


 , Alikhan Asoyan 1, Olga Maltseva 2, Alexander Orekhov 3
, Alikhan Asoyan 1, Olga Maltseva 2, Alexander Orekhov 31 Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, 125315 Moscow, Russia
2 Institute of Experimental Medicine, 197022 Saint Petersburg, Russia
3 Institute for Atherosclerosis Research, 121609 Moscow, Russia
Abstract
Mast cells, traditionally recognized for their roles in allergy and host defense, have recently been implicated in the pathogenesis of atherosclerosis. Their strategic localization in vascularized tissues and capacity to release a wide array of bioactive mediators position them as crucial contributors to both early and advanced stages of plaque development. This review summarizes the current understanding of mast cell functions in vascular homeostasis and immunity, with a special focus on their mechanistic involvement in atherogenesis and their potential as therapeutic targets in atherosclerosis. We conducted a comprehensive literature review of experimental, preclinical, and clinical studies addressing mast cell biology in the context of atherosclerosis. Particular emphasis was placed on molecular mechanisms, mast cell-derived mediators, and emerging pharmacologic interventions. Mast cells promote key atherogenic processes, including endothelial dysfunction, low-density lipoprotein (LDL) retention, monocyte recruitment, foam cell formation, fibrous cap thinning, and plaque rupture. Mechanistically, this involves the release of proteases (chymase and tryptase), histamine, and proinflammatory cytokines (e.g., tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6)). Additionally, mast cells contribute to caspase-1-mediated IL-1β production, which activates NF-κB signaling cascades leading to enhanced inflammatory cytokine production and adhesion molecule expression. Therapeutic strategies targeting mast cell activation, degranulation, metabolic activity, and specific receptors have demonstrated efficacy in preclinical models. Emerging approaches include dual protease inhibitors, personalized therapies guided by mast cell phenotyping, and advanced delivery systems. Mast cells are significant drivers of atherogenesis and plaque destabilization. Targeting mast cell-specific pathways represents a promising avenue for therapeutic intervention in atherosclerosis and the prevention of acute cardiovascular events.
Keywords
- mast cells
- atherosclerosis
- plaque instability
- chymase
- tryptase
- cardiovascular inflammation
- targeted therapy
Atherosclerosis involves the development of fibro-fatty plaques within arterial walls. This pathological process leads to atherosclerotic disease. The disease is characterized by hemodynamically significant arterial narrowing that restricts blood flow to tissues and organs [1]. The coronary, carotid, cerebral, mesenteric, renal, and lower extremity arterial beds are typically affected by atherosclerosis, which can occur in any arterial region. Critical disorders like stroke, myocardial infarction (MI), coronary artery disease (CAD), mesenteric ischemia or ischemic colitis, and peripheral arterial disease (PAD) of the lower extremities are clinically caused by atherosclerosis in these routes. Comorbid conditions such as dyslipidemia, hypertension, obesity, and diabetes affect the risk of atherosclerotic disease development [2].
The inflammatory component of atherosclerosis involves complex molecular
pathways, including the caspase-1-mediated formation of interleukin-1
Conventional treatment for atherosclerosis typically involves a combination of lifestyle changes and pharmacologic interventions. Lifestyle changes include adopting a heart-healthy diet low in saturated and trans fats, engaging in regular physical activity, maintaining a healthy weight, and quitting smoking [4]. Pharmacologic treatment may include statins to lower low-density lipoprotein (LDL) cholesterol, antiplatelet drugs such as aspirin to prevent blood clots, and antihypertensive drugs to control blood pressure [5]. In more severe cases, surgical procedures such as angioplasty and stenting or coronary artery bypass grafting may be required to improve blood flow to the heart [6]. The goal of conventional treatment is to slow the progression of atherosclerosis, reduce the risk of cardiovascular events, and manage the symptoms associated with the condition. The conducted studies have shown that immune cells play a fundamental role in the development of atherosclerosis and chronic inflammation. Therefore, immunotherapy targeting immune cells is expected to become a new method in the treatment of atherosclerosis.
As members of the hematopoietic lineage, mast cells serve as essential immune system cells. Maturated mast cells do not normally move through the circulation. However, stem cell factor and other cytokines cause mast cell precursors to move into tissues and develop into mast cells. Mast cells are found all over the organism and are crucial to the pathophysiology of some illnesses as well as the maintenance of numerous physiological processes [7]. This review aims to highlight the various pathways of mast cell involvement in the pathogenesis of atherosclerosis and potential therapeutic approaches to target these pathways.
The bone marrow produces monocytes, which are transient blood cells. While in blood vessels, they have the ability to release inflammatory cytokines and change into long-lived macrophages. The functions that various macrophage subtypes perform in the development of plaque vary. When intima-resident macrophages consume more lipids and generate foam cells, plaques start to grow. Plaques require ongoing blood flow and a monocyte inflow to progress, which encourages the development of plaque macrophages [8].
