





 , Zhihao Luo 1, Weixi Yuan 1, Qingshuang Zou 2, Li Wang 1, Yan He 1, Yao Shen 3, Xiaosha Wen 2, Shang Chen 4, Quan Liu 2, Dixian Luo 2, Zifen Guo 1,*
, Zhihao Luo 1, Weixi Yuan 1, Qingshuang Zou 2, Li Wang 1, Yan He 1, Yao Shen 3, Xiaosha Wen 2, Shang Chen 4, Quan Liu 2, Dixian Luo 2, Zifen Guo 1,*
1 Pharmacy and Pharmacology, University of South China, 421001 Hengyang, Hunan, China
2 Laboratory Medicine Center, Shenzhen Luohu Hospital Group, The Third Affiliated Hospital (The Affiliated Luohu Hospital) of Shenzhen University, Shenzhen University, 518001 Shenzhen, Guangdong, China
3 School of Medicine, Hunan University of Chinese Medicine, 410208 Changsha, Hunan, China
4 Hunan Provincial University Key Laboratory of the Fundamental and Clinical Research on Functional Nucleic Acid, Hunan Provincial Key Laboratory of the Research and Development of Novel Pharmaceutical Preparations, The First Clinical College, Changsha Medical University, 410219 Changsha, Hunan, China
Abstract
Breast cancer is the most prevalent malignant tumor among women worldwide. Its progression is driven, in part, by mitochondrial metabolic dysregulation, which can also contribute to therapeutic resistance. Although targeting mitochondrial metabolism offers new opportunities for treatment, significant therapeutic challenges remain. These include metabolic heterogeneity among subtypes and individual patients, drug resistance arising from metabolic plasticity, and suboptimal clinical translation of metabolic therapies. This review systematically synthesizes the mitochondrial metabolic mechanisms underlying different breast cancer subtypes, emphasizing the spatial network regulatory functions of mitochondrial metabolism. It further critically evaluates combined therapeutic strategies targeting metabolic vulnerabilities. By integrating current research limitations with emerging breakthroughs, we outline novel therapeutic frameworks to advance the development of precision medicine approaches focused on mitochondrial metabolism.
Graphical Abstract
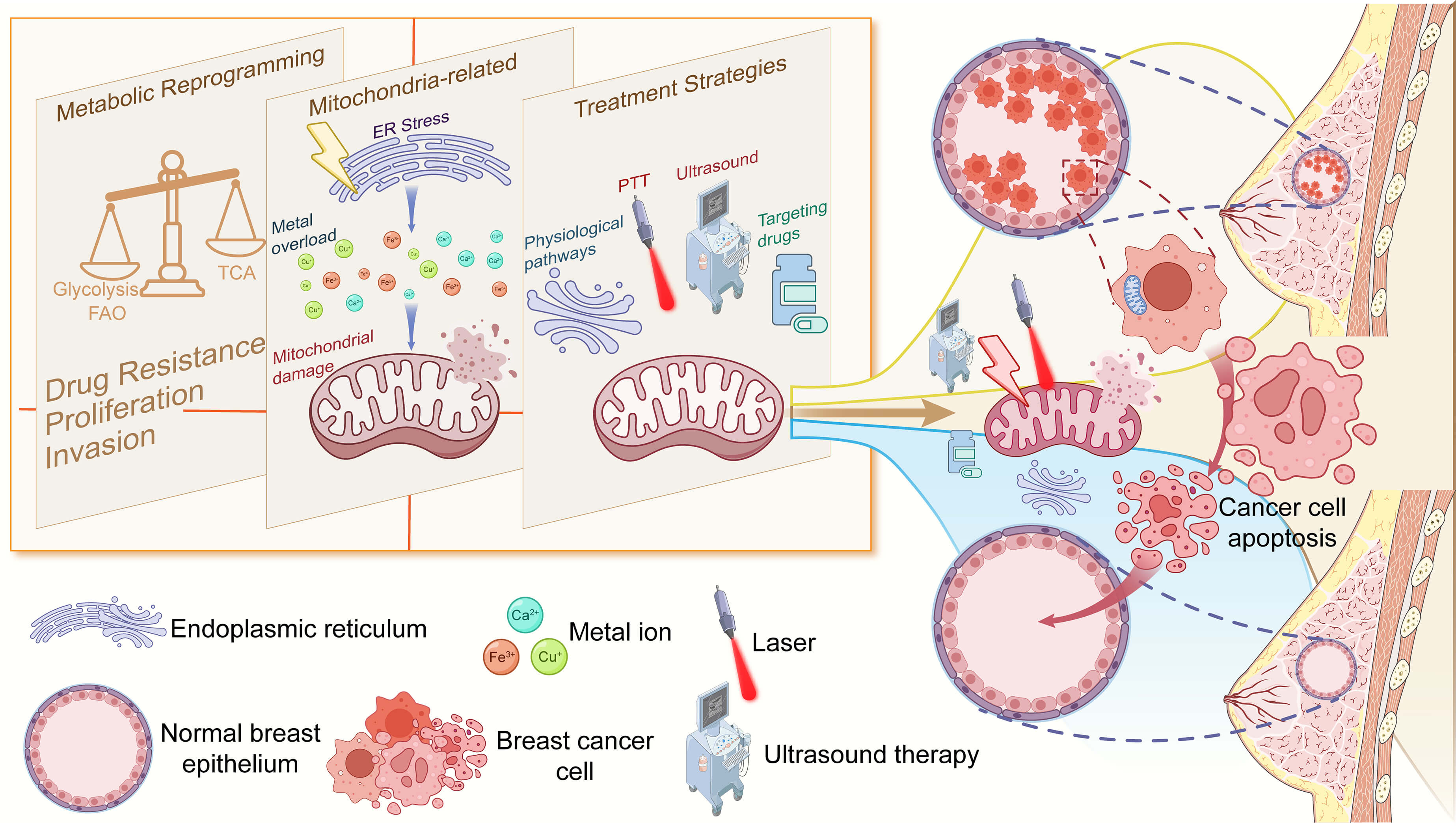
Keywords
- mitochondria
- energy metabolism
- breast cancer
- oxidative stress
- targeted therapy
Breast cancer remains the most prevalent malignancy and the leading cause of cancer-related mortality among women globally, with its molecular heterogeneity posing persistent therapeutic challenges. Current molecular subtyping categorizes breast cancer into distinct entities—Luminal A, Luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, and triple-negative breast cancer (TNBC)—which each exhibit unique metabolic dependencies and clinical trajectories [1]. Among these subtypes, Luminal A breast cancer is generally associated with a favorable prognosis and is highly sensitive to endocrine therapy. In contrast, while Luminal B breast cancer often retains hormone receptor expression, it typically has higher proliferative activity, has a poorer prognosis than Luminal A, and frequently has a diminished or less durable response to endocrine therapy [2]. HER2-enriched breast cancer is defined as breast cancer in which the HER2 protein is overexpressed. Although contemporary targeted therapies have significantly improved survival outcomes, this subtype has a relatively high risk of invasion and metastasis [3]. Conversely, TNBC is characterized by high invasiveness, a propensity for metastasis, and a lack of established therapeutic targets. These features contribute to complex treatment challenges and significantly reduced survival [4]. The metabolic heterogeneity between subtypes presents challenges for traditional therapies; however, this situation has gradually improved with the development of targeted drugs and the rise of precision medicine. For instance, hormonal therapy and HER2-targeted agents have demonstrated significant efficacy in patients with hormone receptor (HR)+ and HER2+ breast cancer. Nevertheless, resistance to chemotherapy, tumor heterogeneity, and adaptation to the tumor microenvironment remain the primary challenges in the treatment of breast cancer [5]. These challenges underscore the urgent need to decipher the metabolic underpinnings of breast cancer progression, particularly mitochondrial reprogramming, which governs cellular energetics, redox homeostasis, and therapeutic vulnerability across subtypes.
