



1 Food and Nutrition Major, Division of Integrative Biosciences, Myongji University, 17058 Yongin, Republic of Korea
Abstract
Diabetes mellitus leads to chronic, multi-organ complications, most notably diabetic nephropathy, peripheral neuropathy, and retinopathy. While hyperglycemia serves as the initiating insult, disease progression involves a complex interplay of molecular mechanisms, including oxidative stress, mitochondrial dysfunction, inflammation, and impaired antioxidant defenses. This focused review examines how these shared pathways contribute to organ-specific damage and how they are reflected in experimental mouse models. Key regulatory networks—including nuclear factor kappa B (NF-κB), transforming growth factor-β (TGF-β), protein kinase C (PKC), the advanced glycation end product (AGE)–receptor for AGE (RAGE) axis, and nuclear factor erythroid 2-related factor 2 (Nrf2)—link metabolic stress to fibrosis, vascular dysfunction, and neural injury. Mitochondrial dysfunction is also a commonly shared pathological feature across affected tissues. To investigate these mechanisms in vivo, this review outlines the characteristics of widely used mouse models—streptozotocin (STZ)-induced, Akita mice (harboring the Ins2Akita mutation), db/db, and Black and Tan Brachyury (BTBR) ob/ob—in relation to specific diabetic complications. STZ-induced and Akita mice effectively model hyperglycemia-induced injury, while db/db and BTBR ob/ob mice recapitulate insulin resistance, dyslipidemia, and systemic inflammation. We describe how each model reflects distinct pathogenic features—such as TGF-β–mediated podocyte loss in nephropathy, aldose reductase activation and mast cell dysfunction in neuropathy, and PKC-dependent pericyte apoptosis in retinopathy. Therapeutic strategies targeting these conserved molecular pathways—including Nrf2 activation, NF-κB inhibition, or mitochondrial restoration—have demonstrated efficacy across multiple models. By aligning pathophysiological mechanisms with appropriate experimental systems, this review provides a practical framework for selecting preclinical tools and developing multi-targeted interventions to prevent or slow the progression of diabetic complications.
Keywords
- diabetes complications
- diabetic nephropathies
- diabetic neuropathies
- diabetic retinopathy
- oxidative stress
- mitochondria
- glycation end products, advanced
Diabetes mellitus (DM) is a chronic metabolic disorder characterized by persistent hyperglycemia resulting from insulin deficiency or resistance. Over time, DM leads to progressive multi-organ damage, with major complications including diabetic nephropathy (DN), diabetic peripheral neuropathy (DPN), and diabetic retinopathy (DR) [1].
The economic burden of DM is profound. In 2022, the United States alone incurred
These complications also place an escalating burden on healthcare systems. DN treatment costs far exceed those of uncomplicated DM and increase as the disease advances [4, 5]. DPN adds substantially to healthcare utilization, especially among patients with painful neuropathy [6]. DR affects an estimated 22% of individuals with DM and is projected to affect over 160 million people worldwide by 2045, posing a major public health challenge [7].
Given the clinical and socioeconomic impact of diabetic complications, a clearer understanding of their molecular pathogenesis is urgently needed. We examine the shared and organ-specific molecular mechanisms that drive DN, DPN, and DR. We also describe the features and applications of commonly used experimental mouse models, including streptozotocin (STZ)-induced, Akita (carrying the Ins2Akita mutation), db/db, and Black and Tan Brachyury (BTBR) ob/ob mice. By exploring their relevance to specific diabetic complications, we aim to offer a practical overview for selecting mouse models suited to mechanistic studies and therapeutic development.
A comprehensive literature search was conducted using the PubMed database (https://pubmed.ncbi.nlm.nih.gov/) to identify relevant studies on mouse models of diabetic complications. The search terms included combinations of the following keywords:
“mouse model”, “diabetes”, “diabetic complications”, “diabetic nephropathy”, “diabetic neuropathy”, “diabetic retinopathy”, “type 1 diabetes”, and “type 2 diabetes”.
Boolean operators (e.g., AND, OR) were used to enhance the sensitivity and specificity of the search. The search focused on titles and abstracts to identify studies relevant to experimental mouse models used in the study of diabetic complications. To ensure the quality and relevance of the selected literature, the following inclusion and exclusion criteria were applied.
• Peer-reviewed original articles or reviews;
• Studies focusing on mouse models of diabetes or diabetic complications;
• Articles examining pathophysiological mechanisms, functional assessments, or therapeutic interventions in diabetic nephropathy, neuropathy, or retinopathy.
• Studies published in non-English languages;
• Conference abstracts, letters to the editor, or reports lacking primary data;
• Articles not directly related to mouse models of diabetic complications;
• Duplicate publications or studies lacking a clear methodology.
The pathogenesis of diabetic complications extends beyond hyperglycemia and involves a complex network of interrelated mechanisms. Key contributors include oxidative stress, mitochondrial dysfunction, chronic inflammation, and the accumulation of advanced glycation end products (AGEs), all of which drive progressive, tissue-specific injury [8, 9, 10, 11].
Although DN, DPN, and DR present with distinct clinical features, they share overlapping pathogenic pathways. At the same time, each complication involves tissue-specific mechanisms that contribute to its unique histopathological profile. Understanding both the common and divergent pathways is essential for the development of effective therapeutic strategies. Targeting shared mechanisms may yield systemic benefits, whereas organ-specific approaches allow for more tailored interventions.
The following section outlines four major, interconnected molecular mechanisms involved in DN, DPN, and DR, with a focus on both shared and tissue-specific elements.
Despite the diverse clinical manifestations of diabetic complications across organ systems, they share fundamental molecular mechanisms that drive progressive tissue injury. Understanding these common pathogenic pathways is critical for developing unified therapeutic strategies capable of addressing multiple complications simultaneously. This section highlights five key mechanisms implicated across organ systems: chronic inflammation, oxidative stress, impaired antioxidant defenses, mitochondrial dysfunction, and purinergic signaling. These interconnected pathways are summarized in Fig. 1.
