



 , Vy Thuy Nguyen 1,†, Kim Tran Thien Duong 1, Huy Gia Truong 2, Duong Nguyen Thuy Le 1, Quan Minh Mai Le 3, Thu Ngoc Trinh 1, Tin Hoang Nguyen 2,4,*
, Vy Thuy Nguyen 1,†, Kim Tran Thien Duong 1, Huy Gia Truong 2, Duong Nguyen Thuy Le 1, Quan Minh Mai Le 3, Thu Ngoc Trinh 1, Tin Hoang Nguyen 2,4,*
1 Oral Diagnosis and Periodontology Department, Faculty of Odonto-Stomatology, Can Tho University of Medicine and Pharmacy, 900000 Can Tho, Vietnam
2 Department of Physiology, Faculty of Medicine, Can Tho University of Medicine and Pharmacy, 900000 Can Tho, Vietnam
3 Faculty of Odonto-Stomatology, College of Health Sciences, Nam Can Tho University, 900000 Can Tho, Vietnam
4 Department and Institute of Physiology, College of Medicine, National Yang Ming Chiao Tung University, 112304 Taipei, Taiwan
†These authors contributed equally.
Abstract
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease defined by exocrine gland infiltration and systemic involvement. The management of pSS is hampered by three persistent challenges: seronegativity, heterogeneity, and delayed diagnosis. Up to one-third of patients lack anti-Sjögren’s-syndrome-related antigen A/B (SSA/SSB) autoantibodies, contributing to misclassification and delayed recognition. Recent studies have expanded the autoantibody repertoire, identifying novel targets such as anti-D-aminoacyl-tRNA deacylase 2 (DTD2), anti-retroelement silencing factor-1 (RESF1), and anti-calreticulin (CALR), as well as multiplex panels including anti-salivary protein-1 (SP-1), anti-parotid secretory protein (PSP), and anti-carbonic anhydrase VI (CA6). These can detect disease before conventional seroconversion, thus offering diagnostic value for seronegative cases. The greatest challenge remains early detection, as the current reliance on biopsy and late-appearing serologies overlooks subclinical disease. In this context, non-invasive fluid biomarkers are transformative, with salivary and tear fluid proteomics (β2-microglobulin, clusterin, matrix metalloproteinase-9), exosomal micro ribonucleic acid (miRNAs), and metabolomic fingerprints providing sensitive indicators of glandular dysfunction and immune activation. When combined with machine learning, integrated multi-omics panels can achieve diagnostic accuracies comparable to biopsy while enabling prognostic stratification. Emerging approaches also leverage artificial intelligence (AI) to refine biomarker discovery and clinical translation. AI-assisted ultrasonography enables reproducible quantification of glandular inflammation, while the application of integrative AI models to multi-omics datasets can identify biomarker signatures with superior predictive accuracy. Such tools have the potential to accelerate early diagnosis, automate risk prediction, and guide precision therapeutics in real time. The future use of biomarker panels in clinical practice should reduce the time to diagnosis, thereby facilitating the anticipation of risk and the provision of therapy based on the underlying cause. In this review, we describe how pSS exemplifies some of the problems inherent in contemporary autoimmunity. This multifaceted and diverse condition is now well-positioned to benefit from integrative, biomarker-driven methodologies, which should lead to improved patient outcomes.
Keywords
- BAFF
- anti-Ro/SSA
- anti-La/SSB
- DNA methylation
- miRNA
- dry mouth
- dry eyes
- salivary gland ultrasonography
- artificial intelligence
- multi-omics
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease characterized by chronic lymphocytic infiltration of exocrine glands, most notably the salivary (Fig. 1) and lacrimal glands, leading to sicca symptoms such as dry eyes and dry mouth [1]. PSS predominantly affects middle-aged women (female-to-male ratio ~9:1), with a prevalence of approximately 0.1–0.2% among adults [2, 3, 4]. In addition to dysfunction of the exocrine glands, it frequently presents with a variety of complications outside the glands, such as interstitial lung disease (ILD), renal tubular acidosis, peripheral neuropathy, vasculitis, and B-cell lymphoma (Fig. 2), demonstrating the significant clinical heterogeneity of pSS [5]. This broad spectrum of presentations complicates both diagnosis and therapeutic decision-making, with experts identifying heterogeneity as a major barrier to progress in research and clinical care [5].
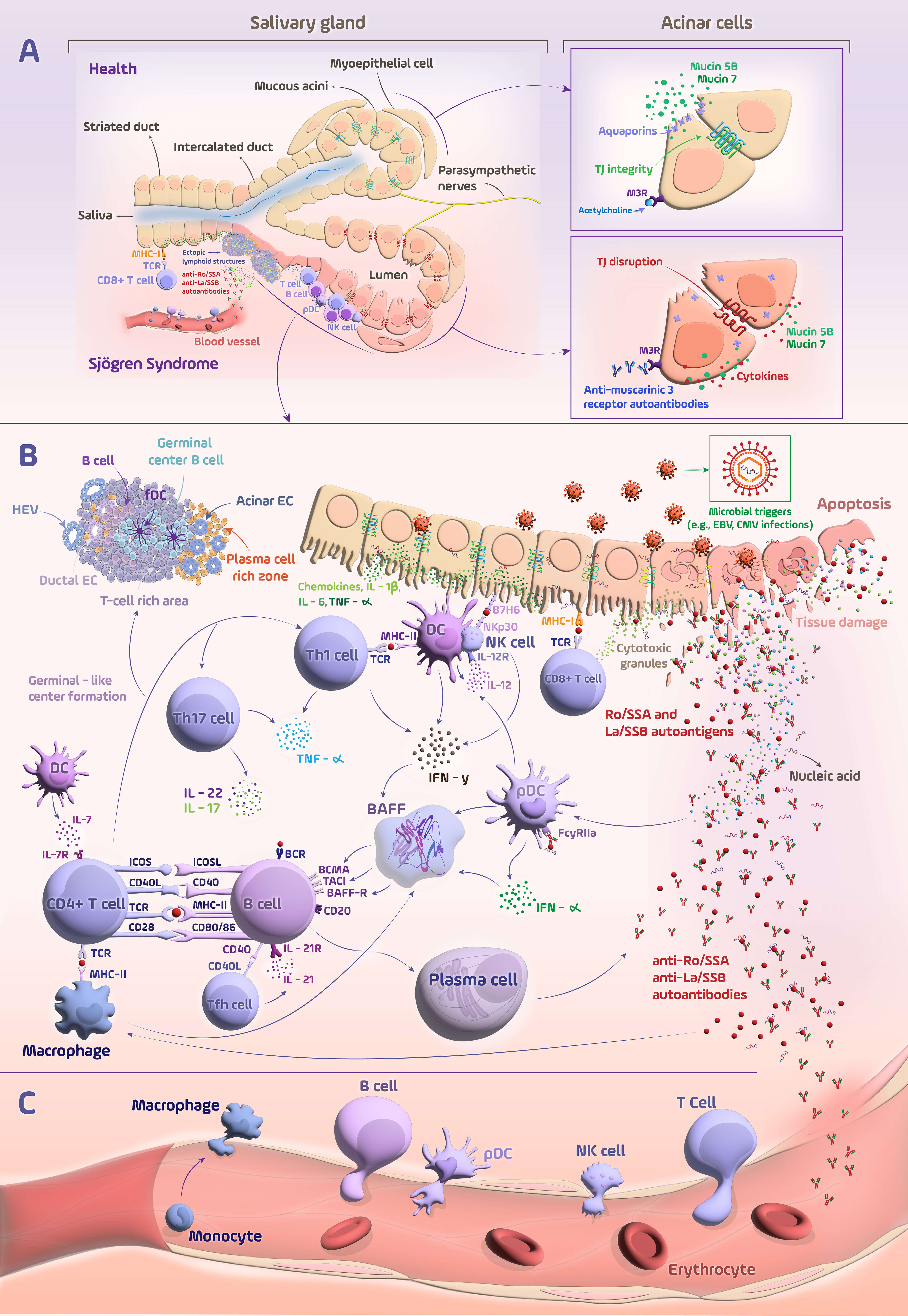 Fig. 1.
Fig. 1.
