


 , Binchuan Wang 1,†, Jie Yang 1, Lishang Liao 1,*
, Binchuan Wang 1,†, Jie Yang 1, Lishang Liao 1,*
1 Department of Neurosurgery, Affiliated Hospital of Traditional Chinese Medicine, Southwest Medical University, 646000 Luzhou, Sichuan, China
†These authors contributed equally.
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder primarily affecting the geriatric population, characterized by progressive cognitive impairment and behavioral abnormalities. Due to the absence of effective disease-modifying therapies, AD imposes a substantial burden on patients and their families. The etiology and pathogenesis of AD have not been fully elucidated; multiple pathological alterations have been implicated, including the deposition of β-amyloid (Aβ) plaques, abnormal tau phosphorylation, and neuroinflammatory responses. These pathological changes contribute to neuronal damage, synaptic dysfunction, and neuronal death, ultimately leading to brain atrophy. Recent studies have identified PANoptosis as a critical regulatory mechanism of programmed cell death that influences the pathological progression of AD through multiple pathways, including modulation of Aβ plaque deposition and regulating neuroinflammatory responses. However, the precise mechanisms of these effects remain unclear. This review aims to comprehensively analyze recent research findings, focusing on the regulatory role of PANoptosis in AD, exploring the specific manifestations of the intricate network of cell death regulation in AD pathogenesis. By providing a systematic overview of emerging findings, this review offers new insights into the pathogenesis of AD and highlights potential directions for the development of targeted therapeutic strategies.
Keywords
- PANoptosis
- Alzheimer’s disease
- cell death
- network mechanism
With the accelerating global aging process, the incidence of neurodegenerative diseases has been rising significantly. Among them, Alzheimer’s disease (AD) has become the most common form of dementia in the elderly, imposing a substantial burden on both society and families [1, 2]. According to statistical data from the World Health Organization, the global number of AD patients is increasing by several million annually and is expected to continue rising in the coming decades. The clinical characteristics of AD primarily manifest as progressive decline in multiple cognitive and behavioral functions, including memory, orientation, and language abilities. This deterioration severely impairs patients’ ability to perform daily activities independently, leading to a sharp decline in their quality of life [3]. Meanwhile, the long-term caregiving needs impose a substantial economic burden and psychological stress on patients’ families, posing a significant challenge to healthcare resources. The pathogenesis of AD is highly complex, involving intricate interactions among genetic, environmental, and neurobiological factors. Despite extensive research efforts over the years, the pathogenesis of AD remains incompletely understood. This lack of clarity has resulted in the absence of effective therapeutic strategies in clinical practice. Currently available drugs, such as cholinesterase inhibitors (e.g., donepezil, galantamine) and N-Methyl-D-Aspartate (NMDA) receptor antagonists (e.g., memantine), can only provide symptomatic relief to a certain extent but fail to fundamentally halt disease progression [4]. Therefore, exploring the pathogenesis of AD and finding new therapeutic targets and strategies has become an important issue that urgently needs to be addressed in the field of neuroscience.
The study of cell death mechanisms has long been a central focus in the field of medicine. Over the past few decades, apoptosis and pyroptosis have garnered extensive attention and have been the subject of in-depth research. Apoptosis, also known as programmed cell death, is a genetically regulated and orderly cell death process that plays a crucial role in physiological and pathological processes, including multicellular organism development, tissue homeostasis, and immune defense [5]. Pyroptosis, on the other hand, which is mainly mediated by the activation of inflammatory bodies and plays a key role in infectious diseases and inflammatory related diseases [6]. In recent years, PANoptosis has emerged as a novel form of cell death, gradually attracting the attention of researchers. PANoptosis is an inflammatory form of programmed necrosis that integrates features of apoptosis, pyroptosis, and necroptosis. It is activated through the assembly of the PANoptosome protein complex [7, 8]. Currently, the role of PANoptosis has been partially explored in various diseases, including cancer, autoimmune disorders, and infectious diseases, yielding significant research advancements [9, 10]. However, the role of PANoptosis in AD, a neurodegenerative disorder, remains insufficiently explored. Therefore, investigating the function and underlying mechanisms of PANoptosis in AD is of paramount importance. This will help us to comprehensively and deeply understand the pathogenesis of AD from a new perspective, fill the gap in cell death mechanism research in this field, and reveal the intrinsic relationship between pan apoptosis and AD pathological processes. Furthermore, it is expected to provide a solid theoretical basis for the development of new treatment strategies for AD, bring more effective treatment methods for AD patients, and improve their clinical efficacy.