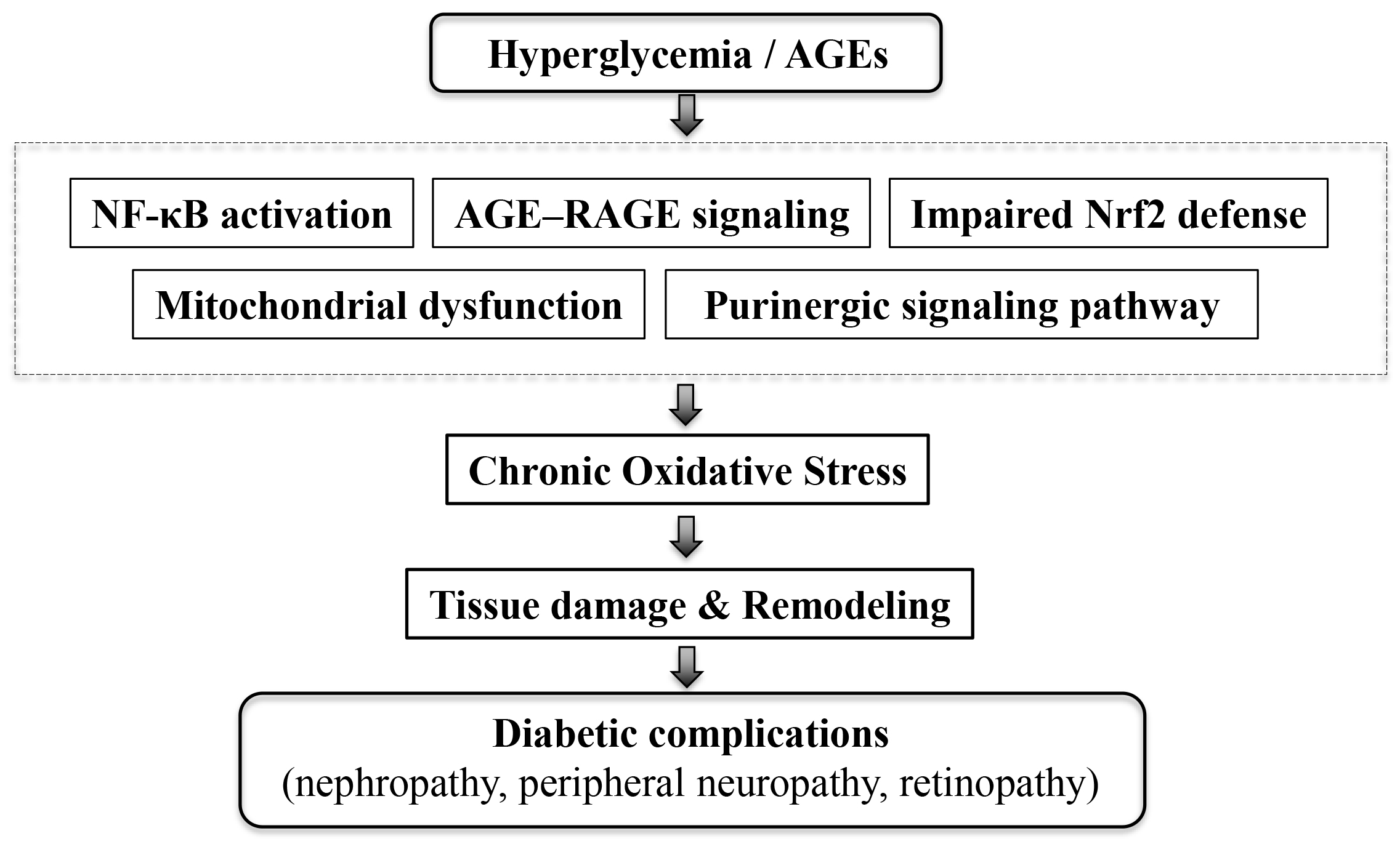 Fig. 1.
Fig. 1.
Schematic representation of hyperglycemia-induced pathways
leading to diabetic complications. Hyperglycemia and AGEs activate multiple
signaling pathways, including NF-
NF-
NF-
Targeting inflammation yields promising therapeutic outcomes. Pharmacologic
inhibition of NF-
AGE–RAGE signaling is a pivotal mechanism connecting chronic hyperglycemia to oxidative stress and inflammation [20]. AGEs interact with RAGE on target cells, activating downstream Mitogen-activated protein kinase (MAPK) pathways and inducing pro-inflammatory cytokine production [21, 22]. RAGE expression is markedly upregulated in diabetic lesions, and genetic polymorphisms in RAGE influence susceptibility to complications [23, 24].
This pathway contributes to both systemic and tissue-specific pathology. RAGE activation promotes expression of vascular endothelial growth factor (VEGF) and recruits inflammatory cells [25], sustaining chronic inflammation and driving pathological tissue remodeling. It is also linked to autonomic dysfunction and peripheral nerve injury. Specific AGE profiles—reflecting glycolytic dysfunction, lipid peroxidation, and glucotoxicity—correlate with impaired function in multiple organ systems [26].
Therapeutic targeting of AGE–RAGE interactions is multifaceted. Inhibitors of AGE formation, such as pyridoxamine, reduce tissue damage; combination therapies further enhance efficacy [27]. Natural compounds and herbal formulations have shown potential in lowering AGE levels, boosting endogenous antioxidant defenses, and modulating apoptosis-related pathways [28]. RAGE-deficient models exhibit preserved tissue integrity and reduced inflammation, reinforcing the clinical relevance of this pathway [29].
The Nrf2 pathway is a critical regulator of antioxidant responses and cellular redox balance. In DM, this pathway becomes dysfunctional, impairing the transcription of cytoprotective genes and exacerbating oxidative damage [30, 31]. Under normal conditions, oxidative stress induces Nrf2 translocation to the nucleus; however, chronic hyperglycemia disrupts this process [32].
Under high-glucose conditions, Nrf2 activity undergoes dynamic regulation.
Antioxidant gene expression is transiently suppressed, followed by partial
compensation despite ongoing accumulation of reactive oxygen species (ROS). At
the same time, elevated NF-
Mitochondrial dysfunction is a unifying pathogenic feature in diabetic complications, linking hyperglycemia to cellular injury through impaired energy metabolism, oxidative stress, and disrupted organelle dynamics [40, 41]. Fragmented mitochondria, an imbalance in adenosine triphosphate/adenosine diphosphate (ATP/ADP) ratio, and defective mitophagy are observed across affected tissues. Reduced expression of NAD+-dependent deacetylase sirtuin-3 (SIRT3) further exacerbates mitochondrial damage [42].
Mitochondrial stress generates ROS and releases mitochondrial deoxyribonucleic
acid (mtDNA), which activates innate immune responses and contributes to fibrosis
[43]. Synergistic effects of hyperglycemia and hypertension further compromise
mitochondrial integrity, increasing superoxide and depleting glutathione [44].
Matrix metalloproteinases (MMP)-2 and MMP-9 aggravate this dysfunction via
increased membrane permeability [45, 46]. Downregulation of peroxisome
proliferator-activated receptor gamma coactivator 1-alpha (PGC-1
Targeting mitochondrial quality control offers therapeutic promise. SIRT3
overexpression restores mitophagy through the Forkhead box O3a
(FoxO3a)–PTEN-induced kinase 1 (PINK1)–Parkin pathway [48], while AICAR, an AMP
analog, activates PGC-1
Purinergic signaling has recently been recognized as an important contributor to
the pathogenesis of DM and its complications, but it remains underexplored in
most preclinical models. Extracellular nucleotides (e.g., ATP) and nucleosides
(e.g., adenosine) act through P2 and P1 receptors to regulate inflammation,
oxidative stress, and fibrosis, thereby exacerbating microvascular and
macrovascular injury [51, 52]. In metabolic regulation, purinergic pathways
modulate pancreatic
Diabetic complications involve distinct yet interconnected pathophysiological mechanisms that lead to progressive organ damage.
DN is a major complication of DM and the leading cause of end-stage renal
disease worldwide. It is characterized by progressive damage to glomerular,
tubular, and interstitial compartments, ultimately resulting in renal failure
[57]. DN pathogenesis involves a combination of fibrotic signaling and podocyte
injury, with transforming growth factor-
TGF-
Podocyte injury is a hallmark of DN and contributes directly to proteinuria. The
slit diaphragm—comprising nephrin, podocin, and other structural proteins—is
compromised in DM, leading to foot process effacement and podocyte loss [64, 65].