Immunopathogenic mechanisms underlying primary Sjögren’s
syndrome. (A) Structural and epithelial alterations in the salivary glands of
patients with pSS. In healthy glands, mucous acini, myoepithelial cells, and
ductal segments (intercalated and striated ducts) coordinate saliva secretion
through intact TJs, polarized expression of aquaporins, mucin 5B, and mucin 7,
and proper parasympathetic muscarinic signaling. In pSS, TJ breakdown, cytokine
overproduction, and anti-M3R autoantibodies impair salivary secretion and barrier
integrity. T cells, B cells, and APCs drive autoantibody production and
inflammation, causing epithelial damage and salivary dysfunction. (B)
Dysregulated immune activation and epithelial-immune crosstalk in the salivary
glands of pSS patients. Microbial triggers initiate epithelial stress and TJ
disruption within the salivary glands, leading to the release of autoantigens
(Ro/SSA, La/SSB). This early phase activates innate immune cells, such as DCs,
macrophages, and NK cells, which release cytokines like IL-1
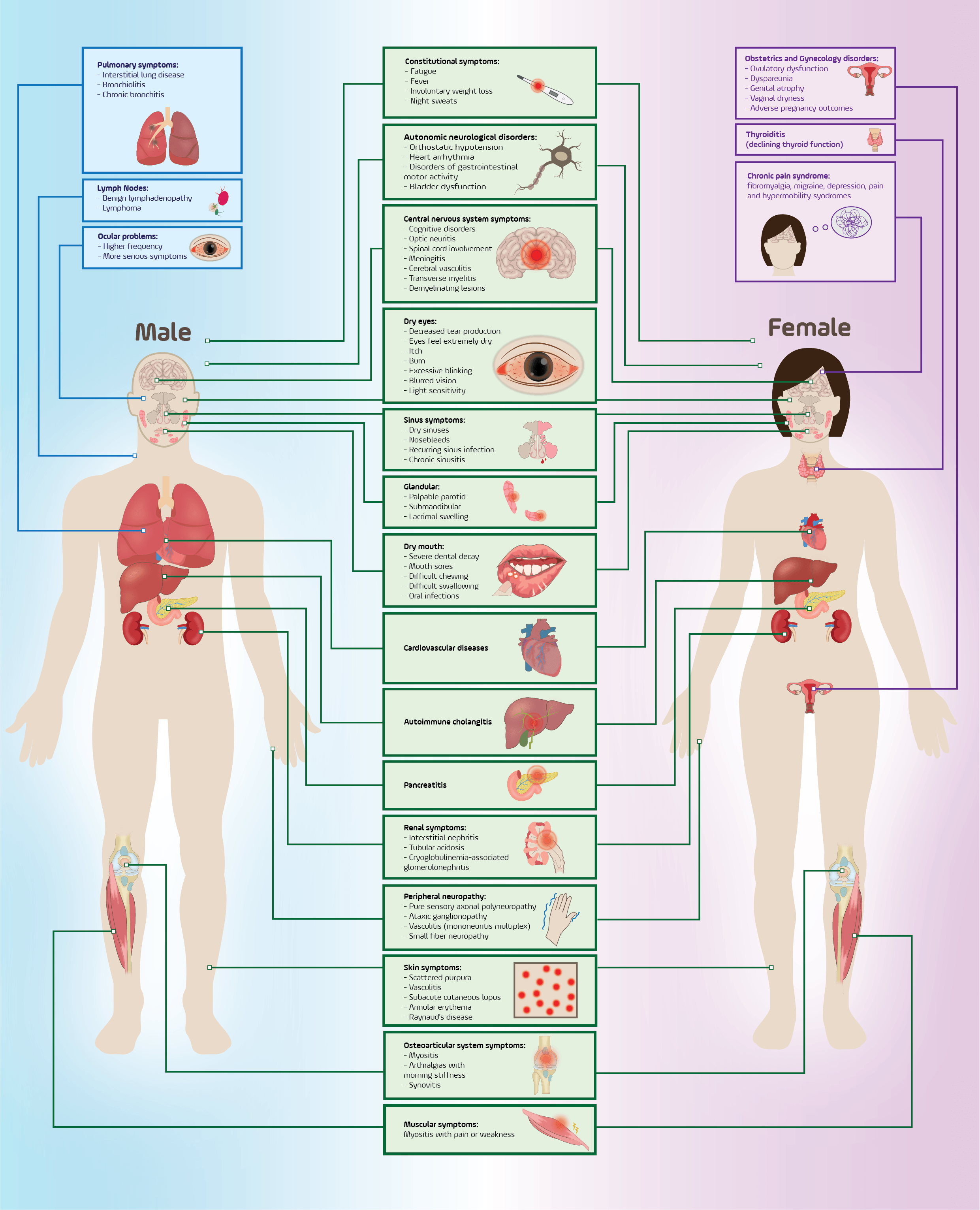 Fig. 2.
Fig. 2.
Sex-based differences and multisystem manifestations in primary Sjögren’s syndrome. The constitutional symptoms are common to both sexes and highlight organ-specific differences. Male patients more frequently develop pulmonary disease, lymphoma, and severe ocular involvement, whereas female patients more commonly present with thyroid dysfunction, chronic pain syndromes, and gynecological complications. Shared features include glandular dysfunction, neuropathies, renal involvement, and autoimmune-mediated systemic inflammation. This graphical work was created with Inkscape 1.4.2 (https://inkscape.org).
Currently, diagnosis of pSS relies on a combination of clinical features and objective criteria, such as those outlined in the 2016 classification system proposed by the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) [6, 7, 8]. These include evidence of autoimmunity, typically indicated by the presence of anti-Ro/SSA (Sjögren’s-syndrome-related antigen A) and anti-La/SSB (Sjögren’s-syndrome-related antigen B) autoantibodies, or positivity for rheumatoid factor (RF), as well as histopathological findings of focal lymphocytic sialadenitis (FLS) from a minor salivary gland biopsy. Although these criteria achieve high specificity (95%) and sensitivity (96%) [6], their application in routine clinical practice remains limited. Serologic markers are absent in approximately one-third of patients, and minor salivary gland biopsy, while informative, is invasive and subject to sampling variability [9, 10]. Moreover, sicca symptoms and other non-specific features, such as fatigue, arthralgia, and chronic cough, often overlap with numerous other conditions [11], leading to diagnostic delay or misclassification in many patients [12]. These challenges have driven an urgent need to identify novel, non-invasive, and more specific biomarkers for pSS. Traditional tools such as anti-SSA/SSB serologies and histopathology remain valuable, but are limited by low early sensitivity, lack of disease specificity, and procedural invasiveness [11]. Recent advances in molecular profiling and multi-omic technologies have opened new avenues for biomarker discovery. For example, autoantibodies that target salivary gland proteins expressed early in pSS, such as anti-salivary protein 1 (SP-1), anti-parotid secretory protein (PSP), and anti-carbonic anhydrase VI (CA6), have been found in early-stage or seronegative cases and may enhance diagnostic sensitivity [11]. In parallel, noninvasive sampling of saliva and tears has enabled “liquid biopsy”-based approaches, with cytokine profiling, transcriptomic signatures, and proteomic/metabolomic analyses revealing promising indicators of disease activity and subtype differentiation [13, 14].
Building on these advances, the field has entered a transformative phase marked by high-throughput biomarker discovery. High-density peptidome arrays combined with machine learning have identified 45 novel autoantibodies since 2021 [15]. Notably, multi-marker panels such as anti-DTD2 (D-aminoacyl-tRNA deacylase 2) and anti-RESF1 (retroelement silencing factor-1) demonstrate strong predictive value in seronegative patients and correlate with histopathologic gland damage [16]. Other combinations, such as immunoglobulin G (IgG) antibodies targeting formin-binding protein 4 (FNBP4), small nuclear ribonucleoprotein polypeptide C (SNRPC), and C-C motif chemokine ligand 4 (CCL4), or IgA antibodies against small ubiquitin-like modifier 2 (SUMO2) and 2′-5′-oligoadenylate synthetase 3 (OAS3), have shown specificities of up to 97% in pSS cohorts with early-stage or systemic disease [15]. Concurrently, genetic and epigenetic markers, including interferon-induced protein 44-like (IFI44L) hypomethylation and gene panels comprising hes family bHLH transcription factor 4 (HES4), otoferlin (OTOF), tetratricopeptide repeat domain 21A (TTC21A), and zinc finger CCHC-type containing 2 (ZCCHC2), have also shown potential, especially in ethnically diverse and seronegative populations [17, 18]. However, several challenges remain, including the need for standardized protocols, validation across independent cohorts, and careful interpretation of biomarker heterogeneity in relation to clinical phenotypes [19]. This review synthesizes recent advances in molecular biomarker research for pSS, with a focus on compartment-specific discoveries spanning serum autoantibodies, circulating cytokines and chemokines, salivary and tear proteomics, and transcriptomic, epigenetic, and metabolomic signatures. By aligning these emerging biomarkers with clinical presentation and disease course, we aim to highlight how a precision-medicine framework can improve early diagnosis, risk stratification, and personalized therapeutic decision-making in this complex autoimmune disorder.
pSS is a complex autoimmune disorder that is triggered in genetically predisposed individuals by environmental factors such as viral infections. It involves a coordinated interaction between epithelial cells and both the innate and adaptive immune systems (Fig. 1). Rather than just being passive targets, salivary gland epithelial cells (SGECs) actively initiate the immune response by expressing immunomodulatory features, including toll-like receptors (TLRs), major histocompatibility complex (MHC) molecules, and co-stimulatory ligands.
In the early stages of pSS, the salivary glands are primarily infiltrated by CD4+ T cells. As the condition becomes chronic, B cells become more prominent, forming clusters that resemble ectopic germinal centers (GCs). A key factor in the reduced saliva production and inflammation is dysfunction of the epithelial barrier, in particular, the tight junctions (TJs) and ion channels (Fig. 1A). The breakdown of this barrier exposes autoantigens, like Ro/SSA and La/SSB, which in turn drive a continuous autoimmune response [20, 21, 22].
Plasmacytoid dendritic cells (pDCs) accumulate in inflamed salivary tissue and are a major source of type I interferon (IFN-I), contributing to a persistent “interferon signature” that is detectable both locally and systemically [23]. These IFNs enhance the production of B-cell survival mediators such as B-cell activating factor (BAFF) and drive the recruitment of lymphocytes via chemokine signaling, thereby linking innate immune sensing with adaptive immune activation (Fig. 1B). Type I IFNs activate the downstream Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, which plays a pivotal role in this process and has emerged as a therapeutic target in ongoing clinical trials [23, 24].
Diverse T cell subsets contribute to pSS, with Th1 and Th17 cells driving early
inflammation via interferon gamma (IFN-
BAFF is secreted by epithelial and immune cells and sustains autoreactive B-cell survival (Fig. 1B). This leads to hypergammaglobulinemia, RF production, and the generation of autoantibodies that enter the systemic circulation (Fig. 1C), including anti-SSA/SSB and emerging markers (anti-SP-1, anti-CA6), some of which appear years before clinical onset [26]. Ectopic GCs within glands also increase the risk of lymphoma, especially mucosa-associated lymphoid tissue (MALT) lymphomas [27, 28].
Genetic susceptibility, including variants in human leukocyte antigen (HLA)-DR, interferon regulatory factor 5 (IRF5), and STAT4, can synergize with viral exposures such as Epstein-Barr virus (EBV) or cytomegalovirus (CMV) to initiate the loss of immune tolerance. Epigenetic alterations, such as abnormal deoxyribonucleic acid methylation (DNAm) and dysregulated micro ribonucleic acid (miRNA) profiles, amplify proinflammatory pathways. Moreover, circulating exosomal miRNAs (miR-142-3p, miR-1290) are emerging as both mechanistic indicators and promising non-invasive biomarkers [29, 30, 31].
Early detection of pSS remains a major challenge, particularly in seronegative
individuals or those without classic extra-glandular involvement. Traditional
markers such as anti-SSA/SSB antibodies often appear later in the disease course,
and are entirely absent in up to 30%–50% of patients, necessitating more
sensitive and non-invasive tools for early-stage recognition [16]. Early
epithelial damage and immune infiltration in the salivary glands initiate
molecular changes, such as the release of
Several novel autoantibodies have demonstrated value in detecting pSS prior to traditional anti-SSA/SSB seroconversion. Antibodies targeting SP-1, PSP, and CA6 appear earlier in disease and may complement or substitute the classical serologies, especially in seronegative individuals [32]. Together, these autoantibodies form a promising panel for identifying early-stage SS, offering the ability to detect disease onset prior to traditional serologic conversion [33, 34]. Anti-CA6 correlates more closely with ocular involvement, while anti-SP-1 and PSP better reflect salivary dysfunction [33, 35]. These antibodies are especially valuable in the screening of younger patients or those with subtle glandular symptoms.