In the pathological process of AD, apoptosis, as a form of programmed cell
death, has been extensively studied and is recognized as playing a crucial role
in the disease’s pathogenesis [11]. The study has shown that neuronal apoptosis
occurs in the brains of AD patients, which is closely associated with multiple
pathological factors [4]. Firstly, the accumulation of
In a word, the mechanisms of apoptosis in AD are highly complex and
multifaceted, involving A
Pyroptosis, as an inflammatory form of cell death, plays a significant role in
the pathological progression of AD. Recent studies have shown that A
This shows, pyroptosis plays a significant role in the pathological progression
of AD. Its interaction with A
In the pathological process of AD, necroptosis, as a regulated form of cell
death, has been demonstrated to play a crucial role. Under the influence of
various stress factors, neurons in the brains of AD patients activate the
necrotic apoptotic signaling pathway. This process is primarily mediated by
Receptor-interacting protein kinase 1 (RIPK1), Receptor-interacting protein
kinase 3 (RIPK3), and the pseudokinase Mixed lineage kinase domain-like protein
(MLKL). Ultimately, it leads to cell membrane rupture, the release of cellular
contents, and the subsequent induction of inflammatory responses and tissue
damage [24]. Recent studies have further elucidated the specific mechanisms of
necroptosis in AD. For example, abnormal tau protein phosphorylation has been
identified as a key trigger of necroptosis [24] AD mouse models, the
presence of tau pathology has been shown to significantly activate necroptosis,
whereas this activation is not observed in models expressing only A
The occurrence of necroptosis not only exacerbates neuronal damage but also
further promotes the development of neuroinflammation by releasing inflammatory
factors [28, 29]. This inflammatory response interacts with A
Therefore, necroptosis plays a crucial role in the pathological mechanisms of
AD, and its interaction with A
In AD research, the crosstalk and regulatory interactions among apoptosis,