The ability to precisely react to triggers in the local microenvironment to either eliminate or prolong inflammation is one of the traits that sets macrophages apart [9]. Macrophage morphologies are classified into two groups (M1 and M2) based on cytokine-induced in vitro characteristics. The production of pro-inflammatory cytokines and host defense depend on M1 macrophages, which are pro-inflammatory macrophages. M2 macrophages have been shown to lower inflammation and promote tissue repair [10]. Both traditionally and alternatively activated subgroups of macrophages have been found to adhere to plaque in both human and murine atherosclerotic lesions. Prolonged inflammation triggers macrophage mortality and, in the absence of efficient efferocytosis, leads to the buildup of apoptotic cells and debris, which facilitates the formation of a necrotic plaque core. M2 macrophage- representing cells are located in stable plaque locations in human diseases, whereas proinflammatory marker- expressing macrophages are observed in unsteady, rupture-prone places [11].
The most prevalent type of leucocytes is neutrophils, which release a number of inflammatory mediators, including pentraxin 3, matrix metalloproteinases (MMPs), reactive oxygen species, and myeloperoxidases (MPOs). Pentraxin 3 stimulates the activation of complement and leukocyte adherence by binding to C1q. Cathelicidin is secreted by triggered neutrophils. It promotes the adhesion of monocytes and other myeloid cells either directly or through receptor binding. The azurophilic, particular, gelatinase, and secretory granules that are found inside neutrophils and developed during their development are all known to exacerbate atherosclerosis. Proteasessuch as gelatinase (MMP-9) and MMP-25, which are found in azurophilic granules, break down the extracellular matrix and encourage the lysis of the fibrous plaque cap [12]. Type IV collagen, which is essential to the endothelial cell membrane, is broken down by MMP-9 in conjunction with MMP-2. Defensins found in azurophilic granuleslinked to dysfunction of endothelial cells, chemokine attraction, and deprived cholesterol utilization are also releasedby neutrophils [13].
In the early phases of atherosclerosis, T lymphocytes, monocytes, and macrophages are the first cells to be attracted. The same chemokines that cause monocyte adherence also drive T lymphocyte recruitment. Depending on whether they are CD4 + or CD8 + T cells, all T cells express CD3 +, the T cell receptor (TCR) complex, and other co-receptors. Atherosclerosis is mainly triggered by CD4 + T cells, which are the source of T helper cells differentiation. Several Th subtypes (Th1, Th2, Th9, Th17, Th22, follicular helper T cells (TFH), and CD28 + T cells) or Treg subtypes develop from CD4 + T cells after antigen presentation [14]. Th1 and Th2 subgroups prevail in the atherosclerotic plaque, Th1 is the predominant type with a demonstrated atheroprotective effect. The influence of other lymphocyte types such as Th2, Th9, Th17 and Th22 has not been fully recognized [14]. CD44, a signaling marker that separates naive T cells from memory cells and effector counterparts, is one of the markers displayed on T cells that shows the clonal formation of these cells upon contact with antigens. It also plays a significant role in infarct recovery. Mice lacking CD44 exhibited enhanced leukocyte adherence to the infarct zone with noticeable collagen deposition, as well as extended production of inflammatory cytokines. CD4 + T cells, which function as autoantigens with the activation of the adaptive immune system, can be activated by antigens such as LDL cholesterol and apolipoprotein B [15].
Both normal and diseased arteries contain B cells, which are crucial components
in atherosclerosis. While follicular B cells and innate triggers of B cell
activation have been demonstrated to encourage atherosclerosis by inducing the
Th1 adaptive immune system, B1 and marginal zone B cells are believed to offer
defense against atherosclerosis [16]. Inflammatory events promote the aggregation
of B lymphocytes and the development of arterial tertiary lymphoid tissue when B
cells are recruited to the vascular adventitial microenvironment. These lymphoid
organs include a much smaller proportion of CD19 + CD11b– and CD19 + CD11b+ B
cells, which are the two main kinds of B cells. By inhibiting T cell responses,
the latter B cell subtype seems to have protective benefits [17]. Three primary
methods are used by B cells to induce atherogenesis: cytokine release,
immunoglobulin synthesis, and antigen presentation. The immune response in
atherosclerosis is counterbalanced by the production of antigen- specific
antibodies, which activate a range of cells by binding to the FcR receptor,
neutralizing oxidized lipoproteins, and initiating the complement pathway.
