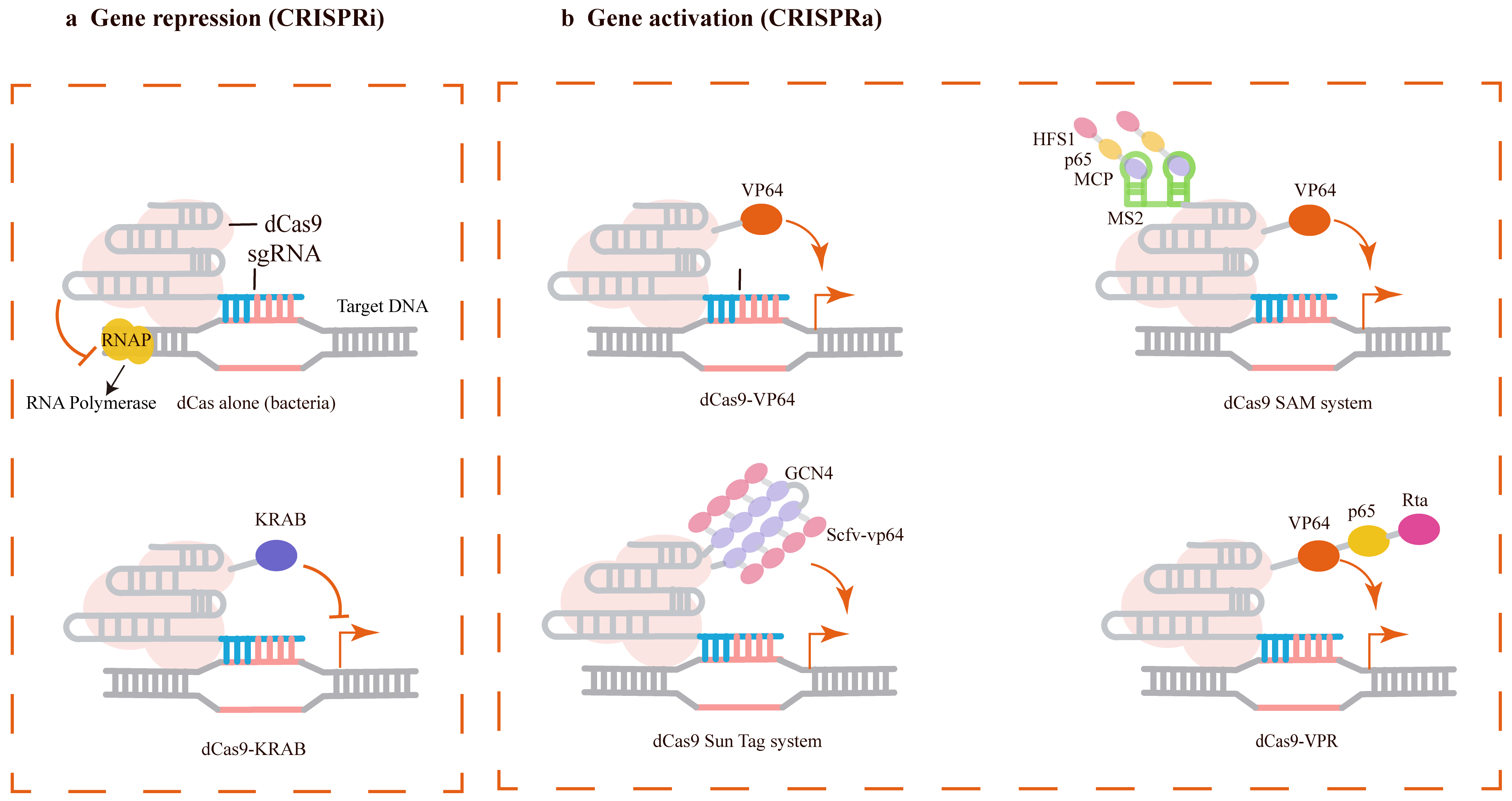 Fig. 2.
Fig. 2.
Sequence-specific gene regulation mediated by nuclease-deactivated Cas9 (dCas9). (a) CRISPRi (interference) strategies: Transcriptional repression is achieved by dCas9 alone through steric hindrance of RNA Polymerase (RNAP) in bacteria or by a dCas9-KRAB fusion for enhanced efficacy. (b) CRISPRa (activation) strategies: Transcriptional activation is accomplished via direct fusion of dCas9 to an activator domain (e.g., VP64) or by multi-activator recruitment systems, including SAM (recruiting multiple transcriptional activators using the synergistic activation mediator), SunTag, and VPR (VP64-p65-Rta). Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
In addition to transcriptional repression, transcriptional activation can be achieved in mammalian cells by fusing activators such as VP64 and p65AD to dCas9 (Fig. 2b) [10, 45, 49, 50]. These fusion proteins increase expression levels of corresponding host genes. Given the importance of transcriptional activation for studying gene function, this system has been continuously improved to achieve higher levels of activation. More advanced systems, including the dCas9 SAM (recruiting multiple transcriptional activators using the synergistic activation mediator) system, dCas9 SunTag system, and dCas9-VP64-p65-Rta (VPR) system, have since been developed [51, 52, 53].
The CRISPR/Cas9 system is regarded as the latest generation of gene editing tools [54]. It is of great significance to clarify how the sgRNA binds to the Cas9 protein and targets the target DNA. A comprehensive understanding of the sgRNA’s structural architecture is not merely descriptive, but fundamental to its rational engineering. The precise three-dimensional arrangement of its loops, stems, and linkers defines both the constraints and opportunities for embedding exogenous functional RNA elements, such as aptamers and ribozymes. Therefore, deconstructing the sgRNA into its core components and evaluating their individual contributions to Cas9 function is a critical prerequisite for informed design. This knowledge allows researchers to strategically target tolerant regions for modifications while avoiding disruptions to the structural integrity essential for activity. The following detailed structural analysis serves to map these “engineering handles” within the sgRNA scaffold.
The sgRNA is composed of sequences derived from both crRNA and tracrRNA, which are linked via an engineered tetraloop (Fig. 3a) [55]. Its architecture features a crRNA segment partitioned into a 20-nucleotide guide region and a 12-nucleotide repeat region, alongside a tracrRNA segment that includes a 14-nucleotide anti-repeat region and three distinct stem loops [56, 57]. Crystallographic analysis shows that upon binding target DNA, the sgRNA adopts a T-shaped structure. This architecture is constituted by a guide:target heteroduplex, a repeat:anti-repeat duplex, and three stem loops (Fig. 3a,c). A single nucleotide (Adenine) serves as the connector between the repeat:anti-repeat duplex and stem loop 1, while a 5-nucleotide single-stranded linker (purple) bridges stem loops 1 and 2 [55].
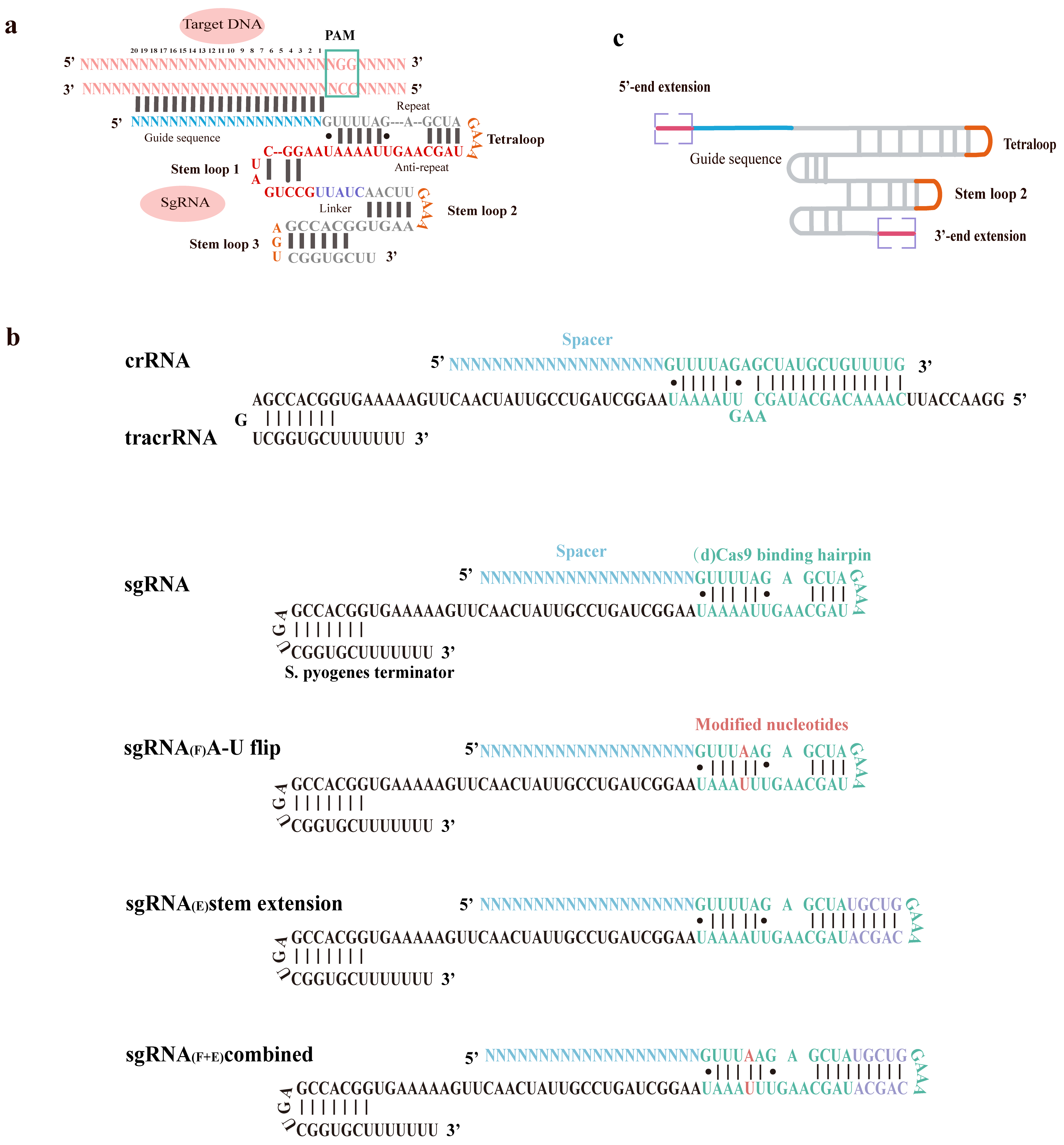 Fig. 3.
Fig. 3.
sgRNA engineering for enhanced functionality and specificity. (a) Structure of the single guide RNA (sgRNA): Target DNA Complex. This schematic illustrates the complex formed between the sgRNA and its target DNA. The crRNA component is divided into a sky-blue guide sequence and a repeat sequence. The tracrRNA is shown in red, with its linker segment highlighted in violet. Target DNA and the tetraloop are colored pink and yellow, respectively. Black lines represent Watson-Crick base pairs. (b) Schematic of the natural crRNA and tracrRNA components, the initial sgRNA and engineered sgRNAs. Depicted are three optimized sgRNA designs: sgRNA(F) with an A-U pair flip; sgRNA(E) with a 5 bp extension of the hairpin; and sgRNA(F+E), a combination of both modifications. (c) Primary sgRNA engineering regions for functional modifications. Four key regions are commonly modified for sgRNA engineering by incorporating aptamers or ribozymes: the 5′-terminus, 3′-terminus, tetraloop and stem loop 2. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
These findings demonstrate that stem loop 1 is indispensable for assembling a functional Cas9-sgRNA complex, while stem loops 2 and 3 play auxiliary roles in stabilizing the complex and boosting sgRNA stability, thereby enhancing its activity in vivo. To evaluate the contribution of each structural element of the sgRNA to Cas9 function, Hiroshi Nishimasu et al. [55] engineered and assayed multiple sgRNA variants containing mutations in the repeat:anti-repeat duplex, stem loops 1 through 3, and the interconnecting linker between stem loops 1 and 2. Experimental results show that stem loops 2 and 3, along with the linker, are permissive to extensive mutagenesis. In contrast, the integrity of the repeat:anti-repeat duplex and stem loop 1 is crucial for Cas9’s activity (Fig. 3a). Furthermore, the sgRNA sequence overall exhibits considerable tolerance to a wide array of mutations (Fig. 3c, reconstructed sgRNA). Collectively, these findings underscore the critical role of Cas9’s structure-specific recognition of the repeat:anti-repeat duplex [55].