Early in pSS, immune-mediated changes disrupt the epithelial and acinar cell
function of the salivary glands, leading to altered secretion before the
occurrence of significant tissue destruction. Key early markers include increased
levels of innate immune proteins (such as lactoferrin), changes in mucin
glycosylation, and elevated electrolyte concentrations, particularly sodium and
chloride, reflecting both epithelial channel dysfunction and ongoing inflammation
[36, 37]. Importantly, even in morphologically intact glands, acinar cells from
pSS patients can show impaired calcium signaling due to deficits in type 3
inositol 1,4,5-triphosphate (IP3) receptors, leading to reduced fluid secretion
and explaining why secretory dysfunction can precede overt glandular damage [38].
These molecular and functional alterations often manifest before measurable
decreases in saliva flow, making them valuable indicators for pre-clinical or
early diagnosis. Salivary analysis offers a direct window into glandular immune
activity.
High-throughput proteomic studies have transformed early detection and risk stratification in pSS by identifying multi-protein biomarker panels with robust diagnostic performance. A validated salivary panel consisting of complement factor B (CFB), clusterin (CLU), and neutrophil elastase (NE) has shown excellent accuracy with an area under the curve (AUC) up to 0.93 in distinguishing pSS from controls, particularly when combined with anti-SSA serology and Schirmer’s test [42]. Each protein reflects a distinct pathological axis: CFB is associated with complement activation, CLU with tissue stress and cytoprotection, and NE with neutrophil-mediated inflammation, providing a multidimensional view of early glandular injury [43, 44]. Saliva-based proteomic profiles also differentiate pSS from non-autoimmune sicca and healthy individuals, typically showing upregulation of inflammatory proteins and downregulation of secretory components [43, 44]. In addition, a proteomic study of extracellular vesicles (EVs) from saliva and plasma has identified disease-specific proteins, such as members of the S100 family and neutrophil-related markers, thereby enhancing diagnostic accuracy and enabling potential stratification [45]. Tear fluid proteomics further supports non-invasive screening, with biomarkers such as matrix metalloproteinase-9 (MMP-9) and lipocalin-1 showing high sensitivity and specificity for pSS diagnosis [46].
Salivary metabolomics has revealed early biochemical disturbances in pSS, providing sensitive indicators of subclinical immune activation. Among these, dipeptides such as phenylalanyl-alanine indicate altered amino acid and tryptophan metabolism, consistent with systemic inflammation and metabolic reprogramming during early disease [47]. Other salivary metabolites, such as tryptophyl-isoleucine and oxidative stress markers like 8-hydroxyadenine, can further distinguish pSS from non-autoimmune sicca, highlighting the role of altered amino acid and oxidative pathways in early disease [47]. Broader metabolomic profiling confirms that pathways including tryptophan, tyrosine, and aspartate metabolism are significantly altered in pSS, correlating with inflammatory injury and immune response [47, 48]. In labial salivary gland tissue, a metabolomic signature that combines kynurenine (an immune-metabolite linked to anti-SSA autoimmunity) with specific phospholipids (reflecting lymphocytic infiltration) shows robust differentiation of early pSS from other sicca causes [48]. These findings underscore the value of metabolomic markers, especially those linked to immune cell metabolism and oxidative stress, as non-invasive tools for early detection and risk flagging in SS.
Exosome-derived miRNAs in saliva and glandular tissues are emerging as promising, non-invasive biomarkers for the early detection and screening of pSS. Panels including miRNAs such as let-7b-5p and miR-1290 have emerged as non-invasive biomarkers that mirror early epithelial stress and immune cell activation, supporting their potential role in preclinical disease detection [49]. Multiple studies have confirmed that miRNA expression is significantly altered in pSS across various tissues and fluids, including saliva, tears, conjunctival cells, and immune cell subsets [50, 51]. Key miRNAs, such as let-7 family members, miR-146a, miR-155, miR-181a, miR-30b-5p, and miR-130a, are consistently deregulated in pSS and target pathways involved in immune regulation, inflammation, and glandular damage [52]. For example, miR-146a and miR-155 are upregulated in peripheral blood and glandular tissues, correlating with disease activity and inflammatory cytokine production [52]. Downregulation of miR-130a and miR-708 in DCs and glandular tissues is associated with increased cell activation and the release of pro-inflammatory cytokines [53]. These miRNAs are stable, easily detected in exosomes, and reflect the underlying pathophysiology of early glandular inflammation, making them attractive candidates for early screening and risk flagging in clinical practice.
Approximately one-third of pSS patients present without detectable anti-Ro/SSA or anti-La/SSB autoantibodies. This subgroup is commonly referred to as “seronegative”, with the patients often facing diagnostic delays or misclassification due to the absence of conventional markers and the variable presentation of sicca symptoms, fatigue, or neuropathic features [16, 42]. In the absence of anti-SSA/SSB, seronegative patients often exhibit local antibody responses, such as anti-M3 muscarinic acetylcholine receptor (M3R) or anti-calreticulin (CALR). These are produced within ectopic GCs forming at sites of epithelial-immune crosstalk (Fig. 1A).
A study has revealed that anti-DTD2 and anti-RESF1 autoantibodies have been identified in seronegative pSS patients, where they strongly associate with histopathologic evidence of glandular infiltration, highlighting their potential to complement tissue-based diagnosis [16]. These autoantibodies correlate with lymphocytic infiltration in the labial gland, thus potentially substituting for histological confirmation in selected cases. Similarly, auto-antibodies against CALR, identified in both seropositive and seronegative patients, target SGECs and are linked to elevated inflammatory markers (erythrocyte sedimentation rate, IgG subclasses, fibrinogen) and systemic features like weight loss and vasculitis [42]. The local expression of CALR correlates with immune cell infiltration, making it both a diagnostic and a mechanistic marker.
Glandular autoantibodies such as anti-M3R and anti-SP-1/PSP/CA6 are particularly useful in seronegative individuals. They are frequently found in younger or early-stage patients prior to the appearance of SSA/SSB, and correlate with glandular dysfunction such as reduced saliva flow and elevated focus score [32, 54, 55]. For example, although anti-M3R IgG is not part of the classification criteria, it is negatively correlated with salivary flow rates and positively correlated with histopathological focus scores, indicating more severe glandular inflammation and acinar damage in antibody-positive patients [56].
Salivary exosome-derived miRNAs are highly stable and accessible in seronegative patients. These miRNAs likely reflect local immune activation and epithelial stress independent of systemic seropositivity, offering an orthogonal diagnostic axis. Beyond miRNAs, long non-coding RNAs (CTA-250D10.23, GABPB1-AS1) and circular RNAs (has_circ_001264) also show promise in stratifying seronegative cases. These RNAs demonstrate differential expression profiles in glandular tissue and blood, correlating with C-X-C motif chemokine ligand 13 (CXCL13) levels and B-cell hyperactivity [57, 58, 59, 60].
Despite negative serology results, seronegative pSS patients often show
proteomic and metabolic shifts. For example, a diagnostic panel of CFB, CLU, and
NE performs well in early-stage and seronegative pSS [42]. Metabolomic studies
further identify kynurenine and phenylalanyl-alanine as potential indicators of
immune dysregulation, with the former reflecting IFN-
The clinical course of pSS varies widely, from glandular-limited disease to systemic complications and lymphoproliferative disorders. Prognostic biomarkers have therefore become essential for stratifying patients by disease severity, extra-glandular involvement, and risk of progression. The chemokine CXCL13 is consistently associated with higher scores in the EULAR Sjögren’s syndrome disease activity index (ESSDAI), systemic manifestations, and the development of MALT lymphoma. Its overexpression in salivary glands and elevated serum levels correlate with GC-like structures and lymphoid infiltration. A serum cutoff of 123.45 pg/mL for CXCL13 has been suggested to identify patients at increased risk of lymphoma [62].
The IFN-I axis remains a dominant pathway in pSS pathogenesis. Patients with a high interferon-stimulated gene (ISG) signature exhibit more active systemic disease, increased autoantibody titers, and reduced response to B-cell-targeted therapies [63, 64, 65]. Corresponding epigenetic features, such as hypomethylation at IFN-responsive loci [e.g., IFI44L, interferon-induced protein with tetratricopeptide repeats 1 (IFIT1)], serve as stable indicators of heightened immune activation. A DNAm-based IFN score identifies pSS patients with earlier onset, high serologic activity, and increased lymphoma susceptibility [17, 66].
BAFF is another critical molecule linked to disease progression. Elevated BAFF expression in serum, salivary glands, and epithelial cells reflects chronic immune stimulation and correlates with autoantibody positivity and systemic inflammation [67, 68]. Clinical trials of BAFF blockade have shown promising results, with baseline BAFF levels predicting therapeutic response [69]. Elevated CXCL13 and BAFF levels reflect the persistence of organized lymphoid structures within glandular tissues, which serve as niches for lymphoproliferative risk (Fig. 1B) and may contribute to extra-glandular dissemination via peripheral immune cell trafficking (Fig. 1C).
Serological markers other than SSA/SSB are increasingly being used for phenotype
stratification. Anti-IFI16 correlates with high focus scores, hyperglobulinemia,
and GC formation [70], while anti-cofilin-1, anti-
Recent advances in high-throughput profiling technologies have enabled a more holistic approach to biomarker discovery in primary pSS, moving beyond isolated molecular features toward integrated, multi-parameter models. Multi-omics analyses now allow integration of biomarkers across compartments, linking local epithelial stress (salivary proteomics) (Fig. 1A), immune gene expression patterns in affected tissue (Fig. 1B), and circulating inflammatory profiles (Fig. 1C).
Transcriptomic profiling has revealed reproducible immune expression patterns in pSS, most notably the enrichment of type I ISG CLUs. Stratification based on IFN-high versus IFN-low expression identifies patients with distinct immunological activity, systemic features, and differential treatment response [72, 73]. The clinical relevance of this signature is further enhanced by the integration of DNAm data, particularly hypomethylation at IFN-responsive loci such as IFI44L and IFIT1. This reflects sustained pathway activation and correlates with systemic manifestations and lymphoma risk [17, 66]. Combinatorial models have emerged as a next-generation diagnostic approach.