Through costimulatory markers including CD40, CD86, CD80, or MHC-II, B
lymphocytes and Th cell lymphocytes engage in arterial tertiary lymphoid tissue
[18]. The effector activity of T cells is ensured by this contact between B and T
cells. Research demonstrates the presence of plasma cells that secrete anti-LDL
immunoglobulins targeting oxidation-specific epitopes, which trigger protective
antibodies and reduce plaque. The fact that B cells carry the B cell receptor
(BCR) antigen and TLRs implicated in both innate and adaptive responses at the
same time, however, makes studying B cells in atherosclerosis much more
challenging. By boosting inflammatory cytokines and chemokines, stimulation of
the TLR in atherosclerosis encourages the formation of plaque [19]. It is unclear
exactly which endogenous or foreign antigens activate BCR and/or TLRs.
Additionally, B lymphocytes release cytokines that either delay the development
of atherosclerotic plaque (IL-2, IL-4, IL-10) or induce it (tumor necrosis
factor-alpha (TNF-
While macrophages, neutrophils, T cells, and B cells each contribute distinct functions to atherosclerotic progression as outlined above, mast cells represent a unique immune cell population that combines and amplifies many of these individual functions. Unlike other immune cells that typically exhibit specialized roles, mast cells serve as multifunctional orchestrators capable of simultaneously promoting lipid accumulation, enhancing inflammatory cell recruitment, mediating matrix degradation, and driving plaque destabilization through their diverse secretory profile. This multifaceted nature, combined with their strategic tissue localization and rapid response capabilities, positions mast cells as particularly significant contributors to atherosclerotic pathogenesis, justifying their dedicated examination in the following sections.
Mast cells contribute to the regulation of numerous physiological processes, including vasodilation, angiogenesis, and host defense against bacterial and parasitic infections. Additionally, they modulate the activity of various cell types, such as dendritic cells, macrophages, T and B lymphocytes, fibroblasts, eosinophils, endothelial, and epithelial cells. This regulatory versatility is attributed to their capacity to synthesize and release a broad spectrum of bioactive molecules, including histamine, proteases, prostanoids, leukotrienes, heparin, as well as diverse cytokines, chemokines, and growth factors. Consequently, mast cells influence the functional status of multiple tissues and organs. Among their most extensively studied roles are their contributions to vascular and bronchial homeostasis. Furthermore, mast cells are implicated in the regulation of bone development, remodeling, and mineral balance [20].
Mast cells are recognized as key promoters of angiogenesis [21]. They secrete
numerous pro-angiogenic mediators, such as vascular endothelial growth factor
(VEGF), basic fibroblast growth factor (bFGF), transforming growth factor-beta
(TGF-
Mast cells play a pivotal role in maintaining immune homeostasis. Due to their strategic localization in the skin and mucosal surfaces, they act as frontline defenders against external antigens [22]. They are particularly important in preserving the equilibrium of intestinal microbiota [23]. Given the constant exposure of the gastrointestinal tract to microbial and dietary antigens, epithelial barriers require immune support. Mast cells, via ATP-mediated signaling, facilitate the differentiation of follicular helper T cells, thereby contributing to IgA production and microbial homeostasis in the gut [23].
Mast cells are integral to both innate and adaptive immune responses. They can
detect pathogenic stimuli through direct interaction with microbes or by
recognizing pathogen-associated molecular patterns (PAMPs) via pattern
recognition receptors such as Toll-like receptors (TLRs) and complement receptors
[24]. Upon engagement with specific antigens, mast cells initiate the release of
inflammatory mediators aimed at pathogen clearance. The nature of the immune
response depends on the specific TLR involved. For instance, activation of TLR2
by Gram-positive bacteria or mycobacteria leads to IL-4 secretion, whereas TLR4
engagement by lipopolysaccharides from Gram- negative bacteria induces the
release of pro-inflammatory cytokines (e.g., TNF-
Mast cells support bacterial elimination by increasing vascular permeability,
promoting edema, and recruiting immune effector cells such as eosinophils,
neutrophils, and natural killer (NK) cells. They also directly produce
antimicrobial peptides including cathelicidins, defensins, and psidins. In
antiviral defense, mast cells enhance cytotoxic responses by recruiting CD8+ T
cells and promoting the release of type I interferons (IFN-
In the context of adaptive immunity, mast cells are capable of processing and
presenting antigens via MHC class I and II molecules [28]. They also activate
dendritic cells, which serve as critical antigen-presenting cells. Upon
stimulation through Toll-like receptor 7 (TLR7), mast cells secrete IL-1 and
TNF-
Mast cells contribute to early atherogenesis by facilitating the infiltration of low-density lipoprotein (LDL) particles into the arterial intima. Histamine, acting through H1 receptors on endothelial cells, increases vascular permeability, enhancing LDL entry and promoting plaque formation in hypercholesterolemic models [29]. Despite mast cells being primary sources of histamine, other cells, such as endothelial cells also produce it, implying that not all histamine-driven effects are mast cell-specific. Exceptions include unique mast cell mediators like heparin and tryptase. Tryptase, for instance, can compromise the endothelial barrier to LDL via activation of protease-activated receptor-2 (PAR-2) [29].