As illustrated in Fig. 3b, while optimized sgRNA designs (e.g., with an A-U flip or hairpin extension) exhibit enhanced binding affinity to Cas9 and target DNA, their core architecture is conserved. Consequently, the selection of a specific guide RNA structure for experimental applications can be made flexibly based on the specific requirements of the study [58]. Crystallographic analysis reveals that the nucleotides of Stem loop 1 and stem loop 3 interact directly with the Cas9 protein, whereas the tetraloop and the -GAAA- motif of stem loop 2 are exposed on the protein surface (shown in Fig. 3c). Consequently, the insertion of aptamer or ribozyme sequences into these two structural elements does not interfere with the inherent function of the sgRNA, while simultaneously allowing the incorporation of regulatory modules. Two additional sites for sequence addition are the 5′- and 3′- ends of the sgRNA. Extending the 3′- end has no significant impact on sgRNA function [59]. However, at the 5′- end, the 20 nucleotides preceding the protospacer adjacent motif (PAM) are responsible for target gene recognition; shortening this guide sequence to less than 17 nucleotides significantly impairs the sgRNA’s ability to bind target DNA [60]. While excessive extension of the 5′- end can influence the sgRNA’s efficiency in binding its target DNA sequence, it does not completely abolish its activity [61, 62]. Therefore, researchers primarily focus on these four regions for engineering sgRNA structure by introducing aptamers or ribozymes: 5′- end extension, 3′- end extension, and replacement of the tetraloop and the -GAAA- sequence in stem loop 2.
Table 1 lists common RNA aptamers and ribozymes (Ref. [29, 32, 33, 59, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81]). The listed RNA aptamers (such as theophylline, tetracycline, and 3-methylxanthine) and ribozymes (including hammerhead ribozyme and Csy4 substrate) represent a versatile toolkit for the precise regulation and functional expansion of synthetic RNA systems, particularly sgRNA. These elements enable external control by responding to specific small-molecule ligands to modulate RNA activity or structure, and facilitate the recruitment of effector proteins (e.g., via MS2 and PP7) for tasks like imaging, localization, or translational control. Ribozymes introduce self-cleaving activity for processing RNA transcripts or engineering genetic circuits. Collectively, they are pivotal components for constructing sophisticated, programmable biological tools.
| Aptamer or Ribozyme name | Structure | Sequence (5′-3′) | Specific response substance | Application | Reference |
| Theophylline aptamer | 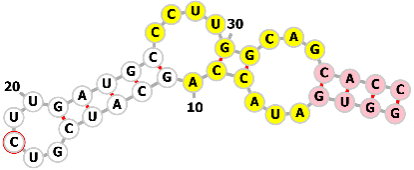 |
GGUGAUACCAGCAUCGUCUUGAUGCCCUUGGCAGCACC | Theophylline | Genome editing | [63, 64, 65, 66] |
| CRISPRa | |||||
| CRISPRi | |||||
| Riboswich | |||||
| Theophylline-dependent ribozyme | 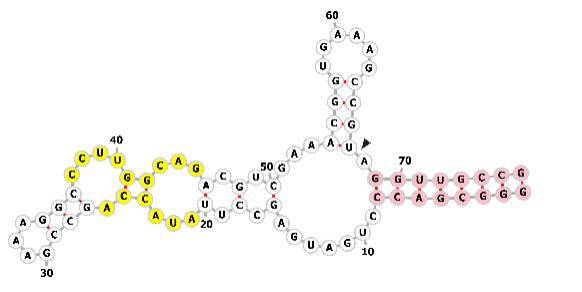 |
GGGCGACCCUGAUGAGCCUUAUACCAGCCGAAAGGCCCUUGGCAGACGUCGAAACGGUGAAAGCCGUAGGUUGCCG | Theophylline | Genome editing | [67, 68, 69] |
| CRISPRa | |||||
| CRISPRi | |||||
| Tetracycline aptamer | 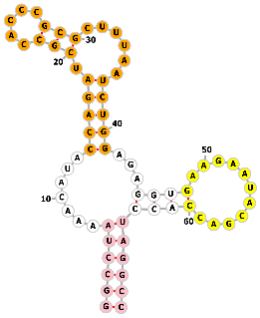 |
Sequence1: GGCCUAAAACAUACCAGAUCGCCACCCGCGCUUUAAUCUGGAGAGGUGAAGAAUACGACCACCUAGGCC | Tetracycline | CRISPRa | [59, 70, 71] |
| CRISPRi | |||||
| Riboswich | |||||
 |
Sequence2: GGCCUAAAACAUACCCAGACGCUUUAUCUGGGAGAGGUGAAGAAUACGACCACCUAGGCC | ||||
| Guanine aptamer | 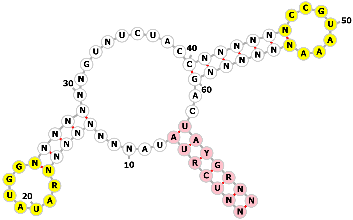 |
Sequence1 (universal): NNUCRUAUANNNNNNNNRAUAUGGNNNNNNNGUNUCUACCNNNNNNCCGUAAANNNNNNGACUAYGRNN | Guanine | None | [68] |
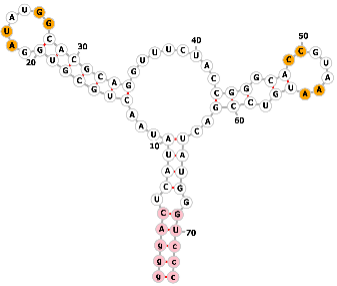 |
Sequence2: GGGACUCAUAUAACUGCGUGGAUAUGGCACGCAGGUUUCUACCGGGCACCGUAAAUGUCCGACUAUGGGUCCC | ||||
| Guanine-dependent ribozyme | 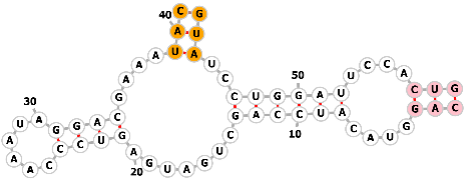 |
Sequence1: CAGGUACAUCCAGCUGAUGAGUCCCAAAUAGGACGAAAUACGUAUCCUGGAUUCCACUG | Guanine | Genome editing | [68, 72, 73] |
| CRISPRa | |||||
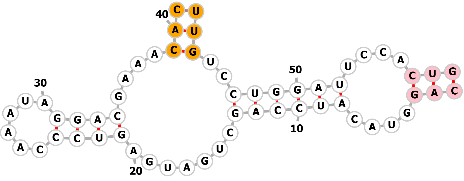 |
Sequence2: CAGGUACAUCCAGCUGAUGAGUCCCAAAUAGGACGAAACACUUGUCCUGGAUUCCACUG | ||||
| 3-methylxanthine aptamer | 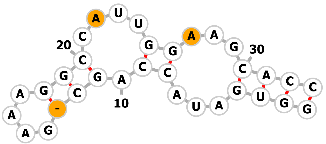 |
Sequence1: AUACCAGC-GAAAGGCCAUUGGAAG | 3-methylxanthine | Genome editing | [74] |
 |
Sequence2: AUACCAGCCGAAAGGCCAUUGGCAG | ||||
| S-adenosyl methionine aptamer | 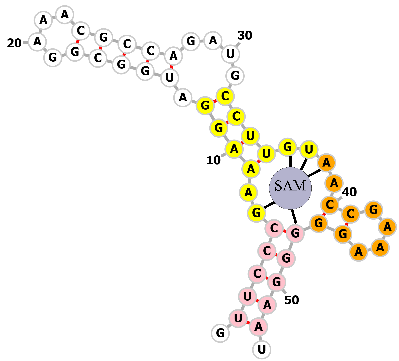 |
GUUCCCGAAAGGAUGGCGGAAACGCCAGAUGCCUUGUAACCGAAAGGGGGAAU | S-adenosyl methionine (SAM) | CRISPRa | [75] |
| MS2 aptamer |  |
ACAUGAGGAUUACCCAUGU | MS2 | chromatin imaging | [29, 76] |
| PP7 aptamer |  |
GGCACAGAAGAUAUGGCUUCGUGCC | PP7 | ||
| S1m aptamer | 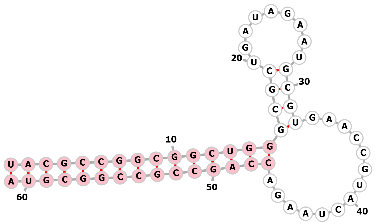 |
UACGCCGGCGGCUGGGCGCUGAUAGAAUGCGUGAACCGUACUAAGACCAGCCGCCGGCGUA | streptavidin | Genome editing | [77, 78] |
| Hammerhead ribozyme (HHR) | 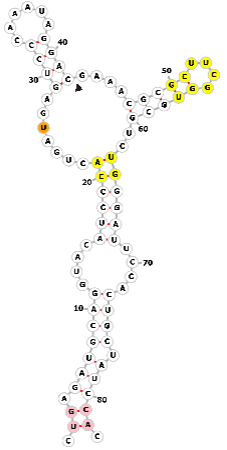 |
CUGAGAUGCAGGUACAUCCCACUGAUGAGUCCCAAAUAGGACGAAACGCGCUUCGGUGCGUCUGGGAUUCCACUGCUAUCCAC | None | Genome editing | [32, 33, 79] |
| CRISPRa | |||||
| CRISPRi | |||||
| Riboswich | |||||
| Csy4 RNA | 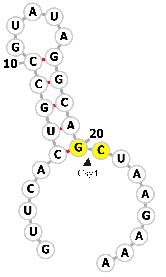 |
GUUCACUGCCGUAUAGGCAGCUAAGAAA | Pseudomonas aeruginosa endoribonuclease (Csy4) | Genome editing | [80, 81] |
The CRISPR-Cas9 system is widely used for gene editing, transcriptional regulation, and imaging due to its ability to target any DNA sequence via the sgRNA. However, most existing regulation methods focus on controlling the Cas9 protein, resulting in synchronous regulation of all target genes and lacking the capability for independent spatiotemporal control of multiple genes [82, 83, 84]. Kale Kundert et al. [66] proposed engineering the sgRNA itself to achieve ligand-dependent activation or inhibition of CRISPR function, thereby providing a new tool for studying complex biological processes (Fig. 4a).
