




 , Xuehan Yan 1,†, Zheng Wang 1,*
, Xuehan Yan 1,†, Zheng Wang 1,* , Zizhen Zhang 1,*
, Zizhen Zhang 1,*1 Department of Gastrointestinal Surgery, Renji Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 200127 Shanghai, China
†These authors contributed equally.
Abstract
DNA methylation is a key epigenetic modification catalyzed by DNA methyltransferases (DNMTs) and predominantly occurs at cytosine-phosphate-guanine (CpG) islands, which are often located in gene promoter regions. Hypermethylation of CpG islands within gene promoters can silence tumor suppressor gene expression, thereby disrupting normal cellular functions, including maintenance of genomic stability and regulation of cell growth, and contributing to tumor initiation and progression. In contrast, global hypomethylation may promote genomic instability and oncogene activation. This review discusses the molecular mechanisms underlying DNA methylation and evaluates its functional and clinical significance in colorectal and gastric cancers, with emphasis on its potential application as a noninvasive biomarker for diagnosis.
Keywords
- DNA methylation
- DNA demethylation
- gastrointestinal cancer
- clinical biomarker
DNA methylation is a crucial epigenetic modification that regulates gene expression by adding methyl groups to cytosine bases in Cytosine-Phosphate-Guanine (CpG) islands. In cancer, this process becomes dysregulated: tumor suppressor genes often undergo promoter hypermethylation, leading to their silencing, while global hypomethylation can promote genomic instability [1]. These alterations play a pivotal role in tumor initiation and progression. Gastrointestinal cancers, including gastric and colorectal cancer, frequently exhibit such aberrant methylation patterns. This review systematically explores the molecular mechanisms of DNA methylation and demethylation, focusing on their roles in gene silencing and gastrointestinal tumorigenesis. Furthermore, it highlights the growing clinical significance of DNA methylation as sensitive biomarkers for early detection, prognosis, and monitoring of colorectal and gastric cancers, underscoring their potential in advancing cancer diagnostics and personalized medicine.
The chemical essence of DNA methylation lies in an enzymatic process catalyzed
by DNA methyltransferases (DNMTs), whereby a methyl group (-CH₃) is covalently
attached to the fifth carbon (C5) of a cytosine base, resulting in the formation
of 5-methylcytosine (5-mC) [2, 3, 4]. This transformation can be summarized by a
simplified chemical equation: cytosine + S-adenosylmethionine (SAM)
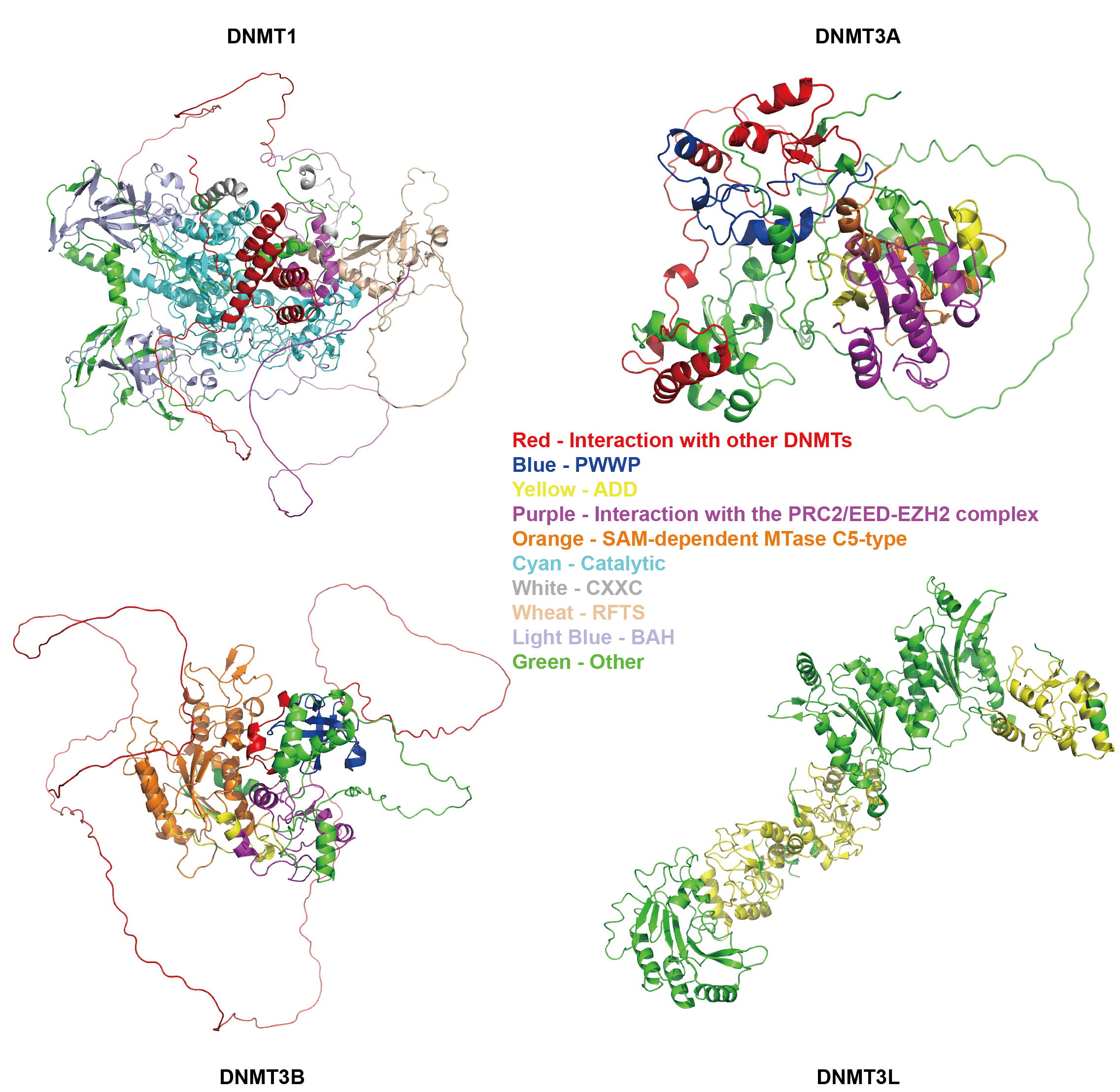 Fig. 1.
Fig. 1.
Structure of DNMTs. PWWP, Pro-Trp-Trp-Pro domain; ADD, ATRX-DNMT3-DNMT3L domain; PRC2, Polycomb Repressive Complex 2; EED, Embryonic Ectoderm Development; EZH2, Enhancer of Zeste Homolog 2; SAM, S-AdenosylMethionine; CXXC, CXXC zinc finger domain; RFTS, Replication Focus Targeting Sequence; BAH, Bromo-Adjacent Homology domain. The image was obtained from AlphaFold (https://alphafold.com/) and visualized using PyMOL (https://www.pymol.org).