Urinary mRNA levels of podocyte markers such as nephrin, podocin, synaptopodin,
Wilms tumor 1 (WT-1), and alpha-actinin-4 are significantly elevated in patients
with biopsy-confirmed DN. These elevations correlate with proteinuria severity
and renal function decline, while WT-1 expression reflects the extent of
tubulointerstitial fibrosis [66]. A key mechanism of podocyte injury involves
dysregulation of the transient receptor potential canonical 6 (TRPC6) channel
[67]. TGF-
Together, these findings highlight TGF-
DPN is one of the most common and debilitating complications of diabetes, affecting nearly half of individuals with type 1 and type 2 DM [71]. Its prevalence rises with disease duration, exceeding 50% in patients with DM for over 10 years [72]. DPN contributes significantly to morbidity, including lower-extremity amputations and chronic neuropathic pain [73]. Following amputation, life expectancy averages just two years, underscoring the severe clinical and socioeconomic burden [74].
DPN most frequently presents as distal symmetric polyneuropathy, with sensory symptoms such as paresthesia, burning pain, and hypersensitivity, typically beginning in the feet and progressing proximally [75]. With disease progression, motor deficits emerge, leading to muscle weakness, atrophy, gait disturbances, and impaired coordination that interfere with daily activities [71]. The pathogenesis of DPN involves multiple interconnected mechanisms, prominently including metabolic dysregulation and immune system dysfunction.
A key metabolic driver of DPN is the polyol pathway. Under hyperglycemic conditions, aldose reductase (AR)—the rate-limiting enzyme—converts glucose to sorbitol in a Nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reaction [76]. This process leads to sorbitol accumulation, NADPH depletion, and increased ROS production, promoting oxidative stress [77]. Mouse studies support this mechanism; AR overexpression in Schwann cells exacerbates motor nerve conduction velocity (MNCV) deficits in diabetic and galactosemic mice, despite similar polyol accumulation, suggesting oxidative stress—not sorbitol per se—as the primary pathogenic factor [78]. Transgenic mice expressing human AR exhibit worsened DPN phenotypes, including increased sorbitol, decreased MNCV, and nerve fiber atrophy. These effects are reversed by AR inhibition [79].
Beyond metabolic perturbations, emerging evidence reveals that immune cell dysfunction plays a crucial role in DPN pathogenesis. Recent single-cell transcriptomic analyses have identified mast cells as critical mediators in DPN development. In STZ-induced diabetic mice, hyperglycemic conditions enhance glucose transporter type 3 (GLUT3)-mediated glucose uptake in mast cells. This triggers extracellular signal-regulated kinases 1 and 2 (ERK1/2) activation and mechanistic target of rapamycin (mTOR) hyperactivity, leading to metabolic dysregulation that drives mast cell degranulation and pro-inflammatory mediator release. Ultimately, this disrupts the neural microenvironment [80].
These findings demonstrate that DPN results from interconnected metabolic and immunological disturbances. AR-induced oxidative stress and mast cell–mediated inflammation converge to impair peripheral nerve integrity under hyperglycemic conditions. This dual-pathway mechanism highlights the need for therapeutic strategies that target both metabolic dysfunction and immune activation.
DR constitutes a leading cause of blindness in working-age adults. It features progressive retinal microvascular damage and neurodegeneration. This microvascular complication affects both the retinal vasculature and neural retina, ultimately leading to vision loss [81, 82]. DR progresses through distinct stages, from an early non-proliferative stage to an advanced proliferative stage. The early stage features pericyte loss, microaneurysms, retinal capillary leakage, and acellular capillaries. As the condition advances, the proliferative stage becomes marked by retinal neovascularization and fibrous tissue proliferation [83]. The pathogenesis of DR involves multiple interconnected pathways, including protein kinase C (PKC) activation, VEGF-mediated angiogenesis, and oxidative stress-induced cellular dysfunction.
PKC activation plays a central role in the pathogenesis of diabetic retinopathy
through distinct isoform-specific mechanisms [84]. Pericytes are essential for
maintaining blood vessel integrity and regulating endothelial cell growth;
however, in the diabetic retina, pericyte loss leads to the formation of
microaneurysms and acellular capillaries [85]. A clinical study evaluated the
therapeutic efficacy of an oral PKC
PKC activation, in addition to directly affecting pericytes, also contributes to retinal ischemia through vascular dysfunction. In advanced diabetic retinopathy, this ischemia triggers compensatory but pathological angiogenesis mediated by VEGF. VEGF becomes upregulated in response to retinal ischemia from capillary dropout, ultimately driving proliferative diabetic retinopathy. The Ins2Akita mouse model reveals retinal neovascularization by 9 months, developing new blood vessels and preretinal neovascular tufts accompanied by increased retinal VEGF levels [89]. Therapeutic interventions targeting the VEGF pathway have shown promise in experimental models. Inhibition of the VEGF pathway reduced the severity of retinal damage in STZ-induced rat models [88]. Additionally, the glucagon-like peptide-1 (GLP-1) analogue exendin-4 exhibited protective effects by downregulating placental growth factor (PLGF) and VEGF expression via the ERK and protein kinase B (AKT/PKB) signaling pathways, while preserving blood–retinal barrier integrity [90].
These findings support a mechanistic model in which hyperglycemia-induced PKC activation impairs pericyte survival, leading to retinal ischemia and triggering VEGF-driven neovascularization—key events in the progression of DR.
The pathophysiological mechanisms underlying diabetic complications have been extensively studied using a range of mouse models, including genetic models (e.g., db/db, Ins2Akita), chemically induced models (e.g., STZ), and diet-induced models. These systems offer genetic tractability and experimental reproducibility. Research using these models has revealed that diabetic complications result from both common metabolic disturbances and organ-specific vulnerabilities [91]. Crucially, the choice of model profoundly affects how diabetic complications manifest and progress. Models vary in the degree of hyperglycemia, insulin resistance, obesity, and associated metabolic disruptions [92], creating a complex experimental landscape where disease mechanisms differ in tissue specificity and translational relevance. Selecting an appropriate model is therefore essential for accurate interpretation of preclinical findings and for guiding therapeutic development.
The following sections provide a complication-specific overview of widely used mouse models. We highlight their pathological features, underlying mechanisms, and relevance to human disease.
The pathophysiology of DN involves complex and interconnected molecular
mechanisms. These include profibrotic TGF-
| Model (Type of DM) | Key characteristics | Renal pathology | References |
| STZ-induced (Type 1) | Chemically induced pancreatic |
Moderate albuminuria, glomerular hyperfiltration; mesangial matrix expansion, increased glomerular surface area, tubular cell damage (no nodular lesions); |
[93, 94, 95, 96, 97, 98, 99] |
| Ins2𝐴𝑘𝑖𝑡𝑎 (Type 1) | Spontaneous Ins2 mutation |
Variable DN severity: DBA/2 background = severe albuminuria; C57BL/6 background = mild albuminuria; mesangial expansion; modulated by genetic factors (eNOS, ACE, Nrf2, Keap1) | [38, 100, 101, 102, 103] |
| OVE26 (Type 1) | Transgenic (FVB background) model with severe early-onset DM; exhibits strongest DN phenotype among type 1 DM models | Severe progressive albuminuria, hyperfiltration |
[104, 105] |
| db/db (Type 2) | Lepr mutation |
Phenotype severity depends on genetic background (Albuminuria, mesangial matrix expansion, GBM thickening, etc.) | [106, 107, 108, 109] |
| BTBR ob/ob (Type 2) | Leptin-deficient ob/ob on BTBR background; severe DM with hyperinsulinemia, insulin resistance, hyperlipidemia; accelerated DN progression | Early podocyte loss & proteinuria (8 wks); mesangial expansion (10 wks); advanced DN by 18–22 wks: massive proteinuria, mesangial sclerosis with nodular features, GBM thickening, arteriolar hyalinosis, mesangiolysis, interstitial fibrosis; |
[110, 111, 112, 113] |
| STZ + ApoE−/− (Hyperglycemia-hyperlipidemia combined) | Combines STZ diabetes with ApoE deficiency |
Severe albuminuria, glomerular & tubulointerstitial injury, interstitial fibrosis, |
[114, 115, 116, 117] |
ACE, angiotensin-converting enzyme; ApoE, apolipoprotein E; BUN, blood urea
nitrogen; DM, diabetes mellitus; eNOS, endothelial nitric oxide synthase; GBM,
glomerular basement membrane; GFR, glomerular filtration rate; HO, heme
oxygenase; NF-
STZ-treated C57BL/6 mice represent a commonly used model of type 1 DM. However, they exhibit relatively mild DN and typically require additional insults to manifest robust pathology. These additional insults include uninephrectomy or endothelial nitric oxide synthase (eNOS) deletion [93, 94]. The nephropathy phenotype can be enhanced through combinations with advanced oxidation protein products or high-protein diets, exacerbating albuminuria and tubulointerstitial injury [95].