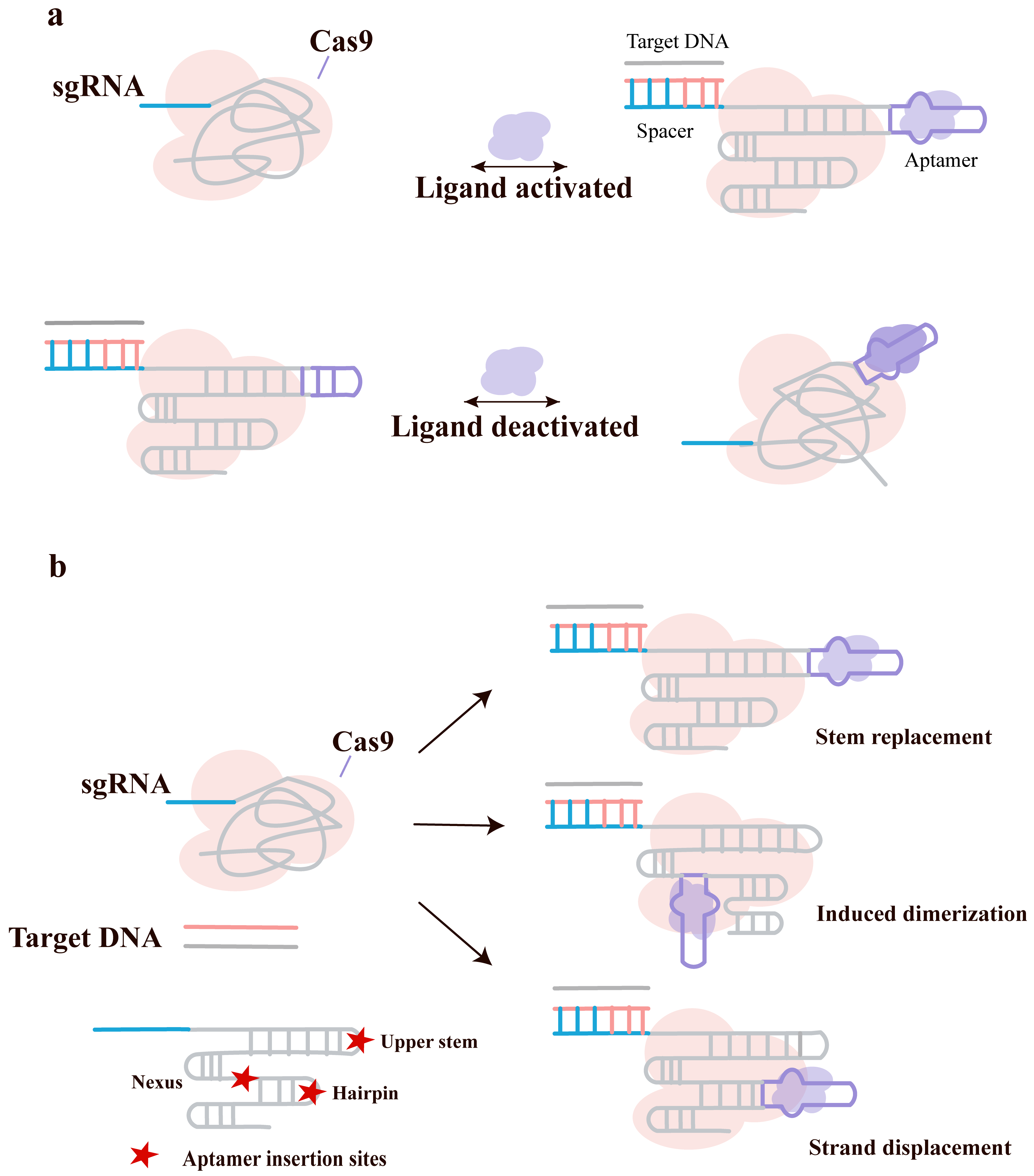 Fig. 4.
Fig. 4.
Engineering ligand-responsive sgRNAs through the modular integration of small-molecule aptamers. (a) Schematic depicting the design strategy to stabilize the functional Cas9/sgRNA/target DNA complex either in the presence (top) or absence (bottom) of a specific small-molecule ligand. (b) Potential sites for aptamer insertion within the sgRNA scaffold, with structural domains annotated as defined with upper stem, nexus and hairpin with proposed methods for the covalent attachment of the aptamer to the sgRNA. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
They inserted theophylline aptamers into multiple regions of the sgRNA (such as
the upper stem, linker regions, and hairpin structures) as shown in Fig. 4b,
employing three connection strategies: replacing part of the stem structure,
splitting the sgRNA and connecting it with aptamers, and designing
strand-displacement structures. Through in vitro DNA cleavage assays,
they screened 10 sgRNA designs responsive to theophylline, nine of which were
ligand-activated and one was ligand-inhibited. Using a CRISPRi-based fluorescent
reporter system and fluorescence-activated cell sorting, they identified ligRNA variants that activate or inhibit in the presence of theophylline: ligRNA+
denotes activation of CRISPRi when the ligand is present, while ligRNA–
denotes inhibition of CRISPRi when the ligand is present. Both exhibited a
dynamic range of
The proposed response mechanisms for these engineered sgRNAs include: ligand binding stabilizing the active conformation and promoting Cas9 binding to DNA (ligRNA+); and ligand binding potentially affecting the base-pairing status of A-U in stem loop 3, thereby interfering with Cas9 function (ligRNA–). Both designs exhibited a linear response to theophylline concentration changes within a certain range, with fast response times (from several minutes to over ten minutes)—significantly faster than the residence time of dCas9 on DNA. Furthermore, the designs were effective for most tested spacer sequences, indicating that ligRNAs can be used to regulate most genes. Successful application was demonstrated for endogenous genes in E. coli (e.g., the lac operon) and in another bacterium, Pseudomonas aeruginosa, confirming the broad applicability of this strategy. Additionally, by replacing the aptamer (e.g., with 3-methylxanthine or thiamine pyrophosphate), independent regulation of different genes was achieved, enabling temporal and reversible control of gene expression programs within the same cell.
Meanwhile, the team led by Iwasaki et al. [85] proposed and validated a novel small-molecule-regulated sgRNA for achieving time- and dose-dependent control of CRISPR-Cas9 gene editing in E. coli. Their strategy involved inserting a small-molecule-binding aptamer into the tetraloop region of the sgRNA, creating an “aptamer-gRNA” (agRNA) (Fig. 5a) [85]. Specifically, by randomizing the internal loop and adjacent helical regions, they constructed an agRNA library and employed an in vivo screening strategy to isolate agRNA variants with low background and high inducibility as shown in Fig. 5b. Using a survival selection system based on the galK gene, along with positive and negative selection cycles, they enriched for agRNAs exhibiting theophylline dependence. After multiple rounds of screening, several highly efficient and inducible agRNAs (such as A9 and GU19) were identified and their applicability across different genomic loci was verified (Fig. 5c).
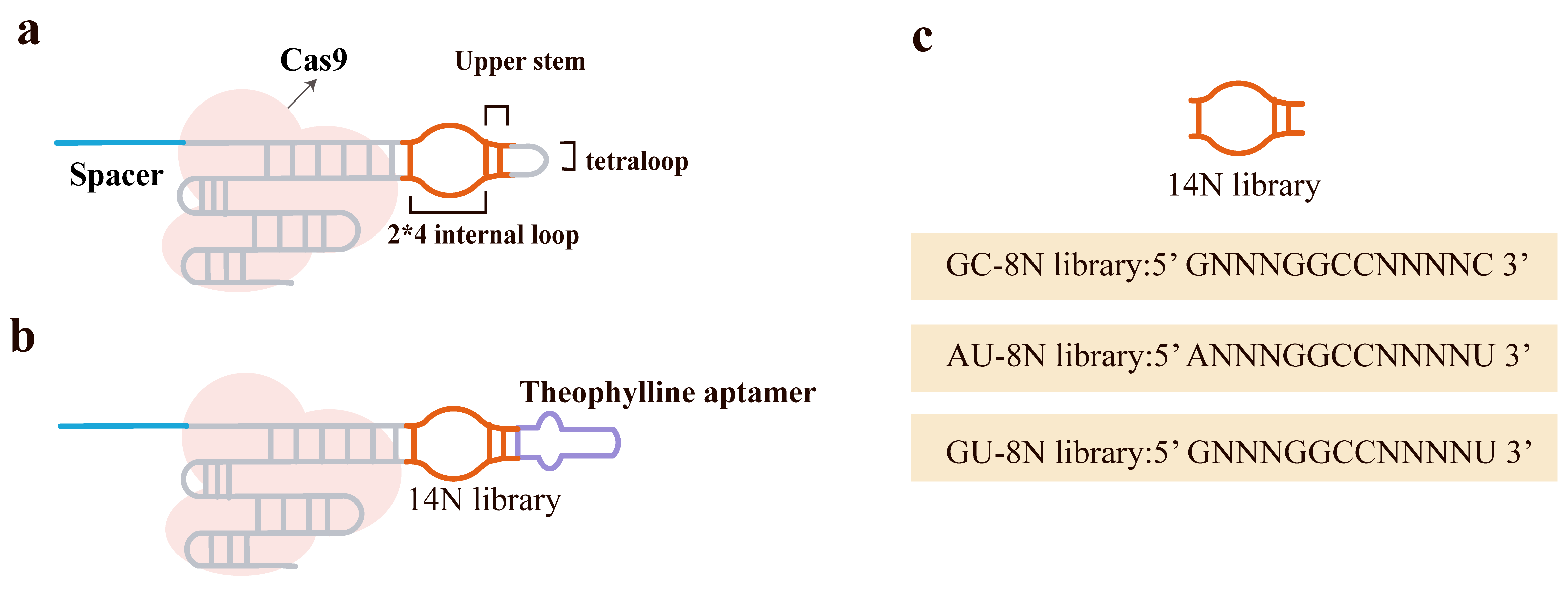 Fig. 5.
Fig. 5.
Design, construction, and functional screening of aptamer-gRNA
(agRNA) libraries. (a) The theophylline aptamer was inserted into the sgRNA
scaffold, replacing the native tetraloop that connects the guide and tracrRNA
segments. (b) An agRNA library was constructed by randomizing the 2
The agRNAs were activated upon addition of theophylline or 3-methylxanthine (3MX), significantly enhancing editing efficiency. This efficiency increased with higher concentrations of the small molecule and longer exposure times. By introducing point mutations to convert the theophylline aptamer into a 3MX aptamer, they further expanded the diversity of the regulatory toolbox. This strategy improved transformation and editing efficiency: the use of agRNAs increased transformation efficiency by 104-fold while maintaining editing efficiency as high as 80%. In the presence of non-functional sgRNAs, the agRNA system significantly reduced the enrichment of non-edited cells, thereby improving the representation of edited libraries. The study also provided insights into the mechanism of small-molecule regulation of CRISPR/Cas9: Electrophoretic mobility shift assay (EMSA) experiments indicated that the binding of agRNA to Cas9 was not affected by the small molecule, suggesting that regulation occurs at the stage of DNA recognition and cleavage, not during ribonucleoprotein (RNP) assembly. Furthermore, in vitro cleavage assays showed that the small molecule enhanced the nuclease activity of Cas9, although the induction range was narrower than observed in vivo, hinting at the significant influence of the cellular environment on tool performance. Finally, by designing a plasmid carrying agRNAs responsive to both theophylline and 3MX regulation, they achieved multi-locus, sequential editing in a single experiment through the induction of different small molecules, overcoming the issue of cell death associated with multiplexed editing.
Under non-viral delivery conditions, the CRISPR-Cas9 system often results in a high proportion of imprecise edits (such as insertions/deletions, or indels) rather than the desired precise homology-directed repair (HDR). Conventional methods involving the co-delivery of ribonucleoprotein (RNP) complexes with donor templates frequently suffer from uneven delivery and low editing efficiency. Carlson-Stevermer et al. [78] designed a modular RNA aptamer-streptavidin strategy termed Simplex (Fig. 6a). Its core design is the S1m-sgRNA, created by inserting the S1m RNA aptamer into specific stem-loop structures of the sgRNA (the Tetraloop, Stem loop 2, and the 3′ extension), enabling high-affinity binding to streptavidin as shown in Fig. 6b. Streptavidin acts as a bridge to connect the S1m-sgRNA to biotinylated molecules. By 5′-terminal biotin modification single-stranded oligodeoxynucleotides synthesized through DNA Synthesizer (ssODNs, donor templates for HDR), a complete RNP complex comprising the sgRNA, Cas9 protein, and single-stranded oligodeoxynucleotide (ssODN) is formed. Simplex can be pre-assembled in vitro into unified nanoparticles, ensuring the co-delivery of the RNP and its donor template in a defined stoichiometric ratio.