The catalytic mechanism proceeds through three well-defined steps. To begin with, a cysteine residue within the active site of DNMT acts as a potent nucleophile, attacking the C6 position of the cytosine ring to generate an enzyme-substrate covalent intermediate [7, 8, 9, 10]. After that, this covalent bond alters the electron distribution within the pyrimidine ring, increasing the nucleophilicity of the C5 position [11], which attacks the electrophilic methyl carbon of SAM. The methyl group is thereby transferred from SAM to the C5 atom of cytosine, forming the core structure of 5-mC [10, 12, 13, 14]. In the end, a general base within the active site (such as a glutamate residue) abstracts a proton from the C5 position of the nascent 5-mC, cleaving the covalent bond at C6 and releasing the product, 5-mC, into the DNA duplex. Meanwhile, the DNMT enzyme is restored to its original state, ready for another catalytic cycle [10, 12].
DNMTs are a critical family of enzymes that primarily catalyze the process of DNA methylation, which involves the transfer of a methyl group from SAM to cytosine bases in DNA, resulting in the formation of 5-mC [15]. This process plays a central role in epigenetic regulation. Based on their distinct functions and mechanisms, DNMTs can be classified into three main categories: maintenance methyltransferases, de novo methyltransferases and auxiliary regulatory factors [9, 16, 17] (Fig. 1). These members work in concert to ensure the accurate execution of DNA methylation in biological processes such as gene expression and genomic stability.
2.1.2.1 Maintenance Methyltransferases – DNMT1
The primary function of DNMT1 is to accurately maintain and transmit DNA methylation patterns to daughter cells during DNA replication, a mechanism that relies on its specific recognition and catalytic activity toward hemi-methylated DNA [18]. After DNA replication, the parental strand retains its original methylation marks, while the newly synthesized daughter strand remains unmethylated, resulting in a transient hemi-methylated state. DNMT1 specifically recognizes these hemi-methylated CpG sites and uses the methylated parental strand as a template to catalyze the methylation of the corresponding cytosine residues on the daughter strand, thereby restoring full methylation. This process constitutes the basis of “cellular memory” in epigenetic inheritance, ensuring the stable transmission of gene expression patterns—such as the silencing or activation of tissue-specific genes—across cell generations, and maintaining the silent state of critical functional genes like tumor suppressor genes [19]. Furthermore, in female somatic cells, DNMT1 is involved in sustaining the stable silencing of one X chromosome, underscoring its essential role in epigenetic regulation [20].
2.1.2.2 De Novo Methyltransferases – DNMT3A/DNMT3B
DNMT3A and DNMT3B are de novo DNA methyltransferases responsible for establishing novel DNA methylation patterns at previously unmethylated CpG sites during early embryonic development and germ cell formation [21, 22, 23]. Unlike maintenance methyltransferases, they operate independently of pre-existing methylation templates [24]. Their catalytic activity directly methylates unmodified DNA, with targeting specificity guided by their structural domains and interactions with partner proteins [25, 26]. The targeted recruitment of DNMT3A/3B to specific genomic loci is facilitated by multiple mechanisms, for example, their Pro-Trp-Trp-Pro (PWWP) domain recognizes H3K36me3—a histone mark enriched in transcriptionally active regions—while specific transcription factors anchor them to several gene promoters [27]. Furthermore, the PWWP domain of DNMT3B is essential for its interaction with the transcription factor ZHX1 (which contains a homeobox motif), and this interaction potentiates DNMT3B-mediated transcriptional repression [28]. Additionally, during gametogenesis, they collaborate with DNMT3L to establish parent-of-origin-specific methylation marks at imprinting control regions (ICRs) [29, 30]. Biologically, DNMT3A and DNMT3B play essential roles in embryogenesis and cellular differentiation by establishing methylation landscapes that guide pluripotent stem cell fate and lineage commitment [21, 22]. They also contribute to genomic stability by silencing transposable elements and retrotransposons through methylation, thereby preventing mutagenic transposition [31].
2.1.2.3 Auxiliary Regulatory Factors – DNMT3L
DNMT3L is an epigenetic regulatory factor that, although lacking catalytic activity itself, functions as a key auxiliary protein that significantly enhances and modulates the methylation functions of DNMT3A and DNMT3B [29, 30]. Mechanistically, DNMT3L forms stable complexes with DNMT3A or DNMT3B, inducing conformational changes that greatly increase their affinity for the methyl donor SAM, thereby effectively promoting catalytic efficiency [29, 30]. Biologically, DNMT3L is highly expressed during gametogenesis, where it is essential for establishing correct parent-of-origin-specific methylation patterns at ICRs [29, 30].
According to UniProt (https://www.uniprot.org/), we analyzed the structures of DNMTs and presented in Fig. 1, where each color represents a distinct functional domain.
Both DNMT1 and DNMT3A/B contain specific domains that mediate interactions with other DNMTs, enabling them to form complexes and exhibit functional synergy [32]. Furthermore, the C-terminal of these enzymes all harbor a conserved SAM - dependent methyltransferase domain, which constitutes the molecular basis for their DNA methyltransferase activity. Additionally, the presence of domains that facilitate interaction with the Polycomb Repressive Complex 2/Embryonic Ectoderm Development-Enhancer of Zeste Homolog 2 (PRC2/EED-EZH2) complex provides structural support for their recruitment to specific genomic loci and the execution of their catalytic functions [33].
Fundamental differences in domain architecture between DNMT1 and DNMT3A/B dictate their distinct roles in epigenetic regulation. DNMT1 contains unique Replication Focus Targeting Sequence (RFTS) and Bromo-Adjacent Homology (BAH) domains, which collectively confer its characteristic maintenance DNA methylation activity. The RFTS domain mediates autoinhibition by occupying the DNA-binding channel of the catalytic center, which is relieved specifically after DNA replication when Ubiquitin-like with Plant HomeoDomain finger (PHD) and RING Finger Domains 1 (UHRF1) and other proteins recognize the hemimethylated DNA signal [34, 35]. This ensures that DNMT1 specifically methylates hemimethylated CpG sites, enabling accurate copying of the parental methylation pattern to the daughter strand. The BAH domain mediates protein-protein interactions, contributing to the stabilization of DNMT1 at the DNA replication fork [36]. Moreover, its CXXC zinc finger (CXXC) domain specifically binds to unmethylated CpG dinucleotides, positioning an auto-inhibitory linker between the DNA and the active site, thereby ensuring that only hemimethylated CpG sites undergo methylation [8, 34].
In contrast, DNMT3A and DNMT3B are equipped with distinct targeting modules—the ATRX-DNMT3-DNMT3L (ADD) and PWWP domains—enabling them to perform de novo methylation [37, 38]. The ADD domain recruits DNMT3A/B to the promoters of genes destined for transcriptional silencing by recognizing the unmethylated state of H3K4me0 [38], while the PWWP domain specifically recognizes H3K36me2/3; particularly in DNMT3B [39]. This dual recognition system allows DNMT3A/B to establish novel DNA methylation patterns in specific functional regions of the genome, such as promoters and gene bodies, based on distinct histone modification signals, thereby precisely establishing the epigenetic landscape during embryonic development and cell differentiation [9, 37].