Functionally, STZ-induced diabetic mice exhibit moderate albuminuria and glomerular hyperfiltration in early stages. The Animal Models of Diabetic Complications Consortium defined criteria for validating murine models require greater than 50% decline in glomerular filtration rate (GFR) over the lifetime of the animal and greater than 10-fold increase in albuminuria compared with controls for that strain at the same age and gender [96].
Histologically, STZ-treated mouse strains show moderate mesangial matrix expansion, increased glomerular surface area, and tubular cell damage but lack nodular lesions [97].
Therapeutic target validation in STZ models emphasizes glycemic control and antioxidant interventions. Moreover, the involvement of both RAGE-dependent and RAGE-independent mechanisms highlights the complexity of AGE-mediated renal injury. Targeting AGEs directly, as shown by the efficacy of alagebrium even in RAGE-deficient mice, suggests that AGE-lowering therapies may offer additive benefits beyond RAGE blockade alone [98]. Recombinant human bone morphogenetic protein-7 significantly inhibits glomerular hypertrophy and tubulointerstitial fibrosis by restoring expression of Ski-related novel protein N via activation of Smad1/5. This suppresses partial epithelial-to-mesenchymal transition and ECM accumulation in diabetic kidneys [99].
Akita mice carry a spontaneous Ins2 gene mutation causing
pancreatic
Genetic background significantly influences disease severity in Akita mice. DBA/2 background mice manifest severe hyperglycemia and pronounced renal dysfunction including significant albuminuria. In contrast, C57BL/6 Akita mice show mild albuminuria and limited mesangial expansion [102]. Various Akita-based strain studies have revealed marked differences in albumin-to-creatinine ratios, establishing new models for DN research [103]. Genetic modifications further modulate nephropathy severity. Bradykinin receptor, eNOS, or Angiotensin-converting enzyme (ACE) deficiency exacerbates kidney injury, while interventions like ketogenic diet can reverse diabetic nephropathy [101]. Additionally, Nrf2 knockout (Akita::Nrf2-/-) worsened diabetic kidney disease, while Nrf2 induction (Akita::Keap1FA/FA) provided protection through antioxidant mechanisms [38].
OVE26 transgenic mice exhibit severe early-onset type 1 DM within weeks
of birth. They manifest comprehensive DN features including progressive
albuminuria, hyperfiltration followed by GFR decline, glomerular enlargement,
mesangial matrix expansion, tubulointerstitial fibrosis, and glomerular basement
membrane thickening [104]. The FVB genetic background proves critical for disease
severity, as these mice exhibit the highest albuminuria among diabetic models.
Crossing to C57BL6 or DBA2 backgrounds reduces albuminuria 17-fold through
susceptibility loci on chromosomes 9, 11, 13, and 19 that account for
The db/db mouse constitutes one of the most extensively used models for investigating type 2 DM and its renal complication. This model harbors a G-to-T point mutation in the leptin receptor gene (Lepr), resulting in leptin resistance, hyperphagia, obesity, and persistent hyperglycemia. Consequently, db/db mice manifest hallmark features of DN, including albuminuria, mesangial matrix expansion, and glomerular basement membrane thickening [106, 107].
The severity of the diabetic phenotype in db/db mice varies markedly
depending on their genetic background. On the C57BLKS/J background,
db/db mice exhibit severe hyperglycemia accompanied by progressive
degeneration of pancreatic islet cells. This leads to reduced insulin secretion
and body weight loss by 5–6 months of age. This strain also displays increased
sensitivity to
To model severe DN, the eNOS-deficient db/db mouse (eNOS-/-db/db) was generated on the C57BLKS/J background. These mice manifest a constellation of advanced DN features, including severe albuminuria, hypertension, arteriolar hyalinosis, mesangiolysis, nodular mesangial expansion, and tubulointerstitial fibrosis. This closely resembles the pathology seen in advanced human DN [108].
Among available models, the DBA/2J db/db mouse exhibits the most aggressive DN phenotype. It features an approximately 50-fold increase in the albumin-to-creatinine ratio by 12 weeks of age. These mice also show glomerulosclerosis, foot process effacement, and marked glomerular basement membrane thickening [109], making them particularly valuable for studying advanced DN pathogenesis.
The BTBR ob/ob mouse model represents a valuable tool in DN research. It offers a robust system that closely mirrors human diabetic kidney disease progression. This model combines the leptin-deficient ob/ob mutation with the BTBR genetic background, resulting in severe type 2 DM accompanied by hyperinsulinemia, insulin resistance, hypercholesterolemia, and elevated triglyceride levels [110]. Unlike traditional mouse models that exhibit limited renal involvement, the BTBR ob/ob mouse manifests a full spectrum of renal lesions that align with both early and advanced stages of human DN [111].
Disease progression follows a predictable timeline. Podocyte loss and
proteinuria emerge by 8 weeks, followed by mesangial expansion around 10 weeks.
Between 18–22 weeks, advanced DN features become evident, including marked
proteinuria,
A key advantage lies in the accelerated disease progression, enabling interventional studies within shorter timeframes compared to other models [112]. MicroRNA sequencing analyses have identified upregulation of 99 microRNAs linked to inflammatory and fibrotic pathways that mirror human DN. This supports the model’s translational relevance for mechanistic studies and therapeutic development [111, 113].
While traditional diabetic nephropathy models focus on hyperglycemia alone,
clinical diabetic kidney disease often involves multiple metabolic abnormalities.