Activated mast cells also release chemokines that attract monocytes and
stimulate endothelial cells to express adhesion molecules, further promoting
monocyte transmigration into the subendothelial space [30]. In vitro,
chymase from mast cells has been shown to cleave the C-terminal region of
apolipoprotein A-I (apoA-I), impairing its anti- inflammatory properties.
Truncated apoA-I fails to inhibit TNF-
Within the intima, heparin-containing mast cell granules bind to apoB-100 on LDL via electrostatic interactions. Chymase then proteolytically modifies apoB-100, destabilizing LDL particles and promoting their aggregation into larger complexes akin to those observed in atherosclerotic lesions. This aggregation increases cholesterol accumulation within the plaque [32].
Histamine increases endothelial permeability to High-Density Lipoprotein (HDL)
particles. Pre
Continual LDL influx and foam cell formation, compounded by inadequate cholesterol efflux to dysfunctional HDL, lead to macrophage retention and eventual cell death within the plaque [35]. Mast cell-derived histamine can trigger macrophage apoptosis, contributing to necrotic core expansion and plaque enlargement [36].
As atherosclerotic plaques enlarge and reduce arterial lumen diameter, disturbed
blood flow induces endothelial dysfunction and detachment. Mast cells residing
beneath the endothelium may release tryptase and chymase, which degrade the
basement membrane and promote endothelial cell apoptosis, exacerbated by
TNF-
Thinning of the fibrous cap is a critical event in plaque vulnerability. It is
driven by diminished collagen synthesis due to smooth muscle cell senescence and
apoptosis, and by increased matrix degradation via metalloproteinases (MMPs)
[40]. Chymase induces apoptosis in smooth muscle cells by degrading fibronectin
and inhibits collagen synthesis through both TGF-
Mast cells serve as crucial orchestrators of inflammatory cascades in
atherosclerotic plaques through complex interactions with multiple immune cell
populations [21]. Upon activation by oxidized LDL or complement components, mast
cells release a diverse array of inflammatory mediators, including leukotrienes,
prostaglandins, and platelet-activating factor, which collectively amplify the
local inflammatory response [44]. These lipid mediators promote additional
recruitment of inflammatory cells, including T lymphocytes and dendritic cells,
establishing a self-perpetuating cycle of inflammation [45]. Mast cell-derived
IL-6 and IL-1
Recent evidence demonstrates that mast cells contribute to atherosclerosis progression through metabolic reprogramming of the plaque microenvironment and enhancement of oxidative stress pathways [49]. Activated mast cells undergo glycolytic reprogramming, leading to increased lactate production and local acidification of the atherosclerotic milieu. It impairs endothelial function and promotes smooth muscle cell proliferation [50]. Mast cell-derived reactive oxygen species (ROS), particularly superoxide and hydrogen peroxide, directly oxidize LDL particles within the plaque, creating highly atherogenic oxidized LDL that is preferentially taken up by macrophages [51]. Furthermore, mast cells secrete myeloperoxidase, which catalyzes the formation of hypochlorous acid and chlorotyrosine-modified proteins, contributing to endothelial dysfunction and plaque instability [52]. The release of histamine also upregulates NADPH oxidase expression in vascular smooth muscle cells, perpetuating ROS production and promoting a pro-oxidant environment [53]. Mast cell granules contain significant amounts of heparin sulfate proteoglycans, which can bind and concentrate oxidized phospholipids, creating localized zones of enhanced oxidative stress that accelerate atherosclerotic progression [54]. This metabolic dysregulation extends to cholesterol homeostasis, as mast cell-derived enzymes can interfere with ATP-binding cassette transporter function, further impairing reverse cholesterol transport [55].
Mast cells undergo metabolic shifts similar to those observed in tumor-associated macrophages, characterized by enhanced glycolytic activity that drives lactate production and creates an acidic microenvironment that promotes inflammatory mediator release [50]. This metabolic reprogramming not only affects mast cell function but also influences neighboring immune cells. The lactate produced by glycolytically active mast cells can directly impact macrophage polarization, promoting their differentiation toward the pro-inflammatory M1 phenotype while simultaneously enhancing foam cell formation.
Furthermore, mast cell-derived reactive oxygen species (ROS) directly impact macrophage lipid metabolism by promoting lipid peroxidation and enhancing the uptake of oxidized LDL, thereby accelerating foam cell formation [51]. The cross-regulation extends to T cell metabolism, where mast cell-derived inflammatory mediators can influence T helper cell differentiation and metabolic programming, creating a self-perpetuating cycle of metabolic dysfunction and immune activation.
These findings illustrate how mast cell metabolic changes create a complex network of metabolic and immunological interactions that synergize to drive immune dysregulation in atherosclerosis, representing potential targets for therapeutic intervention.