DNMT3L possesses an ADD domain structurally similar with that of DNMT3A/B but lacks intrinsic catalytic activity. It interacts with DNMT3A/B via its ADD domain, stabilizing its conformation and thereby functioning as a crucial auxiliary factor that significantly enhances the de novo methyltransferase activity [29, 30, 37].
DNA methylation and demethylation are dynamically opposing processes that occur concurrently. Demethylation proceeds through two distinct mechanisms: passive and active (Fig. 2 Passive demethylation & Active methylation) [40, 41, 42].
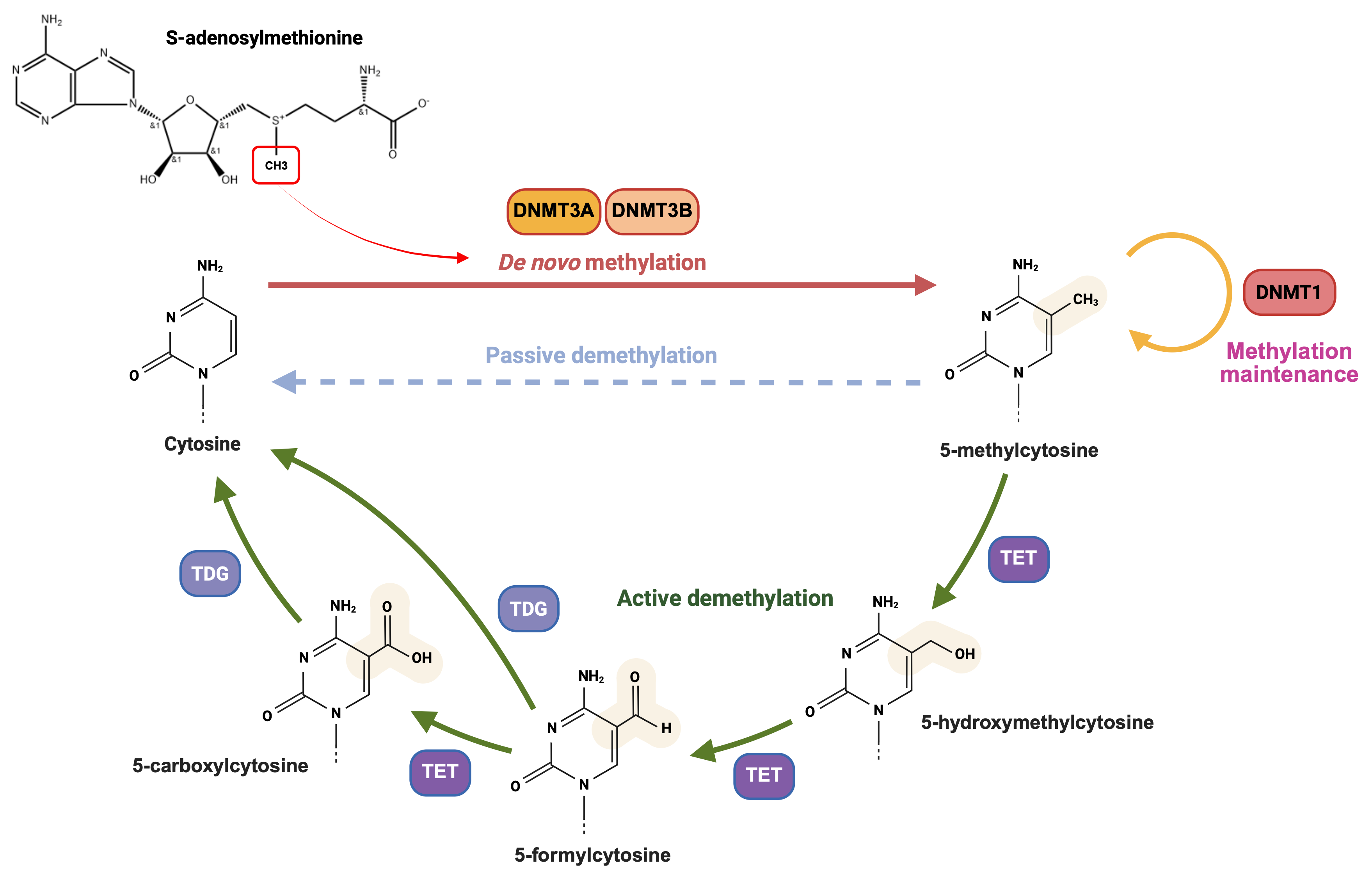 Fig. 2.
Fig. 2.
Chemical mechanism of DNA methylation and demethylation. DNA methylation and demethylation are concurrent dynamic processes. Methylation, catalyzed by DNA methyltransferases, covalently adds a methyl group to form 5-methylcytosine. Demethylation occurs via passive or active mechanisms. Passive demethylation dilutes methylation during DNA replication without chemical removal, while active demethylation enzymatically removes methyl groups via TET - mediated oxidation and TDG mediated - base excision repair throughout the cell cycle. DNMT, DNA methyltransferase; TET, ten-eleven-translocation enzyme; TDG, thymine-DNA glycosylase. Created in BioRender. (2025) https://BioRender.com/dveowg0.
Passive demethylation is a cell division-dependent process that leads to the dilution of methylation levels during DNA replication due to incomplete maintenance of methylation patterns, without involving direct chemical removal of the methyl group [16, 42]. In contrast, active demethylation enables rapid and specific removal of methyl groups at any stage of the cell cycle. Active demethylation is primarily mediated by the Ten-eleven-translocation (TET) dioxygenase family and can be divided into two sequential biochemical stages: The first stage involves TET-mediated iterative oxidation, during which TET enzymes progressively catalyze the oxidation of 5-mC to generate 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and finally 5-carboxylcytosine (5-caC) [43, 44, 45, 46]. The second stage entails base excision repair (BER), which consists of recognition/excision and repair/replacement steps by thymine-DNA glycosylase (TDG) [44, 45, 47].