The STZ-induced diabetic ApoE-/- mouse model addresses this limitation by
combining hyperglycemic and hyperlipidemic conditions. This more closely mimics
the complex metabolic environment of human diabetes [114, 115]. This
dual-pathology model reveals enhanced susceptibility to renal injury compared to
either STZ-induced diabetes or ApoE deficiency alone. The combined metabolic stress results in severe albuminuria, glomerular and tubulointerstitial damage,
increased fibrosis, and elevated expression of profibrotic markers including
TGF-
Multiple therapeutic interventions have shown renoprotective efficacy in this
complex model. Avosentan reduced albuminuria and glomerular pathology through
suppression of profibrotic and inflammatory gene expression [115]. Omapatrilat, a
dual inhibitor of ACE and neutral endopeptidase, showed superior renoprotection
compared to ACE inhibition alone [116]. Additionally,
The pathogenesis of DPN involves multifactorial mechanisms, including chronic hyperglycemia, dyslipidemia, oxidative stress, mitochondrial dysfunction, and inflammation. These factors collectively contribute to peripheral nerve injury [71]. Given the heterogeneous features of DPN across diabetic mouse models, careful selection remains essential. Models must recapitulate the relevant molecular and pathological mechanisms for translational relevance [73]. Table 2 (Ref. [73, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137]) provides an overview of commonly used mouse models of DPN, together with their defining characteristics and related neuropathology.
| Model (Type of DM) | Key characteristics | Neurological pathology | References |
| STZ-induced (Type 1) | Rapid onset (4–8 wks); reduced sensory & motor NCV; early thermal hypoalgesia; initial hyperalgesia |
Small fiber dysfunction; selective loss of peptidergic and nonpeptidergic nociceptors; compressed temporal progression of DPN vs. human; thermosensory circuit alterations | [73, 118, 119, 120] |
| Ins2𝐴𝑘𝑖𝑡𝑎 (Type 1) | Spontaneous mutation; lean, insulin deficient + insulin resistant; autonomic & sensory neuropathy; progressive NCV reduction; hypoalgesia, tactile allodynia | Sympathetic autonomic neuropathy: neuritic dystrophy, swollen axons, ~1/3 neuronal loss in celiac ganglia by 8 mo; perikaryal abnormalities (membranous aggregates, mitochondrial dysfunction); sensory neuropathy with IENF loss, impaired DRG responses | [121, 122, 123, 124, 125] |
| Alloxan-induced (Type 1) | Chemical destruction of pancreatic |
Ultrastructural abnormalities: demyelination/remyelination, axonal degeneration/regeneration, onion bulb formations; unique: crystalline deposits, glycogen accumulation in axonal mitochondria, Lafora-like inclusions; pathology dependent on hyperglycemia (less direct neurotoxicity vs. STZ) | [126, 127, 128] |
| db/db (Type 2) | Obese, insulin resistant, hyperglycemic; progressive sensory loss; IENF loss by 18 wks; biphasic neuropathy (metabolic |
Schwann cell apoptosis, CD3+ T-cell infiltration in sciatic nerve; progressive axonal atrophy, impaired axonal transport; metabolic phase reversible with insulin, neuronal phase refractory | [129, 130, 131] |
| BTBR ob/ob (Type 2) | Severe early-onset neuropathy; NCV deficits by 9 wks, IENF loss by 13 wks; sex-specific differences (females: preserved IENF despite neuropathy) | Small fiber demyelinating neuropathy in adipose tissue; Schwann cell gene dysregulation; neuroinflammation as central mechanism | [132, 133, 134] |
| Diet-induced (Type 2) | High-fat diet (54%) induces neuropathy with impaired glucose tolerance; B6-wild type most consistent phenotype; reversible with diet normalization | Large-fiber neuropathy; obesity, hyperinsulinemia, dyslipidemia, oxLDL elevation; pathways: lipid metabolism, calcium signaling, inflammation; pathology independent of hyperglycemia | [135, 136, 137] |
IENF, intraepidermal nerve fiber; NCV, nerve conduction velocity; oxLDL, oxidized low-density lipoprotein; STZ, streptozotocin.
STZ-induced type 1 DM mouse models serve as widely used platforms to investigate the pathophysiology of DPN and to validate potential therapeutic targets. These models exhibit relatively rapid onset of neuropathic symptoms following STZ administration. Within 4 to 8 weeks post-injection, mice typically show reduced sensory and motor nerve conduction velocities. These changes occur alongside thermal hypoalgesia and diminished tactile sensitivity—hallmarks of early small fiber dysfunction [73].
Behaviorally, these mice initially present with hyperalgesia, which gradually transitions to hypoalgesia during the chronic phase. However, this temporal progression becomes considerably compressed compared to the course of human DPN [118]. Advanced fluorescent labeling techniques have further revealed that peptidergic and nonpeptidergic nociceptive neurons exhibit differential susceptibility to diabetic injury. Specifically, peptidergic fiber loss occurs as early as 4 weeks post-STZ. This precedes the degeneration of nonpeptidergic fibers at approximately 8 weeks [119]. The selective correlation between early behavioral deficits and peptidergic fiber loss suggests that these neurons may drive early neuropathic symptoms [119].
Given the thermal sensory deficits observed in STZ-induced models, careful behavioral assessment proves critical. Traditional thermal avoidance tests, such as the plantar assay, consistently detect thermal hypoalgesia but may fail to capture the full complexity of thermosensory dysfunction. Recent studies employing thermal gradient behavioral assays have identified altered temperature preference patterns in diabetic mice that are distinct from those observed in transient receptor potential vanilloid 1 (TRPV1)-deficient animals, despite comparable thermal avoidance behaviors [120]. These findings emphasize the utility of complementary thermal behavior paradigms in uncovering mechanisms underlying thermosensory deficits in DPN [120].
Beyond chemically induced models, spontaneous genetic models such as the Akita mouse offer an additional platform for investigating the development and progression of DPN. The Akita mice exhibit milder signs of neuropathy, potentially due to lower circulating insulin levels and the absence of obesity. This represents a pure type 1 DM phenotype [121]. However, despite their lean phenotype, Akita mice manifest marked insulin resistance—shown by an ~80% reduction in glucose infusion rate during hyperinsulinemic-euglycemic clamps. This results from decreased glucose uptake in skeletal muscle and brown adipose tissue, as well as impaired hepatic insulin action [122]. This combination of insulin deficiency and insulin resistance renders the model particularly relevant for studying diabetic complications involving both hormonal and metabolic disturbances [122].
Notably, Akita mice exhibit pronounced diabetic sympathetic autonomic neuropathy, with characteristic neuritic dystrophy in prevertebral ganglia. This includes prominently swollen axons and dendrites that progressively worsen over 2 to 8 months of diabetes [121]. Progressive neuronal loss occurs, with approximately one-third of neurons lost in the celiac ganglia by 8 months. This becomes accompanied by distinctive perikaryal abnormalities, including membranous aggregate accumulation and mitochondrial dysfunction [121]. In addition to autonomic features, Akita mice also manifest significant sensory neuropathy. This features impaired mechanical and thermal nociception and substantial intraepidermal nerve fiber loss [123]. Electrophysiological analyses reveal reduced action potential discharge in mechanonociceptors and markedly impaired heat responsiveness in dorsal root ganglion neurons, while cold-sensitive and low-threshold A-fiber functions remain largely intact [123]. By 16 weeks of age, these mice exhibit reduced sensory nerve conduction velocities, thermal and mechanical hypoalgesia, and tactile allodynia, with further deterioration in some measures by 20 weeks [124].
The extended survival and consistent pathological manifestations establish Akita mice as a valuable model for studying both autonomic and sensory components of DPN [121]. Moreover, their responsiveness to a broad range of therapeutic interventions—including complete reversal with insulin therapy, partial improvement with inhibiting poly(ADP-ribose) polymerase, and selective effects with erythropoietin-derived peptides—highlights their utility in evaluating mechanism-based treatments and dissecting distinct pathogenic pathways involved in DPN [123, 124, 125].