Mitochondria orchestrate a triad of core processes—the tricarboxylic acid
cycle (TCA), oxidative phosphorylation (OXPHOS), and fatty acid
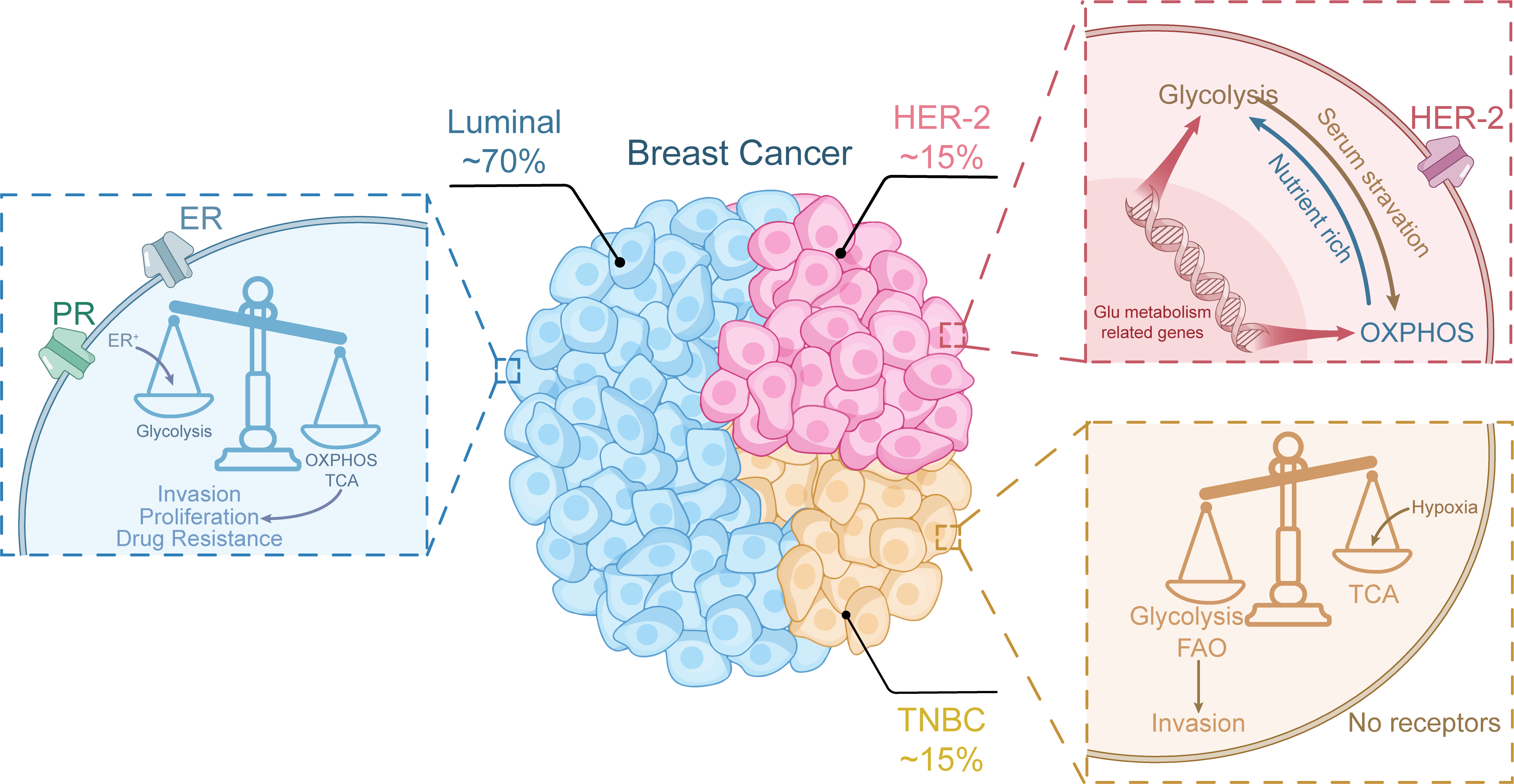 Fig. 1.
Fig. 1.
Metabolic differences of breast cancer cells with different
subtypes. OXPHOS, oxidative phosphorylation; FAO, fatty acid
This review systematically explores the mechanisms of mitochondrial metabolism and subtype-specific regulation in breast cancer progression. It highlights the interconnections between mitochondrial metabolic reprogramming, homeostatic regulation, and spatial networks, while also identifying therapeutic advances that target mitochondrial metabolism. By elucidating these mechanisms, we aim to provide a rationale for developing subtype-specific therapeutic strategies that address cell-autonomous metabolic defects, thereby advancing the individualized treatment process for breast cancer.
Mitochondrial metabolic pathways are central to the progression of breast cancer and act as dynamic regulators of bioenergetics, biosynthesis, and redox adaptation. The metabolic reprogramming observed in breast cancer transcends the traditional Warburg effect, involving significant remodeling of mitochondrial pathways, including the TCA cycle, OXPHOS, and FAO [12]. These pathways function as energy-generating systems and are intricately connected to oncogenic signaling, epigenetic regulation, and microenvironmental interactions, enabling tumors to sustain proliferation, evade therapeutic interventions, and metastasize.
The TCA cycle, also known as the Krebs cycle or citrate cycle, serves as a central metabolic hub in mitochondrial bioenergetics, facilitating adenosine triphosphate (ATP) synthesis through OXPHOS while reducing equivalents [nicotinamide adenine dinucleotide (NADH) and flavine adenine dinucleotide, reduced (FADH2)] for electron transport chain activity. In breast cancer, the dynamic rewiring of the TCA cycle is a hallmark of metabolic adaptation, enabling neoplastic cells to meet heightened bioenergetic demands, sustain proliferative signaling, and maintain redox homeostasis within the tumor microenvironment (TME). In addition to its canonical role in energy production, emerging evidence highlights the diverse functions of the TCA cycle in epigenetic regulation, the provision of biosynthetic precursors, and adaptation to stress across various breast cancer subtypes.
Breast cancer subtypes exhibit distinct patterns of TCA cycle utilization that
are influenced by receptor status and oncogenic drivers. Estrogen
receptor-positive (ER+) tumors preferentially depend on oxidative
metabolism, wherein glucose oxidation predominates over glycolysis to supply
energy for TCA cycle activity and OXPHOS [13, 14]. ER
The activity of the TCA cycle in breast cancer varies across different stages.
Comparative analyses indicate that compared with that in primary tumors, TCA
cycle activity is significantly elevated in metastatic breast tumors,
particularly in pulmonary lesions [22]. Here, pyruvate carboxylase (PC)-mediated
anaplerosis plays a crucial role in maintaining oxaloacetate pools, which are
essential for meeting bioenergetic demands under fluctuating nutrient
availability [23]. Furthermore, metastatic cells exhibit a dynamic ability to
switch between glycolytic and OXPHOS states through modulation of the
HIF-1
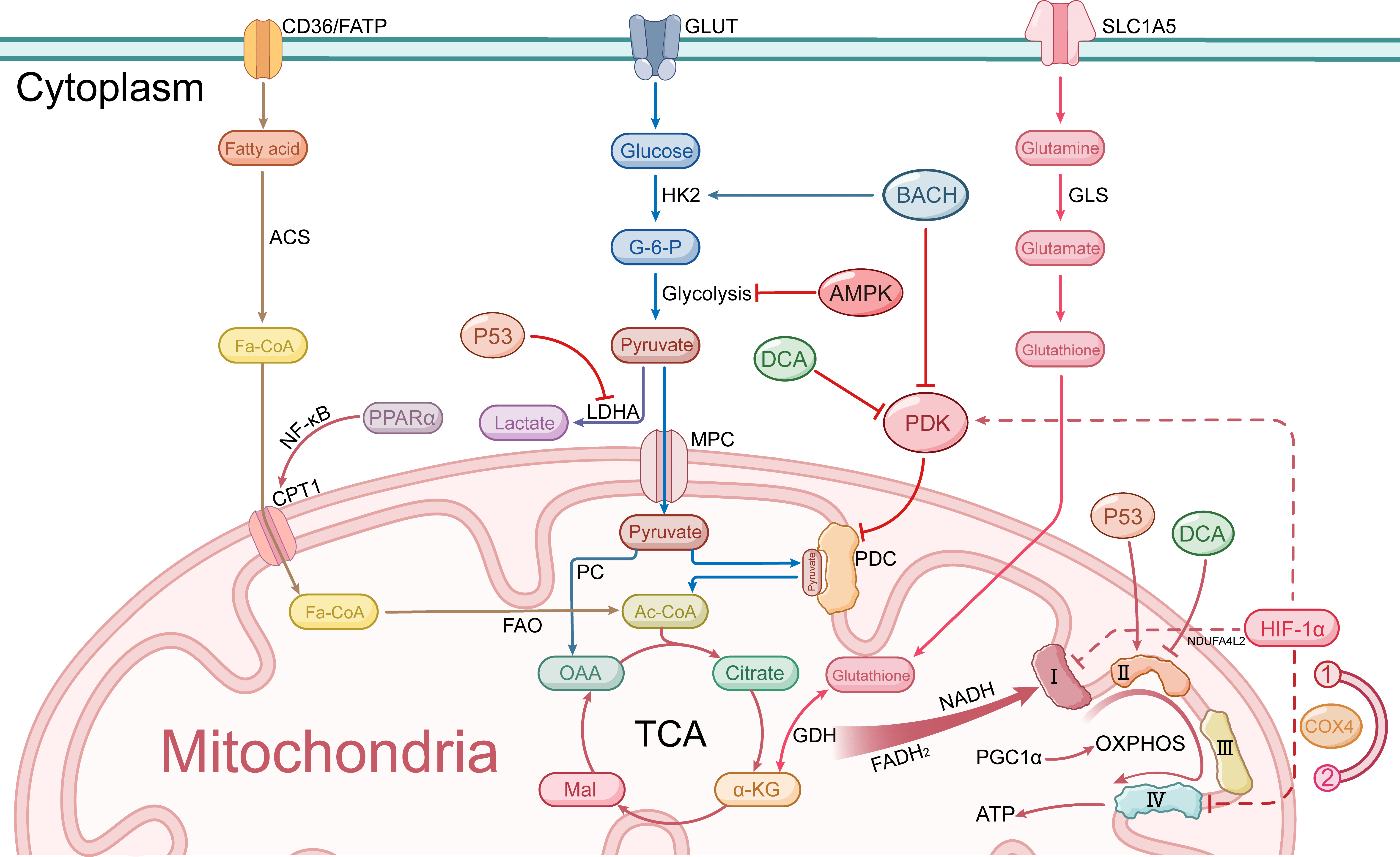 Fig. 2.
Fig. 2.