Similarly, metabolomic signatures derived from saliva and gland tissue have
demonstrated potential for composite diagnostics. The tryptophan-kynurenine
pathway, a marker of IFN-
The pursuit of novel biomarkers in pSS continues to evolve toward frontier technologies and exploratory targets that extend beyond conventional serology and transcriptomics. Novel biomarkers such as exosomal RNAs and tear-derived cytokine panels are emerging as indicators of local epithelial stress and immune activation (Fig. 1B), while animal models faithfully replicate these processes in early glandular lesions (Fig. 1A).
Exosomes are nanoscale extracellular vesicles that carry proteins, lipids, and nucleic acids. They are increasingly recognized as being stable, cell-specific reporters of disease activity. In pSS, salivary- and serum-derived exosomes have been found to contain distinct proteomic and RNA cargos, reflecting epithelial stress, B-cell activation, and immune dysregulation [75, 76]. Proteomic analyses have revealed enrichment of epithelial-derived proteins associated with ferroptosis, while exosomal miRNAs (e.g., let-7b-5p) and circular RNAs (e.g., has_circ_001264) demonstrate diagnostic specificity and correlation with immune cell infiltration [49, 58]. Exosomes are accessible via non-invasive sampling and remain stable under standard storage conditions, making them attractive for future point-of-care diagnostics.
The tear film represents another promising matrix for biomarker discovery,
particularly in ocular-dominant phenotypes of pSS. Beyond classical tests like
Schirmer’s test and ocular staining, multiplex tear proteomics has revealed a
constellation of upregulated cytokines (IL-1
Insights from animal models continue to inform studies on early-phase biomarker
identification. In IL14
Transcriptomic evidence highlights the extensive immune dysregulation lurking
beneath glandular-only symptoms. In a multi-omic analysis of
Beyond transcriptomes, proteomic and cellular data confirm that sicca-dominant patients often mount the same immune responses that are classically associated with systemic involvement. For example, elevated serum levels of IFN-induced chemokines, such as CXCL10/interferon gamma-induced protein 10 (IP-10) and CXCL11, and antiviral proteins like MX1 are detectable in many glandular-only pSS cases [84]. These circulating biomarkers are tightly correlated with salivary gland IFN activity and lymphocytic infiltration, providing a window into “hidden” glandular inflammation [84]. B-cell hyperactivity is another hallmark, and patients with pure sicca symptoms frequently exhibit high titers of RF or anti-Ro/La antibodies, reflecting an ongoing adaptive immune response. BAFF, a cytokine crucial for B-cell survival, is significantly elevated in the blood and salivary tissue of pSS patients, even in the absence of systemic lesions [85, 86]. BAFF levels correlate with autoantibody titers and global disease activity, indicating that clinical dryness does not equate to immunological calm. On the contrary, patients with the highest BAFF concentrations show exuberant B-cell activation and may even develop lymphoid follicles in their salivary glands [85, 87]. Such ectopic GC-like structures can arise in so-called “limited” disease and are known precursors to lymphoma, a serious systemic complication [85]. Type I and II IFNs themselves consistently drive BAFF production by monocytes and glandular cells [86, 88, 89], creating a feed-forward loop of B-cell stimulation even in patients without clinically obvious extra-glandular symptoms.
Recent epigenetic data further substantiate the presence of immunopathological changes from the onset of disease, irrespective of phenotype. DNA methylation profiling of blood from pSS patients identified IFN-regulated genes (IFI44L, MX1, IFIT1) that were hypomethylated and therefore presumably transcriptionally poised. This was observed across different patient subgroups, including those with predominantly sicca manifestations [17, 74]. Such epigenetic alterations were not seen in healthy controls, thus reinforcing the notion of an intrinsic “ready-to-activate” state of the immune system in pSS patients generally. In essence, the molecular groundwork for systemic autoimmunity is laid even in glandular-focused disease. The traditional dichotomy of “mild sicca” versus “severe systemic” is blurred in light of findings that IFN pathway genes show a similar hypomethylation status in patients with only dry eyes as in those with multi-organ involvement [74]. As a result, two individuals with similar tear and saliva gland symptoms might be at opposite ends of a molecular activity spectrum: one with minimal immune perturbation, and the other with high IFN-BAFF signaling. This unseen heterogeneity is masked by the overlapping phenotypes. Fig. 1 illustrates the concept further. Sex-based clinical differences and multisystem manifestations can intersect in complex ways, underscoring that outward phenotype alone (male with female, or sicca-only with systemic) often fails to predict the underlying immune landscape.
Collectively, these insights recast “glandular-limited” pSS as part of a
trajectory of pathogenic activity rather than as a benign or interim stage. In
fact, longitudinal data show that most pSS patients will eventually develop
extra-glandular complications over time [85]. The magnitude of subclinical immune
activation may be what differentiates patients who progress from those who remain
sicca-only. For example, a high IFN gene signature in blood is associated with
greater disease activity and higher odds of systemic features (vasculitis,
arthritis), whereas patients who lack this signature tend to have more restricted
disease [85]. Intriguingly, a predominant type II IFN (IFN-
PSS exhibits one of the strongest female predispositions among autoimmune diseases, with ~90% of patients being women [79, 92]. This profound sex bias influences not only the disease prevalence but also clinical patterns, with female patients more often developing comorbid thyroid autoimmunity and chronic pain syndromes. Although fewer in number, male pSS patients have higher rates of severe extra-glandular complications such as ILD and lymphoma [92]. As shown in Fig. 2, these differences underscore how sex-based factors fundamentally shape the pSS immune landscape, warranting their consideration in prognostic stratification. Indeed, male sex is associated with elevated lymphoma risk, especially in the presence of biomarkers like RF and cryoglobulins, whereas females tend to mount stronger B-cell autoimmune responses [92]. The mechanistic underpinnings for this “molecular immune switch” between the sexes involve an interplay of genetic (sex chromosome) dosage, hormonal milieu, and epigenetic regulation.
The X chromosome carries numerous immune-regulatory genes that are relevant to pSS, including TLR7, CD40 ligand (CD40LG), forkhead box P3 (FOXP3), and interleukin 2 receptor subunit gamma (IL2RG) [93]. A key genetic distinction is that females possess two X chromosomes, one of which is normally inactivated, while males have only one. Certain X-linked genes can escape X-inactivation, resulting in biallelic expression in females [94, 95]. This includes TLR7 (encoding Toll-like receptor 7 at Xp22) and CD40LG, both of which are crucial for antiviral and B-cell signaling. TLR7 overexpression can amplify TLR pathways and downstream IFN-I production, a signature immune pathway in pSS [93, 94, 95]. Female pSS patients have higher TLR7 expression and TLR7 hypomethylation in their immune cells compared to men, correlating with elevated IFN-I activity [94, 95]. The net effect is a “double dose” of pro-autoimmune gene expression in females when X-linked immune genes are variably expressed from both X copies [96]. Epidemiological data support this gene-dosage model: women with an extra X chromosome (47, XXX) have about a 2.5-3-fold higher prevalence of pSS than 46, XX women, and an estimated 40-fold greater risk than 46, XY men. In other words, each additional X copy confers substantial autoimmune susceptibility. This X-linked dosage effect appears to be a stronger risk factor for pSS than any single-gene polymorphism, accounting for the marked female predominance of the disease [97]. Notably, a similar enrichment of 47, XXX karyotypes is seen in systemic lupus, reinforcing the paradigm that X chromosome “load” is directly tied to autoimmune risk [90, 98]. X-linked genes that escape inactivation [CD40LG, C-X-C motif chemokine receptor 3 (CXCR3), IL2RG] are significantly hypomethylated and overexpressed in the salivary gland tissue of pSS patients [23], suggesting that both genetic dose and epigenetic derepression of the X chromosome contribute to female-biased immune activation.
Sex hormones further tilt the immune balance, with estrogens generally promoting
humoral immunity. For example, 17
Beyond DNA sequence, sex differences in epigenetic regulation can affect the immune dysregulation observed in pSS. Genome-wide methylation studies have revealed that pSS patients exhibit significant DNA hypomethylation at numerous immune genes relative to healthy controls [23, 99]. Notably, many of these demethylated loci are ISGs and X-linked genes, implicating both the IFN pathway and the X chromosome in disease pathogenesis [30]. Hypomethylation leads to overexpression of the corresponding genes, effectively acting as an epigenetic amplifier of immune activation. For instance, the prominent ISG IFI44L is significantly upregulated in the B cells of pSS patients, and its promoter is concurrently hypomethylated [17]. Higher IFI44L expression (and lower methylation) correlates with disease activity indices and immunoglobulin levels, highlighting IFI44L as both a molecular effector and a candidate biomarker of the pSS IFN signature. More broadly, an “IFN methylation score”, based on the degree of demethylation of IFN-regulated genes, has been used to define patient subsets in pSS [30]. Patients with the most severe systemic disease show pronounced hypomethylation of IFN genes and CLU in epigenetic analyses [23]. Intriguingly, several X-linked immune genes, such as CD40LG, IL2RG, and Wiskott-Aldrich syndrome (WAS), are among those hypomethylated in pSS salivary glands [23], suggesting the convergence of sex chromosome effects and epigenetics. Female-biased factors such as X-escape and estrogen can influence epigenetic marks. For example, estrogen receptor signaling recruits chromatin-modifying enzymes that induce DNA demethylation at specific promoters. Lifelong exposure to estrogens, in tandem with genetic X-dose effects, is likely to skew the epigenome of immune cells in females toward an autoimmune-prone state.
PSS exhibits striking heterogeneity, prompting efforts to classify patients into clinical CLUs based on symptomatology and immune endotypes based on underlying molecular profiles. Clinical CLUs identified by data-driven analyses often distinguish patients with predominant sicca symptoms (dryness with fatigue/pain) from those with significant systemic involvement [108]. For example, a large international cohort study defined four symptom-based CLUs: low symptom burden, dryness-dominant with low pain/fatigue, dryness with high pain, and high symptom burden (HSB) across all domains [108]. Notably, patients in the HSB CLU reported severe fatigue and pain, but paradoxically had milder objective disease (fewer organ complications, normal lab markers) than other CLUs [108, 109]. Therefore, self-reported symptom severity does not always mirror immunological activity. In fact, HSB patients are often heavily treated with immunosuppressants, despite the absence of high levels of serologic or inflammatory markers, highlighting a divergence between symptomatic CLUs and classical immune features [108].