A summary diagram of the role of mast cells in atherosclerosis is presented in Fig. 1.
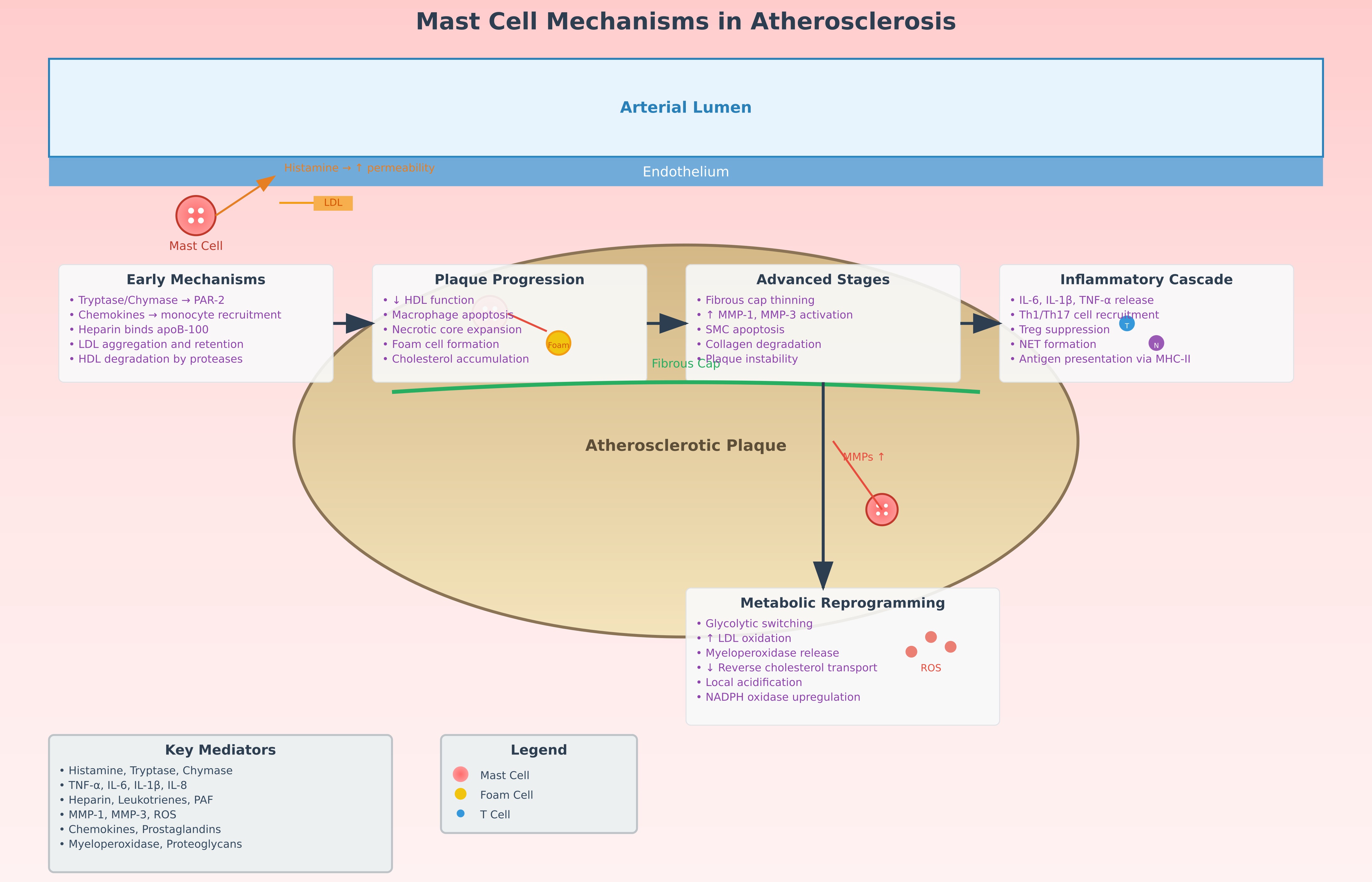 Fig. 1.
Fig. 1.
Scheme of the influence of mast cells on the pathogenesis of
atherosclerosis. This schematic diagram illustrates the multifaceted role of
mast cells in atherosclerosis pathogenesis. (1) Early atherogenic events show
mast cells releasing histamine that increases endothelial permeability, allowing
LDL infiltration into the arterial wall. (2) Mast cell-derived tryptase and
chymase promote LDL aggregation and foam cell formation while degrading
protective HDL particles. (3) Inflammatory mediator release (TNF-
Immune cells have been linked to damage, remodeling, and healing in a number of cardiovascular disorders during the past few decades, according to a substantial amount of clinical and experimental findings [56]. The physiology of cardiac lymphocytes is becoming more understood, but efforts to use this information therapeutically in patients with cardiovascular illnesses have so far been very unsuccessful [57].