Mitochondrial metabolic pathways in breast cancer cells. FATP,
fatty acid transport protein; GLUT, glucose transporter; SLC1A5, solute carrier
family 1 member 5; ACS, acyl-CoA synthetase; HK2, hexokinase 2; BACH, BTB domain
and CNC homolog 1; GLS, glutaminase; G-6-P, glucose-6-phosphate; AMPK,
AMP-activated protein kinase; P53, tumor protein p53; DCA, dichloroacetate; PDK,
pyruvate dehydrogenase kinase; LDHA, lactate dehydrogenase A; MPC, mitochondrial
pyruvate carrier; PPAR
In recent years, considerable attention has been given to altering TCA cycle flux through the modulation of pyruvate metabolism. BTB domain and CNC homolog 1 (BACH1), a transcription factor that is highly expressed in breast tumors, inhibits pyruvate dehydrogenase (PDH) activity by transcriptionally activating pyruvate dehydrogenase kinase (PDK). This action limits the entry of pyruvate into mitochondria and enhances the Warburg effect [25]. Similarly, the therapeutic agent miR-211, which targets pyruvate metabolism, shifts the cellular phenotype toward a proglycolytic state by depleting PDK4, thereby increasing the susceptibility of cancer cells to mitochondrial respiratory inhibition [26]. In contrast, the modulation of AMPK activation leads to the phosphorylation of PDH, thereby restoring TCA cycle flux and promoting cancer metastasis by enabling cancer cells to adapt to metabolic and oxidative stress. These findings suggest a dual role for metabolic checkpoints in balancing biosynthetic and catabolic demands during metastasis [22] (Fig. 2). In chemotherapy-resistant cells, the TCA cycle is among the pathways that are most significantly upregulated, indicating that these cells have evolved a more pyruvate-dependent TCA cycle metabolic pathway, which is closely associated with reduced sensitivity to chemotherapy [17]. Targeting the TCA cycle may help overcome the mitochondrial adaptations observed in chemotherapy-persistent triple-negative breast cancer (TNBC), restoring sensitivity to therapeutic agents.
As a nexus of metabolic, epigenetic, and biosynthetic regulation, the TCA cycle serves as a critical linchpin in breast cancer progression. However, further research is necessary to fully elucidate the alterations in TCA cycle metabolic signals and their implications for the treatment of mitochondrial metabolism in breast cancer and to advance the development of related metabolic therapeutic agents. The clinical translation of interventions aimed at the TCA cycle is hindered by the metabolic heterogeneity observed among various breast cancer subtypes. To address this challenge, future studies should focus on tailored therapeutic strategies informed by the metabolic specificity of these distinct subtypes.
OXPHOS, a mitochondrial process that couples electron transport chain (ETC) activity with ATP synthase-mediated proton gradient utilization, is a critical mechanism for energy production in eukaryotic cells. In cancer biology, dysregulated OXPHOS has emerged as a hallmark of metabolic reprogramming, challenging the classical Warburg effect by demonstrating its indispensable role in tumor adaptation and therapy resistance. While aerobic glycolysis predominates ATP synthesis in many malignancies, accumulating evidence highlights OXPHOS as a key metabolic driver across breast cancer subtypes, particularly under microenvironmental stress and therapeutic pressure [27, 28].
Like that of the TCA cycle, the oxidative phosphorylation activity of different
subtypes also varies significantly with metabolic preference. In HER2+ BC,
17
Genetic, epigenetic, and microenvironmental factors influence OXPHOS plasticity.
Mutations in the breast cancer type 1 susceptibility protein (BRCA1) induce
mitochondrial dysfunction and aberrant activation of OXPHOS, fostering energy
dependency and malignant progression [35] (Fig. 2). Chronic inflammation and
environmental toxins exacerbate the risk of breast cancer by impairing
p53-mediated mitochondrial quality control, which leads to OXPHOS dysfunction and
genomic instability [36]. The age-associated decrease in mitochondrial
respiration further synergizes with host–environment interactions to promote
breast cancer pathogenesis (Fig. 2). Additionally, the hypoxic microenvironment
and dynamic changes in ROS also regulate OXPHOS. HIF-1
Numerous treatments for breast cancer involve the regulation of oxidative
phosphorylation. Among these, targeting electron transport chain complexes is a
significant strategy for modulating oxidative phosphorylation. Coptisine, a
natural isoquinoline alkaloid, selectively inhibits complex I in TNBC, leading to
metabolic crisis and apoptosis [28]. Similarly, disruption of the
ShcA-PGC-1
OXPHOS-related signaling pathways play subtype-specific roles in tumor progression, metastasis, and treatment resistance. Their regulation by oncogenic signals, tumor suppressors, and microenvironmental cues highlights the need for precise targeting strategies. A comprehensive understanding of the interactions among these pathways could facilitate the development of targeted therapies to restore metabolic balance and improve patient outcomes.
FAO, a mitochondrial process that breaks down long-chain fatty acids via
Distinct FAO activities are observed across breast cancer subtypes, reflecting
their divergent metabolic demands. In estrogen ER+ breast cancer, FAO is
significantly upregulated in cells that are resistant to endocrine therapy [9, 43, 44]. Mechanistically, the activation of FAO stimulates the Src signaling
pathway, which drives resistance to antiestrogen therapies. Similarly, basal-like
breast cancers exhibit elevated FAO levels to sustain rapid proliferation [45].
TNBC, the most FAO-dependent subtype, has unique regulatory mechanisms: CD24-low
TNBC cells activate PPAR
Given the importance of FAO in mitochondrial metabolism in breast cancer, exploring its regulatory mechanism is essential. The rate-limiting enzyme carnitine palmitoyl transferase 1A (CPT1A), which governs mitochondrial fatty acid import, has emerged as a central regulator. Its overexpression facilitates metabolic adaptation under stress conditions [48]. Pharmacological inhibition of CPT1A by quercetin suppresses FAO, depletes ATP, and elevates ROS levels, ultimately inducing apoptosis in TNBC [49] (Fig. 2). Beyond enzymatic control, tumor suppressor networks modulate FAO: inactivation of the retinoblastoma (RB) protein upregulates CPT1A, enhances FAO-driven secretion of the chemokine CCL2, and recruits immunosuppressive myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), fostering a protumorigenic microenvironment [50]. These findings highlight CPT1A as both a metabolic modulator and an immune modulator in breast cancer.
FAO metabolism has significant advantages in terms of environmental constraints, particularly in TNBC, where metabolic reorganization prioritizes fatty acid catabolism to meet high energy demands [9]. This metabolic pathway also preserves mitochondrial membrane integrity, counteracting apoptosis under therapeutic stress [33, 47]. Under nutrient deprivation, breast cancer cells utilize FAO for survival: mesencephalic astrocyte-derived neurotrophic factor (MANF) stabilizes parkin RBR E3 ubiquitin protein ligase (PRKN)-mediated mitophagy to sustain FAO during glucose starvation [51], whereas snail family transcriptional repressor 1 (SNAI1) suppresses Acetyl-CoA carboxylase 2 (ACC2), thereby reducing malonyl-CoA levels to relieve CPT1A inhibition and activate FAO [52]. Furthermore, the loss of retinoic acid receptor responder 1 (RARRES1) enhances fatty acid synthesis (FAS), providing substrates for FAO to adapt to nutrient-poor environments [53] (Fig. 2). These adaptive mechanisms establish FAO as a vulnerability in energy-restricted tumors.
Current evidence suggests that FAO is a multifaceted driver of breast cancer progression, influencing metabolic flexibility, therapeutic resistance, and immune evasion. Targeting key nodes such as CPT1A or upstream regulators may disrupt FAO-driven malignancy. Combinatorial strategies that inhibit FAO alongside Src signaling or immune checkpoint pathways show promise in preclinical models. Future investigations should prioritize further elucidating the interactions between FAO, tumor microenvironmental factors, and epigenetic regulators to advance the precision treatment of FAO-dependent breast cancer subtypes.
Primary breast cancer cells exhibit extensive metabolic heterogeneity, with distinct breast cancer subtypes and treatment backgrounds presenting divergent metabolic phenotypes. In endocrine-resistant ER+ breast cancer, fibroblast growth factor 1 (FGF1) activates ER signaling independent of estradiol, driving glycolytic shifts while suppressing mitochondrial metabolism. Adipocyte-derived FGF1 associated with obesity exacerbates this pathway [54]. HER2+ breast cancer cells overexpressing NDUFA4L2 exhibit impaired mitochondrial complex I activity and increased glycolysis, conferring resistance to trastuzumab through HER2 mitochondrial relocalization and ROS suppression [55] (Fig. 1). Metabolic phenotypic distinctions are most pronounced between ER+ breast cancer and TNBC. Owing to the robust metabolic plasticity of TNBC, more metabolic intermediates accumulate in TNBC than in ER+ breast cancer (Fig. 3). These intermediates serve as substrates and cofactors, thereby promoting TNBC epigenetics. Diverse oncoproteins, including EGFR, AR, VEGF, and PD-L1, are highly expressed, further increasing malignant characteristics [56, 57, 58, 59, 60, 61, 62] (Fig. 3).