In contrast, immune endotyping of pSS has revealed subgroups defined by
molecular drivers such as IFN-I signaling, B-cell activation, and epigenetic
modifications. Roughly half of pSS patients demonstrate a prominent IFN-regulated
gene signature (“IFN-high” endotype) characterized by upregulation of ISGs in
blood and tissue [110]. The IFN-high endotype correlates strongly with
seropositivity (anti-SSA/SSB antibodies) and higher systemic disease activity
[110, 111], aligning with clinically “active” disease CLUs. Indeed, a recent
multi-cohort study integrating transcriptomes found that circulating
IFN-
Several high-profile analyses support this interplay. McCoy et al. [108] observed that symptom-based CLUs with high pain/fatigue did not correlate with elevated inflammatory markers, emphasizing a “neuro-immune” or nociceptive dimension to pSS that is distinct from classic autoimmune endotypes. In contrast, molecular CLU of pSS patients by multi-omics data (transcriptomics and DNAm) yielded four robust endotypes, of which one had minimal immune activation, and the others were defined by strong IFN-driven inflammation, T cell/B cell activation, and cytokine excess [115, 116]. Interestingly, the clinically mild “seronegative” patients likely populate the low-immune CLU, whereas patients with high IFN scores, lymphopenia, and hypergammaglobulinemia populate the immune-active CLUs [117]. Thus, partial alignment can be found when considering clinical CLUs versus immune endotypes. Groups with systemic complications generally exhibit the corresponding immune signatures (IFN, BAFF/CXCL13, etc.), whereas those defined by patient-reported sicca symptoms or fatigue may diverge, reflecting mechanisms (neuroimmune pain, glandular dysfunction) that are not captured by standard immune biomarkers [108].
The pattern of organ involvement in pSS, or organ tropism, often serves as a clinical proxy for the dominantly active immune axis. In essence, the specific organs targeted by pSS can reflect how the immune system is misfiring. Patients with disease confined to exocrine glands (salivary and lacrimal) and presenting primarily with sicca symptoms tend to have a more localized autoimmune response. In contrast, patients with extra-glandular manifestations (arthritis, vasculitis, lung or kidney involvement) usually harbor a broader systemic immune activation [110]. For example, systemic vasculitic features such as palpable purpura, skin ulcers, or glomerulonephritis almost invariably indicate the presence of circulating immune complexes (RF-IgG complexes), cryoglobulins, and hypocomplementemia [118, 119, 120], pointing to a B-cell-driven immune complex axis. Accordingly, patients with cutaneous vasculitis in pSS are often those with high titers of RF, hypergammaglobulinemia, elevated BAFF levels, and an IFN-I gene signature that perpetuates B-cell activation [110]. Clinically, this corresponds to the classic “high-risk” pSS phenotype that is prone to systemic damage and lymphoma. Indeed, retrospective analyses have shown that individuals who develop lymphoma often have a history of organ-specific complications such as vasculitis, parotid gland enlargement, or lymphadenopathy, together with serological hallmarks of B-cell hyperactivity [anti-Ro/SSA, low complement component 4 (C4), etc.] [114].
Conversely, patients whose organ tropism is limited to the glands (salivary/lacrimal) and have no major systemic involvement often lack those immune signatures. These sicca-dominant patients may be seronegative and have normal complement levels, suggesting only modest engagement of the adaptive immune axis. Their pathology may be driven more by local tissue-specific factors (glandular epithelial stress and local T/B cell foci) than systemic IFN surges [114]. This concept is supported by epigenetic data. For instance, patients with no systemic manifestations exhibit higher DNAm (quiescence) at IFN-regulated gene loci such as IFI44L and IFIT1, whereas those with multisystem disease show pronounced hypomethylation (activation) at these loci in immune cells [17]. In other words, the lack of organ spread correlates with a silenced IFN axis. Clinicians intuitively use organ tropism as a guide. PSS patients who present solely with dry eyes and mouth (no arthritis, no vasculitis) are often thought to have a more benign immunological profile, whereas those with lung lesions or neuropathy raise suspicion of an “immune-high” state needing systemic therapy.
Specific organ involvement can map to particular immune pathways. For example,
arthritis in pSS tends to be associated with higher IL-21 levels and a skewing of
Tfh-B cell interactions, analogous to rheumatoid-like inflammation of joints
[113]. A 2021 study found that pSS patients with active synovitis had
significantly elevated IL-21 and IL-22 levels, correlating with altered B and T
cell subsets (lower memory B cells, higher CD4+ T cell activation) [113]. This
suggests the Th17/Tfh axis drives articular disease. In contrast, pulmonary
involvement (such as ILD) in pSS often co-occurs with high titers of SSA/Ro and
RF, implicating the immune-complex/IFN axis. Interestingly, the
IL-14
Sex-based differences in organ tropism also reflect underlying immune axis
biases. Recent research has identified TLR7, an X-linked innate sensor, as a key
driver of female-biased manifestations in pSS [122]. In a murine model of pSS,
female mice exhibited more severe nephritis and hypergammaglobulinemia than male
mice, whereas the males had more dacryoadenitis (lacrimal gland inflammation)
[122]. Knocking out TLR7 protected females from systemic inflammation
and reduced renal disease and autoantibodies, but intriguingly led to worse
glandular disease in males [122]. This indicates that TLR7-driven IFN signaling
disproportionately favors certain organ-specific diseases in females (kidney,
systemic autoimmunity), again linking an organ (kidney) with an immune axis
(TLR7-IFN
As our understanding in this field deepens, pSS phenotypes are starting to be redefined not just by their clinical presentation, but also by the biomarker profiles that drive them. The ultimate goal is to integrate serologic, genomic, and molecular markers into the clinical taxonomy of pSS to yield biomarker-defined phenotypes that can guide personalized therapy. This approach stems from the recognition that traditional phenotypes (sicca-only versus systemic pSS) are quite broad and can miss critical distinctions in the underlying immunopathology [108]. For instance, two patients may both have “high systemic activity” clinically, but one might be IFN-high and the other IFN-low, with very different responses to therapy [110]. The incorporation of biomarkers such as the IFN-I signature, BAFF levels, or unique epigenetic marks is likely to resolve such differences.
A prime example is the IFN-I signature. Rather than treating pSS as one disease,
investigators now stratify patients by IFN status, leading to novel insights in
the research context. Trutschel et al. [110] showed that an
IFN-
Beyond individual markers, composite biomarker scores are emerging to capture the multidimensional immune landscape of pSS. One promising tool is the DNAm-based IFN score, which assesses epigenetic activation of the IFN pathway. In a recent study, the DNAm IFN score was calculated from the methylation level of several ISGs [radical S-adenosyl methionine domain-containing 2 (RSAD2), IFIT1, IFI44L]. This was elevated in 59% of pSS patients and was strongly associated with earlier disease onset, seropositivity (Ro/La antibodies), hypergammaglobulinemia, low C4, and the likelihood of concurrent or future lymphoma [66]. Importantly, this epigenetic score provides a stable, blood-based biomarker that can define a clinically relevant phenotype. Patients with a high DNAm IFN score represent the classic, high-risk immune-active phenotype, whereas those with a low DNAm IFN score belong to the more benign phenotypes. The authors propose the adoption of such an IFN score for patient stratification in trials and for monitoring disease activity [66]. Along similar lines, high-throughput proteomics and machine learning are being harnessed to generate multivariate biomarker panels. Berry et al. [124] identified distinct serum protein networks corresponding to clinical subtypes of pSS. By overlaying proteomic data on their previously defined clinical CLUs, they could pinpoint which pathways were enriched in each subtype [e.g., IL-6 signaling, metabolic stress regulators such as Forkhead box protein O1 (FOXO1) and BTB domain and CNC homolog 1 (BACH1)] [124]. Strikingly, when they re-analyzed a clinical trial of tocilizumab (IL-6 blockade) by stratifying patients according to these biomarker-defined subtypes, only the subset with the strongest IL-6 signature showed significant improvement in fatigue [124]. Had the trial been restricted to IL-6-high patients, the therapy may have been found to be effective for that particular phenotype, highlighting the power of biomarker-defined phenotyping.
Another frontier is the incorporation of novel biomarkers such as
IL-14
Composite endpoints and phenotypic classifications are being refined to ensure that clinical trials enroll more homogeneous patient subsets [123]. In this regard, the Composite of Relevant Endpoints for Sjögren’s Syndrome proposal incorporates both patient-reported and biological measures. Ultimately, the aim is that pSS patient care will be guided by a biomarker panel. For example, if Patient A is “IFN-high, BAFF-high, epithelial autoantibody-positive”, indicating an IFN-I/B-cell axis phenotype, they will be treated with an IFN inhibitor or B-cell therapy. If Patient B is “seronegative, IFN-low, high pain”, indicating a neuropathic sicca phenotype, they will be treated with symptomatic and neuro-modulatory therapies, rather than aggressive immunosuppression. Even within classic phenotypes like “SSA-positive” pSS, additional biomarkers such as specific DNAm signatures or CXCL13 levels may further define whether the patient is on a trajectory toward lymphoma or remains a benign variant [66].
Taken together, these advances highlight a paradigm shift in which pSS can no longer be adequately described by glandular versus systemic phenotypes alone. Instead, distinct immunobiological endotypes are emerging, each defined by a dominant pathway and a characteristic biomarker profile. By aligning patients with endotypes such as IFN-high, B-cell activation-high, or metabolic dysregulation, researchers can move beyond symptom clusters and toward mechanistic subgroups [63, 74, 124]. Table 1 (Ref. [17, 48, 61, 63, 73, 74, 84, 110, 112, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141]) summarizes the current evidence by presenting the major endotypes, their key biomarkers, and the typical clinical outcomes.
| Endotypes | Core biomarkers | Clinical outcomes |
| IFN-high | IFI44, MX1 [73], IFI44L [17], IFI27 [126], Siglec-1 [127], CXCL10 [128], IFN- |
High ESSDAI, multi-organ activity, seropositive, hyper-IgG, mild cytopenia; |
| B-cell activation-high | CXCL13 [130], BAFF/TNFSF13B [131], IgG, RF, anti-Ro/SSA, anti-La/SSB [84], MZB1 [132], XBP1 [133] | B-cell-driven ESSDAI domains (LN, skin, kidney, neuro); |
| Metabolic dysregulation | Fatigue/pain-predominant phenotype; high symptom burden despite limited ESSDAI discrimination [140, 141] |
IFN, interferon; IFI44, interferon-induced protein 44; MX1,
myxovirus resistance 1; IFI44L, interferon-induced protein 44-like;
IFI27, interferon alpha inducible protein 27; Siglec-1,
sialic-acid-binding Ig-like lectin 1; CXCL, C-X-C motif chemokine ligand;
OAS, 2′-5′-oligoadenylate synthetase; BAFF, B-cell activating factor;
TNFSF13B, tumor necrosis factor superfamily member 13B; IgG,
immunoglobulin G; RF, rheumatoid factor; SSA, Sjögren’s-syndrome-related
antigen A (Ro); SSB, Sjögren’s-syndrome-related antigen B (La);
MZB1, marginal zone B and B1 cell-specific protein; XBP1, X-box
binding protein 1; lysoPC, lysophosphatidylcholine; ESSDAI, EULAR Sjögren’s
Syndrome Disease Activity Index;
Anti-Ro/SSA and anti-La/SSB autoantibodies remain central to SS diagnosis, but
their clinical utility has been refined by advances in biomarker discovery and
testing methodologies [18, 142]. Anti-Ro/SSA generally exhibits higher
sensitivity than anti-La/SSB but slightly lower specificity in the serologic
diagnosis of pSS (Table 2, Ref. [15, 27, 34, 143, 144, 145, 146, 147, 148, 149]). The specimen matrix
primarily affects sensitivity rather than specificity, with saliva testing
markedly reducing anti-SSA/SSB detection compared with serum while maintaining
similarly high specificity. These autoantibodies are often detected by
immunodiffusion or enzyme-linked immunosorbent assay (ELISA) methods [150, 151].