The regulatory mechanisms governing inflammatory responses in atherosclerosis
involve complex signaling networks. MAPK and Tyro3 signaling pathways work
together or in parallel to reduce or “ameliorate” the production of
IL-1
The function of mast cells in cardiac fibrosis and the best way to pharmacologically target these cells have been the subject of numerous investigations. These cells might exhibit pro-fibrotic, anti-fibrotic, or neutral impacts in cardiac fibrosis, according to inconsistent findings from experiments conducted in various animal models [59]. There could be a number of causes for these contradictory findings. First, the frequency of all mast cells has been examined, but the great differences between human and murine mast cells have not been taken into consideration [60, 61].
Second, it is important to take into account the restrictions of any mouse model of mast cell insufficiency that is at present accessible [62, 63, 64]. Recent single-cell transcriptomic analyses have confirmed substantial interspecies variability in mast cell populations, particularly regarding receptor expression profiles, mediator release patterns, and tissue-specific phenotypes [59, 60]. These differences may partially explain why findings in mouse models do not always replicate human pathophysiology, especially in cardiovascular contexts. For instance, human cardiac mast cells differ significantly from those found in rodent hearts in terms of their protease content and degranulation patterns [65]. Additionally, the frequency and distribution of mast cell subsets vary considerably between species, with human tissues containing distinct populations that may not have direct murine counterparts [62]. Mast cell ablation is seldom 100% successful, and other cell deficits may also be present in murine models of mast cell shortage [66]. Additionally, the mast cells seen in human cardiac tissue are not the same as those found in rodent hearts. Finally, medications such as disodium cromoglycate and nedocromil, which are believed to disrupt mast cell functions and have also been used to study the significance of mast cells in coronary fibrosis [60].
Some medications that particularly target human mast cell stimulation or IgE
have been developed or introduced in recent years. Immunoglobulin E with Fc
Epsilon Receptor I (IgE–Fc
These clinical insights highlight the therapeutic promise of anti-IgE approaches for atherosclerosis and related cardiovascular disorders, moving beyond the preclinical evidence to demonstrate real-world potential. However, dedicated cardiovascular outcome trials specifically designed to evaluate omalizumab’s efficacy in atherosclerotic disease prevention are still needed to establish definitive clinical recommendations. Human mast cell-mediated diseases are presently being studied with lidalizumab, a second-generation humanized anti-IgE monoclonal antibody showing a greater affinity for IgE [67].
For the treatment of human diseases mediated by mast cells, a number of novel medication types that have the ability to decrease tissue mast cells population are very desirable. Furthermore, by focusing on intracellular signaling pathways such as spleen tyrosine kinase (SYK), bruton tyrosine kinase (BTK), and janus kinase (JAK), a number of promising small molecular weight drugs have been demonstrated to prevent mast cell stimulation [68]. Mast cell degranulation is inhibited by SYK inhibitors and aerosolized SYK antisense oligodeoxynucleotides. Leukotriene emission from mast cells is likewise inhibited by SYK inhibitors [69].
Mast cells include a number of inhibitory receptors, including Siglec-8, Siglec-6, CD200R, CD300a, and FcRIIb, which may be targets for atherosclerosis treatment. In humanized mice, anti-Siglec-8 antibody inhibited anaphylaxis and human mast cell activity that was dependent on IgE and non-IgE [70]. A humanized monoclonal antibody against Siglec-8 called lentelimab has demonstrated encouraging outcomes in a number of illnesses mediated by mast cells. Lirentelimab decreased the quantity of tissue mast cells [71].
Recent advances in understanding mast cell-specific proteases have opened new therapeutic avenues for atherosclerosis management through targeted enzyme inhibition [72]. Chymase inhibitors, such as chymostatin and TEI-F00806, have demonstrated significant potential in reducing plaque progression by preventing collagen degradation and smooth muscle cell apoptosis in experimental models [73]. These compounds specifically target the chymotrypsin-like activity responsible for angiotensin II formation and fibronectin cleavage, thereby preserving fibrous cap integrity [74]. Tryptase inhibitors, including nafamostat mesylate and APC 366, have shown efficacy in reducing endothelial permeability and preventing LDL infiltration into the arterial wall [75]. Additionally, dual protease inhibitors that simultaneously target both chymase and tryptase are being developed to provide comprehensive protection against mast cell-mediated plaque destabilization [76]. The metabolic reprogramming of mast cells in atherosclerotic environments has also emerged as a therapeutic target, with glycolysis inhibitors such as 2-deoxyglucose showing promise in reducing mast cell activation and inflammatory mediator release [77]. Furthermore, targeting mast cell-derived myeloperoxidase through specific inhibitors like 4-aminobenzoic acid hydrazide (4-ABAH) has demonstrated potential in reducing oxidative stress and LDL oxidation within atherosclerotic plaques [78].