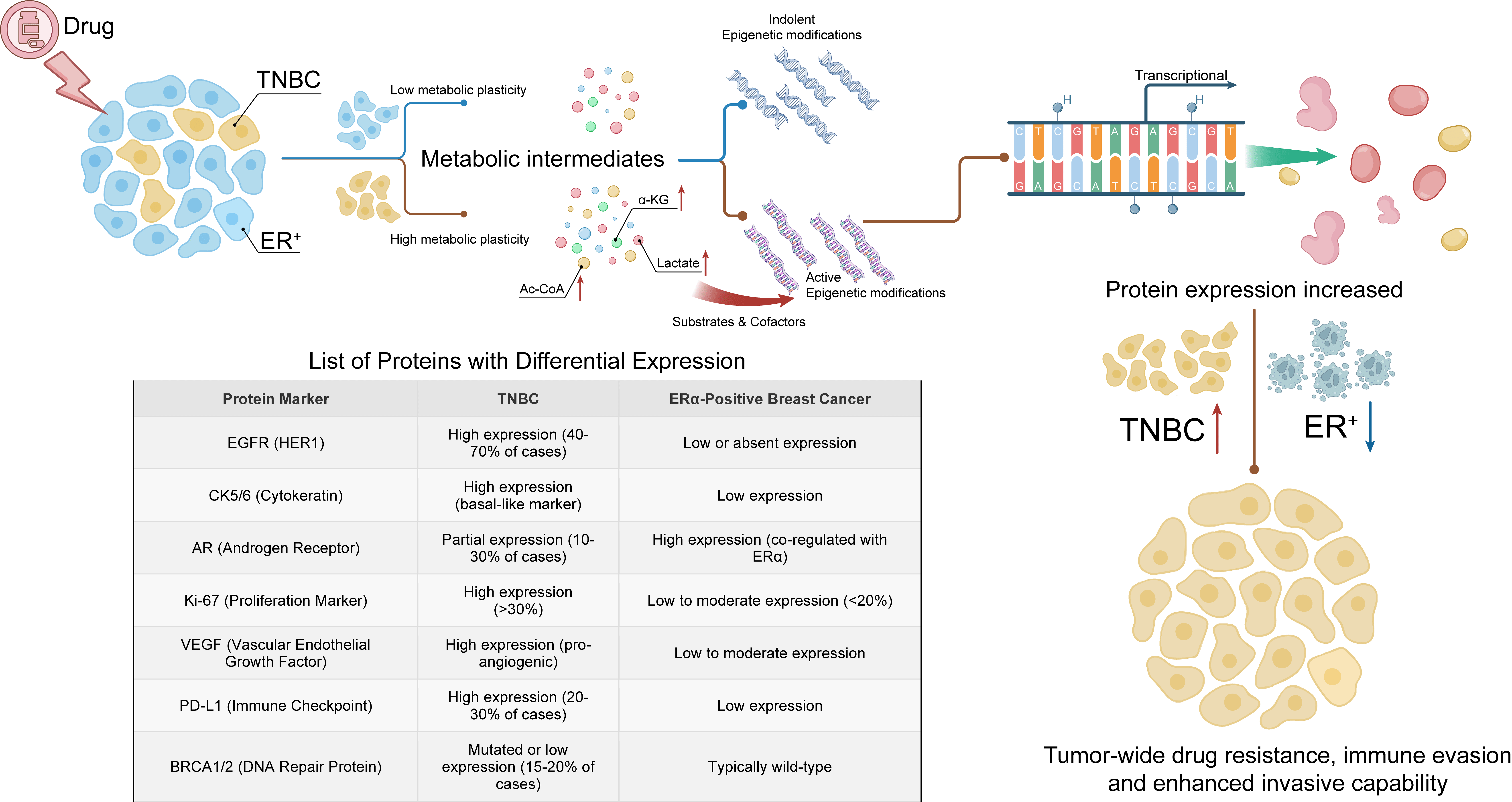 Fig. 3.
Fig. 3.
Metabolic plasticity of subtypes of breast cancer. The robust
metabolic plasticity of TNBC endows it with greater accumulation of metabolic
intermediates compared to ER+ breast cancer. These intermediates serve as
substrates and cofactors that facilitate the epigenetic process in TNBC,
consequently enabling the expression of a diverse array of oncogenic proteins and
enhancing invasion, therapy resistance, and immune evasion capabilities. Red
upward arrow: Enhanced drug resistance, immune evasion, and invasion in TNBC;
Blue downward arrow: Reduced in ER+. TNBC, triple-negative breast cancer;
ER+, estrogen receptor-positive; Ac-CoA, acetyl-CoA;
Organ-specific metastasis further diversifies metabolic dependencies. Compared
with bone or lung metastatic cells, liver-metastatic breast cancer cells display
increased glycolytic flux and decreased mitochondrial metabolism. This adaptation
is correlated with heightened HIF-1
These discrepancies fundamentally stem from tumor heterogeneity and microenvironmental dependence. Even within identical subtypes, metabolic preferences differ across cell lines. Second, dynamic microenvironmental changes—including changes in metastatic site, nutrient availability, and hypoxia severity—reshape metabolic phenotypes. Moreover, disparities between cell line models and clinical specimens impact the generalizability of conclusions. Consequently, future research must prioritize metabolic dynamics and context dependency, analyze reprogramming trajectories, and avoid oversimplified subtype generalizations. The development of clinically relevant models and the performance of subtype-stratified clinical studies represent promising approaches. These directions will not only resolve current research contradictions but also strengthen the theoretical foundation for the clinical translation of mitochondrial metabolism-targeted therapies.
As the primary energy hubs of eukaryotic cells, mitochondria govern cellular bioenergetics and engage in dynamic cross-talk with other organelles. These interorganellar interactions—particularly between mitochondria and the endoplasmic reticulum (ER), lysosomes, and lipid droplets—play critical regulatory roles in mitochondrial metabolism, tumor progression, and therapeutic resistance in breast cancer.
The ER, a central organelle for protein folding, lipid biosynthesis, and calcium (Ca2+) storage, forms specialized membrane contact sites with mitochondria known as mitochondria-associated ER membranes (MAMs) [64]. These junctions facilitate bidirectional communication, enabling coordinated regulation of Ca2+ transfer, lipid metabolism, and stress responses (Fig. 4).
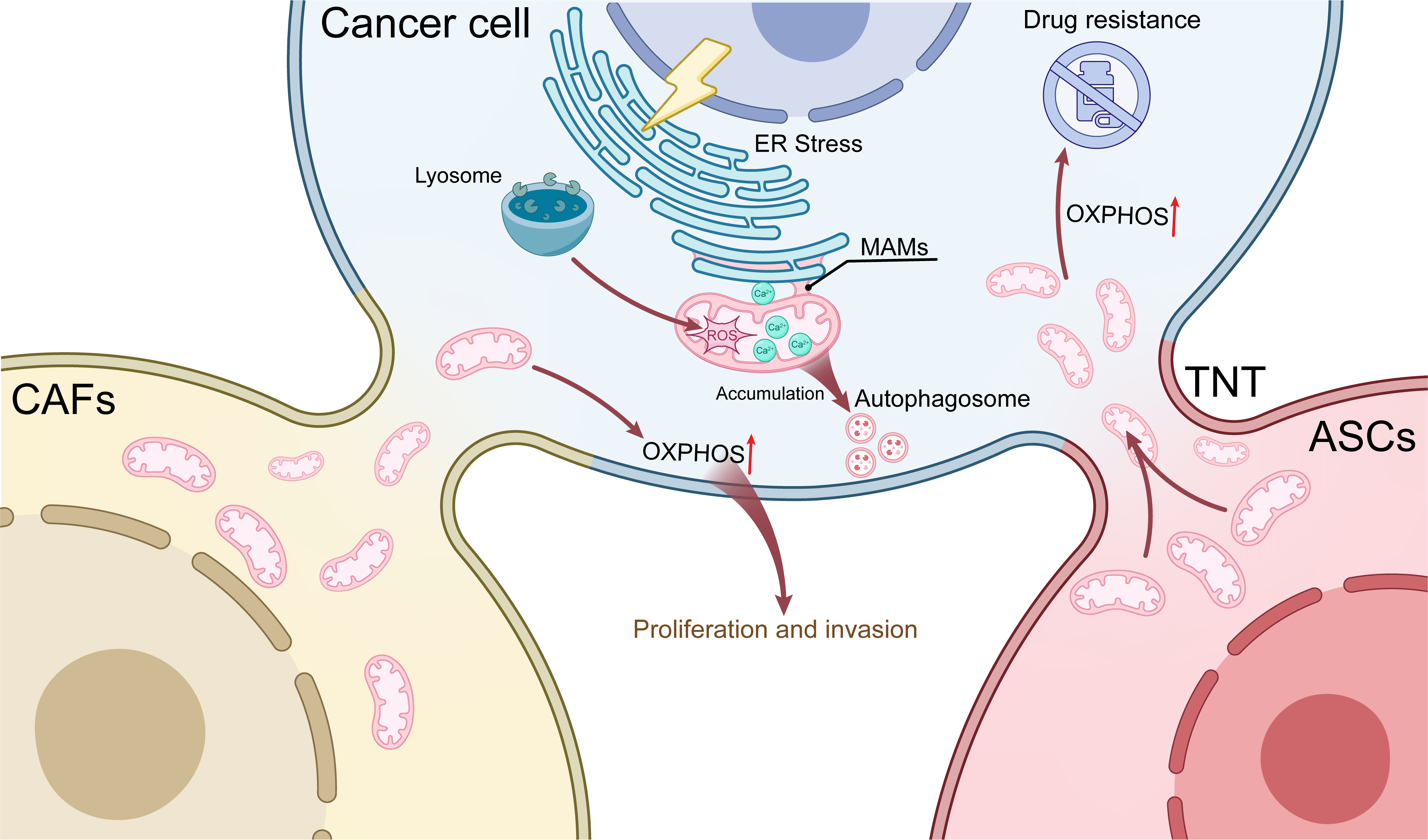 Fig. 4.