Alloxan-induced type 1 DM shares many pathophysiological features with
STZ-induced models but represents an alternative chemical approach for inducing
Longitudinal studies in alloxan-diabetic rats have documented extensive ultrastructural abnormalities, including demyelination and remyelination, axonal degeneration and regeneration, and characteristic onion bulb formations formed by proliferating Schwann cells [127]. Unique pathological features observed in this model include crystalline deposits within vessel walls and endoneurium, glycogen accumulation in axonal mitochondria, and axoplasmic inclusions resembling Lafora bodies. These findings suggest that alloxan-induced neuropathy involves complex metabolic disruption affecting axons, Schwann cells, and microvasculature [127].
Importantly, comparative studies suggest that alloxan may offer certain advantages over STZ in isolating hyperglycemia-mediated mechanisms. While STZ induces mechanical hypersensitivity in both hyperglycemic and normoglycemic animals—likely via direct neurotoxic effects involving nociceptive neurons and TRPV1 modulation—alloxan-induced mechanical sensitization occurs only under hyperglycemic conditions [128]. This distinction highlights alloxan’s potential to more selectively model glucose-dependent neuropathic changes, reducing confounding from direct chemical neurotoxicity.
The C57BL/KsJ db/db mouse constitutes a well-established model of type 2 DM that reproduces key metabolic features such as hyperglycemia, insulin resistance, and obesity. This makes it suitable for investigating DPN in the context of metabolic syndrome. Longitudinal studies in db/db mice show a characteristic progression of DPN from early functional impairments to subsequent structural degeneration, closely mirroring the human disease trajectory [129].
Functional deficits, including progressive sensory loss and electrophysiological impairments, emerge during early-to-mid disease stages. These become accompanied by reduced intraepidermal nerve fiber density (IENFD) as early as 18 weeks of age [129]. As the disease advances, evident structural alterations—such as Schwann cell apoptosis and CD3+ T-cell infiltration in the sciatic nerve—highlight the evolving neuropathology. This progression reflects a transition from metabolic disturbance to established neurodegeneration [129].
Notably, db/db mice exhibit a biphasic pattern of neuropathy progression [130]. An initial “metabolic” phase, which remains responsive to insulin therapy, becomes followed by a “neuronal” phase featuring progressive axonal atrophy and impaired axonal transport of acetylcholinesterase, with peak severity occurring around 180 days of age. During this later stage, neuropathy becomes refractory to insulin treatment but shows partial responsiveness to ganglioside therapy, reflecting a fundamental shift from systemic metabolic disturbance to intrinsic neuronal pathology. This biphasic progression may parallel the degenerative phases observed in human DPN, providing valuable insights into the timing and nature of therapeutic intervention windows [130].
Mechanistically, db/db mice display unique pathogenic features that distinguish them from other diabetic models. Importantly, Na+,K+-ATPase activity in sciatic and optic nerves remains unchanged across disease stages (50–280 days), suggesting that this pathway does not contribute significantly to neuropathy development in this model [131]. Additionally, the absence of AR staining in nerve tissue indicates minimal involvement of the polyol pathway, a mechanism commonly implicated in other diabetic complications [131]. These findings suggest that DPN in db/db mice may arise through alternative, model-specific biochemical pathways.
The leptin-deficient BTBR ob/ob mouse constitutes a robust model of type 2 DM that exhibits early-onset and severe DPN. These mice manifest nerve conduction deficits by 9 weeks and intraepidermal nerve fiber loss by 13 weeks, making them particularly valuable for studying accelerated DPN progression [132].
An emerging area of interest involves the role of adipose tissue innervation. BTBR ob/ob mice exhibit small fiber demyelinating neuropathy in subcutaneous white adipose tissue, along with dysregulated Schwann cell gene expression, mirroring changes seen in obese humans [133]. These findings reveal that adipose neuropathy, driven by Schwann cell dysfunction, may contribute to metabolic impairments and represents a historically underrecognized component of DPN.
Sex-specific studies have further validated the model’s utility. Female BTBR ob/ob mice manifest DPN similar to males, with comparable motor and sensory deficits but preserved IENFD, suggesting sex-dependent variations in small fiber involvement [134]. Although both sexes show similar metabolic profiles, males exhibit higher triglyceride levels, indicating subtle metabolic differences. Transcriptomic analyses of sciatic nerve and dorsal root ganglia confirm that inflammatory pathways become dysregulated in both sexes, reinforcing neuroinflammation as a central mechanism in DPN [134].
Diet-induced mouse models provide a valuable platform for studying DPN, particularly in the context of metabolic syndrome and prediabetes, where neuropathy can manifest independently of marked hyperglycemia. In a comparative study involving BKS, B6, and BTBR genotypes, mice fed a 54% high-fat diet (HFD) manifested large-fiber neuropathy and impaired glucose tolerance, with B6-wt mice showing the most consistent phenotype, including obesity, hyperinsulinemia, dyslipidemia, and elevated oxidized low-density lipoproteins [135]. Notably, dietary reversal after 16 weeks of high-fat feeding completely normalized both neuropathy and metabolic parameters, suggesting that lipid and inflammatory pathways, rather than hyperglycemia alone, may serve as primary drivers of neuropathy in metabolic syndrome [135].
Mechanistic studies using these models have revealed converging pathways involving lipid metabolism, calcium signaling, and inflammation, reinforcing their value for therapeutic screening and for elucidating the multifactorial pathogenesis of obesity-associated neuropathy [136, 137]. Given the increasing global prevalence of obesity and metabolic syndrome, and the high incidence of neuropathy as a complication, these models offer critical insights into non-glucose–centric therapeutic strategies [136].
Mouse models play a critical role in DR research, providing an accessible platform for exploring disease mechanisms and testing new treatment approaches. While they do not fully replicate the complexity of advanced human DR—particularly the proliferative stage involving both vascular and neural complications [138]—mice are especially valuable due to their suitability for genetic modification. Although neuroretinal involvement in diabetes is increasingly recognized, vascular lesions remain the hallmarks of DR, and not all retinal vascular lesions observed in diabetic patients have been successfully reproduced in diabetic mice [139]. Nevertheless, the development of transgenic and knockout mouse lines has further deepened our mechanistic understanding by enabling detailed analyses of gene- and cell type-specific contributions to disease progression [140]. Table 3 (Ref. [74, 85, 89, 138, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158]) summarizes various mouse models used in diabetic retinopathy (DR) research, along with their key characteristics and associated retinal pathologies.
| Model (Type of DM) | Key characteristics | Retinal pathology | References |
| STZ-induced (Type 1) | Induced with STZ | Pericyte loss, acellular capillaries, retinal thickening, microaneurysms, transient neuronal apoptosis followed by normalization | [74, 85, 138, 141] |
| eNOS-deficient STZ (Type 1) | eNOS−/− C57BL/6J mice | Vascular leakage, gliosis, acellular capillaries, basement membrane thickening, aggressive form of DR with earlier vascular issues | [142] |
| Ins2𝐴𝑘𝑖𝑡𝑎 (Type 1) | Spontaneous hyperglycemia by 2 months | Pericyte loss, vascular leakage, microaneurysms, retinal neovascularization, progressive retinal changes from early to late DR, RPE dysfunction | [89, 143, 144, 145] |
| Db/db (Type 2) | Leptin receptor-deficient, obese mice | Pericyte loss, microvascular damage, gliosis, neurodegeneration, inflammatory signaling, chronic interferon- |
[146, 147, 148, 149, 150] |
| BTBR ob/ob (Type 2) | Leptin-deficient, obese mice | Retinal dysfunction, inner retinal thinning, gliosis, neuroinflammatory changes precede vascular degeneration | [151, 152] |
| Diet-induced (Type 2) | Varying fat compositions and/or sucrose supplement, accelerated model combining with STZ | Retinal nerve infarcts, vascular leakage, reduced vascular density | [153, 154, 155, 156, 157, 158] |
eNOS, endothelial nitric oxide synthase; RPE, retinal pigment epithelium; STZ, Streptozotocin.