pyroptosis, and necroptosis have garnered extensive attention from research
teams. The emerging study continues to reveal the complex roles of these cell
death mechanisms in disease progression [31]. For instance, Caspase-8 acts as a
pivotal regulatory factor in cell death, functioning as a cellular compass to
facilitate apoptosis, necroptosis, or pyroptosis based on its post-translational
modifications and the specific cell type involved. Moreover, Majerníková et
al. [32] discovered that the abnormal accumulation of A
These studies provide critical experimental evidence, demonstrating that the
crosstalk and regulatory interactions among apoptosis, pyroptosis, and
necroptosis in AD are not merely theoretical hypotheses. Key pathological factors
such as A
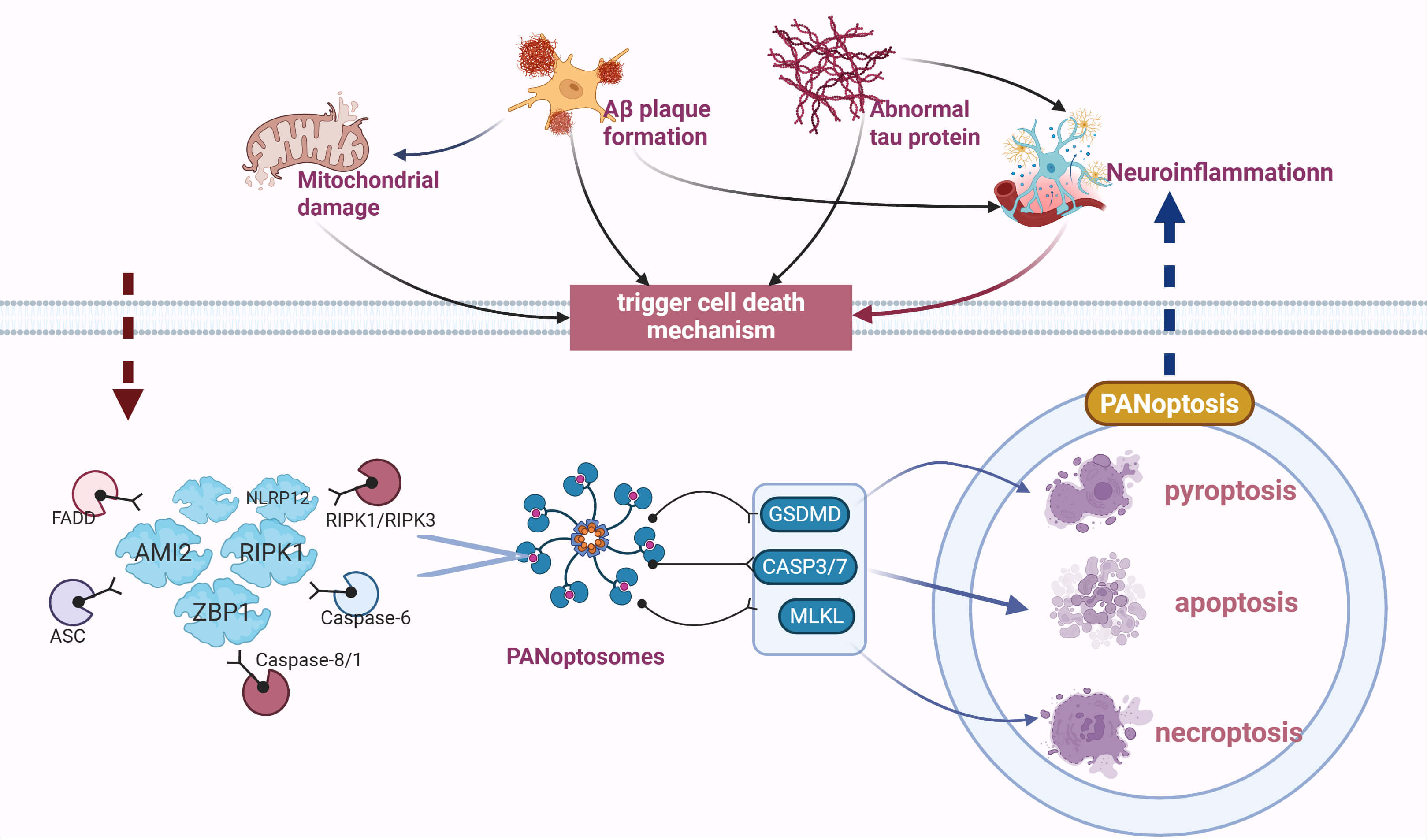 Fig. 1.
Fig. 1.
PANoptosis regulatory network in Alzheimer’s disease (AD). This study first
reveals the interconnections and mutual influences among four core pathological
factors in AD —
PANoptosis is a unique form of regulated inflammatory cell death mediated by PANoptosomes, a concept first proposed by Malireddi et al. [38]. In 2019, this phenomenon was described as a cell death mechanism that cannot be fully explained by any single traditional programmed cell death pathway [38]. PANoptosomes, a multi - protein complex serving as the molecular platform for PANoptosis, integrate key molecules from three programmed cell death pathways—pyroptosis, apoptosis, and necroptosis—forming a shared activation platform. Depending on their functions, PANoptosomes consist of specific sensors (e.g., Z-DNA binding protein 1 (ZBP1), RIPK1, Interferon-inducible protein AIM2 (AIM2)), adapters (e.g., apoptosis-associated speck-like protein containing a CARD (ASC), Fas-associated via death domain (FADD)), and effectors (e.g., RIPK1, RIPK3, caspase-8, caspase-1). Their assembly is triggered by upstream signals, ultimately leading to lysosomal cell death. Based on their distinct characteristics, we have identified PANoptosis-related molecules and their potential therapeutic value in AD (Table 1).