Fig. 4.
Network regulation of mitochondrial metabolism. Mitochondria interact with key organelles such as lysosomes and the endoplasmic reticulum to influence malignant features. Additionally, breast cancer cells acquire functional mitochondria from CAFs and ASCs via TNTs, thereby enhancing their metabolic fitness and invasive potential, upregulating OXPHOS, and promoting therapy resistance. Red upward arrow: Increased OXPHOS activity. TNT, tunneling nanotube; CAFs, cancer-associated fibroblasts; ASCs, adipose-derived stem cells; MAMs, mitochondria-associated endoplasmic reticulum membranes; ER, endoplasmic reticulum; OXPHOS, oxidative phosphorylation.
Emerging evidence indicates that ER–mitochondria coupling enhances Ca2+ shuttling, which directly modulates mitochondrial ATP synthesis by activating key enzymes in the TCA cycle and OXPHOS. This metabolic synergistic collaboration supports the high bioenergetic demands of proliferating breast cancer cells [65]. However, sustained ER stress triggers the unfolded protein response (UPR), disrupting mitochondrial membrane potential and increasing ROS production, ultimately driving apoptosis [66]. Laminin subunit gamma 2 (LAMC2), an ER-resident protein, mitigates ER stress by promoting ER‒mitochondria interactions, increasing tumor cell survival under stress conditions [67].
Ca2+ dysregulation serves as a pivotal mediator of ER-mitochondria interactions. Cancer cells preferentially take up Ca2+ released from the ER through MAMs to meet energy requirements, but Ca2+ overload induces mitochondrial dysfunction and apoptosis (Fig. 4). Cancer cells enhance the connection of MAMs through the dynamic regulation of the ER-mitochondrial tether protein Rab32, which leads to Ca2+ homeostasis by affecting the expression of the Ca2+ uptake protein UCP2. Therefore, regulating the expression of both can trigger mitochondrial Ca2+ overload-driven cell death [65]. Similarly, curcumin (CUR) disrupts mitochondrial Ca2+ homeostasis by promoting ER Ca2+ release while inhibiting efflux pathways, leading to Ca2+ overload and the activation of proapoptotic cascades in breast cancer cells (BCCs). This mechanism offers tumor-specific therapeutic potential when combined with chemotherapy [68, 69]. Furthermore, ER-derived Ca2+ flux regulates mitochondrial metabolic plasticity, enabling BCCs to adapt to nutrient deprivation and chemotherapeutic challenges. These findings highlight Ca2+ signaling as a dual-edged sword in maintaining metabolic homeostasis and driving oncogenic progression [70, 71].
Given that intimate mitochondria‒endoplasmic reticulum coupling is mediated by Ca2+ signaling, Ca2+ represents a promising therapeutic target for anticancer treatment. Future research should focus on disrupting Ca2+ homeostasis through the modulation of its uptake and transport mechanisms, thereby inducing endoplasmic reticulum stress and promoting tumor apoptosis when combined with mitochondrial-targeted therapies.
Lysosomes, the primary degradative compartments, cooperate with mitochondria to regulate autophagy, redox balance, and metabolic reprogramming in cancer. Key lysosomal regulators, including NPC intracellular cholesterol transporter 1 (NPC1), glycogen phosphorylase L (PYGL), and tbc1 domain family member 5 (TBC1D5), orchestrate this interplay (Fig. 4).
NPC1, a cholesterol transporter localized to lysosomes, ensures mitochondrial membrane integrity by facilitating cholesterol efflux. NPC1 deficiency induces lysosomal cholesterol accumulation, mitochondrial fragmentation, and impaired mTOR signaling, all of which suppress TNBC aggressiveness [72]. Conversely, PYGL, a glycogen phosphorylase, links lysosomal glycogenolysis to mitochondrial metabolism. PYGL inhibition disrupts mitochondrial–lysosomal colocalization, impairing mitophagy and exacerbating metabolic stress in glioblastoma, a mechanism that is likely conserved in breast cancer [73]. In doxorubicin-resistant BCCs, lysosomal‒mitochondrial cross-talk promotes drug resistance via ROS-mediated mitophagy. MtROS activate autophagy-lysosomal pathways to eliminate damaged mitochondria, a survival mechanism that is attenuated by the mtROS scavenger mitoTEMPO [74]. Therapeutic strategies exploiting this axis include TPGS/CS-modified nanocarriers, which leverage lysosomal hyaluronidase to release chemotherapeutics and disrupt mitochondrial function, achieving synergistic cytotoxicity in multidrug-resistant tumors [75].
By modulating interorganellar metabolic crosstalk between mitochondria and lysosomes to disrupt mitochondrial metabolic homeostasis and compromise mitophagy while increasing mitochondrial targeting efficacy, this therapeutic strategy merits rigorous investigation.
Lipid droplets (LDs), which are dynamic lipid storage organelles, interact with mitochondria to balance FAO and energy production [76]. In TNBC, cub domain-containing protein 1 (CDCP1) reprograms lipid metabolism by depleting cytoplasmic LDs and enhancing mitochondrial FAO, fueling metastatic dissemination. Cotargeting CDCP1 and acyl-CoA-synthetase ligase (ACSL) reverses this metabolic adaptation, highlighting the LD–mitochondria interplay as therapeutic vulnerability [46].
Caspase-1 further links lipid metabolism to mitochondrial function through the
cleavage of PPAR
These findings illustrate how lipid droplets collaborate with mitochondria to drive metabolic flexibility, stress adaptation, and immune evasion in breast cancer. Targeting this mechanism and compromising tumor metabolic adaptability and plasticity by disrupting mitochondrial-lipid droplet metabolic synergy represents a promising adjuvant therapeutic strategy.
The tumor microenvironment comprises dynamic interactions between malignant cells and diverse stromal components, including immune cells, fibroblasts, vascular endothelial cells, and the extracellular matrix. Beyond chemical signaling, physical intercellular communication—particularly mitochondrial transfer—has emerged as a critical mechanism modulating tumor progression, metastasis, and therapeutic resistance. Mitochondrial transfer involves the intercellular exchange of intact mitochondria or mitochondrial components via tunneling nanotubes (TNTs), extracellular vesicles (EVs), or direct membrane contact. This process enhances cancer cell bioenergetics and profoundly influences proliferation, migration, and drug resistance (Fig. 4). Recent studies have elucidated key mechanisms and functional consequences of mitochondrial crosstalk within the TME [11].
First, adipose-derived stem cells (ASCs) facilitate multidrug resistance (MDR)
in breast cancer through mitochondrial donation. Coculture experiments have
revealed that ASCs transfer functional mitochondria to BCCs via TNTs under
hypoxic conditions. This transfer augments OXPHOS-driven ATP production,
upregulates ABC transporter activity, and reduces HIF-1
Similarly, the metastatic potential of CAFs is enhanced through mitochondrial exchange. CAFs establish TNT-mediated connections with breast cancer cells, delivering functional mitochondria that increase the amount of OXPHOS-derived ATP. This metabolic shift significantly enhances 3D cancer cell migration, independent of glycolytic ATP modulation. Intriguingly, artificial OXPHOS activation fails to replicate this promigratory effect unless glycolysis remains unperturbed, underscoring the precision of CAF-driven metabolic symbiosis [79].
Mitochondrial hijacking extends to immune evasion. Tumor cells exploit TNTs to acquire mitochondria from immune cells, depleting immune metabolic resources while inducing their proliferation (Fig. 4). This transfer impairs immune cell activation and promotes tumor immune escape. Pharmacological inhibition of TNT assembly combined with immune checkpoint blockade significantly reduces tumor growth in immunocompetent models, highlighting the therapeutic potential [80].
Mitochondrial transplantation further modulates therapeutic responses. Introducing healthy mitochondria into MDA-MB-231 cells enhances cisplatin-induced apoptosis in mitochondrial-dysfunctional subpopulations but paradoxically increases invasion in intact cells. Conversely, dysfunctional mitochondrial grafts shift metabolism toward glycolysis, attenuating proliferation unless OXPHOS is restored [81].
Finally, horizontal mtDNA transfer via CAF-derived EVs drives endocrine resistance. Hormone therapy-resistant (HTR) breast cancer cells acquire host-derived mtDNA, reactivating OXPHOS and enabling escape from metabolic dormancy. This EV-mediated mtDNA transfer preferentially enriches cancer stem-like cells, linking mtDNA acquisition to therapeutic evasion [82].
Mitochondrial transfer orchestrates metabolic adaptation, immune evasion, and therapeutic resistance in cancer. Targeting intercellular mitochondrial trafficking—through TNT inhibition, EV modulation, or mitochondrial transplantation—represents a promising strategy to disrupt tumor–stromal metabolic symbiosis and improve therapeutic outcomes.
Collectively, these studies indicate that the network regulation of mitochondrial metabolism involves exploitable metabolic vulnerabilities that are amenable to therapeutic intervention. Targeting the mitochondrial spatial network represents a promising paradigm for next-generation precision strategies to overcome tumor metabolic plasticity, therapeutic resistance, and metastatic progress.