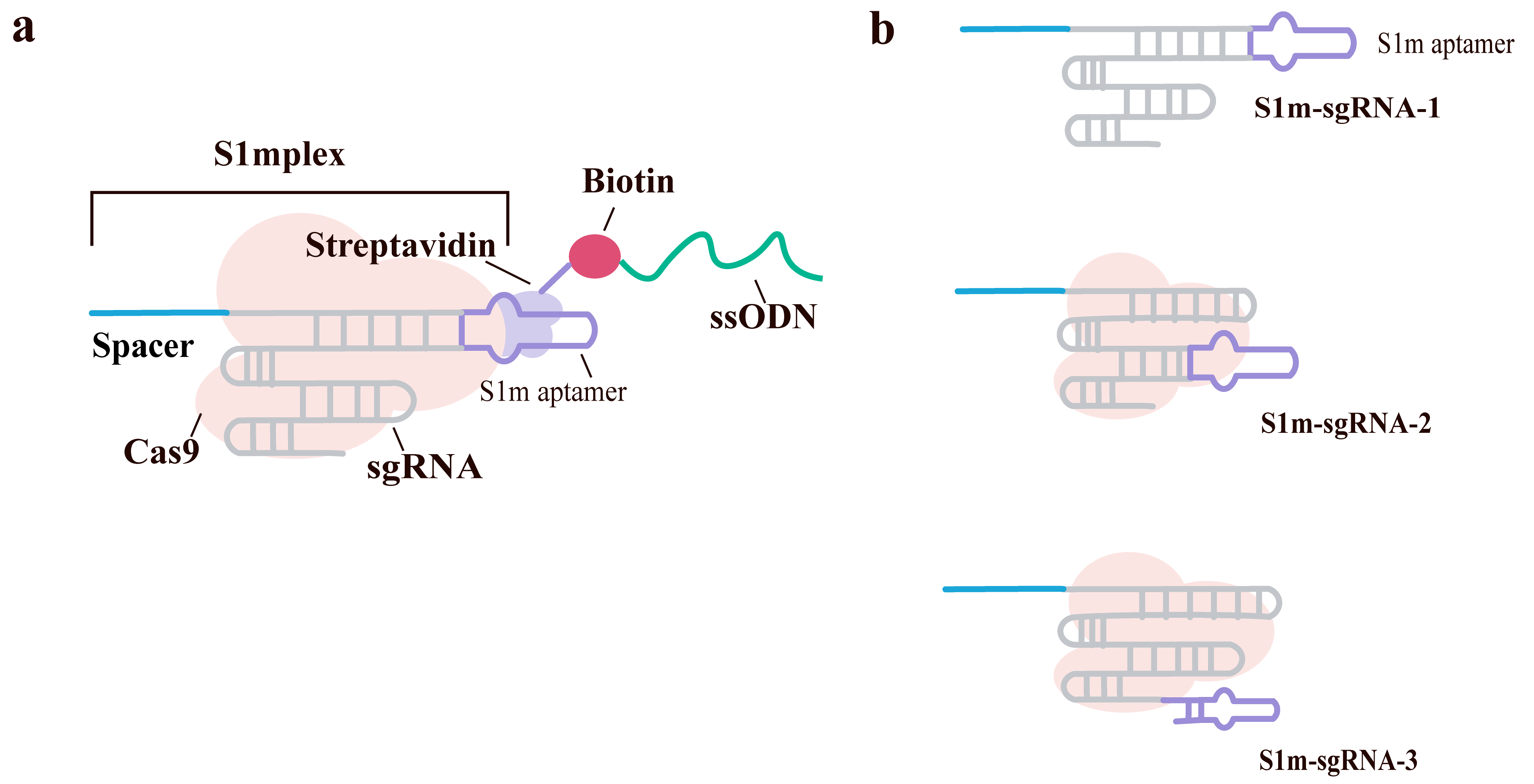 Fig. 6.
Fig. 6.
Design of S1mplex donor complexes for SpCas9-mediated HDR. (a) Schematic of the pre-assembled ssODN-S1mplex, a multi-component complex comprising SpCas9 protein, an S1m-modified sgRNA, streptavidin, and a single-stranded oligodeoxynucleotide (ssODN) donor template. The S1m aptamer, inserted into a specific stem-loop of the sgRNA, recruits streptavidin, which in turn binds to a biotinylated ssODN. This complex is designed to enhance homology-directed repair (HDR) at the target site. (b) Sequence and secondary structure of the individual S1m-sgRNAs. The protospacer sequence (blue) determines genomic targeting, while the S1m loop (purple) facilitates streptavidin binding. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
The primary advantage of this strategy is the significant enhancement of the precise-to-imprecise editing ratio. Across various human cell lines (such as HEK293T and hPSCs) and multiple genomic loci (e.g., BFP, EMX1, GAA), Simplex increased this ratio by up to 18-fold, achieving nearly 4:1 in some cases. Notably, Simplex also demonstrated robust and highly precise editing capability when correcting the pathogenic GAA mutation. Experiments using dynamic light scattering, EMSA, and confocal microscopy confirmed that Simplex complexes remain stable both in vitro and inside cells, and facilitate the co-localization of Cas9 and the donor template within the nucleus. Furthermore, Simplex can be loaded with biotinylated molecules such as quantum dots or fluorophores for applications like fluorescence-activated cell sorting (FACS) to enrich successfully edited cells. Using different colored fluorescent markers, multiplexed gene editing was achieved, allowing for the isolation of highly pure edited clones via FACS. Analysis of multiple potential off-target sites showed no significant detection of off-target editing.
The team led by Lin et al. [65] developed a simple, versatile, and non-invasive regulatory strategy—SMART-sgRNA as shown in Fig. 7a. This approach leaves the original sgRNA’s guide function intact but incorporates a blocking motif (complementary to a stem region of the sgRNA, forming a stable duplex that prevents its binding to Cas9) and a triggering motif (an aptamer sequence, such as the theophylline RNA aptamer, introduced at the 3′ end of the sgRNA). In the absence of the small molecule, the blocking motif binds the sgRNA, rendering the system inactive. Upon addition of the small molecule, the aptamer undergoes a structural change, disrupting its interaction with the blocking motif and releasing the functional sgRNA, which can then bind Cas9 and activate gene editing (Fig. 7b) [65].
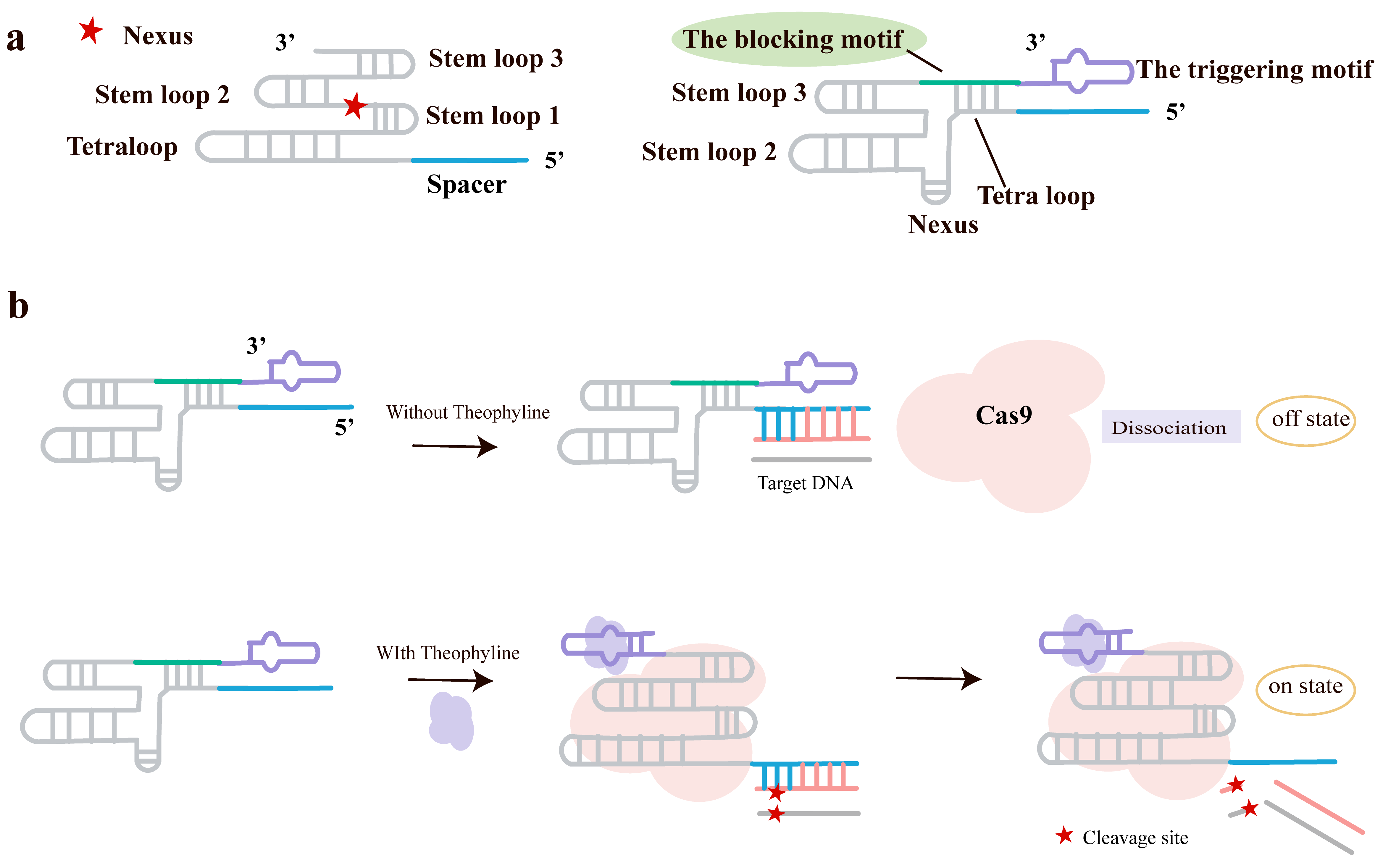 Fig. 7.
Fig. 7.
Allosteric control of Cas9 by SMART-sgRNAs. (a) Components of the SMART-sgRNA. The chimeric RNA is created by 3′-end extension of the original sgRNA with two functional modules: a blocking sequence complementary to the sgRNA stem regions, and an allosteric aptamer serving as the triggering motif. (b) Working model of conditional activation. State I (OFF): The blocking motif hybridizes with the sgRNA, occluding the Cas9 binding site. State II (ON): Ligand-binding to the aptamer induces a conformational switch, through competitive hybridization, that dissociates the blocking motif and restores the functional sgRNA structure for Cas9 complex formation and DNA cleavage. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
In vitro experiments optimized the lengths of the blocking motif and the aptamer structure, identifying sgB18-A30-S7 as achieving the optimal balance between blocking efficiency and theophylline-induced activation efficiency. Furthermore, in HEK-293T cells, using sgB18-A30-S7 to regulate luciferase gene expression successfully demonstrated theophylline-dependent activation of gene editing, consistent with the in vitro results. Applying this strategy to both the luciferase and TurboRFP genes required only swapping the guide sequence (the first 20 nucleotides) of the sgRNA, without the need to redesign the blocking structure, highlighting its broad generality.
SMART-sgRNA provides a simple, universal, and efficient method for temporal control of CRISPR-Cas9. This strategy does not alter the Cas9 protein and achieves regulation merely by extending the sgRNA sequence, thereby maximally preserving its native function. In the future, incorporating different aptamers could enable responsiveness to various small molecules (e.g., metabolites, drugs), making SMART-sgRNA suitable for diverse applications including multiplexed gene regulation, drug screening, and chromosome imaging.