| Molecules | Mechanism | Pathological significance | Therapeutic value |
| ZBP1 | Perceive Z-DNA to activate pan-apoptosis | ZBP1 inhibitors; Traditional Chinese medicine intervention | Medium risk, further study and verification are required |
| RIPK1 | The RIPK1/RIPK3/MLKL complex is activated through the TNF- |
RIPK1 inhibitors (Nec-1, GSK-872); Traditional Chinese medicine intervention | High risk, but research has shown its therapeutic potential |
| RIPK3 | Forming a complex with RIPK1 to recruit MLKL | RIPK3 inhibitor (GSK’2118436); Traditional Chinese medicine intervention | Medium risk, further study and verification are required |
| AIM2 | The RIPK1/3 complex releases mitochondrial DNA (mtDNA) and activates inflammatory caspases | Indirect regulation by NLRP3 inhibitors; intervention with traditional Chinese medicine | Low risk, but limited research; requires further verification of its direct association with AD |
| ASC | Serves as an adapter protein that bridges NLRP3 and caspase-1 | Angiotensin-converting enzyme inhibitors; Traditional Chinese medicine intervention | High risk, further study is needed |
| FADD | Interacts with RIPK1 to form the death-inducing signaling complex (DISC) | No specific inhibitor exists | Low risk, but little research |
| Caspase-1 | Activated by NLRP3 inflammasome, it cleaves IL-1 |
Caspase-1 inhibitor (VX-765); Traditional Chinese medicine intervention | Moderate risk, supported by animal studies |
| Caspase-3 | Activated by A |
Caspase-3 inhibitors (Z-VAD-FMK, IDN-6556); Traditional Chinese medicine intervention | High risk, treatment strategies need to be carefully designed |
| Caspase-8 | Interacts with RIPK1 to regulate necroptosis; may be involved in pyroptosis | Caspase-8 inhibitor (Z-IETD-FMK); Traditional Chinese medicine intervention | Moderate risk, supported by some animal studies |
| NLRP3 | Identify A |
NLRP3 inhibitors (MCC950, BMS-986020); Traditional Chinese medicine intervention | Moderate risk, requiring balance between anti-inflammatory and immunomodulatory effects; supported by clinical trials |
| ADAR1 | Inhibit ZBP1-mediated apoptosis | ADAR1 activator; Traditional Chinese medicine intervention | Low risk, but little research |
| TAK1 | Upstream regulation of RIPK1 activity | TAK1 inhibitors; Traditional Chinese medicine intervention | Low risk, but little research |
ZBP1, Z-DNA binding protein 1; Z-DNA, Z-shaped nucleic acid; RIPK1,
Receptor-interacting protein kinase 1; RIPK3, Receptor-interacting protein kinase
3; MLKL, Mixed lineage kinase domain - like protein; TNF-
Firstly, as one of the sensors, ZBP1 functions as an innate immune receptor
capable of recognizing Z-shaped nucleic acid (Z-DNA) structures generated by
viruses or hosts. This recognition activates immune signaling pathways and
regulates cell death and inflammatory responses. The hallmarks of ZBP1-PANoptosome include the activation of Caspase-1, Caspase-3, Caspase-8, and
the phosphorylation of MLKL. Studies have demonstrated that when stimulated by
Influenza A Virus (IVA), ZBP1 is activated as a specific recognition receptor,
triggering the assembly of PANoptosome complexes [39, 40], ZBP1 also induces
necroptosis by activating the RIPK3-MLKL signaling axis (RIPK3 phosphorylates
MLKL, which damages the cell membrane) [41], the RIPK1-PANoptosome is mainly
composed of RIPK1, ASC, Caspase-1, Caspase-8, and fas-associated protein
with death domain (FADD). Professor Malireddi’s team [38] found that the deletion
or functional inactivation of the TAK1 gene can trigger the assembly of the RIPK1-PAnoptosome, which can serve as the main regulatory switch of the RIPK1-PANoptosome [38] has shown that in the TNF/TNF receptor 1 (TNFR1) signaling
pathway, RIPK1 activates the NF-
In addition to the above, studies have also found that NLR family pyrin domain
containing 12 (NLRP12) drives the activation of inflammasomes and PANoptosomes,
cell death, and inflammation in response to heme plus pathogen-associated
molecular patterns (PAMPs) or TNF. Toll-like receptor 2/4 (TLR2/4) induces NLRP12
expression via the Interferon Regulatory Factor 1 (IRF1)-mediated signaling
pathway, leading to the formation of inflammasomes and subsequently inducing the
maturation of IL-1
PANoptosis is closely associated with various diseases, including infectious diseases, cancer, neurodegenerative disorders, and cardiovascular diseases. Due to its highly pro-inflammatory nature, PANoptosis plays a crucial role in the onset and progression of diseases, making it a potential therapeutic target.