Metabolic reprogramming is a hallmark of cancer progression and is classically characterized by enhanced aerobic glycolysis (the Warburg effect) and dysregulation of OXPHOS. Although mitochondrial function often remains intact in tumors, cancer cells preferentially utilize glycolysis to rapidly generate ATP and biosynthetic precursors while concurrently secreting lactic acid to remodel the tumor microenvironment (TME). Breast cancer, however, exhibits profound metabolic heterogeneity. Single-cell resolution imaging reveals coexisting glycolytic and OXPHOS-dependent subpopulations within tumors that are dynamically regulated by PI3K signaling, actin cytoskeleton remodeling, and spatial confinement effects [14, 83]. In vivo metabolic profiling further reveals that, compared with nonmetastatic 67NR tumors, metastatic 4T1 tumors display elevated glycolytic flux and mitochondrial membrane potential (MMP), underscoring a link between metabolic plasticity and metastatic potential [84].
The therapeutic implications of these metabolic characteristics are becoming
increasingly evident, driving the development of interventions targeting key
enzymes involved in aerobic glycolysis and gene therapy. For instance, mutations
in the critical tumor suppressor gene TP53 disrupt mitochondrial metabolism,
intensify glycolytic dependence, and sensitize tumors to agents such as
2-deoxyglucose [85]. PDK1 acts as a key glycolytic gatekeeper, phosphorylating
PDH to divert pyruvate away from the TCA cycle, thereby promoting glycolysis
[40]. The PDK inhibitor DCA can restore mitochondrial function and,
synergistically with metformin, induce apoptosis while reducing lactic acid
accumulation [41]. Combining inhibitors targeting both glycolysis and
mitochondrial metabolism represents a promising strategy to simultaneously
suppress both pathways, disrupt compensatory mechanisms, and induce metabolic
catastrophe. For example, the GLUT1 inhibitor BAY-876 blocks glycolysis, and its
combination with the mitochondrial agent closalamine inhibits compensatory
metabolic pathways, leading to metabolic collapse [86]. Furthermore,
dual-targeting approaches, such as concurrently inhibiting the CAF-TGF-
Emerging evidence increasingly suggests that OXPHOS serves as the primary energy source in many tumors, especially in chemotherapy-resistant malignancies, which contradicts the classical metabolic characteristics of tumors. Cemile demonstrated that OXPHOS inhibitors trigger mitophagy via oxidative stress-induced mitochondrial dysfunction, thereby reducing the metastatic potential of chemotherapy-resistant TNBC cells. These findings indicate that compared with their chemotherapy-sensitive counterparts, chemotherapy-resistant triple-negative breast cancer cells rely more strongly on OXPHOS for energy production [88]. Consequently, future therapeutic strategies should focus on combining OXPHOS inhibitors with conventional anticancer drugs for the treatment of chemotherapy-resistant breast cancer.
ROS, which are primarily produced through mitochondrial OXPHOS, serve as important mediators of cellular homeostasis and stress responses. Physiological ROS modulate redox signaling, but excessive accumulation induces oxidative stress, disrupts genomic stability, and promotes tumor progression. In breast cancer, metabolic adaptation enables cancer cells to balance ATP production with ROS mitigation by promoting glycolysis. Paradoxically, elevated ROS levels may also drive mitochondrial dysfunction, genomic instability, and metastatic dissemination, highlighting the context-dependent role of ROS in tumorigenesis [84, 89].
HIF-1
Mitochondrial quality control mechanisms further regulate ROS homeostasis. Caveolin-1 (Cav-1) modulates mitochondrial dynamics through interactions with Mfn2 and Drp1; Cav-1 deficiency promotes ROS accumulation and chemotherapy resistance via monocarboxylate transporter 4 (MCT4)-driven lactate shuttling [94, 95]. Similarly, ULK1-mediated mitophagy eliminates damaged mitochondria under ROS stress, whereas MANF stabilizes PRKN to restore mitochondrial integrity during glucose starvation, linking ROS adaptation to metabolic plasticity [51, 96].
Given the close connection between ROS and metabolism, many therapeutic approaches aim to alter ROS levels by disrupting metabolic balance. Among these, strategies that increase ROS levels—the use of chemotherapy drugs, nanomaterials, or targeted agents—induce ROS overload, exceeding cellular antioxidant capacity to trigger apoptosis, ferroptosis, or immunogenic cell death (ICD). However, this approach faces limitations, including potential damage to normal tissues, difficulty in the precise control of ROS levels, and the risk that chronic ROS exposure may induce drug resistance or metastasis.
Targeting redox balance represents a viable therapeutic strategy for increasing ROS levels. Breast cancer cells dynamically maintain ROS equilibrium through glutathione (GSH)-dependent systems. GK-1 disrupts redox balance by reducing the GSH/GSSG ratio and increasing H2O2 levels, inducing mitochondrial uncoupling and apoptosis in TNBC92 cells. Obesity-associated tumors adapt through FAO-dependent YAP signaling; AP signaling alleviates FAO-induced oxidative stress by transcriptionally upregulating the expression of SOD and GPX4 [97]. Nanotherapeutic strategies exploit redox vulnerabilities by depleting GSH and amplifying ROS through peroxidase-like activity, thereby enhancing the efficacy of radiotherapy [98]. Another therapeutic strategy involves the reduction of ROS levels in cancer cells through the use of antioxidants or ROS-generation inhibitors to diminish proliferation, metastasis, and therapeutic resistance or to restore sensitivity to endocrine therapy. Wang and Zhang [99] reported that 4–10 Gy X-ray radiotherapy reduces breast cancer cell viability independently of ROS. Reducing ROS levels prevent the ability of cisplatin to decrease cell viability and induce cell death but does not impair the cytotoxic effects of X-irradiation. These findings indicate that antioxidants may mitigate the ROS-related side effects of radiotherapy without compromising its efficacy.
ROS signaling in breast cancer constitutes a double-edged sword. A comprehensive understanding of the mechanisms governing ROS fluctuations is crucial for breast cancer prevention and treatment. Precise strategies targeting ROS regulators or amplifying oxidative stress must consider tumor subtypes, microenvironments, and treatment history. Future research should prioritize the exploration of effective ROS-modulating strategies and their combination with immunotherapy or metabolic inhibitors to overcome treatment resistance.
In recent years, targeted mitochondrial therapy has emerged as a prominent cancer treatment approach because of its high specificity, favorable therapeutic outcomes, and minimal adverse effects. Mitochondria contain multiple therapeutic targets, including the MPTP, various ionic components, ROS, and mitochondrial dynamics. These critical components are closely associated with metabolic dysregulation in breast cancer cells, making them effective targets for cancer intervention through the induction of mitochondrial dysfunction. Therefore, the development of precise therapeutic strategies that target these mitochondrial components can optimize treatment efficacy while maintaining cellular homeostasis.
Mitochondrial targeting has been widely studied in different types of breast cancer cells, and targeted therapy can be conducted through metabolic heterogeneity among different types of cells. Metastatic cells exhibit heightened reliance on mitochondrial respiration and OXPHOS. Pharmacological inhibition of extracellular signal-regulated kinase 1/2 (ERK1/2) signaling disrupts OXPHOS-dependent metabolic adaptations, effectively suppressing metastatic progression and tumor growth [100].
Endocrine therapy resistance in ER+ breast cancer is mechanistically linked to metabolic reprogramming. Activation of the AMPK-FAO-OXPHOS axis enhances cancer cell survival under metabolic stress. Conversely, inhibition of carnitine palmitoyl transferase 1 (CPT1) or FAO restores endocrine sensitivity by impairing energy flexibility [7]. In addition, combining the mitochondrial inhibitor everolimus with ONC201/tic10, a dual-acting drug that inhibits OXPHOS while activating integrated stress response pathways, was found to target mitochondrial respiration, highlighting its potential as a metabolic reprogramming strategy to overcome drug resistance in ER+ breast cancer [101]. Previously, ONC201 monotherapy administered orally at 625 mg demonstrated no significant clinical activity in recurrent/refractory metastatic breast cancer. A key limitation was the absence of companion diagnostics: tumors were not screened for mitochondrial caseinolytic protease P (ClpP) expression or mitochondrial dependency, resulting in the enrollment of nondependent patients. Furthermore, suboptimal drug delivery efficiency further constrained the therapeutic efficacy, underscoring the need to address interpatient metabolic heterogeneity and biomarker deficiencies [102] (Table 1, Ref. [102, 103, 104, 105, 106, 107, 108]).
| Target | Type of study | Results | Reference |
| Oxphos inhibitor ME-344 | Phase 0/I clinical trial, HER2-patients | Significant biological antitumor activity | [103] |
| Oxphos inhibitor Metformin | Phase II clinical trial | Metformin targets FAO, affecting drug combinations | [104] |
| Mitochondrial biogenesis ONC201 | Phase II clinical trial, Recurrent/Refractory Metastatic Breast Cancer | ONC201 monotherapy did not induce objective responses but had an acceptable safety profile | [102] |
| The immune co-enhancer Apt-LPR | Phase III, in combination with pdt | Released antigens to activate immunity and reduce immune escape | [105] |
| mTOR inhibitor Rapamycin | In vitro, the Human breast CSC cell line | mTOR is a crucial effector of Sal-induced ferroptosis | [106] |
| FAO inhibitor CP4 | In vitro and in vivo, TNBC cell lines and models | Resulting in remarkable tumor regression | [107] |
| Targeting STAT1-mediated ROS scavenger drug |
In vitro and in vivo, BC cell lines and models | Significantly increased the therapeutic potential of several complex I inhibitors in oncology | [108] |
Emerging mitochondrial-targeted interventions in HER2-associated breast cancer have demonstrated considerable therapeutic potential. Targeted sonodynamic therapy using piezoelectric nanoparticles (PGd@tNBs) has demonstrated therapeutic potential for HER2-positive breast cancer. Ultrasound-triggered reactive oxygen species (ROS) generation disrupts mitochondrial membrane potential, inducing apoptosis through mitochondrial dysfunction [109]. In contrast, HER2-negative tumors lack classic oncogenic drivers but exhibit metabolic vulnerabilities. A randomized phase 0/I trial has revealed that combining bevacizumab with the mitochondrial inhibitor ME-344 results in this dependency. Bevacizumab first induces vascular normalization to reoxygenate tumors and shift their metabolism toward mitochondrial respiration. ME-344 cells subsequently inhibit oxidative phosphorylation, leading to a 23.4% reduction in Ki67 expression and significantly attenuation of tumor activity [103] (Table 1).