The complex phenotypes of eukaryotic cells are regulated by signaling pathways and decision-making circuits, but there has been a lack of efficient and programmable tools for constructing artificial signal connections. Researchers aim to develop synthetic devices capable of sensing diverse biological signals and regulating gene expression to reprogram cell behavior. The team led by Liu et al. [59] proposed and validated a new synthetic biology tool based on the CRISPR-Cas9 system—the “signal conductor”—which enables the sensing and processing of external or internal biological signals and regulates the expression of endogenous genes. Its design principle involves integrating signal-responsive riboswitches into the 3′-end of the sgRNA, allowing conformational changes upon binding specific signals (such as small molecules or proteins), thereby activating or inhibiting dCas9/dCas9-VP64-mediated regulation of target genes as shown in Fig. 8a,b. In the absence of a signal, the guide region of the sgRNA is sequestered by an antisense stem structure, preventing DNA binding. Conversely, in the presence of a signal, the signal molecule binds to the aptamer, releasing the guide region to bind target DNA and subsequently regulate transcription [59].
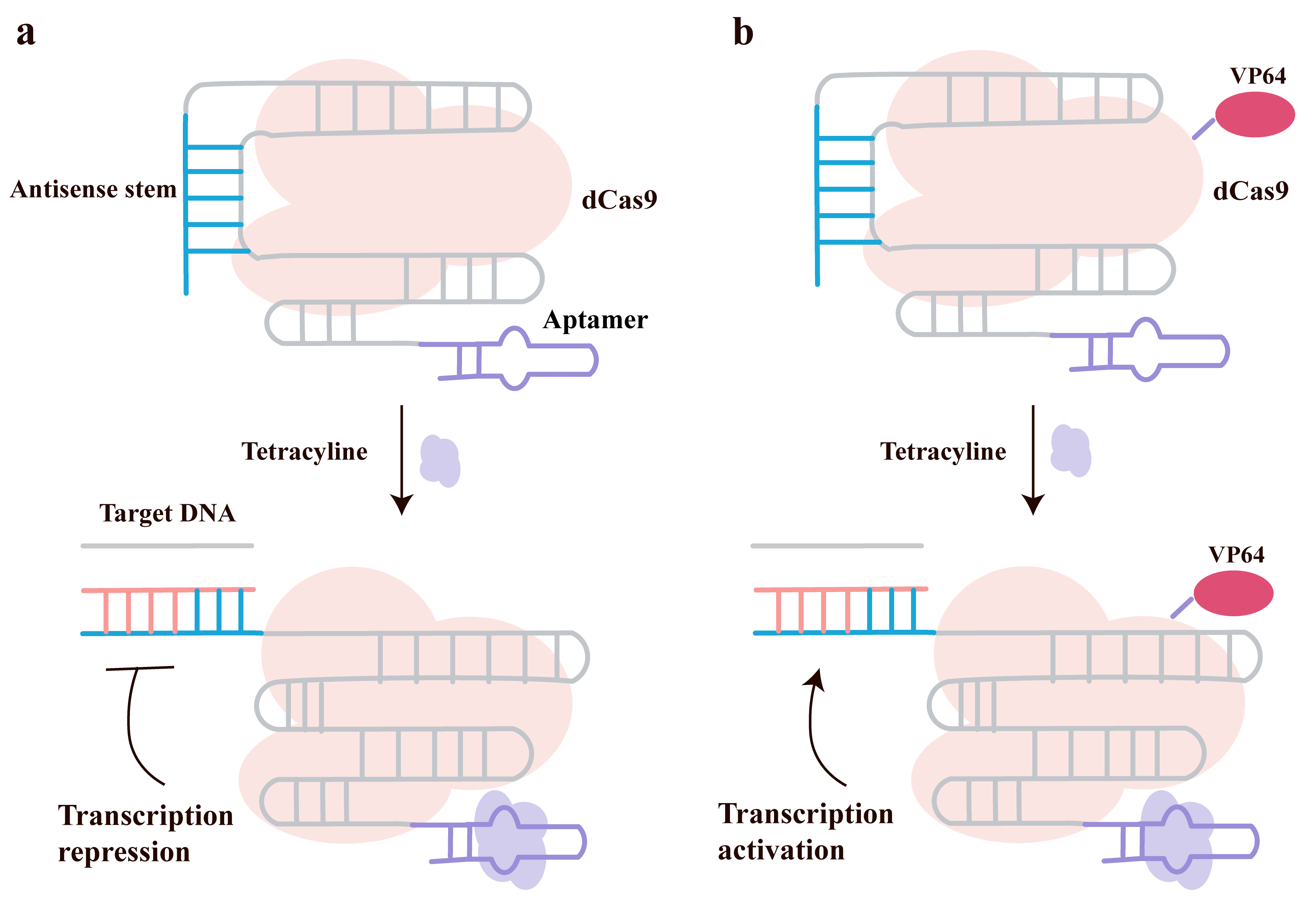 Fig. 8.
Fig. 8.
CRISPR-Cas9-based signal transducer for programmable gene regulation. (a) Signal-induced gene repression: A redesigned sgRNA, incorporating an aptamer stem (purple), remains in an ‘off’ state when tetracyline is absent. In this state, its guide sequence (blue) is sequestered by an antisense stem. Upon binding tetracyline, a strand-displacement mechanism switches the sgRNA to the ‘on’ state, freeing the guide sequence to direct a dCas9 protein to the target gene and repress expression. (b) Signal-induced gene activation: The same conformational switching principle is applied to turn on production. In the presence of tetracyline, the sgRNA is activated via strand displacement, enabling a dCas9-VP64 fusion protein (with an activator domain) to bind the target DNA and activate transcription. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
In their study, the team successfully used small molecule signals such as
tetracycline and theophylline to regulate the expression of genes like vascular
endothelial growth factor (VEGF) in HEK-293T cells, demonstrating dose dependence
and a high dynamic range, thereby achieving regulation of endogenous genes. By
combining different signal conductors, they implemented all basic logic gate
functions, including NOT, AND, OR, NAND, NOR, XOR, and XNOR, showcasing the
capability for logical operations in mammalian cells. Furthermore, they
constructed an artificial bidirectional connection between the
This strategy features a modular design (using sgRNA as a universal scaffold for signal sensing and gene regulation) and high flexibility, enabling responses to multiple signals (small molecules, proteins) and bidirectional (activation/inhibition) regulation. Additionally, complex logic functions can be achieved with a single sgRNA without the need for multi-layer circuits. It holds strong potential for clinical applications, such as cancer therapy and cell fate reprogramming.
The team led by Liu et al. [86] proposed a ligand-inducible gene regulation strategy based on the combination of a theophylline aptamer and the CRISPR/Cas9 system, achieving precise control of gRNA function through synergistic allosteric regulation. The authors introduced a minimally invasive structural modification strategy: inserting the theophylline aptamer into non-Cas9-recognized but structurally critical regions of the gRNA—the tetraloop and stem loop 2. They designed a communication module (CM) as a structural bridge between the gRNA and the aptamer (Fig. 9a). This module is unstable in the absence of the ligand but stabilizes upon ligand binding, thereby regulating gRNA activity (Fig. 9b). Structural analysis ensured that the modifications did not affect Cas9/gRNA complex formation but only affected its DNA-binding capability [86].
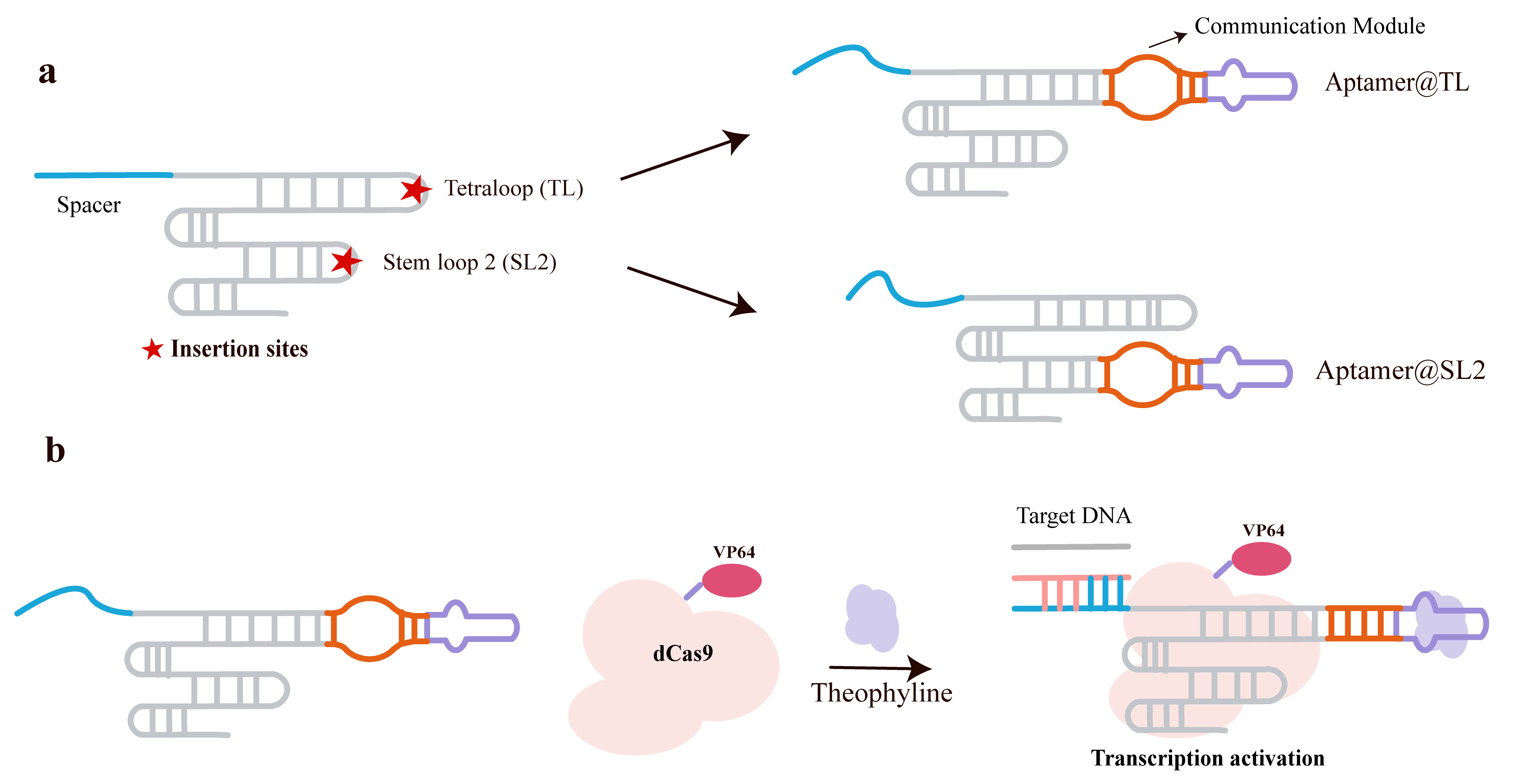 Fig. 9.
Fig. 9.