2.2.2.1 TET
The TET enzyme family, including TET1/2/3, plays a key role as dioxygenases in
the active DNA demethylation process. Their primary function is to catalyze the
stepwise oxidation of 5-mC, successively generating 5-hmC, 5-fC and 5-caC
[43, 48]. Structurally, both TET1 and TET3 contain a CXXC zinc finger domain that
specifically recognizes and binds to unmethylated CpG islands, thereby recruiting
these enzymes to specific genomic regions such as gene promoters [48]. Due to a
chromosomal inversion, TET2 has lost its CXXC domain (which is now encoded by
the IDAX gene) and relies on interactions with other proteins for its
targeting mechanism [49]. The catalytic function of all TET enzymes depends on
the C-terminal dioxygenase domain, which uses Fe2+ and
| Process participation | Enzyme | General description | Key structural domains | Primary biological functions | Citations |
| Methylation | DNMT1 | Maintenance Methylation | RFTS, CXXC, BAH, Catalytic Domains | Propagates methylation patterns during DNA replication, ensuring epigenetic memory and cellular identity inheritance. | [8, 9, 15, 18, 19, 20, 34, 35, 36] |
| DNMT3A/3B | De novo Methylation | PWWP, ADD, Catalytic Domains | Establishes novel methylation patterns de novo; critical for embryogenesis, silencing transposons, and genomic imprinting. | [9, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 37, 38, 39] | |
| DNMT3L | Auxiliary Regulatory Factors | Similar with DNMT3A/3B but lacks catalytic domain | Enhances the catalytic activity and specificity of DNMT3A/3B, particularly during germ cell development. | [9, 29, 30, 37] | |
| Demethylation | TET | Active Demethylations Initiation | CXXC (TET 1/3), IDAX (TET2), Dioxygenase Domain | Initiates active DNA demethylation by oxidizing 5-mC to 5-hmC, 5-fC, and 5-caC. | [43, 44, 45, 46, 48, 49] |
| TDG | Glycosylase | Uracil-DNA Glycosylase Domain | Completes active demethylation by excising TET oxidation products (5-fC/5-caC), initiating BER to restore an unmodified cytosine. | [44, 45, 47, 50, 51, 52, 53, 54] |
DNMT, DNA methyltransferases; TET, Ten-eleven-translocation; TDG, thymine-DNA glycosylase; RFTS, Replication Focus Targeting Sequence; CXXC, CXXC zinc finger domain; BAH, Bromo-Adjacent Homology domain; PWWP, Pro-Trp-Trp-Pro domain; ADD, ATRX-DNMT3-DNMT3L domain; 5-mc, 5-methylcytosine; 5-hmc, 5-hydroxymethylcytosine; 5-fC, 5-formylcytosine; 5-caC, 5-carboxylcytosine; BER, base excision repair.
2.2.2.2 TDG
TDG is a key glycosylase involved in the BER pathway and plays a central role in
the final step of active DNA demethylation [45, 47]. Specifically, glycosylases
such as TDG recognize 5-fC and 5-caC and hydrolyze the N-glycosidic bond between
the aberrant base and the deoxyribose sugar, resulting in an
apurinic/apyrimidinic (AP) site [50]. Subsequently, AP endonuclease 1 (APE1)
cleaves the phosphodiester backbone at the AP site [51]. DNA polymerase
(typically Pol
CpG islands, which are often located in promoter regions, play a critical role in the initiation of transcription. Methylation of these CpG-rich regions can disrupt the assembly of the transcription initiation complex, leading to failure of transcriptional activation and consequent silencing of the associated gene [55]. Mechanistically, DNA methylation-mediated gene silencing operates through two principal modes: direct physical interference with transcription factor binding, and indirect repression via recruitment of methyl-binding proteins and histone modifiers that promote a transcriptionally inactive chromatin state (Fig. 3) [15].
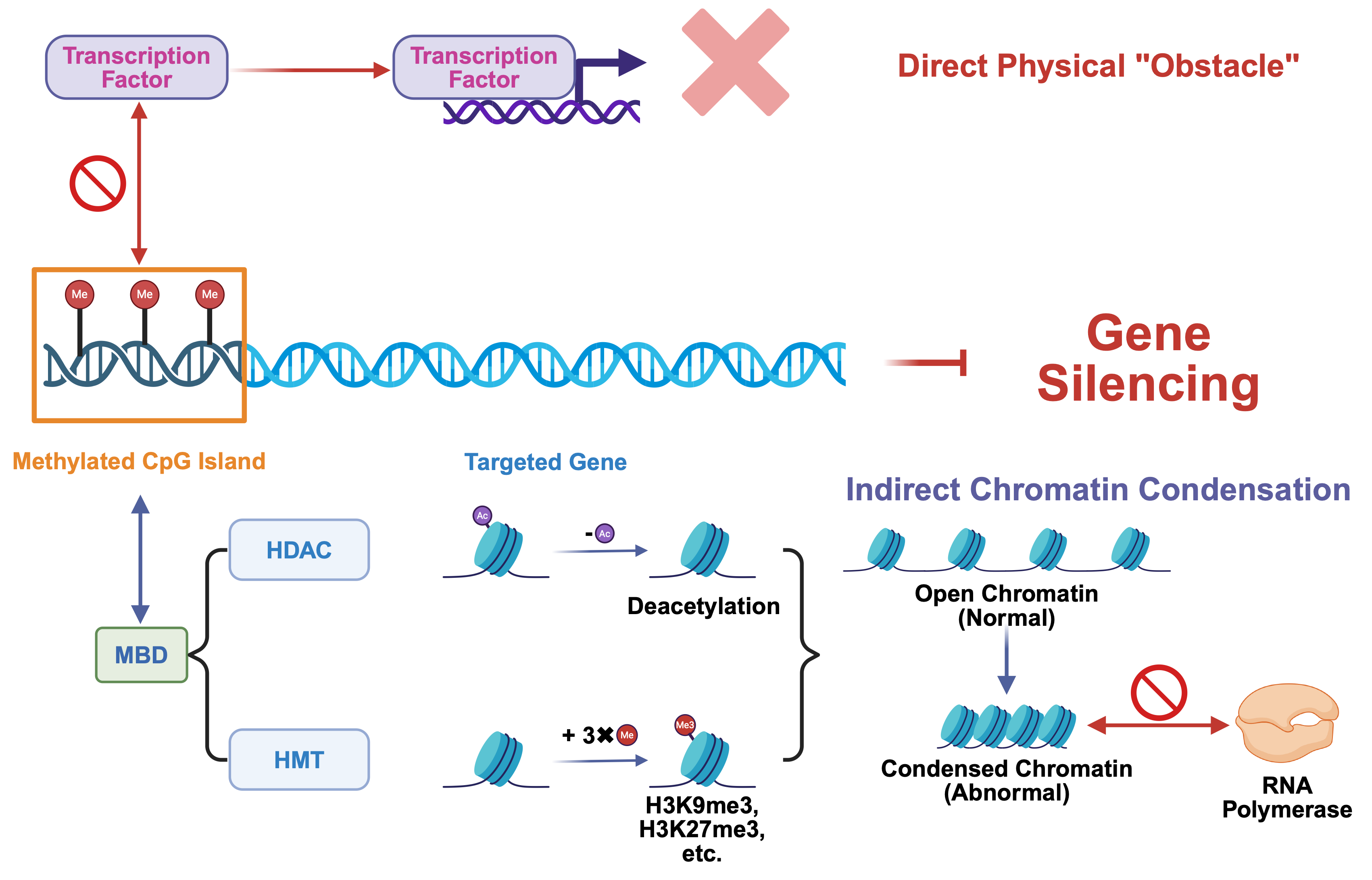 Fig. 3.
Fig. 3.
Mechanism of DNA methylation in gene silencing. Gene silencing via DNA methylation occurs through two primary mechanisms: direct physical blockade of transcription factor access and indirect repression mediated by methyl-binding proteins and histone modifiers, which collectively establish transcriptionally inactive chromatin. CpG island, Cytosine-Phosphate-Guanine island; MBD, methyl-CpG-binding domain protein; HDAC, histone deacetylase; HMT, histone methyltransferase. Created in BioRender. (2025) https://BioRender.com/o6r8csp.