The heterogeneity of mast cell populations and their diverse roles in atherosclerosis progression necessitate personalized therapeutic approaches based on individual patient characteristics and disease stage [79]. Combination therapies targeting multiple mast cell pathways simultaneously have shown enhanced efficacy compared to single- agent approaches in preclinical studies [80]. The co-administration of histamine receptor antagonists (H1 and H2 blockers) with protease inhibitors has demonstrated synergistic effects in reducing both acute inflammatory responses and chronic plaque progression [81]. Novel therapeutic combinations include the use of omalizumab with JAK inhibitors to simultaneously block IgE-mediated activation and cytokine signaling pathways [82]. Precision medicine approaches utilizing mast cell phenotyping and functional assessment are being developed to identify patients most likely to benefit from specific mast cell-targeted interventions [83]. Biomarker-guided therapy based on serum tryptase levels, chymase activity, and specific mast cell-derived cytokine profiles is emerging as a strategy to optimize treatment selection and monitoring [84]. Additionally, the development of nanoparticle-based drug delivery systems specifically targeting mast cells within atherosclerotic plaques offers the potential for localized therapy with reduced systemic side effects [85]. These targeted nanocarriers can be engineered to respond to the specific microenvironmental conditions within plaques, such as low pH and high protease activity, ensuring selective drug release at sites of mast cell accumulation [86].
Potential mast cell-targeted therapeutic strategies for atherosclerosis are summarized in (Table 1A, Ref. [59, 60, 63, 64, 65, 75]; Table 1B, Ref. [62, 63, 66, 67, 68, 69]; Table 1C, Ref. [62, 70, 71, 74, 75, 76, 77, 78, 79]).
| Target/Pathway | Therapeutic Agent | Mechanism of Action | Effect | Development Stage |
| IgE-Mediated Activation Inhibitors | ||||
| IgE–FcεRI interaction | Omalizumab | Monoclonal antibody binding free IgE, preventing mast cell activation | Reduces cardiac remodeling, prevents AngII-induced hypertension | FDA Approved [63, 64] |
| IgE–FcεRI interaction | Ligelizumab | Second-generation humanized anti-IgE antibody with higher IgE affinity | Enhanced mast cell deactivation, potential superior cardiovascular protection | Clinical Trials [59] |
| Intracellular Signaling Inhibitors | ||||
| SYK (Spleen Tyrosine Kinase) | SYK inhibitors | Blocks mast cell degranulation and leukotriene release | Reduces mediator release, prevents plaque inflammation | Preclinical [60, 65] |
| BTK (Bruton Tyrosine Kinase) | BTK inhibitors | Inhibits mast cell activation via FcεRI pathway | Prevents mast cell-mediated plaque destabilization | Preclinical [60] |
| JAK (Janus Kinase) | JAK inhibitors | Blocks cytokine signaling in mast cells | Reduces cytokine production, limits plaque progression | Clinical Trials [60, 75] |
This section presents therapeutic agents targeting IgE- mediated activation
pathways and intracellular signaling cascades in mast cells. Target/Pathway
column identifies the specific molecular mechanism being inhibited. Therapeutic
Agent lists the drug name or class. Mechanism of Action describes how the agent
works at the cellular/molecular level. Effect summarizes the therapeutic outcome
observed in studies. Development Stage indicates the current status of clinical
development with relevant reference citations. FDA, Food and Drug Administration;
IgE–Fc
| Target/Pathway | Therapeutic Agent | Mechanism of Action | Effect | Development Stage |
| Inhibitory Receptor Agonists | ||||
| Siglec-8 receptor | Anti-Siglec-8 antibody | Prevents IgE- and non-IgE-dependent activation | Inhibits degranulation, reduces anaphylaxis risk | Preclinical [62] |
| Siglec-8 receptor | Lirentelimab | Humanized antibody depleting mast cells in tissues | Reduces mast cell number and inflammation | Clinical Trials [66] |
| Mast Cell Protease Inhibitors | ||||
| Chymase | Chymostatin, TEI-FO0806 | Specific inhibition of chymase | Preserves fibrous cap, reduces plaque vulnerability | Preclinical [63, 67] |
| Tryptase | Nafamostat mesylate, APC 366 | Inhibits tryptase, reduces endothelial permeability | Maintains endothelial integrity, prevents LDL infiltration | Preclinical [68] |
| Dual targeting | Dual inhibitors | Simultaneous chymase and tryptase inhibition | Broad protection from mast cell-mediated damage | In development [69] |
This section focuses on therapeutic strategies utilizing inhibitory receptor agonists and specific protease inhibitors. Inhibitory Receptor Agonists target cell surface receptors that naturally suppress mast cell activation. Mast Cell Protease Inhibitors specifically block the enzymatic activity of key proteases (chymase, tryptase) that contribute to plaque destabilization. Each entry provides comprehensive information about mechanism, therapeutic effects, and current development status. LDL, low-density lipoprotein.