Synergistic regulation of guide RNA (gRNA) by integrating a theophylline aptamer into permissive sites of the CRISPR-Cas9 complex. (a) Secondary structure of a conventional single-guide RNA (sgRNA). Regions located outside the Cas9 protein binding pocket are marked with stars. Engineering of theophylline-responsive gRNAs by replacing the boxed regions with the theophylline aptamer (core scaffold in purple), generating two distinct designs (aptamer@TL and aptamer@SL2). The communication modules (CMs), which facilitate interplay between the gRNA and the aptamer, are indicated by yellow boxes. (b) Mechanism of the synergistic gRNA regulation employing the aptamer@TL design. Theophylline binding shifts the aptamer from a disordered, inactive state to a structured, active one, enabling the communication module to assemble a functional Cas9/gRNA/DNA complex. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
In in vitro binding experiments, the theophylline aptamer was introduced into the tetraloop and stem loop 2 positions, testing six CMs of different lengths. CM-3 at the tetraloop position and CM-2 and CM-3 at the stem loop 2 position showed the best responsiveness. Dose-dependent experiments demonstrated that ternary complex formation increased with theophylline concentration, showing clear dose dependence. Subsequent intracellular gene regulation experiments were conducted in HEK293T cells using the CRISPRa system to activate endogenous genes (such as ASCL1 and CXCR4). Theophylline treatment significantly enhanced gene expression (from ~3-fold to ~22-fold) with concentration-dependent regulation. Although some background leakage was observed, the dynamic regulatory range was broad, proving the strategy’s effectiveness in living cells.
The elegance of this synergistic allosteric design lies in achieving ligand control over CRISPR/Cas9 function through minimal modifications to non-core regions of the gRNA. It offers strong generality (the design is not target-sequence-specific and can be extended to other genes) and high expandability (besides the theophylline aptamer, other RNA aptamers can be similarly incorporated for multiplexed ligand control). Additionally, it enables dose-dependent gene regulation both in vitro and in cells, providing a new tool for precise gene editing and synthetic biology.
The team led by Liu et al. [87] developed a novel inducible CRISPR-dCas9 transcription activation system that enables quantitative detection and imaging of the endogenous metabolite S-adenosylmethionine (SAM) in living cells through small molecule-mediated split aptamer assembly (Fig. 10a). The strategy involves splitting the SAM aptamer into two fragments: Frag1 inserted into the tetraloop region of the gRNA, and Frag2 inserted into the loop region of the MS2 RNA motif. When SAM is present, as shown in Fig. 10b, the two fragments reassemble, recruiting the MCP-VPR transcription activator to the dCas9-gRNA complex, thereby activating the expression of downstream reporter genes (such as the near-infrared fluorescent protein iRFP670).
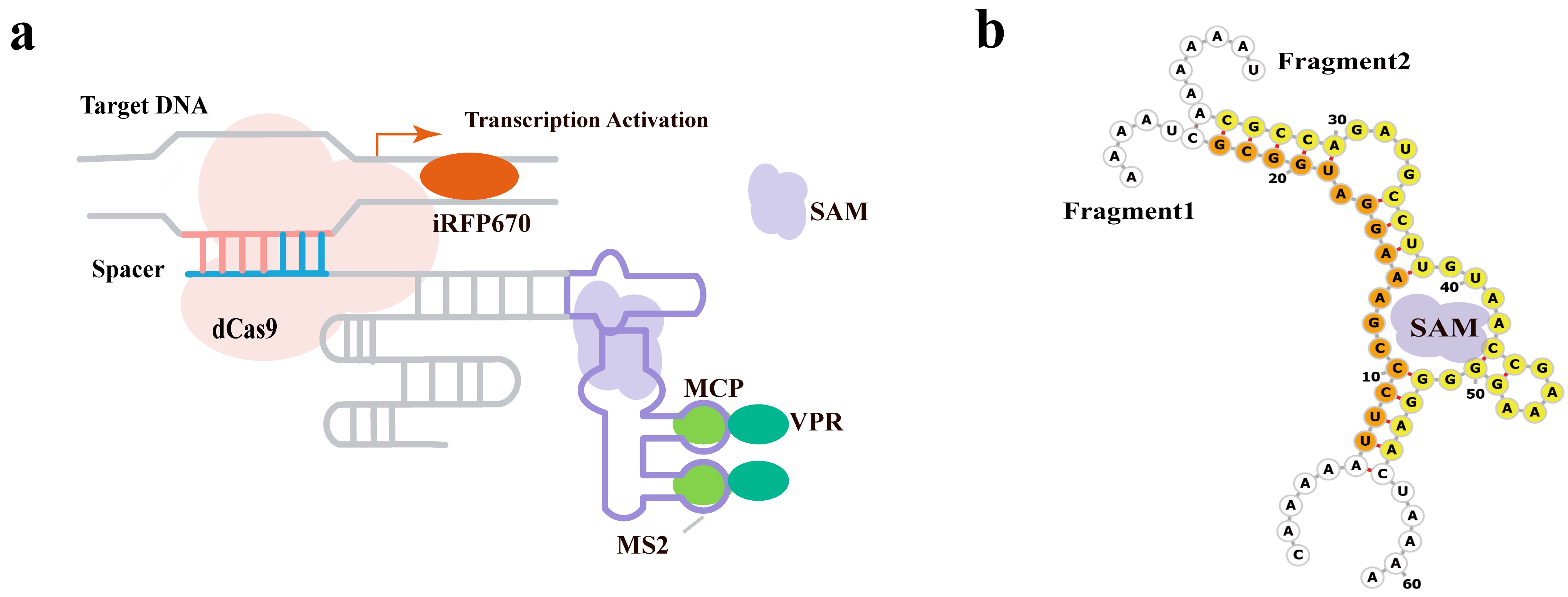 Fig. 10.
Fig. 10.
A SAM-inducible transcription activation system based on small molecule-mediated split-aptamer assembly. (a) Schematic of split-aptamer-based inducible Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) transcription system for SAM imaging. (b) Partial sequences for fragment 1 and fragment 2. This design enables live-cell small-molecule sensing by reconstituting the CRISPR-dCas9 activator complex in a ligand-dependent manner. The system components are spatially separated in the absence of S-adenosyl methionine (SAM): one fragment of a split SAM aptamer is integrated into the gRNA, while the other is attached to MS2 RNA arrays that bind an MCP-fused transcription activator. This separation results in minimal background reporter expression. Upon SAM binding, the split aptamer reassembles, tethering the activator to the dCas9-gRNA complex and driving robust expression of the fluorescent reporter gene for imaging. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
The team first validated the feasibility of split aptamer assembly in vitro. Then, they constructed a signal-amplified transcription system in living cells: by increasing the number of MS2 repeats and target sites (TS), the fluorescence signal was significantly enhanced. The system successfully activated iRFP670 expression in HEK293T cells with very low background expression. SAM-induced transcription activation experiments showed that the system is highly specific to SAM, and its activity could be enhanced by exogenous SAM or inhibited by the SAM synthesis inhibitor cycloleucine. Dynamic response experiments indicated that the system responds to SAM changes within 2–4 hours. The system was used to quantitatively detect endogenous SAM levels, successfully imaging SAM in different cell lines (HEK-293T, HeLa, HepG2, MCF-7), with results consistent with UPLC–MS/MS quantitative data. Additionally, the team investigated the synthesis mechanism of intracellular SAM: Small interfering RNA (siRNA) knockdown experiments showed that MAT2A is the primary enzyme for SAM synthesis in HEK293T. Overexpression of either MAT1A or MAT2A increased SAM levels, with MAT1A being more efficient. Epigenetic drugs (SAHA and 5-azacytidine) upregulated MAT1A messenger RNA (mRNA), thereby increasing SAM levels in cancer cells.
This study is the first to apply the split aptamer assembly strategy to the CRISPR-dCas9 system, achieving inducible, quantitative, and dynamic imaging of the small molecule metabolite SAM. The system offers advantages such as low background, high specificity, and scalability, providing a new tool for studying the regulation of gene expression by endogenous metabolites. In the future, by replacing other aptamers, the system can be extended to detect and regulate more small molecules or metabolites.
Tang et al. [68] proposed and validated a new strategy for regulating the activity of the CRISPR-Cas9 system using small molecules. The research team designed a structure called agRNA, as shown in Fig. 11, which embeds an aptazyme—a combination of a self-cleaving ribozyme and an aptamer—into the sgRNA. The core design involves introducing an RNA sequence complementary to the spacer region at the 5′-end of the sgRNA, forming a secondary structure that inhibits sgRNA function in the absence of a ligand, known as the blocking sequence. Additionally, when a specific small molecule (such as theophylline or guanine) is present, the aptazyme is activated, undergoes self-cleavage, releases the blocking sequence, and thereby restores sgRNA activity [68].
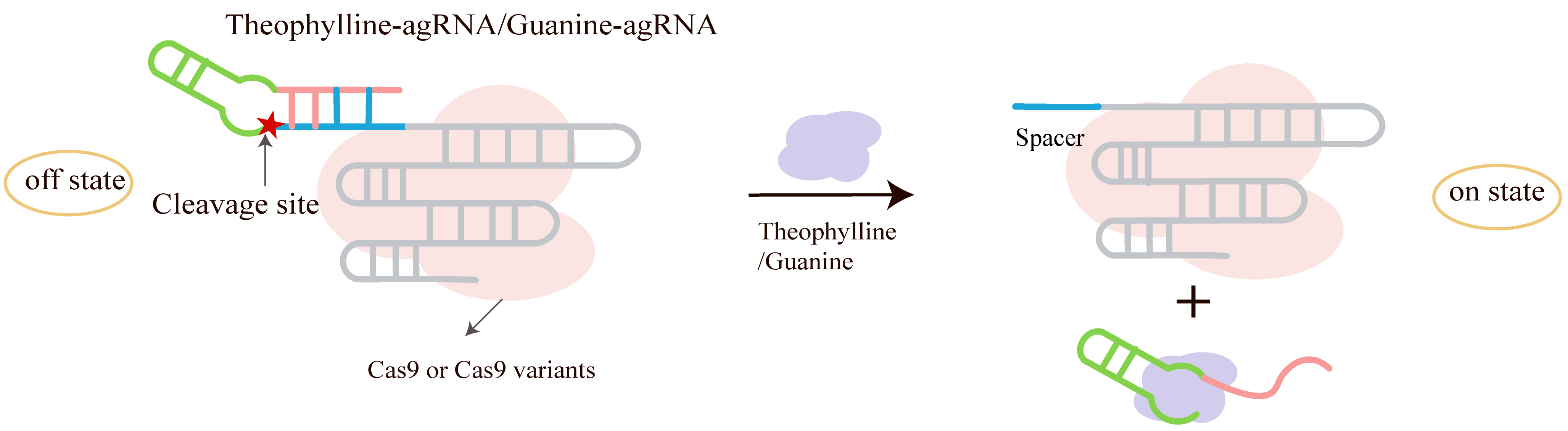 Fig. 11.
Fig. 11.
Engineering ligand control into gRNA activation using a theophylline or guanine-dependent aptazyme. The hammerhead ribozyme was replaced with an aptazyme, a ribozyme fused to a theophylline- or guanine-binding aptamer, to create theophylline-agRNA. This design transduces ligand binding into self-cleavage activity. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
First, they validated the blocking strategy of this design by introducing a complementary blocking sequence into the spacer region of the sgRNA, successfully inhibiting Cas9 cleavage activity. They also compared different blocking positions (spacer region vs. crRNA-tracrRNA binding region) and found that blocking at the 5′-end spacer was most effective. Then, they verified the restoration of activity by the Hammerhead ribozyme. By inserting the hammerhead ribozyme between the blocking sequence and the sgRNA, post-transcriptional self-cleavage was achieved, restoring sgRNA function. A “dead” version (dHHR-bsgRNA) with a mutated ribozyme active center was constructed, confirming that self-cleavage is key to restoring activity. Subsequently, this strategy was successfully applied to achieve small-molecule-regulated genome editing and transcriptional activation. For regulated genome editing, a theophylline-dependent aptazyme was embedded into the sgRNA to construct theophylline-agRNA, enabling small-molecule-dependent Cas9-mediated gene knockout and base editing (using the BE3 system). This was validated at multiple endogenous sites (such as HEK-3 and FANCF), confirming its feasibility and specificity. For regulated transcriptional activation, a guanine-dependent aptazyme was used to construct guanine-agRNA, which, combined with the dCas9-VPR system, achieved small-molecule-dependent transcriptional activation of GFP or RFP genes. Significant fluorescence enhancement was observed in HEK293T cells. Furthermore, the team optimized the strategy by introducing a “bulge” structure into the blocking sequence to improve the activation/inhibition ratio. The universality of agRNA was also validated across different targets and reporter systems.