Isolated anti-SSB positivity was recognized as rare (3.6% of cases) and
non-specific, while anti-SSA cross-reacted with systemic lupus erythematosus
(SLE) [152, 153]. RF and antinuclear antibodies (ANAs) were adjunct markers, but
low specificity limited their standalone diagnostic value [150]. The 2016
ACR/EULAR criteria integrated anti-SSA positivity or labial salivary gland biopsy
(focus score
| Markers/Panels | Cohorts/Settings | Sensitivity (%) | Specificity (%) | References | |
| Conventional SSA/SSB Autoantibodies (Traditional Markers) | |||||
| Single-marker panels | Anti-Ro/SSA | pSS (n = 100), controls (n = 140) | 49.0 | 87.5 | [146] |
| pSS (n = 31), non-pSS (n = 64) | 76.7 | 88.7 | [147] | ||
| pSS (n = 238), controls (n = 135) | 48.0–79.0 | 70.0–100 | [148] | ||
| Anti-La/SSB | pSS (n = 100), controls (n = 140) | 29.0 | 95.0 | [146] | |
| pSS (n = 31), non-pSS (n = 64) | 52.4 | 100 | [147] | ||
| pSS (n = 238), controls (n = 135) | 39.0–44.0 | 100 | [148] | ||
| Multi-marker panel | Anti-Ro/SSA + Anti-La/SSB (combination) | JSS (n = 27), non-JSS (n = 78) | 55.6 | 92.2 | [34] |
| Novel Autoantibody Panels (Emerging/Early Sjögren’s Markers) | |||||
| Single-marker panels | Anti-SP-1 IgG | pSS (n = 60), controls (n = 60) | 58.3 | 70.0 | [143] |
| JSS (n = 27), non-JSS (n = 78) | 3.7 | 85.9 | [34] | ||
| Anti-PSP IgG | pSS (n = 60), controls (n = 60) | 75.0 | 63.3 | [143] | |
| JSS (n = 27), non-JSS (n = 78) | 14.8 | 82.1 | [34] | ||
| Anti-CA6 IgG | JSS (n = 27), non-JSS (n = 78) | 37.0 | 68.0 | [34] | |
| pSS (n = 60), controls (n = 60) | 32.0 | 71.7 | [143] | ||
| Anti-cofilin-1 | pSS (n = 50), pSS/MALT (n = 20), controls (n = 50) | 76.0 | 82.0 | [27] | |
| Anti- |
82.0 | 74.0 | |||
| Anti-RGI2 | 84.0 | 74.0 | |||
| Multi-marker panels | Anti-SP-1+Anti-PSP (combination) | pSS (n = 60), controls (n = 60) | 84.6 | 100 | [143] |
| eSjA (anti-SP-1, CA6, PSP) | JSS (n = 27), non-JSS (n = 78) | 3.7 | 85.9 | [34] | |
| The Sjö® test (anti-SP-1, PSP, CA6, ANA, RF) | Confirmed SS (n = 267), controls (n = 189) | 91.4 | 79.8 | [145] | |
| The Sjö® test (anti-SP-1, PSP, CA6, Ro/SSA, La/SSB, ANA, RF) | Confirmed SS (n = 267), controls (n = 189) | 91.4 | 79.8 | ||
| Confirmed SS (n = 248), controls (n = 143) | 89.9 | 78.7 | [144] | ||
| 3-marker panel (anti-FNBP4, SNRPC, CCL4) | Ro/SSA– SjD (n = 90), HC (n = 118), NSS (n = 44) | 30.0 (Ro/SSA–) | 97.0 (HC), 95.0 (NSS) | [15] | |
| 5-marker panel (anti-FNBP4, SNRPC, CCL4, M3R, KDM6B) | 46.0 (Ro/SSA–) | 95.0 (HC), 84.0 (NSS) | |||
| 8-marker panel (anti-BTBD7, CCL4, M5, HNRNPA1, KDM6B, TMPO, TONSL, and OAS3) | pSS (n = 347), non-pSS (n = 136), controls (n = 123) | 73.0 | 94.0 | [149] | |
| Anti-cofilin-1+Anti- |
pSS (n = 50), pSS/MALT (n = 20), controls (n = 50) | 86.0 | 93.0 | [27] | |
pSS, primary Sjögren’s syndrome; SSA, Sjögren’s-syndrome-related antigen A (Ro); SSB, Sjögren’s-syndrome-related antigen B (La); JSS, juvenile Sjögren’s syndrome; SP-1, salivary protein 1; PSP, parotid secretory protein; CA6, carbonic anhydrase VI; IgG, immunoglobulin G; RGI2, Rho GDP-dissociation inhibitor 2; MALT, mucosa-associated lymphoid tissue; eSjA, early Sjögren’s syndrome autoantibodies; ANA, antinuclear antibody; RF, rheumatoid factor; HC, healthy controls; NSS, non-Sjögren’s syndrome; Ro/SSA–, anti-Ro/SSA-negative (seronegative); FNBP4, formin-binding protein 4; SNRPC, small nuclear ribonucleoprotein polypeptide C; CCL4, C-C motif chemokine ligand 4; SjD, Sjögren’s disease; M3R, M3 muscarinic acetylcholine receptor; KDM6B, lysine (K)-specific demethylase 6B; BTBD7, BTB domain containing 7; M5, muscarinic acetylcholine receptor subtype 5; HNRNPA1, heterogeneous nuclear ribonucleoprotein A1; TMPO, thymopoietin; TONSL, tonsoku-like; OAS3, 2′-5′-oligoadenylate synthetase 3.
The Sjö test is a commercial diagnostic panel that integrates conventional serology (anti-Ro/SSA, anti-La/SSB, ANA, RF) with early, gland-specific autoantibodies (anti-SP-1, anti-PSP, anti-CA6) to enhance early detection of Sjögren’s disease in routine practice. In adult cohorts, Beckman et al. [144] reported a cumulative sensitivity of 89.9% and specificity of 78.7% for the complete panel, establishing robust performance beyond traditional markers alone. Independent confirmation from the EULAR 2016 dataset demonstrated similar accuracy (sensitivity 91.4% and specificity 79.8%), reinforcing external validity across populations and testing settings. Novel biomarkers alone demonstrated specificity of 83.5% but limited sensitivity of 49.8%; however, when combined with conventional anti-SSA/SSB markers (which demonstrated 74.9% sensitivity and 79.8% specificity), the integrated seven-marker panel achieved superior diagnostic performance with cumulative sensitivity of 91.4% and specificity of 79.8%, demonstrating that the novel biomarkers provide genuine complementary diagnostic value rather than redundant testing [145].
Recent developments in high-throughput proteomics and machine learning-based peptidome analysis have facilitated the identification of more than 45 novel autoantibody targets in pSS [15]. Among these, the panel combining FNBP4, SNRPC, and CCL4 demonstrates excellent specificity for distinguishing seronegative Sjögren’s disease (SjD) from healthy controls (97%) and non-Sjögren’s sicca syndrome (95%), particularly in SSA-negative individuals. A five-marker panel adding anti-M3 and anti-lysine (K)-specific demethylase 6B (KDM6B) increases sensitivity to 46% while maintaining 95% specificity versus healthy controls, though specificity decreases to 84% when differentiating from non-Sjögren’s sicca syndrome [15].
Anti-Ro/SSA and anti-La/SSB autoantibodies remain the serologic backbone for pSS, while novel autoantibodies can enhance detection, particularly in early or seronegative disease, without consistently surpassing the performance and specificity of conventional markers (Table 2). Broad clinical adoption of novel autoantibodies and panels will require standardized assays, harmonized cut-offs, and multicenter validation against biopsy and longitudinal outcomes.
Phenotype stratification and prognosis in pSS increasingly rely on autoantibody profiles, with ongoing transition from traditional markers like anti-Ro/SSA and anti-La/SSB to novel serological biomarkers that delineate glandular, systemic, and high-risk subsets. Fig. 3 presents an overview of the progress to date on biomarker discovery in pSS and the translational relevance.