A
Firstly, A
Studies have shown that A
Thus, A
Abnormal tau protein is another key pathological change in AD, and its role in neuronal damage has become a major focus in neuroscience research [66]. Tau protein primarily stabilizes the microtubule network, maintaining neuronal structural integrity and function [67]. Recent studies have shown that abnormal tau protein is closely associated with PANoptosis, involving multiple cell death pathways, including apoptosis, pyroptosis, and necroptosis. These different forms of cell death exhibit a complex, interwoven, and mutually reinforcing relationship in the pathological process of AD [68].
Firstly, excessive phosphorylation of tau protein alters its structure, leading to the aggregation of tau fibrils and the formation of NFTs. These aggregated tau proteins not only disrupt microtubule stability but also interfere with intracellular transport and cytoskeletal organization, ultimately resulting in neuronal dysfunction [69]. Studies have also found that abnormal tau protein aggregation leads to mitochondrial membrane potential loss, impaired ATP synthesis, and disrupted mitochondrial dynamics, resulting in mitochondrial dysfunction. These changes cause intracellular calcium accumulation, triggering cellular stress responses, including endoplasmic reticulum (ER) stress and oxidative stress. Consequently, these stress signals activate cell death pathways, ultimately leading to PANoptosis [70].
Abnormal tau protein aggregation activates intracellular stress-related signaling pathways, such as JNK, p38 MAPK, and ERK, thereby initiating a series of molecular events associated with PANoptosis [71]. Abnormal tau aggregation not only upregulates pro-apoptotic molecules such as Bax and p53 but also downregulates anti-apoptotic molecules such as BCL-2, leading to the activation of the mitochondrial pathway and the initiation of apoptosis [72]. Further studies have also suggested that tau aggregation can disrupt normal autophagic flux, preventing cells from efficiently removing damaged organelles and proteins under metabolic stress, thereby further exacerbating cell death [73].
Additionally, abnormal tau interacts with neuronal autophagy pathways, regulating autophagy-mediated neuronal death through the classical apoptotic pathway [74] tau may also contribute to the neurodegenerative process of AD through non-classical cell death pathways, such as pyroptosis and necroptosis [75]. For example, studies have shown that abnormal tau accumulation can induce necroptosis by activating the RIPK1/RIPK3 signaling pathway. This pathway is activated when cells are exposed to damage, leading to cell membrane rupture and the release of intracellular contents, which in turn exacerbates inflammation and further damages neural tissue [76, 77]. Additionally, abnormal tau may promote pyroptosis by activating the NLRP3 inflammasome.
Function, leading to the accumulation of abnormal proteins within the cell, which in turn activates intrinsic stress responses and induces cell death [78]. Additionally, tau interacts with key signaling pathways, including protein kinase B (AKT)/mTOR, P53, and BCL-2 family proteins. Dysregulation of these pathways may disrupt the balance between cell survival and death, ultimately accelerating the occurrence of PANoptosis [79, 80].