The advantages of this strategy include the ability to respond to different small molecules by changing the aptazyme type, enabling modular design of responsive systems. This system achieves regulation solely through sgRNA engineering without requiring modification of the Cas9 protein, facilitating broad application. Additionally, the strategy is applicable to various CRISPR applications, including gene knockout, base editing, and transcriptional activation. Due to the strong specificity of the aptamer, the system shows weak responses to analogs (such as 3-methylxanthine), demonstrating excellent regulatory specificity.
Ferry et al. [81] proposed a novel, modular inducible CRISPR transcription regulation system, providing a flexible and efficient gene regulation tool for synthetic biology in mammalian cells. The researchers introduced a strategy based on single-guide RNA (sgRNA) engineering, called the “spacer-blocking hairpin (SBH)”: a “back-fold” sequence complementary to the spacer is introduced at the 5′-end of the sgRNA, forming a hairpin structure that prevents the spacer from binding to the target DNA, thereby completely inhibiting CRISPR Transcriptional Regulation (CRISPR-TR) activity (Fig. 12a). By replacing the connecting loop of the SBH with an RNA unit cleavable by specific inducers, conditional activation is achieved, termed iSBH (inducible SBH) (Fig. 12b) [81].
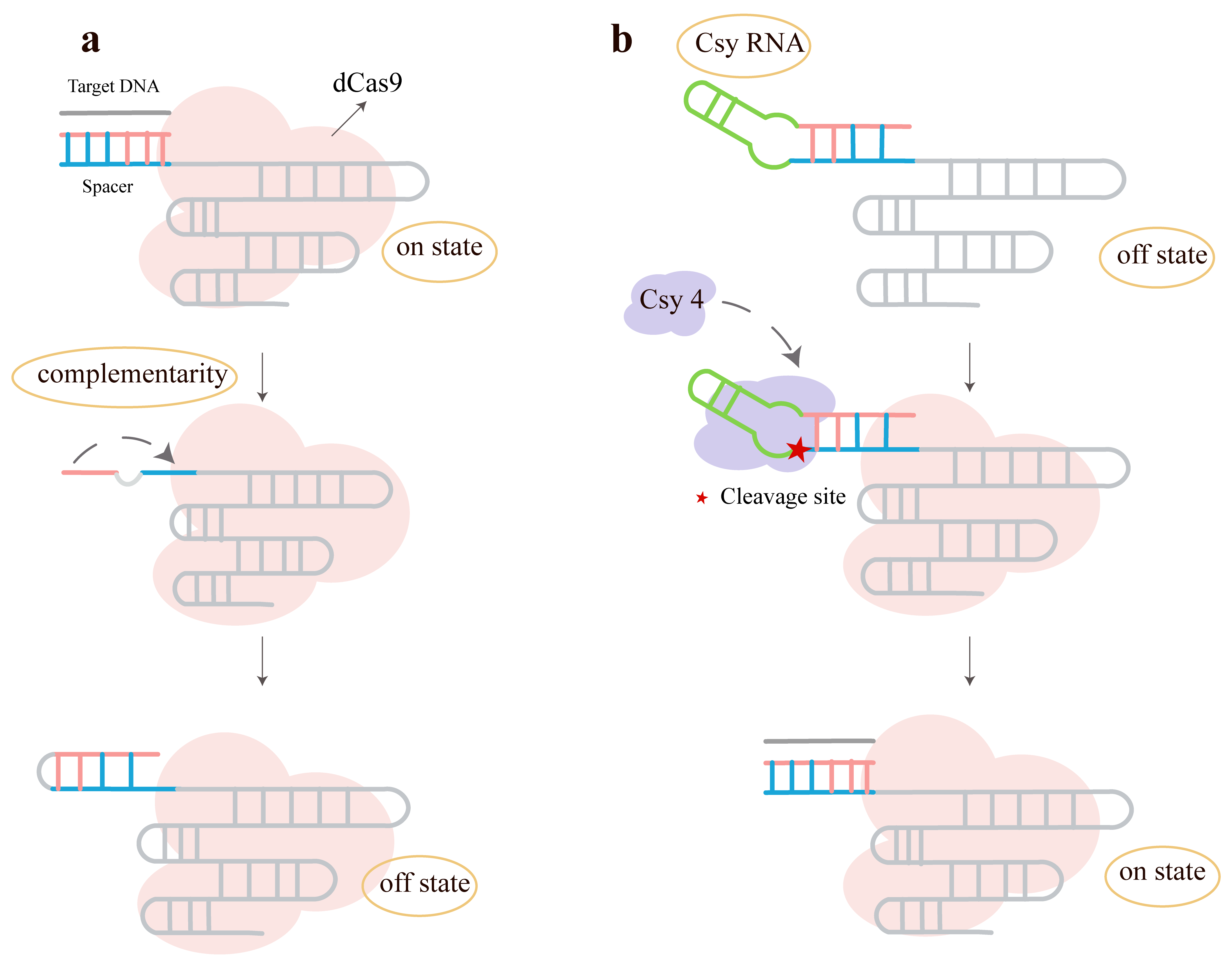 Fig. 12.
Fig. 12.
Transcriptional control using engineered sgRNAs with a spacer-blocking hairpin (SBH). (a) Schematic of the CRISPR Transcriptional Regulation (CRISPR-TR) system for gene modulation. (b) Design principle of allosterically inhibited and inducible SBH-sgRNAs. In the default OFF state, a 5′ extension folds into a spacer-blocking hairpin (SBH), rendering the sgRNA inactive. The inducible SBH design (iSBH, green) replaces the hairpin loop with a conditionally self-cleaving RNA module, which triggers spacer release and restores CRISPR-TR activity upon induction. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
First, they validated the effectiveness of SBH: experiments demonstrated that SBH completely inhibits CRISPR-TR activity. Control experiments (such as mismatched hairpins and random sequences) confirmed that the inhibitory effect depends on base pairing between the back-fold and the spacer. Second, they constructed and optimized iSBH, including protein-responsive iSBH (using RNA endonucleases like Csy4 and Cas6A as inducers to restore sgRNA function by cleaving the SBH structure) and ASO-responsive iSBH (using antisense oligonucleotides (ASOs) to bind the iSBH loop region, recruiting RNase H to cleave the RNA strand, enabling exogenous regulation). Optimization strategies mainly involved adjusting stem stability (e.g., by introducing bulge structures) and the length of residual sequences after cleavage, significantly improving the ON/OFF ratio. Subsequently, the researchers used the iSBH system to successfully construct two basic gene regulation modules: a branching module where a single inducer simultaneously activates multiple target genes, and an orthogonal module where different inducers independently regulate different target genes without cross-interference. These modules were validated in both synthetic reporter genes and endogenous genes (such as HBG1 and IL1B). They also extended the application to ASO- and ribozyme-responsive systems. An online tool, iSBHfold, was developed for automatically designing shared ASO-sensing loops applicable to multiple spacers. By integrating the self-cleaving hammerhead ribozyme (HHR2) into the SBH, self-activation without exogenous proteins was achieved, laying the foundation for future development of small-molecule or protein-responsive systems.
This strategy involves minimal modification to the sgRNA, is compatible with various CRISPR-derived systems (such as gene editing, epigenetic regulation, and base editing), and supports independent regulation of multiple genes. It also offers strong modularity and excellent orthogonality. Additionally, the OFF state exhibits almost no leakage, making it suitable for high-precision regulation scenarios (such as apoptosis switches).
Chen et al. [69] proposed a novel conditional genome editing and gene expression regulation system that regulates the post-transcriptional level of sgRNA under small-molecule control, significantly reducing the off-target effects of the CRISPR-Cas9 system and achieving temporally precise control over gene editing and expression. The authors developed a novel system called Cas9/sgRNA-Aptazyme (AZ): Aptazyme is a small-molecule-dependent self-cleaving ribozyme that self-cleaves in the presence of a ligand (such as theophylline), leading to sgRNA degradation. By inserting the theophylline-dependent aptazyme into the tetraloop, stem loop 2, or both of the sgRNA, three variants were constructed: sgRNA-AZ 1.1 (tetraloop only), sgRNA-AZ 1.2 (stem loop 2 only), and sgRNA-AZ 2.0 (both inserted) (Fig. 13b) [69]. Compared to the original sgRNA as shown in Fig. 13a, the introduction of aptazyme structure does not affect sgRNA activity.
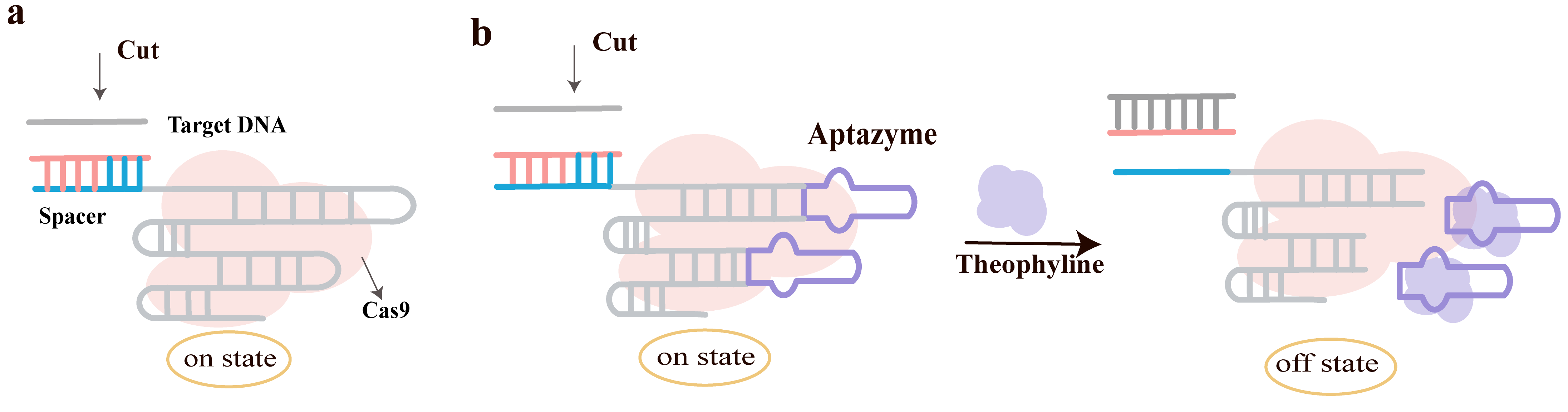 Fig. 13.
Fig. 13.
Engineering ligand-responsive sgRNAs through aptazyme integration. (a) Architecture of the wild-type Cas9/sgRNA complex. (b) Insertion of aptazymes into the tetraloop and stem-loop 2 of the sgRNA scaffold. Upon ligand binding, the embedded aptazymes undergo self-cleavage, resulting in the irreversible inactivation of the sgRNA through backbone degradation. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
In the absence of theophylline, the editing activities of sgRNA-AZ 1.1 and 1.2 were comparable to that of wild-type sgRNA, while sgRNA-AZ 2.0 showed a slight decrease. After adding theophylline, the editing activity of sgRNA-AZ 2.0 significantly decreased, indicating its effectiveness as a small-molecule-controlled gene editing switch. This enables conditional genome editing. Testing with sgRNA targeting EMX1 showed that: the off-target rate for wild-type sgRNA was 15%, while for sgRNA-AZ 2.0 it was 4% without theophylline and 0% with theophylline. The results indicate that timely deactivation of sgRNA activity effectively reduces off-target effects caused by prolonged editing. The system was combined with the dCas9-KRAB transcriptional repression system to target the human PGRN gene promoter. After adding theophylline, the transcriptional repression mediated by sgRNA-AZ 2.0 was lifted, and PGRN promoter activity rapidly recovered, demonstrating the system’s utility for rapid and reversible gene expression regulation, thereby achieving conditional gene expression regulation.