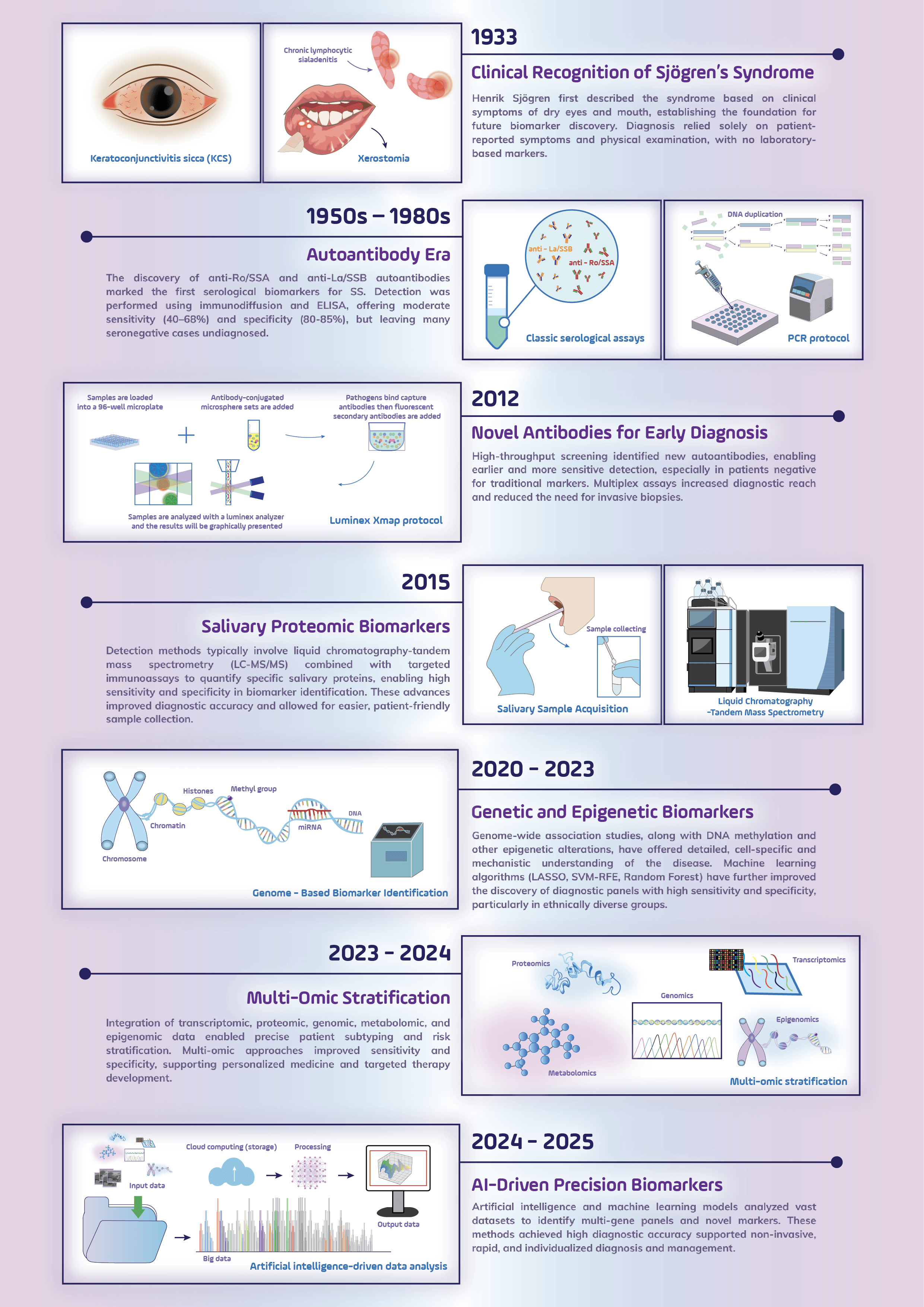 Fig. 3.
Fig. 3.
Evolution of biomarker discovery in Sjögren’s syndrome. Following its clinical recognition in 1933 based on symptoms of dry eyes and mouth, the diagnostic approach to SS has advanced through several key milestones. These include the discovery of autoantibodies (1950s–1980s), novel serologic and salivary protein biomarkers (2012–2015), and the incorporation of genetic and epigenetic insights (2020–2023). Recent integration of multi-omics data (2023–2024) and AI-driven precision models (2024–2025) has enabled earlier, non-invasive, and personalized diagnosis with high accuracy, marking a paradigm shift toward precision medicine. This graphical work was created with Inkscape 1.4.2 (https://inkscape.org). Abbreviations: KCS, keratoconjunctivitis sicca; SS, Sjögren’s syndrome; SSA, Sjögren’s-syndrome-related antigen A (Ro); SSB, Sjögren’s-syndrome-related antigen B (La); ELISA, enzyme-linked immunosorbent assay; DNA, deoxyribonucleic acid; PCR, polymerase chain reaction; LC-MS/MS, liquid chromatography-tandem mass spectrometry; miRNA, micro ribonucleic acid; LASSO, least absolute shrinkage and selection operator; SVM, support vector machines; RFE, recursive feature elimination; AI, artificial intelligence.
Beyond their central role in diagnosis, classic autoantibodies such as anti-Ro/SSA and anti-La/SSB are instrumental in disease stratification in pSS, correlating with severe exocrine gland dysfunction, including reduced salivary flow, higher lymphocytic focus scores, and recurrent parotid swelling [157, 158]. Their presence also reflects systemic B-cell hyperactivity, as evidenced by associations with hypergammaglobulinemia, RF positivity, and increased lymphoma risk [157, 159]. In contrast, seronegative SS patients demonstrate a higher prevalence of ocular dryness (abnormal Schirmer I tests) and ILD, but a lower risk of thrombocytopenia [9]. Other conventional biomarkers, such as ANA and RF, function as nonspecific markers of systemic immune activation and B-cell dysregulation, yet offer limited utility in defining organ-specific risk [157, 160, 161].
These limitations have driven the search for novel autoantibodies that not only support diagnosis but also aid in prognostication. Tissue-specific autoantibodies such as anti-SP-1, anti-CA6, and anti-PSP, initially introduced for early detection, have demonstrated prognostic relevance by identifying ongoing acinar damage and subclinical lymphocytic infiltration, particularly in seronegative patients with persistent glandular dysfunction [162, 163]. Their presence correlates with reduced salivary flow and histological signs of glandular injury, independent of classic serologies, highlighting their role in stratifying patients with localized glandular pathology even in the absence of systemic autoimmunity [33, 162, 164].
Further expanding the biomarker repertoire, anti-DTD2 and anti-RESF1 IgG autoantibodies have emerged as promising non-invasive indicators in seronegative pSS. These markers are significantly associated with labial salivary gland focus score positivity and effectively discriminate pSS from sicca controls [16]. When combined with clinical variables (platelet count, SSB, ANA, RF, and salivary flow), they improve the prediction of abnormal glandular histopathology, potentially reducing the need for invasive biopsies and paving the way toward biomarker-driven, non-invasive diagnostic strategies [165].
In the context of lymphoma surveillance, a salivary autoantibody panel
comprising anti-cofilin-1, anti-
Other autoantibodies have shown potential in systemic disease monitoring. Early work by Liu et al. [166] identified anti-mouse double minute 2 (MDM2) autoantibodies as systemic disease markers in pSS, demonstrating significant positive correlation with ESSDAI disease activity scores and significant negative correlation with circulating complement (C3/C4) and hematologic parameters (platelet count, hemoglobin). More recently, Engelke et al. [15] identified two distinct IgA autoantibody panels with organ-specific associations in pSS. A pulmonary panel [TONSL, IL-6, cathepsin L (CTSL), Fas-associated factor 1 (FAF1), proline-rich 12 (PRR12), and keratin 20 (KRT20)] correlated significantly with pulmonary involvement based on ESSDAI scores, while a salivary gland panel [CCL4, SNRPB, and AE binding protein 1 (AEBP1)] was associated with high-grade lymphocytic infiltration on salivary gland biopsy.
Anti-N-methyl-D-aspartic acid receptor subunit 2 (NR2) antibodies are implicated in neuropsychiatric manifestations, detected in 20% of serum and 12% of cerebrospinal fluid (CSF) samples, correlating with hippocampal atrophy, memory impairment, and depression [167, 168]. Besides, anti-IFI16 antibodies, present in nearly one-third of pSS patients, mark a more aggressive disease phenotype characterized by abnormal Schirmer’s test results, higher salivary gland focus scores, ectopic GC-like structures, hypergammaglobulinemia, and high-titer ANA [70]. Importantly, their prognostic value extends even to SSA/SSB-positive patients, suggesting a potential role in refining risk stratification across serological subsets [70].
Collectively, these findings support the integration of expanded serologic profiles into precision stratification frameworks for pSS, particularly in cases where classic markers fall short in capturing the full spectrum of glandular and systemic manifestations.
Genetic and epigenetic biomarkers have become central to the understanding and
diagnosis of pSS, a complex autoimmune disease defined by exocrine gland
dysfunction and chronic inflammation. Large-scale genome-wide association studies
conducted prior to 2020 established that the strongest genetic predisposition to
pSS resides within the HLA region, particularly alleles such as
HLA-DQA1, HLA-DQB1, and HLA-DRB1 that govern antigen
presentation and immune regulation [169]. Additional non-HLA genes, including
interferon regulatory factor 5 (IRF5), signal transducer and activator
of transcription 4 (STAT4), and TNF
More recent studies published have expanded the biomarker landscape by highlighting the role of epigenetic modifications. Epigenome-wide association studies found pronounced hypomethylation of ISGs such as IFI44L, epithelial stromal interaction 1 (EPSTI1), IFIT1, and MX1, correlating with overactivation of the interferon pathway and disease severity. In particular, IFI44L hypomethylation in B cells drives gene overexpression that serves as a feedback regulator of IFN signaling. Methylation profiling of affected tissues reveals global hypomethylation linked to immune cell infiltration, which can be partially restored following therapy with rituximab [17, 169, 170]. Beyond DNAm, dysregulated miRNAs, including miR-155 and miR-92a-3p, can modulate immune responses and are associated with complications such as lymphoma and atherosclerosis [23, 169]. These miRNAs circulate in blood and saliva, suggesting they could potentially serve as minimally invasive biomarkers and therapeutic targets [171]. Concurrently, metabolomic and proteomic profiling have identified serum metabolites and immune signatures, respectively, that stratify disease subtypes and inform targeted therapies [172, 173].
The integration of multi-omics data and machine learning has further revolutionized biomarker discovery. Metabolomic profiling has identified serum markers such as 2-hydroxypalmitic acid, L-carnitine, and cyclic adenosine monophosphate that distinguish pSS with an AUC value of 1.00, thus implicating amino acid and lipid metabolism in disease pathogenesis and providing non-invasive diagnostic tools [172]. Transcriptomic and proteomic analyses have revealed distinct immune endotypes driven by IFN signaling and metabolic pathways, informing precision therapies such as JAK inhibitors and BAFF blockade [174].
Despite this progress, the diagnosis of pSS faces several significant challenges that impede standardization and broader clinical applicability. A fundamental issue is the technical harmonization of epigenetic biomarkers such as IFI44L hypomethylation. This requires aligning data analysis pipelines across different methylation platforms, such as 450K and EPIC arrays, correcting for batch effects, and implementing uniform pre-analytical protocols that cover sample collection and processing. Without these measures, the variability introduced by platform differences and experimental batches risks compromising diagnostic accuracy [175]. Moreover, because the IFN-related epigenetic signature, including IFI44L hypomethylation, is not exclusive to pSS but also present in other autoimmune diseases such as SLE, multi-marker panels validated in diverse ethnic cohorts are needed to enhance disease specificity [30, 66]. Another challenge lies in translating machine learning-derived diagnostic signatures involving genes such as HES4 and OTOF into routine clinical practice. This requires rigorous external validation, transparent algorithms, and calibration according to the established transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD) + artificial intelligence (AI) guidelines. Compliance with regulatory frameworks such as the Food and Drug Administration (FDA) of the United States oversight of laboratory-developed tests and in vitro diagnostic regulations in the European Union is indispensable. In the absence of compliance with these frameworks, machine learning models risk being rejected by regulatory bodies and clinicians, limiting their clinical utility [176].