In summary, the abnormal tau protein affects neuronal survival not only through the classical apoptotic pathway but also induces various forms of cell death by broadly activating PANoptosis-related signaling molecules. An in-depth investigation into the relationship between abnormal tau and PANoptosis holds significant theoretical value and clinical relevance for elucidating the pathological mechanisms of AD and identifying potential therapeutic targets.
One of the pathological characteristics of AD is the continuous inflammatory
response among neurons. This response not only directly activates immune cells,
triggering local neuroinflammation, but also promotes cell death, particularly
PANoptosis, via multiple signaling pathways [81, 82]. The activation of
inflammatory response. Thereforeically accompanied by the release of inflammatory
cytokines, such as IL-1
Thus, inflammatory cytokines not only directly promote cell death by activating PANoptosis-related signaling pathways but also play a crucial role in PANoptosis through indirect mechanisms, such as oxidative stress, mitochondrial dysfunction, and dysregulation of autophagy. The sustained activation of inflammatory cytokines forms a vicious cycle in the pathological progression of AD: the release of inflammatory cytokines promotes neuronal death, while neuronal death further exacerbates inflammation, thereby accelerating disease progression. Therefore, targeting inflammatory cytokines and their associated signaling pathways may serve as a promising therapeutic strategy for AD treatment in the future.
In summary, PANoptosis is closely associated with neurodegeneration in AD,
particularly in the context of A
The role of PANoptosis in AD is complex and multidimensional. Therefore, its potential therapeutic targets exhibit remarkable diversity and multidimensional characteristics, spanning multiple levels, including PANoptosome core proteins, upstream activation signals, and downstream signaling pathways. Depending on their different characteristics, different potential therapeutic values can be explored (Table 1).
Firstly, the core proteins and signaling pathways involved in PANoptosis provide
potential therapeutic targets for drug development. For example, caspase family
members, particularly caspase-3 and caspase-8, play a key role in the classical
apoptotic process. They not only induce cell death by cleaving intracellular
substrates but also regulate inflammatory responses and cytokine release, thereby
indirectly promoting PANoptosis [95] have indicated that caspase inhibitors may
exert neuroprotective effects against neuronal death in AD. Specifically,
caspase-3 shows considerable activation in AD models, and blocking its activity
can significantly mitigate A
In recent years, monoclonal antibodies and small-molecule inhibitors targeting
TNF-
In addition to these direct regulators of PANoptosis, mitochondrial function and its associated regulatory factors offer a wealth of potential therapeutic targets. Experimental studies have demonstrated that baicalin inhibits PANoptosis in macrophages by blocking mitochondrial Z-DNA formation and ZBP1-PANoptosome assembly, thereby protecting against inflammatory diseases [103], BCL-2 family proteins, such as BCL-2 and Bax, play a critical role in regulating the mitochondrial apoptotic pathway [104, 105] modulating the BCL-2/Bax ratio or directly targeting these proteins with pharmacological interventions, it may be possible to restore apoptotic balance and slow down neuronal death in AD. Additionally, autophagy, an important cellular self-protection mechanism, is closely linked to PANoptosis. In AD, autophagic flux dysregulation can lead to the accumulation of toxic substances within the cell, thereby exacerbating cellular damage [106, 107] modulating autophagy-related molecules such as mTOR and Beclin-1, it may be possible to restore the cell’s ability to clear damaged components, thereby alleviating neurodegenerative lesions [108, 109]. Drugs targeting autophagy regulatory molecules have made significant progress in the preclinical stage [110] and they may provide new therapeutic options for the treatment of AD in the future.
In addition to molecular targets, therapeutic strategies for PANoptosis also include regulating oxidative stress and restoring calcium homeostasis, which are crucial for maintaining intracellular environmental balance [111, 112] such as vitamin E and alpha-lipoic acid have been used in clinical trials for various neurodegenerative diseases. Although the effects have been limited, oxidative stress control remains a promising therapeutic direction [113, 114]. The use of calcium channel antagonists and their analogs helps to reduce the activation of calcium-dependent enzymes, thereby alleviating intracellular toxic responses [115].