This system offers high specificity (by controlling sgRNA lifespan to reduce non-specific cleavage), temporal precision (rapid switching via small-molecule regulation), flexibility (applicable to both gene editing and transcriptional regulation), and simplicity (no need for additional transcription factors or complex regulatory elements).
The spatial organization and dynamic changes of chromatin are crucial for cellular function. Traditional methods, such as fluorescent repressor operator systems (FROS), can label specific loci but are complex to perform and may interfere with endogenous DNA sequences. The emergence of the CRISPR-dCas9 system enables the labeling of endogenous loci without inserting exogenous sequences. However, multicolor labeling typically relies on orthogonal Cas9 proteins from different bacterial species, which have complex and less-studied PAM sequences. The team led by Wang et al. [88] proposed and validated a dual-color CRISPR labeling system based on RNA aptamers for visualizing endogenous genomic sites in living cells.
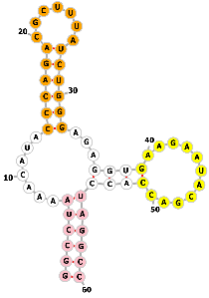
This study developed a dual-color labeling system using only Streptococcus pyogenes Cas9 (SpCas9). By engineering the sgRNA to incorporate MS2 (Fig. 14a) and PP7 (Fig. 14b) RNA aptamers, the system recruits MCP and PCP proteins fused to different fluorescent proteins (EYFP and tagRFP), respectively, enabling dual-color labeling of distinct genomic loci. The study explored two sgRNA design strategies: sgRNA1.0, which adds six MS2 or PP7 hairpin structures at the 3′-end, and sgRNA2.0, which incorporates one aptamer each at the tetraloop and stem loop 2, plus four additional aptamers at the 3′-end, totaling six aptamers.
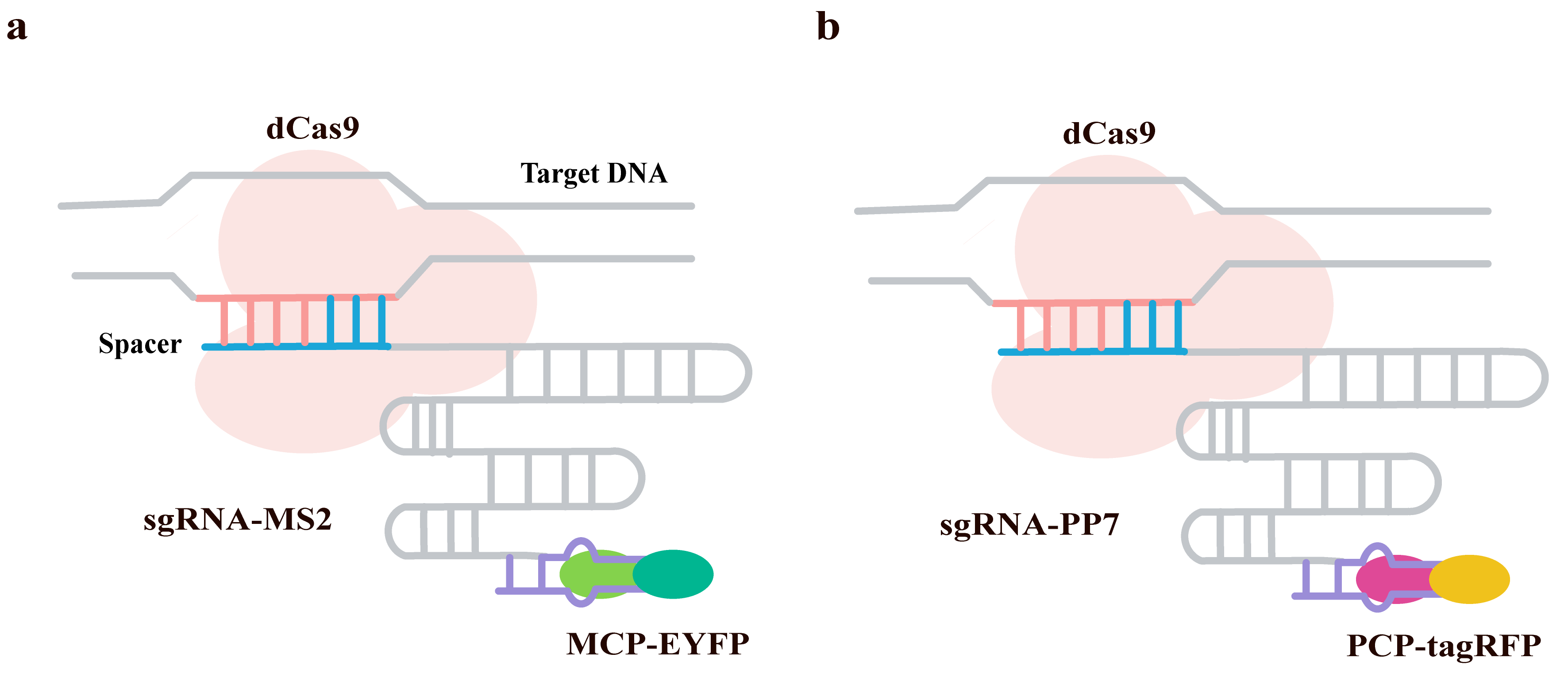 Fig. 14.
Fig. 14.
Two-color CRISPR imaging using orthogonal RNA aptamers. Schematic of the dCas9-based labeling complex, where distinct sgRNAs are engineered to incorporate either (a) MS2 or (b) PP7 RNA hairpins (purple). These hairpins recruit their corresponding coat proteins (MCP-EYFP and PCP-tagRFP), enabling simultaneous two-color detection of genomic loci. DNA and the sgRNA scaffold are shown in grey, with target-specific spacer regions in blue. The dCas9 protein is colored pink. Created with Adobe Illustrator CS6 (Adobe Systems Incorporated, San Jose, CA, USA).
Both sgRNA1.0 and sgRNA2.0 were able to form fluorescent foci corresponding to telomeres in living cells. The sgRNA2.0 design demonstrated a higher signal-to-noise ratio, suggesting that the positioning of the aptamers within the sgRNA influences labeling efficiency. The system successfully labeled telomeric repeats. Immunofluorescence staining (using anti-TRF1 antibody), confirmed that the fluorescent foci indeed corresponded to telomeres. The number of telomeres detected by different labeling methods (MS2, PP7, immunofluorescence) was consistent, verifying the specificity of the labeling. Furthermore, using MS2 to label telomeres and PP7 to label centromeres enabled spatially distinct dual-color labeling, indicating that the MS2 and PP7 systems do not interfere with each other, thus demonstrating good orthogonality of the strategy.
This strategy offers several advantages: it uses only one Cas9 protein (S. pyogenes), whose simple PAM sequence facilitates target site selection; it can be expanded to include more colors (e.g., by introducing other orthogonal aptamers); and it is applicable to non-repetitive sequences by using multiple sgRNAs targeting adjacent regions for labeling. A limitation of the system is that it requires the construction of three fusion proteins (dCas9, MCP, PCP), making cell line construction somewhat complex.
In summary, by integrating functional nucleic acid elements (such as aptamers, ribozymes, and their complexes, aptazymes) into specific structural regions of the sgRNA, researchers have successfully constructed various condition-responsive CRISPR/Cas9 systems. These engineered sgRNAs can respond to a variety of signals, including small molecules (such as theophylline, tetracycline, guanine, etc.), proteins, and endogenous metabolites (such as S-adenosylmethionine), enabling precise spatiotemporal control over functions like gene editing, transcriptional activation/repression, and chromosome imaging. These strategies have not only significantly expanded the applicability of the CRISPR toolkit but also provided powerful new molecular devices for exploring fundamental biological questions and disease treatment [27, 28].
In order to apply the CRISPR/Cas9 system to both prokaryotic and eukaryotic cells, the acquisition of sgRNA is of crucial importance. Extracellularly, engineered sgRNA is mainly obtained through transcription using T7 RNA polymerase. Prokaryotic cells (such as Escherichia coli) can express it using the pJ23119 promoter. In eukaryotic cells (such as HEK 293T or Hela), plasmids are mainly transfected using liposomal reagents, and the cells recognize the U6 promoter in the plasmid, thereby expressing the sgRNA of the relevant sequence. The corresponding delivery system mainly includes the following methods: In prokaryotic cells, the purpose plasmid is mainly transferred into the cells using chemical competent cells, and then the bacteria itself transcribes RNA and translates Cas9-related proteins. In eukaryotic cells, it is mainly through lipid particle transfection reagents that transfer plasmids or RNA or RNA-protein complexes into the cells.
For prokaryotic cells, since plasmids carry antibiotic resistance, selection can be performed based on the different resistance markers carried by various plasmids to ensure that the target plasmid had entered the cells. As a result, the outcomes in prokaryotic cells are relatively uniform. In contrast, eukaryotic cells exhibit greater variability. Due to the limited transfection efficiency of transfection reagents, practical operations often require adjusting experimental conditions (such as reducing the number of cells, increasing the amount of transfected plasmids or RNA, adding more transfection reagents, or extending the transfection time) to enhance experimental outcomes as much as possible. However, the specific experimental conditions still depend significantly on the system. When applying engineered sgRNAs to actual experimental systems, researchers need to explore and optimize the conditions based on their specific circumstances.
Although significant progress has been made in sgRNA engineering, the field still faces several challenges. Firstly, the response efficiency and dynamic range of most current systems that need further improvement to reduce background leakage and enhance induction specificity. Secondly, the tolerance of different sgRNA backbones as well as target gene loci to inserted elements varies, and universal design rules still need to be systematically established and validated. Additionally, for in vivo applications, the stability, delivery efficiency, and potential immunogenicity of engineered sgRNAs are critical factors to consider.
Looking ahead, sgRNA engineering technology is expected to achieve breakthroughs in the following aspects: (1) Development of more diverse high-affinity functional nucleic acid elements to respond to a wider range of physiological or pathological signals; (2) Systematic optimization of the compatibility between the sgRNA backbone and functional modules by combining computational design and high-throughput screening, to achieve higher precision and efficiency in regulation; (3) Deeper integration of condition-responsive CRISPR systems with cutting-edge fields like cell therapy, intelligent drug delivery, and synthetic gene circuits to build smarter and safer theranostic platforms. For instance, “smart” cells capable of sensing specific signals in the tumor microenvironment and triggering therapeutic gene editing could be designed.
Finally, with the deepening understanding of the structure-function relationships of CRISPR systems and continuous advancements in RNA synthesis and modification technologies, the engineering of sgRNAs will become more refined and modular. This will not only drive transformative changes in basic research but also bring unprecedented opportunities for precision medicine, gene therapy, and synthetic biology, ultimately enabling more precise, safer intervention and regulation of human life processes.
WW, LL, and YL conceived and designed the study. XL, QZ, and YL generated the figures and tables and compiled the references. WW, LL, and XL wrote the original draft, which was reviewed and edited by QZ and YL. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Dr. Chongxi Yang from La Trobe University and Ziyi Li from Jihua Laboratory for language editing.
This study was funded by the Fund for Creative Research of The Second People’s Hospital of Foshan (No. 2024B02) and Postdoctoral Initial Foundation of Guangdong Medical University (No. 4SG24185G).
The authors declare no conflict of interest.
During the preparation of this work the authors used Deepseek-R1 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.