Targeted therapy through mitochondrial metabolic heterogeneity in TNBC can effectively avoid the shortage of therapeutic targets for triple-negative breast cancer. TNBCs exhibit unique dependencies on mitochondrial pathways, including CPT1-mediated FAO, for metastatic expansion. Pharmacological CPT1 inhibition reduces ATP production, elevates ROS levels, and suppresses tumor progression. Mitochondrion-targeted ferroptosis inducers amplify oxidative stress by depleting GSH and inducing redox imbalance and apoptosis [107, 110] (Table 1).
Targeted therapy for pyruvate and glutamine metabolism to induce mitochondrial energy stress has also been a novel treatment in recent years. PDH inhibition disrupts glycolytic–TCA cycle coupling, sensitizing hypoxic tumor cells to radiotherapy. Similarly, targeting mitochondrial complex I induces bioenergetic stress by increasing ROS production and synergizing with NQO1 inhibition to overcome therapeutic resistance [42, 108] (Table 1). Glutamine metabolism regulates mitochondrial energy metabolism and REDOX homeostasis and is a key metabolic hub for ferroptosis. GSH depletion strategies and mTOR-mediated iron flux regulation modulate ferroptosis sensitivity. Metformin induces ferroptosis via SLC7A11 destabilization, whereas mTOR inhibitors counteract salinomycin-induced mitochondrial dysfunction, highlighting context-dependent regulatory mechanisms [106, 111] (Table 1).
Targeted therapy involving molecules involved in mitochondrial dynamics is also critical. DRP1-mediated mitochondrial fission supports FAO-dependent energy production in metastatic cells. DRP1 inhibition reduces lipid droplet utilization and impairs metastasis. Conversely, MFN1 overexpression in tamoxifen-resistant cells promotes mitochondrial fusion, and targeting MFN1 restores apoptotic sensitivity [112, 113].
Emerging as a pivotal innovation in precision oncology, mitochondrial nanotargeting has become a burgeoning frontier in cancer therapeutics. Over the past decade, nanoparticle-based delivery platforms have enabled the precise mitochondrial localization of therapeutic agents while minimizing off-target effects and amplifying treatment specificity. Representative systems include triphenylphosphonium (TPP)-conjugated nanoparticles, which leverage mitochondrial membrane potential gradients to achieve higher intramitochondrial drug accumulation compared with conventional carriers [114]. Additionally, combined therapy based on an ionic nanointerference strategy and biomimetic tumor targeting has paved the way for more effective nanotherapeutics. Biomimetic strategies such as platelet membrane-coated Ca2+ nanogenerators (PLT@MCC/CUR) demonstrate tumor-selective cytotoxicity through tumor vasculature-specific membrane fusion, subsequently inducing mitochondrial calcium overload and apoptosis [68].
Mitochondrial metabolic targeting represents a transformative paradigm in breast cancer therapy, addressing subtype-specific vulnerabilities through OXPHOS inhibition, redox modulation, and ferroptosis induction. However, monotherapy efficacy is often inadequate because of intratumoral metabolic heterogeneity. For instance, Rawand Masoud et al. [115] reported significant heterogeneity in OXPHOS rates across patient tumors. Compared with patients with low-OXPHOS tumors, patients with high-OXPHOS tumors demonstrate superior responses to benofamine/gemcitabine combination therapy. Beyond OXPHOS, therapies targeting FAO often underperform because of the lack of specificity of biguanide. Recent clinical studies have revealed that low-dose biguanides activate the Src pathway in patients with breast cancer, promoting proliferation and increasing FAO, whereas high-dose biguanides inhibit this effect. The combination with Src inhibitors shows promise for refractory metastatic TNBC [116].
Metabolic-targeted agents such as CPT1A or Complex I inhibitors have demonstrated efficacy in preclinical models but face significant clinical translation barriers. First, most studies remain confined to cell cultures and animal models, with few advancing to clinical trials. Consequently, many biological findings lack clinical validation. Second, metabolic targets often incur adverse effects, and achieving tumor-specific targeting remains challenging. Many metabolic drugs exhibit suboptimal tumor delivery precision. Finally, individual metabolic differences yield variable therapeutic responses, precluding the universal application of metabolic therapies.
Given disparities between preclinical/clinical outcomes and pervasive metabolic heterogeneity among patients, future efforts should prioritize combined treatment regimens, adopting either pathway plus target synergistic therapy or non-single-target metabolic treatment strategies to overcome drug resistance. It is also possible to optimize nanoparticle-based delivery and verify biomarkers for personalized treatment stratification to achieve precision medicine. In addition, in the face of heterogeneity, metabolic markers should be detected in advance for different patients, and individualized treatment plans should be designed to achieve better therapeutic effects.
As central organelles that govern cellular energy metabolism and apoptosis regulation, mitochondria have emerged as pivotal therapeutic targets for the treatment of breast cancer. Recent advances in mitochondrial-targeted delivery systems—including aptamers, TPPs, and multifunctional nanoparticles—combined with multimodal strategies such as photothermal/photodynamic therapy (PTT/PDT), metabolic inhibition, and immune modulation—have enabled multilayered precision targeting of breast cancer cells. This integrated approach directly induces mitochondrial dysfunction, activates antitumor immunity, and overcomes drug resistance, offering novel approaches for the treatment of aggressive subtypes such as TNBC.
Photodynamic and photothermal therapeutic modalities have recently attracted significant attention as promising strategies for precision oncology. PDT activates photosensitizers (PSs) in response to light at specific wavelengths, inducing the generation of cytotoxic ROS in the presence of molecular oxygen to achieve selective tumor cell destruction. In parallel, PTT uses photothermal conversion materials such as nanometallic particles to absorb near-infrared radiation. The conversion of optical energy into localized hyperthermia enables the direct thermal ablation of malignant tissues. For instance, AS1411 aptamer-modified nanoreactors (IR780/Ce@EGCG/APT) leverage near-infrared (NIR) light to generate localized hyperthermia, amplifying ferroptosis-associated ROS production while disrupting mitochondrial membrane integrity [117] (Table 2, Ref. [105, 117, 118, 119, 120, 121, 122, 123, 124]). Similarly, cuproptosis-inducing CuET@PHF nanoparticles utilize photothermal effects to alleviate tumor hypoxia, thereby enhancing copper ion-mediated toxicity to mitochondrial TCA cycle enzymes, eliminating cancer stem cells (CSCs), and triggering immunogenic cell death [118] (Table 2). Furthermore, the smart nanoplatform BMAEF employs the photothermal-triggered aggregation of gold nanoparticles to synergize with EGTA-mediated Ca2+ chelation, disrupting ion exchange between mitochondria and the Golgi apparatus, which culminates in organelle dysfunction and metabolic collapse [119] (Table 2). These studies highlight that PTT/PDT synergizes with mitochondrial targeting to amplify therapeutic efficacy through physical and biochemical mechanisms that can improve patient outcomes (Fig. 5).