DNA methylation at CpG islands within gene promoter regions directly interferes with the transcriptional initiation process. The core mechanism involves the methyl group added to the cytosine base, which creates a protruding steric hindrance that physically occupies the major groove of the DNA double helix [56]. This region is precisely where many transcription factors must recognize and bind to their specific DNA sequences. Consequently, the methyl “obstacle” directly impedes the ability of critical transcription factors to bind to their target sites [4]. This failure in binding prevents the successful assembly of the transcription initiation complex, ultimately halting the gene transcription process. In essence, methylation effectively blocks the DNA, preventing the transcription factor from engaging and thus directly leading to gene silencing [57].
In addition to direct steric hindrance, DNA methylation exerts a more potent and prevalent gene silencing effect through an indirect mechanism involving the recruitment of repressive complexes and chromatin remodeling. This process initiates when methyl-CpG-binding domain proteins (MBDs), such as MeCP2, recognize and bind to methylated CpG sites, serving as “readers” of the DNA methylation mark [58, 59]. These MBD proteins then act as platforms to recruit chromatin-modifying enzymes, including histone deacetylases (HDACs) and histone methyltransferases (HMTs) [60]. HDACs remove acetyl groups from histones, promoting chromatin condensation [61, 62], while HMTs catalyze the addition of repressive histone marks [62], such as H3K9me3 [63] and H3K27me3. These modifications collectively facilitate the transition from an open, transcriptionally permissive euchromatin state to a tightly packed, transcriptionally inert heterochromatin structure [63]. As a result, the chromatin becomes inaccessible to the transcriptional machinery, leading to stable and heritable gene silencing [15].
DNA hypermethylation of tumor suppressor genes contributes to the carcinogenesis of solid tumors. For example, hypermethylation of tumor suppressor genes such as TP53 leads to their silencing, which is associated with enhanced tumor malignancy and progression [64]. In colorectal neuroendocrine neoplasms (CRNEN), hypermethylation of TREM1 locus (e.g., at site cg04451353) promotes the infiltration of immunosuppressive cell subsets such as CD14+CD16– monocytes, thereby driving tumor progression [65]. In colorectal cancer (CRC), hypermethylation of ZNF671 and ZNF132 is associated with a low Immunoscore and contributes to an immunosuppressive microenvironment by negatively regulating T-cell and macrophage functions [66]. A novel research proves that under chronic Cd exposure, TET2 is inhibited, thus leading hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta (HADHB) low expression to activate the focal adhesion kinase (FAK) signaling pathway and ultimately contribute to CRC progression [67]. While in gastric cancer (GC), hypermethylation of secreted frizzled-related protein 2 (SFRP2) leads to aberrant activation of Wingless-type MMTV integration site family member (WNT) signaling pathway, promoting tumor proliferation and hypermethylation of reprimo (RPRM) attenuates p53 signaling pathway to accelerate its malignant transformation [68]. Besides, hypermethylation of SRY-box transcription factor 21 (SOX21) induces overexpression of the downstream cyclin kinase subunit CDC28 Protein Kinase Regulatory Subunit 2 (CKS2), thereby driving gastric cancer progression [69] (Table 2, Ref. [65, 66, 67, 68, 69, 70, 71, 72, 73]).
| Hyper/Hypo-methylation | Cancer type | Key enzyme change | Gene expression change | Mechanism | Citation |
| Hyper-methylation | CRNEN | / | TREM1 Low | Promotes infiltration of immunosuppressive cell subsets, such as CD14+CD16– monocytes. | [65] |
| CRC | / | ZNF671/132 Low | Correlates with low Immunoscore and negatively regulates T-cell and macrophage functions. | [66] | |
| TET2 Inhibited by Cd | HADHB Low | Activates the FAK signaling pathway and ultimately contributes to CRC progression. | [67] | ||
| GC | / | SFRP2 Low | Activates downstream WNT signaling pathway aberrantly, facilitating tumor proliferation. | [68] | |
| RPRM Low | Attenuates p53 signaling pathway, promoting malignant transformation of cells. | ||||
| DNMT1 Overexpression | SOX21 Low | Induces overexpression of downstream CKS2, driving gastric cancer progression. | [69] | ||
| Hypo-methylation | COAD | / | Lnc00346 High | Upregulates the miR-589-5p/CCL5 axis to enhance tumor progression. | [70] |
| CRC | / | MAGEA High | Triggers aberrant expression of cancer-testis antigens, increasing invasiveness and malignancy. | [71] | |
| / | CCM2 High | Influences the tumor microenvironment via ROCK-mediated endothelial cell senescence. | [72] | ||
| ICAM High | Enhances colorectal cancer metastasis through the Fusobacterium nucleatum-activated ALPK1/NF- |
||||
| GC | TET2 | WTIP High | Inhibits the phosphorylation and kinase activity of AKT. | [73] |
CRNEN, colorectal neuroendocrine neoplasms; CRC, colorectal cancer; GC, gastric
cancer; COAD, colon adenocarcinoma; TREM1, triggering receptor expressed on
myeloid cells 1; ZNF, zinc finger protein gene; TET2,
Ten-eleven-translocation 12; HADHB, hydroxyacyl-CoA dehydrogenase trifunctional
multienzyme complex subunit beta; FAK, Focal Adhesion Kinase signaling pathway;
SFRP2, secreted frizzled-related protein 2; RPRM, reprimo; DNMT1, DNA
methyltransferase 1; SOX21, SRY-box transcription factor 21; Lnc, long
non-coding; MAGEA, melanoma antigen family A; CCM2, cerebral cavernous
malformation 2; ICAM, intercellular adhesion molecule; CD, cluster
differentiation; WNT, Wingless-type MMTV integration site family member; CKS2,
CDC28 Protein Kinase Regulatory Subunit 2; CCL, C-C motif chemokine ligand; ROCK,
Rho-associated coiled-coil containing protein kinase; ALPK1, alpha-protein kinase
1; NF-
In colon adenocarcinoma (COAD), promoter hypomethylation of the long non-coding
RNA Lnc00346 upregulates the miR-589-5p/CCL5 axis, promoting tumor progression
[70]. In CRC, hypomethylation of melanoma antigen family A (MAGEA) results in
aberrant expression of cancer-testis antigens, thus increasing tumor invasiveness
and malignancy [71] and hypomethylation of cerebral cavernous malformation 2
(CCM2) influences the tumor microenvironment via Rho-associated coiled-coil
containing protein kinase (ROCK)-mediated endothelial cell senescence [72]. At
the same time, hypomethylation of intercellular adhesion molecule 1 (ICAM1)
enhances the metastatic capability of colorectal cancer through the
Fusobacterium nucleatum-activated alpha-protein kinase 1/nuclear factor
kappa-B (ALPK1/NF-
Having elucidated the molecular mechanisms underlying DNA methylation and its pivotal role in gene silencing, we now turn our focus to its clinical implications, particularly in the context of colorectal and gastric cancers. The following section synthesizes current evidence and explores the translational potential of DNA methylation biomarkers and therapies in gastrointestinal oncology.