STZ-induced diabetic mice are widely used to model type 1 diabetes-related retinal pathology. After about six months of sustained hyperglycemia, they develop early signs of DR, such as pericyte loss, acellular capillary formation, mild retinal thickening, and occasional microaneurysms [85, 138, 141]. In C57BL/6J mice, transient neuronal apoptosis occurs soon after diabetes onset, accompanied by caspase-3 activation [74]. However, these neural changes normalize over time, and no significant retinal ganglion cell loss is detected even after one year, as confirmed by multiple methods.
The eNOS-deficient STZ model represents a more aggressive variant. In eNOS-/- diabetic mice, vascular leakage appears as early as 3 weeks post-induction, with more rapid and severe DR features such as acellular capillaries, persistent gliosis, and basement membrane thickening [142]. Upregulation of iNOS and total nitric oxide (NO) suggests compensatory changes in NO signaling. These findings highlight the protective role of eNOS-derived NO and make the eNOS-deficient STZ model a valuable tool for studying vascular mechanisms in DR and evaluating therapies targeting endothelial dysfunction.
The Ins2Akita mouse develops spontaneous hyperglycemia by 2 months of age and shows progressive retinal pathology resembling human DR [89]. It is particularly useful for studying early DR features, including vascular leakage, gliosis, and neurodegeneration, typically observed over six-month periods [143]. By 6 months, vascular abnormalities become evident, such as pericyte loss, vascular leakage, and microaneurysm formation [89]. By 9 months, advanced features appear, including retinal neovascularization, new capillary bed formation, and ectopic blood vessels in the outer plexiform layer—findings rarely seen in other diabetic mouse models [89]. These structural changes are accompanied by increased apoptosis and measurable functional decline.
Early retinal pigment epithelium (RPE) dysfunction also emerges with the onset of hyperglycemia, with progressive impairment detected in both neural and RPE responses [144]. Akita mice show poor adaptation to metabolic stress; hypercapnia further impairs inner retinal function without triggering expected increases in retinal blood flow, suggesting dysfunction beyond vascular dysregulation [145]. The model’s ability to develop both early and late DR features, including neovascularization, makes it especially valuable for testing interventions across disease stages.
The db/db mouse model is particularly useful for studying retinal
neurodegeneration, now recognized as an early and possibly independent
contributor to DR pathogenesis [146]. Pericyte loss, a hallmark of early DR, has
been compared between db/db and Akita mice. Both models show
pericyte dropout and gliosis, but differ in inflammatory signaling. In
db/db mice, chronic interferon-
These mechanistic insights have informed several therapeutic strategies that have shown promise in this model. Intravitreal AAV2-mediated SIRT1 overexpression improved both neuronal and vascular retinal function [148]. Additionally, natural compounds such as astragaloside IV (AR inhibitor) [149] and salidroside (oxidative stress modulator) [150] also preserved retinal integrity.
Beyond single-pathway interventions, the db/db model has been further used to examine interactions between hyperglycemia and dyslipidemia. Combined treatment with poloxamer 407 and high glucose aggravated retinal dysfunction more than either condition alone [55]. These observations support the use of this model in dissecting the complex metabolic contributions to diabetic retinopathy.
The ob/ob mouse carries a mutation in the leptin gene, resulting in leptin deficiency. The model has been valuable for studying leptin’s role in retinal disease. In a retinopathy of prematurity model, transgenic mice overexpressing leptin showed increased retinal neovascularization compared to wild-type littermates. In contrast, leptin-deficient ob/ob mice had significantly reduced ischemia-induced neovascularization. This effect is linked to leptin receptor signaling in retinal endothelial cells, where leptin activates VEGF mRNA expression [151].
Longitudinal studies in BTBR ob/ob mice have mapped the progression of DR. Obesity appears by 2 weeks, followed by hyperglycemia at 3 weeks. By 6 weeks, early retinal dysfunction and inner retinal thinning occur. Importantly, neuroinflammatory changes—such as glial activation, leucostasis, and microglial phenotype shifts—precede vascular degeneration and increased permeability [152].
Diet-induced models, particularly high-fat diet (HFD) feeding in C57BL/6J mice, are widely used to study the early stages and progression of type 2 DM and its complications, including diabetic retinopathy (DR). After 12 weeks of HFD feeding, mice typically develop prediabetes or early-stage diabetes with significant retinal deficits. Electroretinography reveals reduced scotopic and photopic responses, indicating impaired retinal sensitivity. At the molecular level, downregulation of key signaling proteins involved in calcium regulation and glucose transport has been observed. Notably, similar molecular changes are seen in human DR retinas, underscoring the translational relevance of this model [153].
For example, extended HFD exposure helps to characterize the temporal progression of DR. Mice fed a 60% fat diet for up to 12 months exhibit neural and vascular abnormalities, including retinal nerve infarcts, vascular leakage, and reduced vascular density, even in the absence of marked hyperglycemia [154]. These findings support the notion that retinal neurodegeneration may precede overt vascular pathology. However, standard HFD-based models often require prolonged feeding periods to induce significant pathology, posing practical limitations. To overcome these time constraints, accelerated protocols have been developed. One approach combines HFD with low-dose streptozotocin (STZ) administration, delivered via osmotic mini-pump, to more rapidly induce type 2 DM. This non-transgenic method enables the study of retinal vascular pathology, including visualization techniques such as fluorescent gelatin vascular casting [155].
Importantly, while HFD-based models are valuable, the diets used in many studies—containing 60% of total caloric intake from fat—far exceed the fat consumption typically observed in human populations, which generally ranges from 28.5% to 46.2% of total daily energy intake [156]. Additionally, growing evidence suggests that both the amount and quality of dietary carbohydrates play a crucial role in the progression of diabetes. Sucrose-rich diets have been shown to worsen hyperglycemia, while fiber-rich or resistant starch diets improve glycemic control by modulating gastric emptying and gut microbiota composition. These findings support the inclusion of carbohydrate composition as a critical factor in model design and interpretation [157]. Therefore, a more recent mouse model has employed a medium-fat diet (e.g., 34.5% of total energy from fat) combined with fructose supplementation in drinking water and low-dose STZ, which more closely mimics Western dietary patterns characterized by excess intake of both fat and sugar. This model better replicates the metabolic and vascular complications of human type 2 DM [158].
Overall, diet-induced models provide a physiologically relevant platform for studying DR progression in the context of type 2 DM. Incorporating more realistic macronutrient compositions, including both fat and carbohydrate sources, may enhance the translational fidelity of these models. Accelerated variants further improve practicality by reducing experimental timeframes, facilitating the investigation of underlying mechanisms and therapeutic interventions.