| Therapeutic intervention | Mechanism of action | Outcome | References |
| IR780/Ce@EGCG/APT | NIR-induced ROS elevation + mitochondrial membrane disruption | Enhances ferroptosis and inhibits primary/metastatic lesions | [117] |
| CuET@PHF | Copper ion toxicity + TCA cycle inhibition | Eliminates cancer stem cells (CSCs) and induces immunogenic cell death | [118] |
| BMAEF | Ion transport blockade + mitochondrial & Golgi dysfunction | Inhibits tumor growth and metastasis, induces tumor cell apoptosis | [119] |
| TPEG-WS2 + FX11 | Ultrasound-triggered ROS + glycolysis inhibition | Induces mitochondrial energy crisis and enhances apoptosis | [120] |
| Shrgcc+pac+car | AMPK inhibition + chemotherapy | Effectively suppresses lung metastasis in TNBC mouse models | [121] |
| CS-HAP@ATO NPs | Mitochondrial function disruption + enhanced calcium overload | Causes cellular swelling, releases inflammatory mediators, and enhances anti-tumor immunity | [122] |
| P-aCD24/CEL+ P/shMFN1P | Mitochondrial fusion inhibition + TAM reprogramming | Polarizes M2→M1 macrophages and enhances immune clearance | [123] |
| DECaNG + Curcumin | Mitochondrial Ca2+ overload induction + thermotherapy | Significantly increases tumor cell apoptosis rate | [124] |
| Apt-LPR+ miR-34a | Unsaturated phospholipid oxidation + inhibitory immune cell activation | Activates systemic anti-tumor immunity and suppresses tumor progression/metastasis | [105] |
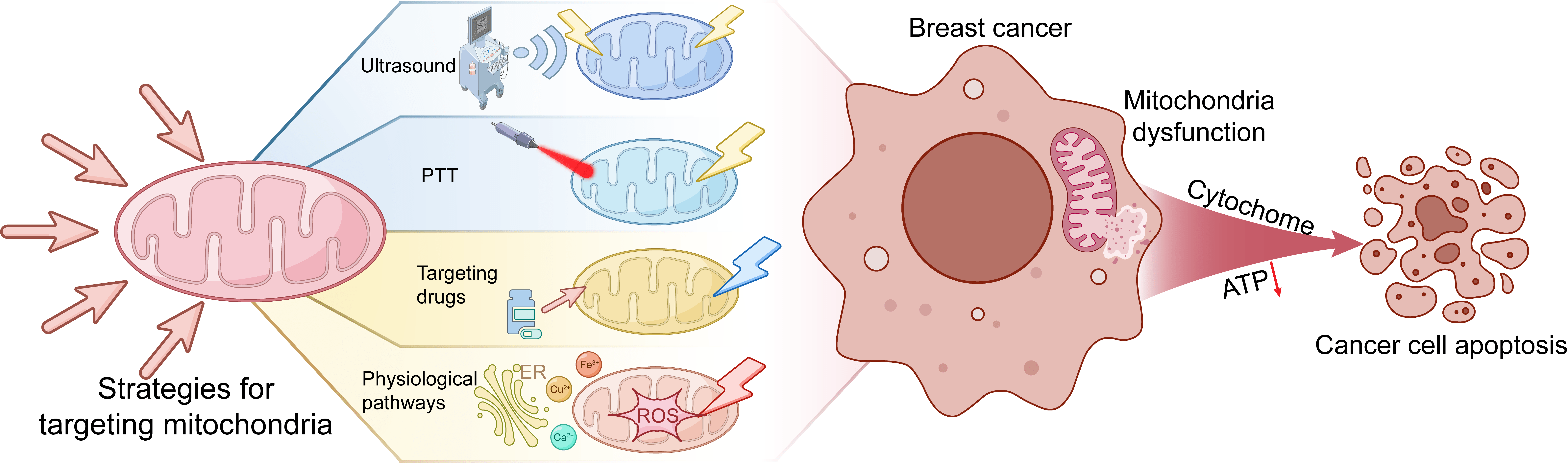 Fig. 5.
Fig. 5.
Targeting mitochondria for breast cancer treatment strategies. Mitochondrial-targeted therapies employ a multimodal strategy to perturb mitochondrial homeostasis, thereby suppressing breast cancer progression through dual targeting of mitochondrial dynamics and metabolic dependencies. Red downward arrow: Reduced ATP level. PTT, photothermal therapy.
Ultrasound therapy combined with metabolic inhibitors is also a highly effective
targeted new therapy. For example, TPP-functionalized WS2 nanosheets
(TPEG-WS2) loaded with the glycolytic inhibitor FX11 generate
ultrasound-triggered piezoelectric reactive oxygen species (ROS) while
suppressing ATP production via blockade of glycolysis, inducing mitochondrial
energy crisis and apoptosis [120] (Table 2, Fig. 5). In addition to improving
the targeting effect and treatment ability by combining new treatment methods,
combination therapy with chemotherapy drugs is also a good choice. In TNBC,
targeting the RGCC/PLK1/AMPK
Recent studies have revealed that combinations with immunotherapy to affect the
immune microenvironment or change the state of immune cells can improve the
effect of targeted killing of tumors and regulate the immunosuppressive tumor
microenvironment. Hydroxyapatite nanoparticles (CS-HAP@ATO) induce mitochondrial
calcium overload, leading to oxidized mitochondrial DNA (OX-mitoDNA) release,
NLRP3 inflammasome activation, and pyroptosis [122] (Table 2). This cascade
converts immunologically “cold” tumors into “hot” tumors through the release
of proinflammatory cytokines such as IL-1
Emerging combination therapy strategies targeting mitochondrial ion homeostasis further expand therapeutic possibilities. The dual-enhanced Ca2+ nanogenerator (DECaNG) utilizes mesoporous copper sulfide to release Ca2+, synergizing with curcumin to inhibit Ca2+ efflux and induce mitochondrial Ca2+ overload. Concurrent NIR-triggered photothermal acceleration of Ca2+ release enhances apoptosis [124] (Table 2, Fig. 5). These approaches demonstrate the efficacy of ion homeostasis regulation in the precise disruption of mitochondrial function.
Mitochondrial bioenergetics plays a pivotal role in the metabolic rewiring of
breast cancer, orchestrating tumor progression through multiple mechanisms. This
review reveals that subtype-specific mitochondrial dependencies emerge from
dynamic interactions between core metabolic pathways and tumor microenvironmental
pressures. Different subtypes of metabolism demonstrate this heterogeneity.
ER+ malignancies exhibit estrogen-OXPHOS codependency. Moreover, TNBC
progression requires FAO‒glutaminolytic coupling. HER2-driven tumors maintain
therapeutic evasion through metabolic adaptability. Notably, metastatic
dissemination is potentiated by mitochondrial plasticity mechanisms, including
PC-mediated anaplerotic flux adjustments and ROS-HIF1
Emerging therapeutic modalities demonstrate advantages against mitochondrial vulnerabilities. CPT1A antagonists, enzyme pathway inhibitors, and ETC interferers have shown selective efficacy in refractory clones. Additionally, metal ion-mediated cell death induction (ferroptosis/cuproptosis) via engineered nanoparticles creates immunogenic vulnerabilities through glutathione depletion and mitochondrial copper dyshomeostasis. Likewise, the mitochondrion-targeted nanoplatforms achieve good therapeutic efficacy when combined with metabolic inhibitors. These approaches collectively address the dual challenges of tumor metabolic heterogeneity and stromal metabolic shielding.
Despite these advances, mitochondrial metabolism-targeted therapies face persistent bottlenecks, including intercellular mitochondrial transfer, intersubtype metabolic heterogeneity, interpatient metabolic variability, resistance due to tumor metabolic plasticity, and clinical translation challenges. These factors collectively compromise monotherapy efficacy. Furthermore, the interplay among metabolic regulatory factors remains incompletely characterized.
To overcome these limitations and enhance therapeutic tractability, future research should pursue the following aspects. First, metabolic dynamics and context dependency should be prioritized. Multiomics approaches can be used to delineate intersubtype and interpatient metabolic differences, analyze reprogramming trajectories, develop early diagnostics and personalized therapies, and establish clinically relevant models. Subtype-stratified clinical trials should be conducted. Second, when targeting specific subtypes, dynamic microenvironmental mechanisms—including the metastatic niche, nutrient status, hypoxia intensity, and ROS thresholds—should be rigorously considered while enhancing oxidative stress-targeting precision. Finally, to circumvent single-agent inefficacy, metabolic inhibitors should be combined with other antitumor modalities (chemotherapy, photothermal therapy, and nanotechnology) and delivery systems should be optimized to improve bioavailability, minimize toxicity, and enhance efficacy. These multiomics diagnostic methods, microenvironment modeling, and combination therapy retain significant optimization potential for personalized implementation. With technological advances and deeper mechanistic insights, precisely targeted mitochondrial metabolism interventions hold promise for delivering more effective breast cancer treatments and ultimately improving patient survival outcomes. and ultimately improving patient survival outcomes.
OXPHOS, oxidative phosphorylation; FAO, fatty acid
QH: Conceptualization, Software, Writing–original draft, Writing–review & editing. ZL: Conceptualization, Writing–review & editing, Data curation. WY: Methodology, Supervision, Writing–review & editing. QZ: Conceptualization, Writing–review & editing, Data curation. LW: Conceptualization, Writing–review & editing, Data curation. YS: Conceptualization, Writing–review & editing. YH: Conceptualization, Resources, Writing–original draft. XW: Conceptualization, Resources, Writing–original draft. SC: Conceptualization, Writing–review & editing. QL: Conceptualization, Supervision, Writing–review & editing. DL: Conceptualization, Funding acquisition, Supervision, Writing–review & editing. ZG: Formal analysis, Supervision, Writing, review & editing. All authors contributed to the conception of the article. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Funded by National Natural Science Foundation of China (NSFC)—General Program (82473279) and Shenzhen Science and Technology Innovation Commission—Basic Research (General Program) (JCYJ20240813114501002).
The authors declare no conflict of interest.
We confirm the use of artificial intelligence-assisted tools during the writing of this article. Artificial intelligence tools (Grammarly, ChatGPT) are only used to enhance language clarity and grammar, check the fluency and professionalism of language, and verify format consistency. It is of vital importance that all intellectual content, explanations and critical scientific reasoning are conceived and executed by the author alone. AI tools play a strict auxiliary role in presenting and improving the content generated by authors.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.