DNA methylation testing has emerged as an ideal biomarker for non-invasive diagnosis and monitoring of gastrointestinal tumors, owing to its high sensitivity, specificity, and stability in body fluids such as peripheral blood [2]. Compared with traditional tissue biopsies, liquid biopsy-based DNA methylation analysis offers distinct advantages including convenient sampling, high reproducibility, and the capacity to reflect tumor heterogeneity. Furthermore, aberrant DNA methylation frequently occurs during early tumorigenesis, making it an optimal target for early cancer screening [74] (Fig. 4).
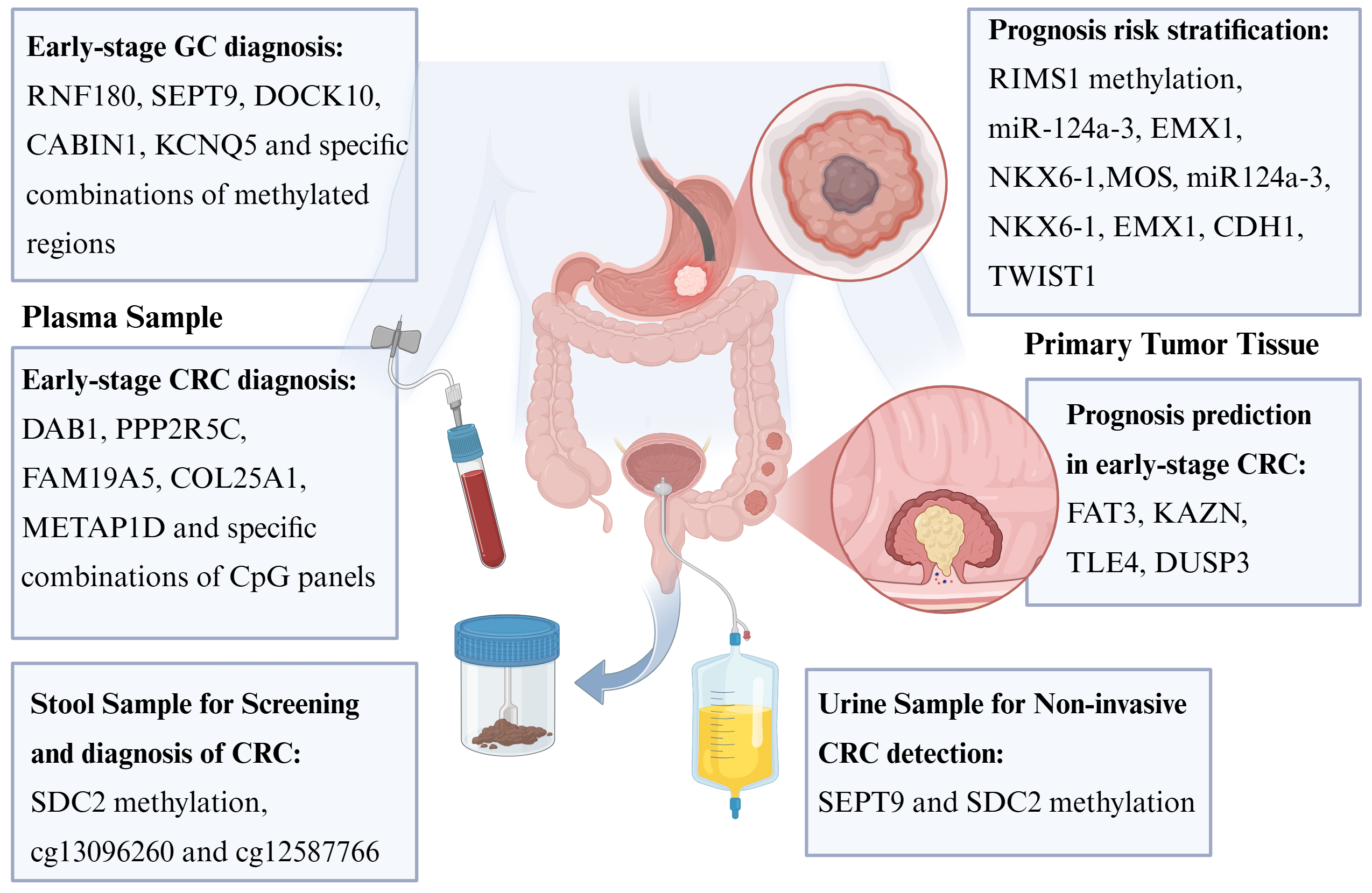 Fig. 4.
Fig. 4.
DNA Methylation in gastrointestinal cancer. DNA methylation plays a significant role in the diagnosis, risk stratification, and prognostic prediction of gastrointestinal tumors. In addition to tumor tissue itself, sources for methylation detection also include plasma samples, stool specimens, and even urine samples. GC, gastric cancer; CRC, Colorectal Cancer. Created in BioRender. (2025) https://BioRender.com/tnownci.
Recent advances in high-throughput sequencing technologies and bioinformatics approaches have enabled the identification of numerous methylation markers associated with colorectal and gastric cancers. Corresponding detection techniques such as methylation-specific PCR (MSP), pyrosequencing, and whole-genome bisulfite sequencing have been developed. The clinical application of these technologies is transforming the diagnostic and therapeutic paradigms for colorectal and gastric cancers, providing novel tools for precision medicine [75, 76, 77].
The following sections will focus on recent advances in clinical applications of DNA methylation in GC and CRC, encompassing early screening, diagnosis, prognostic evaluation, and treatment monitoring (Table 3, Ref. [78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97]), with the aim of informing clinical practice and future research directions.