| Target/Pathway | Therapeutic Agent | Mechanism of Action | Effect | Development Stage |
| Metabolic Interventions | ||||
| Glycolysis | 2-Deoxyglucose | Inhibits glycolytic activation of mast cells | Reduces activation and mediator release | Preclinical [70] |
| Myeloperoxidase | 4-ABAH | Myeloperoxidase inhibitor | Reduces oxidative stress and LDL oxidation | Preclinical [71] |
| Combination & Personalized Therapies | ||||
| Multi-pathway | H1/H2 antagonists + protease inhibitors | Inhibits histamine and protease effects | Synergistic reduction of acute and chronic inflammation | Preclinical [74] |
| IgE + cytokine | Omalizumab + JAK inhibitor | Combines IgE blockade and cytokine inhibition | Comprehensive mast cell control | In development [75] |
| Personalized therapy | Biomarker-guided selection | Therapy based on mast cell phenotype and biomarkers | Optimized selection and monitoring | In development [76, 77] |
| Drug Delivery Systems | ||||
| Plaque-specific | Nanoparticle carriers | pH- and protease- sensitive release | Targeted therapy, reduced systemic toxicity | In development [78, 79] |
| Traditional Stabilizers | ||||
| Membrane stabilization | Cromoglycate, Nedocromil | Inhibits Degranulation via membrane stabilization | Limited evidence for CV protection | FDA Approved [62] |
This final section covers novel metabolic targeting strategies and combination therapeutic approaches. Metabolic Interventions target cellular metabolism pathways in mast cells to reduce their activation and inflammatory mediator production. Combination & Personalized Therapies represent advanced treatment strategies that simultaneously target multiple pathways or utilize personalized medicine approaches. Drug Delivery Systems showcase innovative methods for targeted drug delivery specifically to mast cells within atherosclerotic plaques. ABAH, 4-aminobenzoic acid hydrazide.
Accumulating evidence underscores the pivotal role of mast cells as effector
components in the pathophysiology of atherosclerosis and acute cardiovascular
events. Their pathological influence is mediated through the release of numerous
bioactive substances, including the proteases chymase and tryptase, histamine,
various growth factors, and a spectrum of proinflammatory cytokines and
chemokines such as TNF-
Furthermore, MAPK and Tyro3 signaling pathways are implicated in sepsis,
immunity, and inflammation, working together or in parallel to reduce or
“ameliorate” the production of IL-1
Targeting specific signaling pathways involved in mast cell activation during atherogenesis may offer greater therapeutic precision compared to generalized mast cell stabilization, which could inadvertently suppress their physiological roles in immune defense and tissue repair. Thus, it is critical to evaluate the effects of mast cell inhibitors on reparative processes. Future investigations should focus on identifying the molecular mechanisms responsible for mast cell recruitment to vascular lesions and delineating their cellular communication networks within the atherosclerotic plaque microenvironment. Additionally, detailed phenotypic characterization of plaque-associated mast cells may offer novel insights for the development of targeted therapies aimed at mitigating mast cell-driven plaque destabilization.
Mast cells have emerged as pivotal contributors to the initiation, progression, and complications of atherosclerosis. Their diverse secretory profile, encompassing proteases, cytokines, growth factors, and vasoactive mediators, positions them at the interface of inflammation, tissue remodeling, and vascular dysfunction. This review has highlighted their involvement in promoting endothelial permeability, facilitating LDL accumulation, enhancing leukocyte recruitment, and mediating smooth muscle cell apoptosis and fibrous cap degradation. Collectively, these processes lead to increased plaque vulnerability and heightened risk of acute cardiovascular syndromes.
Therapeutically, the development of mast cell-targeted strategies—ranging from IgE pathway blockers and protease inhibitors to metabolic and receptor-specific modulators—represents a novel and promising direction in cardiovascular medicine. Furthermore, innovative approaches such as personalized treatment guided by mast cell phenotyping and plaque-specific drug delivery platforms may enhance efficacy while minimizing off-target effects.
Future research should aim to clarify the endogenous triggers of mast cell activation in vascular lesions, characterize mast cell heterogeneity within plaques, and validate these therapeutic approaches in clinical settings. Harnessing the pathogenic potential of mast cells through precise pharmacological modulation could significantly improve outcomes for patients at risk of atherosclerotic complications.
AB and AO designed the review plan. AA and OM, performed the data analyses. All authors wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by Russian Science Foundation, grant number 25-15-00082.
The authors declare no conflict of interest. Given his role as the Guest Editor, Alexander Orekhov had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Giordano Pula.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.