House mice (Mus musculus) have long served as key models in biomedical research due to their genetic manipulability, short reproductive cycles, and physiological similarities to humans. However, fundamental interspecies differences limit their translational applicability and must be acknowledged as inherent rather than exceptional [159]. These differences include significant disparities in body size (humans are ~2500 times larger), basal metabolic rate, lifespan, and life history traits such as gestation length and reproductive maturity.
Moreover, environmental exposures—such as diet, microbiota composition, and pathogen exposure—differ markedly and influence disease development and immune responses across generations [159]. In addition, species-specific differences in immune responses, including cytokine profiles and inflammatory resolution pathways, affect the progression of chronic diabetic complications [160, 161]. The influence of sex as a biological variable remains underexplored in most rodent studies, where male mice are typically used. This is in contrast to growing clinical evidence showing that sex differences substantially shape disease onset, risk factor burden, and complication profiles in type 2 DM [162].
Taken together, these physiological, immunological, and sex-based gaps underscore the need for complementary human-based systems—such as organoids, tissue explants, and in vitro models—to enhance translational relevance.
Rodent models have provided valuable insights into the early pathophysiology of DN. STZ-induced and db/db mice consistently develop hyperglycemia and exhibit key features such as albuminuria and mesangial expansion. However, they rarely reproduce advanced renal lesions observed in human DN, including nodular glomerulosclerosis, arteriolar hyalinosis, and interstitial fibrosis [95, 104, 163, 164].
Type 1 DM models—such as STZ-induced and Akita mice—are effective for studying hyperglycemia-driven renal injury, offering reproducible disease onset and glycemic control [93, 94]. Still, additional interventions (e.g., uninephrectomy, high-salt diets, or genetic modifications) are often required to replicate human-like nephropathy severity. In contrast, type 2 DM models such as db/db and BTBR ob/ob mice may provide greater translational relevance, as they naturally exhibit insulin resistance, obesity, dyslipidemia, and chronic inflammation—metabolic disturbances commonly seen in human DN [106, 110]. Their multifactorial pathophysiology offers a more comprehensive platform for evaluating therapeutic strategies targeting the metabolic–renal axis.
Rodent models of DPN typically present with early sensory deficits, including thermal hypoalgesia and tactile allodynia. While some structural abnormalities—such as reduced intraepidermal nerve fiber density and mild myelin thinning—are observed, most models fail to capture the full pathological spectrum of human DPN. Advanced features like Schwann cell loss, demyelination, and axonal degeneration remain poorly replicated, with evidence of overt myelin damage largely absent beyond mild segmental changes [73, 165].
Moreover, many models lack additional risk factors such as hypertension, which significantly contribute to disease progression in humans. The anatomical complexity of the peripheral and autonomic nervous systems, combined with the diversity of underlying pathogenic mechanisms and inconsistent experimental protocols, complicates cross-study comparisons. Given the multifactorial nature of human DPN, there is a clear need for novel models with integrated diabetic, neuropathic, and metabolic profiling, supported by standardized methodologies and translational frameworks [166].
Mouse models are widely used in DR research and have been instrumental in elucidating pathophysiological mechanisms and identifying therapeutic targets. A range of induction methods—including pharmacologic, genetic, environmental, and surgical approaches—have been employed to simulate disease progression [167]. Despite model variability, early DR features such as vascular leakage, glial activation, neuroinflammation, and limited neovascularization are frequently replicated [138].
Nonetheless, major limitations persist. Mouse models rarely develop advanced human DR characteristics, such as microaneurysms and robust neovascularization. These differences may stem from their shorter lifespan and slower disease progression; whereas DR in humans typically develops within 2–5 years of diabetes onset, mice may require 6–18 months to exhibit comparable changes [168]. Moreover, growing evidence suggests that retinal neurodegeneration may precede vascular pathology, challenging the traditional view of DR as a primarily vascular disease [169].
While murine models remain indispensable tools for elucidating the mechanisms of diabetic complications, their translational limitations call for strategic integration with human-relevant platforms and emerging technologies.
This focused review summarizes key pathogenic mechanisms and widely used mouse
models—such as STZ-treated, Akita, db/db, and BTBR
ob/ob—for studying diabetic complications. These models have enabled
detailed investigation of systemic and organ-specific pathologies, including
glomerular sclerosis, retinal neovascularization, and axonal degeneration. They
have also facilitated the identification of conserved molecular targets—such as
NF-
It should be noted that this article is a narrative review, synthesizing key findings without systematic inclusion criteria. Future meta-analyses could build on this foundation by quantitatively evaluating the available evidence. Continued refinement of these models remains essential for advancing our understanding of diabetic complications and informing mechanism-based treatments.
However, significant research gaps remain. Current models often inadequately mimic the temporal progression and heterogeneity of human diabetic complications, particularly in the presence of comorbidities such as obesity, hypertension, and aging. In addition, most rodent models fail to capture the influence of sex differences, environmental exposures, and microbiome dynamics, all of which are increasingly recognized as critical modulators of disease trajectory and therapeutic response. Sex differences, in particular, shape risk factor profiles, complication burden, and therapeutic responses in diabetes, yet remain scarcely considered in preclinical research. To address these limitations, future studies should prioritize the development of physiologically relevant, polygenic, and sex-balanced models that more accurately reflect the multifactorial nature of human DM. Approaches such as the generation of humanized mouse strains, together with cross-species integration of high-throughput omics, spatial transcriptomics, and single-cell technologies, hold particular promise for enabling precise disease mapping and target discovery.
Beyond model refinement, translational challenges must also be proactively addressed. While rodent models replicate key molecular aspects of diabetic complications, fundamental differences in immune function, metabolic regulation, and organ architecture continue to hinder clinical translation. To improve translational relevance, complementary approaches such as patient-derived organoids, ex vivo tissue systems, and computational modeling should be integrated. Incorporating pharmacokinetic and pharmacodynamic analyses early in preclinical studies may also enhance the prediction of clinical outcomes. Ultimately, a multi-pronged strategy that combines advanced animal models, innovative experimental platforms, and strengthened translational pipelines will be essential for the development of effective, mechanism-driven therapies targeting diabetic complications.
AGEs, advanced glycation end products; AKT/PKB, protein kinase B; ALK1, activin receptor-like kinase 1; AR, aldose reductase; ATP/ADP, adenosine triphosphate/adenosine diphosphate; BTBR, Black and Tan Brachyury; BUN, blood urea nitrogen; CCR2, C-C chemokine receptor type 2; CD18, cluster of differentiation 18; DM, diabetes mellitus; DPN, diabetic peripheral neuropathy; DN, diabetic nephropathy; DR, diabetic retinopathy; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; ERK1/2, extracellular signal-regulated kinases 1 and 2; FGF21, fibroblast growth factor 21; FoxO3a, Forkhead box O3a; GFR, glomerular filtration rate; GLP-1, glucagon-like peptide-1; GLUT3, glucose transporter type 3; GRO-
AL is the sole author of this manuscript and was responsible for its conception, literature review, analysis, and writing. AL has prepared, read, and approved the final manuscript, and agrees to be accountable for all aspects of the work.
Not applicable.
I appreciate all the peer reviewers for their valuable opinions and suggestions.
This research received no external funding.
The author declares no conflict of interest.
The author acknowledges the use of a generative AI tool (ChatGPT, GPT-4o version, developed by OpenAI), with use strictly restricted to improving the clarity and readability of the English language in this review. The author is solely responsible for the selection of sources, interpretation of literature, and all conclusions presented.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.