| Study | Sample type | Target | Method | Sample size | Clinical application |
| Gao et al. [78] | Plasma | 161,984 CpG sites | Enhanced linear-splinter amplification sequencing | 2703 (1693 retrospective and 1010 prospective) | Early detection of six cancers (Including CRC) |
| Cai et al. [79] | Tumor tissue; Plasma | 6 DNA methylation markers | Multiplexed qPCR | 108 tissues and 328 plasma samples | Early detection and postoperative surveillance of CRC |
| Long et al. [80] | Plasma | DAB1, PPP2R5C, FAM19A5 | Quantitative methylation-specific PCR | 95 CRC patients and 74 healthy controls | Non-invasive CRC diagnosis |
| Xie et al. [81] | PBMCs | 5 DNA methylation markers | Multi-msqPCR | 1068 participants | Early-stage colorectal cancer diagnosis |
| Zhao et al. [82] | Blood | 149 hypermethylated markers | Bisulfite-free methylation sequencing | 3493 high-risk individuals | CRC screening |
| Mo et al. [83] | Plasma | 6 DNA methylation markers | Multiplex ctDNA methylation qPCR assay | 350 patients | Early detection of CRC recurrence |
| Overs et al. [84] | Plasma | COL25A1 and METAP1D | Methylation-specific digital PCR | 35 CRC patients and 35 healthy donors | CRC screening and follow-up |
| Yu et al. [85] | Primary tumor tissues | FAT3, KAZN, TLE4, DUSP3 | Infinium MethylationEPIC array | 797 patients | Prognosis prediction in early-stage CRC |
| Kim et al. [86] | Stool samples | SDC2 methylation | Real-time PCR | 1124 participants | Early CRC detection |
| Zhao et al. [87] | Stool samples | SDC2 methylation | Not mentioned | 12,106 populations | Community-based CRC screening |
| Cao et al. [88] | Stool samples | cg13096260 and cg12587766 | Quantitative methylation-specific PCR | 758 participants | Screening and diagnosis of precancerous lesions and CRC |
| Bach et al. [89] | Urine supernatant | SEPT9 and SDC2 | Quantitative methylation specific PCR | 92 CRC patients and 63 healthy volunteers | Non-invasive CRC detection |
| Qi et al. [90] | Plasma | 21 methylated regions | Cell-free methylated DNA immunoprecipitation | 150 gastric cancer patients and 100 healthy controls | Early GC detection |
| Nie et al. [91] | Plasma | RNF180, SEPT9 | Specific method not stated | 195 GC and 129 controls | Early detection of GC |
| Ren et al. [92] | Plasma | Mainly DOCK10, CABIN1, KCNQ5 | Genome-scale DNA methylation analysis | 89 GC patient and 82 controls | Non-invasive detection of GC |
| Yamada et al. [93] | Biopsy tissue | RIMS1 methylation | Pyrosequencing | 1624 participants | GC risk stratification |
| Tanaka et al. [94] | Biopsy tissue | miR-124a-3, EMX1, NKX6-1 | Pyrosequencing | 71 patients | Predicting metachronous GC risk |
| Li et al. [96] | Tumor tissue | 610 prognosis-related CpG sites | Specific method not stated | 406 patients | Prognosis risk stratification for GC |
| Kim et al. [95] | Noncancerous gastric mucosa | MOS, miR124a-3, NKX6-1, EMX1, CDH1, TWIST1 | Quantitative MethyLight assay | 245 participants | GC risk assessment in individuals with family history |
| Chen et al. [97] | FF and FFPE GC tissues | GNAS, FCGBP, and CCDC166 | Genome-wide methylation sequencing and qPCR | 47 discovery cohort, 302 model development and 130 validation cohort | Lymph node metastasis diagnosis in early GC |
qPCR, Quantitative Polymerase Chain Reaction; msqPCR, Multiple Methylation-Specific Quantitative PCR; MethyLight, Fluorescence-based Quantitative Methylation-Specific PCR.
As one of the leading causes of cancer-related mortality worldwide, early detection and precise treatment of CRC are crucial for improving patient outcomes. In recent years, DNA methylation-based liquid biopsy techniques have demonstrated remarkable progress in CRC screening, recurrence prediction, and treatment guidance [98, 99], with their clinical utility validated in multiple high-quality clinical trials.
Circulating tumor DNA (ctDNA) methylation markers have shown exceptional performance in early CRC detection and postoperative recurrence prediction. Gao et al. [78] evaluated their independently developed circulating free DNA (cfDNA) methylation detection technology ELSA-seq for screening six common cancers (including CRC), achieving 98.9% specificity and 69.1% sensitivity for CRC detection. Cai et al. [79] analyzed 60 CRC cases, 20 advanced adenomas, and 28 normal tissue samples using targeted bisulfite sequencing (PanSeer), identifying 150 differentially methylated regions (DMRs). Validation in 328 plasma samples demonstrated excellent diagnostic performance (AUC = 0.96 for CRC, AUC = 0.86 for advanced adenomas), leading to the development of ColonAiQ, a multiplex quantitative PCR-based blood test that provides a highly sensitive solution for CRC screening and postoperative monitoring. This PCR-based methylation assay is operationally simple, cost-effective, and particularly suitable for routine clinical implementation.
Moreover, methylation marker panels have shown superior performance compared to single markers [80]. Xie et al. [81] performed whole-genome methylation profiling of peripheral blood mononuclear cells (PBMCs) using microarray, pyrosequencing, and targeted bisulfite sequencing, identifying five DNA methylation markers. Their multiplex methylation-specific quantitative PCR (multi-msqPCR) assay demonstrated outstanding performance in early CRC diagnosis (AUC = 0.91, sensitivity 81.18%, specificity 89.39%) and could predict CRC risk up to two years in advance. Zhao et al. [82] developed ColonSecure, a blood-based test utilizing 149 hypermethylated markers identified through two-stage bisulfite-free sequencing analysis of over 193,000 CpG sites, which showed 86.4% sensitivity (Area Under the Receiver Operating Characteristic Curve (AUROC) = 0.956) for CRC screening, significantly outperforming conventional markers including Carcinoembryonic Antigen (CEA) (45.6%), CRP (39.8%), and CA19-9 (25.2%).
DNA methylation analysis also plays a crucial role in adjuvant chemotherapy decision-making. Mo et al. [83] employed a quantitative PCR method targeting six ctDNA methylation markers to analyze blood samples from 299 stage I-III CRC patients. They found that patients with positive ctDNA one month postoperatively had significantly higher recurrence risk (HR = 17.5), with ctDNA monitoring predicting recurrence 3.3 months earlier than imaging. These findings support ctDNA status-guided personalized adjuvant therapy strategies to avoid overtreatment or undertreatment.
Tissue-based DNA methylation profiling has provided abundant markers for
clinical applications. Overs et al. [84] validated tissue and cell-free
DNA samples from 35 CRC patients and 35 healthy controls using
methylation-specific digital PCR, demonstrating that a combination of COL25A1 and
METAP1D methylation showed 49% sensitivity and 100% specificity (AUC = 1) for
CRC detection. In another study based on colonic mucosal tissue, methylation
markers of FAT3, KAZN, TLE4, and DUSP3 genes
significantly predicted recurrence risk (HR = 2.35–3.20, p
Fecal DNA methylation analysis has also demonstrated advantages in early screening. Kim et al. [86] detected SDC2 methylation in fecal samples from 1124 asymptomatic/high-risk individuals using real-time PCR, achieving 95.0% sensitivity and 81.5% specificity for CRC detection, with a 58.1% detection rate for advanced adenomas, independent of tumor stage (p = 0.864). A real-world study further validated the clinical utility of fecal SDC2 methylation (mSDC2) testing, particularly in settings with limited colonoscopy resource [87]. Cao et al. [88] analyzed 758 fecal samples (including 62 patients with confounding conditions) and six paired tumor/adjacent tissues using methylation-specific PCR, identifying two specific CpG sites (cg13096260 and cg12587766) whose combined diagnostic model showed significant diagnostic value for CRC and precancerous lesions (AUC = 0.89).
Other body fluids may also serve as potential samples for CRC screening. Bach et al. [89] analyzed urine samples from 92 CRC patients and 63 healthy volunteers, demonstrating that a combination of SEPT9 and SDC2 methylation markers could detect 70% of CRC cases (86% specificity), providing the first evidence that urine testing could serve as a novel liquid biopsy approach for non-invasive CRC diagnosis.