The multiple signaling pathways and molecules involved in PANoptosis provide abundant therapeutic targets for AD treatment. By conducting in-depth research into the molecular mechanisms of PANoptosis and developing drugs or interventions targeting these key molecules, we can not only slow down the pathological progression of AD but also effectively protect neurons and alleviate neurodegenerative lesions. As our understanding of PANoptosis mechanisms deepens, we are likely to develop more precise and effective therapies, offering new hope for AD patients.
Although the development of PANoptosis-targeting drugs shows great promise, there are still many challenges in the research and development process. First, drug safety is a significant concern. AD patients often have multiple comorbidities, such as cardiovascular diseases and diabetes, so drugs targeting PANoptosis must consider potential drug interactions and possible side effects. Second, the efficacy and specificity of these drugs need further validation. Since PANoptosis involves multiple signaling pathways and forms of cell death, precisely selecting targets and achieving specific regulation while avoiding damage to normal and off-target cells remains a major challenge in drug development. As AD is a chronic, progressive neurodegenerative disease, the timing and duration of drug treatment are crucial. Therefore, the long-term effects and tolerability of these drugs need to be thoroughly evaluated in clinical trials. In conclusion, the development of PANoptosis-targeting drugs holds the potential to provide new breakthroughs in the treatment of AD.
Current research on PANoptosis in AD encounters several controversies:
First, the specific assembly mechanism of PANoptosome complexes in AD brains
remains unclear. Different studies have shown discrepancies in the interactions
between core components (such as ZBP1 and AIM2). Second, the causal relationship
between PANoptosis and traditional cell-death pathways
(apoptosis/pyroptosis/necroptosis) has not been clarified. Some studies suggest
that it is an independent regulatory mechanism, while other evidence supports the
idea that it may be a synergistic effect under stress conditions. For example,
TAK1 kinase inactivation can both induce RIPK1-dependent PANoptosome assembly
and activate the independent RIPK3-MLKL necroptotic pathway. Additionally,
existing animal models cannot fully simulate the spatiotemporal dynamics of
PANoptosis in AD patient brains, which limits their clinical translational value.
Although the IAV infection model can activate the ZBP1-PANoptosome, it cannot
reproduce the regulatory effects of AD’s characteristic A
Moreover, current research faces multiple challenges: At the experimental
technical level, the dynamic assembly process of the PANoptosome complex is
difficult to monitor in real time, and the spatial resolution of existing super-resolution microscopy techniques in tissue samples is still unable to capture
transient protein interactions. In terms of mechanism analysis, the cross-regulation of multiple signaling pathways increases the complexity of the
mechanism. For example, the NF-
PANoptosis plays an important role in the onset and progression of AD. It is involved in the pathological processes of AD through various mechanisms, such as affecting neuronal survival and function, and promoting the onset of neuroinflammation. However, there are still some differences in the specific roles and mechanisms of PANoptosis in AD across different studies. Future research needs to further clarify the precise mechanisms of PANoptosis in AD to better understand the disease’s pathogenesis. At the same time, there should be an increased focus on studying PANoptosis-related signaling pathways to explore potential therapeutic targets. Moreover, conducting interdisciplinary research and combining basic research with clinical practice will help drive the development of treatment methods for AD. In conclusion, the role and mechanisms of in AD are complex but of significant research importance. Future studies need to integrate different perspectives and findings to advance this field and provide new insights and methods for AD treatment.
AD, Alzheimer’s disease; A
HW & BCW conceptualized the manuscript, defining its theme, direction, and framework. JY contributed to the conception and design of the manuscript. HW & BCW drafted the manuscript. LSL reviewed and revised the manuscript for language, grammatical structure, and logical coherence, collected relevant literature, and offered valuable suggestions for improving certain aspects of the manuscript. All authors reviewed and approved the final version of the manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to all those who assisted us during the writing of this manuscript, as well as all the peer reviewers for their opinions and suggestions. Additionally, we would like to thank BioRender (https://www.biorender.com) for providing the illustration tools used to create the figures in this manuscript.
This work was supported by the Sichuan Medical Association Medical Scientific Research Project (No: S20250012).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.