As the fifth most common cancer and fifth leading cause of cancer death globally, early diagnosis is critical for improving GC patient outcomes. Recent studies have demonstrated significant advantages of DNA methylation markers in GC diagnosis and treatment. Single-cell multi-omics analysis has provided novel insights into GC heterogeneity and DNA methylation characteristics. Bian et al. [100] performed single-cell multi-omics sequencing (scTrio-seq3) on 85 samples from 14 GC patients, identifying hypermethylated promoter regions of TMEM240 and HAGLROS genes and hypomethylated TRPM2-AS and HRH1 genes. The “partial reversion” of methylation levels may suppress abnormal transcription of repetitive sequences, thus reducing antigen presentation properties of cancer cells and consequently making these poorly differentiated cancer cells less recognizable and attackable by the immune system. Building upon these fundamental findings, multiple clinical trials have validated their clinical applications [101].
The development of non-invasive detection techniques represents a major breakthrough in early GC screening. Qi et al. [90] developed an artificial intelligence-based detection technology using cfDNA methylation, which identified 21 GC-specific methylated regions through whole-genome methylation analysis and achieved 88.38% sensitivity and 94.23% specificity in clinical validation, providing a novel approach for non-invasive early GC screening. Additionally, Nie et al. [91] found that even in early gastric cancer, the methylation levels of SEPTIN9 and RNF180 were higher than those in the control group, and the combined panel had a sensitivity of 62.2%, specificity: 84.8%, and AUC of 0.804 for the diagnosis of gastric cancer. Another study [92] employed methylated DNA immunoprecipitation sequencing (MeDIP-seq) to effectively stratify GC patients at different stages, further advancing the clinical application of non-invasive screening technologies. These developments provide valuable data on cfDNA methylation biomarkers for GC and demonstrate the promising potential of cfDNA methylation as a blood-based non-invasive detection method for GC.
Risk stratification following Helicobacter pylori eradication presents another clinical challenge. A multicenter prospective study following 1624 participants after H. pylori eradication found that high methylation levels of the RIMS1 gene strongly predicted GC development, with the high methylation group showing a 470% increased risk (HR = 7.7) compared with the low methylation group [93]. Another study revealed correlations between methylation levels of miR-124a-3 and EMX1 and gastric lesions following H. pylori eradication [94]. A study focusing on familial gastric cancer also found that the methylation level of CDH1 decreased after Hp eradication [95]. These studies provide important evidence for developing personalized surveillance strategies after H. pylori eradication, helping to identify high-risk individuals requiring close follow-up while avoiding unnecessary examinations for low-risk patients.
Although current research on methylation-based prognostic models and treatment response monitoring in GC remains relatively limited, considering the similarities in methylation regulation mechanisms among gastrointestinal tumors, similar approaches may be applicable to GC. In recent years, some studies related to gastric cancer have also revealed the methylation profile of specific gastric cancer types, such as Epstein-Barr Virus-associated gastric cancer [102], and screened out special methylation sites in gastric cancer for subsequent clinical studies [103, 104]. Future studies should explore the value of specific methylation markers in predicting responses to neoadjuvant chemotherapy, targeted therapy, and immunotherapy in GC.
DNA methylation has demonstrated multifaceted clinical value in gastrointestinal cancer management, ranging from early screening to prognostic assessment (Table 3). With the approval of commercial testing products, methylation detection is gradually being implemented in clinical practice. However, current research still has several limitations, including the need for large-scale prospective validation of most markers and the lack of standardized operating procedures and cut-off values. Future multicenter studies are needed to validate the clinical utility of existing markers and explore the combined application of methylation testing with other molecular markers to further improve precision medicine in gastrointestinal oncology.
Despite the considerable potential of DNA methylation testing, current research exhibits notable limitations. Significant heterogeneity in the diagnostic accuracy of reported biomarkers across different populations has been observed, which may be attributed to confounding variables such as ethnic background, environmental influences, and variations in study cohort size and design. Furthermore, the integration of methylation assays into routine clinical practice presents multiple challenges, including the absence of standardized protocols, universally accepted cut-off values, and support from large-scale prospective validation. Issues regarding cost-effectiveness, accessibility of automated platforms, and the definitive translation of results into clinical decision-making pathways remain to be adequately addressed. Future efforts should focus on multi-center, real-world studies to verify the robustness of existing biomarkers and explore their combined application with other molecular markers to advance precision oncology.
This review provides a systematic elaboration of the chemical mechanisms underlying DNA methylation and demethylation, with a focus on the molecular pathways through which DNA methylation mediates gene silencing. It further summarizes recent clinical research advances and application potential of DNA methylation in the diagnosis of gastrointestinal tumors, highlighting its significant translational value from basic research to clinical practice.
As a crucial epigenetic modification, DNA methylation plays a central role in the initiation and progression of colorectal and gastric cancers. For instance, promoter hypermethylation of key tumor suppressor genes can lead to their transcriptional silencing, thereby promoting malignant transformation, uncontrolled proliferation, and enhanced invasiveness—ultimately increasing tumor aggressiveness. Such epigenetic alterations have emerged as important targets in gastrointestinal cancer research and therapeutic development. From a clinical perspective, DNA methylation-based assays demonstrate considerable value due to their non-invasive nature, high sensitivity, and potential for dynamic monitoring (Table 2). They can be applied in multiple aspects of cancer management, including early detection and prediction, risk stratification, prognostic assessment, postoperative monitoring of minimal residual disease and recurrence, as well as evaluation of therapeutic response to chemotherapy or radiotherapy. These applications provide powerful tools for comprehensive cancer care throughout the disease course.
Furthermore, since DNA methylation is catalyzed by enzymes such as DNMTs, and demethylation is mediated by enzymes including the TET family, targeting these enzymes has become a promising strategy for cancer therapy. For example, DNMT inhibitors (e.g., azacitidine and decitabine, DAC) can reduce global methylation levels, reverse gene silencing, and partially restore tumor suppressor function [105, 106]. Similarly, strategies aimed at activating demethylating enzymes (e.g., TET) or their cofactors may help re-establish normal epigenetic states. These interventions seek to restore normal gene expression, suppress malignant phenotypes, and offer novel approaches to overcome chemoresistance and inhibit metastasis [107].
In summary, therapeutic strategies targeting DNA methylation mechanisms are increasingly recognized as vital components of precision oncology, driving the diagnosis of gastrointestinal tumors toward more precise, targeted, and individualized approaches.
Conceptualization, LB, XY, ZW and ZZ; writing-original draft preparation, LB and XY; writing-review and editing, ZW and ZZ. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thanked the use of BioRender.com for the creation of figures in this review.
This work was funded by the National Natural Science Foundation of China (82270552(WZ), 82103510(ZZZ), 82473077(ZZZ)); State Key Laboratory of Systems Medicine for Cancer (KF2205-93(ZZZ)); Shanghai Municipal Education Commission (JWAIZD-6(ZZZ)) and Shanghai Municipal Health Commission (2024ZZ2020).
The authors state they have no financial interests or personal relationships that could have influenced the work presented in this paper.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.