In conclusion, the integration of genetic, epigenetic, and multi-omics data is reshaping the understanding and management of pSS. This integrative approach improves diagnostic precision, enables patient stratification by disease activity, and can reveal novel therapeutic targets. Widespread clinical implementation will require overcoming the various technical, validation, and regulatory challenges. This may be achieved through coordinated multi-center efforts, rigorous standardization, and regulatory harmonization to deliver reliable and reproducible pSS diagnostic tools.
Salivary biomarkers have emerged as promising first-line diagnostic tools for pSS, as they offer several advantages over conventional methods, such as salivary gland biopsy. These biomarkers are derived from a simple, inexpensive, and non-invasive collection of saliva, making them feasible in many clinical settings [177, 178]. Compared to evaluation by histopathological biopsy, assays for salivary biomarkers are less reliant on subjective interpretation from test readers, thus reducing diagnostic variability and increasing reproducibility [145, 179].
Multi-marker salivary panels that include proteins [e.g., tripartite
motif-containing protein 29 (TRIM29), CLU] and cytokines (e.g., IL-4, IL-5) have
demonstrated high diagnostic accuracy, often exceeding 90% sensitivity and
specificity combined. For example, TRIM29 in combination with serum anti-Ro/SSA
antibodies achieved an AUC of 0.995, outperforming biopsies [180]. TRIM29 alone
showed an AUC of 0.88, indicating strong potential for early detection [180].
Conversely,
The high sensitivity of salivary biomarkers makes them particularly valuable in early or preclinical pSS, when symptoms are subtle or absent and traditional serologic tests may be negative [44]. Salivary components reflect localized glandular inflammation and immune dysregulation, and can therefore detect disease activity before overt clinical signs appear. Proteomic analyses have identified elevated levels of inflammatory proteins such as neutrophil elastase and CALR in saliva, correlating with glandular pathology and shedding light on disease mechanisms [179].
Advanced methods like liquid chromatography-tandem mass spectrometry (LC-MS/MS) allow for precise quantification of low-abundance salivary proteins and facilitate the use of multi-analyte panels that improve diagnostic accuracy and address the heterogeneity of pSS [179]. Saliva testing also avoids the invasiveness of biopsies and allows repeated, non-invasive sampling for the monitoring of disease progression [42].
Nevertheless, the clinical adoption of saliva testing faces challenges, such as standardized collection protocols. Protein measurements are markedly influenced by differences between stimulated and unstimulated saliva and by variation in salivary flow rates [182, 183]. Additionally, pre-analytical factors such as sample timing, storage, and contamination risks must be rigorously controlled to ensure reliable results [183]. To enable tailored diagnostic and prognostic applications, the phenotypic and ethnic diversity of pSS requires validation of biomarker panels in varied populations and across disease subtypes, including sicca-dominant, systemic, and lymphoma-associated forms [184].
Other promising strategies include the application of machine learning to
identify interferon pathway-enriched biomarkers such as TTC21A and ZCCHC2,
enabling improved subtype classification and diagnosis [18]. Functional and
molecular docking studies have also identified new therapeutic approaches (e.g.,
Cathepsin S inhibitors) to reduce glandular inflammation [185, 186]. The
development of point-of-care tests for salivary proteins such as TRIM29 and
Tear fluid is increasingly recognized as a valuable source of biomarkers for
systemic disease assessment, particularly in the diagnosis of pSS [14]. Recent
comprehensive analyses have highlighted significant dysregulation in the tear
proteomic profile. For example, an increased ratio of MMP-9 to lactoferrin has
been identified as a distinguishing feature of SS patients [46]. The analysis of
tear biomarkers has progressed substantially since surface-enhanced laser
desorption/ionization time-of-flight mass spectrometry was first used in 2005 to
identify 10 novel biomarkers. Current antibody microarray technology can quantify
multiple markers simultaneously using minimal tear volumes. This is a critical
advance for SS patients with compromised tear production [188]. Notably, the 2016
EULAR classification system for SS incorporates objective measures of ocular
involvement through ocular staining scores (OSS) and Schirmer’s test. However, it
does not integrate molecular signatures, creating a significant gap between
clinical practice and emerging science [189]. Highlighting this point, Akpek
et al. [190] demonstrated that IL-8 levels in tear fluid can
differentiate SS-related dry eye from non-SS dry eye with greater precision than
traditional tests, which showed only limited sensitivity (Schirmer I: 74.6%;
tear breakup time: 61.8%). The diagnostic landscape has further evolved, with
tear ATG5 emerging as a robust biomarker with 94.6% sensitivity and 93.6%
specificity at a cutoff value of
From a methodological standpoint, tear collection techniques have progressed from invasive microcapillary approaches to the now-standard use of Schirmer strips, prevalent in most contemporary studies. These address the challenge of obtaining sufficient tear volume from patients with diminished tear production [192]. Recent innovations include the use of a fluorescence-based photo-detection device to measure lactoferrin levels without antibody dependence, significantly reducing the cost compared to traditional ELISA methods while maintaining diagnostic accuracy [193]. Current implementation strategies involve the use of multi-marker panels organized by function: antimicrobials (lysozyme, lactoferrin), immune regulators (defensins, antibodies), metabolic process components (epidermal fatty acid-binding protein), and inflammatory markers [S100 calcium binding protein A6 (S100A6), MMP-9, cystatin S (CST4)] [194]. In particular, S100A6 appears to be positively correlated with rheumatoid arthritis in SS patients, suggesting utility for identifying overlapping autoimmune conditions. Machine learning approaches are now also being applied to tear proteomics. These can improve diagnostic precision and address concerns about potential limitations of the EULAR 2016 criteria, which many clinicians have argued lack sensitivity for the detection of early-stage disease. Despite these advances, standardization remains a major challenge as protein profiles vary significantly between aqueous-deficient and lipid-deficient phenotypes [57]. As point-of-care testing continues to develop, the integration of tear fluid molecular signatures with clinical findings promises to address the controversial issues between traditional classification systems and modern molecular diagnostics. This may potentially require revision of the EULAR criteria to reflect recent scientific advances [14].
The Schirmer test is a widely utilized diagnostic tool for evaluating tear
production, particularly in patients with suspected keratoconjunctivitis sicca
(KCS), dry eye syndrome, or excessive tear production [195]. This test utilizes
the principle of capillary action, whereby the aqueous component of the tear film
moves along a standardized paper strip. The extent of migration is directly
correlated with the volume of tear production [196]. A Schirmer test result of
Salivary gland dysfunction in SS is evaluated through sialometry, which measures salivary flow, and sialochemistry, which analyzes the composition of saliva. While sialometry detects reduced flow in later disease stages, sialochemistry can identify early biochemical changes in saliva, making it a noninvasive and sensitive tool for early SS diagnosis [201].
4.3.4.1 Sialometry Test
Sialometry serves as a diagnostic tool through two primary approaches: the
collection of whole saliva represents the combined secretion from all salivary
glands, while the collection of glandular saliva isolates the secretion from
specific glands [202]. The whole saliva collection method is widely utilized due
to its simplicity, rapid execution, and lack of need for specialized equipment.
However, from an analytical perspective, whole saliva has limited diagnostic
value, as it does not allow the assessment of individual gland dysfunction or
gland-specific sialochemical alterations [203, 204]. Traditionally, sialometry is
performed by collecting unstimulated whole saliva, where the patient quietly
spits accumulated saliva into a pre-weighed tube over a period of 5–15 minutes.
Alternatively, the collection of stimulated saliva involves chewing paraffin or
gauze to enhance flow, or even isolating parotid gland secretions using suction
cups and gustatory stimulation [205, 206]. The 2016 ACR/EULAR classification
criteria for SS incorporate unstimulated whole saliva flow as one of the weighted
items. One point is assigned for a flow rate of
4.3.4.2 Sialochemistry
Sialochemical analysis of glandular saliva samples can reveal distinct
alterations in electrolyte and protein (enzyme) composition in SS, reflecting the
impact of autoimmune-mediated damage on the secretory cells of individual
salivary glands [210]. Sialometry involves collecting unstimulated whole saliva
(spitting into a pre-weighed tube over 5–15 minutes) or stimulated saliva (using
paraffin or gustatory agents). A flow rate of
The Sjögren’s International Collaborative Clinical Alliance (SICCA) grading
system employs fluorescein and lissamine green (or rose Bengal) to assess ocular
surface damage in SS. The methodology was incorporated into the 2016 ACR/EULAR
classification criteria [142], which considers an OSS
Labial salivary gland (LSG) biopsy remains the definitive histopathologic test
for confirming pSS diagnosis, particularly in patients presenting with sicca
symptoms in the absence of anti-Ro/SSA antibodies [216]. A significant level of
lymphoid infiltration substantially increases the likelihood of lymphoma
development in pSS. The FLS and inflammatory foci count, which quantify
mononuclear cells per 4 mm2, are strongly correlated with both ocular and
serologic manifestations of the disease when assessed using standardized
histopathological protocols such as those established by Daniels et al.
[217]. Beyond FLS assessment, LSG biopsy specimens frequently exhibit additional
morphologic patterns of chronic inflammation, including nonspecific chronic
sialadenitis and sclerosing chronic sialadenitis [218]. LSG biopsy is widely
available and minimally invasive, allowing evaluation of autoimmune
disease-active cells within SS target organs [158]. With a focus score of
However, conventional tissue biopsy exhibits notable limitations, particularly in detecting early or subclinical disease. LSG biopsy primarily identifies advanced glandular lesions and frequently misses early inflammatory changes, potentially leading to delayed diagnoses [61, 226]. Notably, specificity may be overestimated in elderly populations due to age-related fibrosis and nonspecific lymphocytic infiltration, emphasizing the need for careful interpretation [158]. These constraints have prompted investigation of complementary diagnostic approaches. Integrating non-invasive fluid biomarkers with machine learning offers a promising alternative for early and subclinical pSS detection, particularly in heterogeneous populations [227]. Non-invasive biomarkers derived from saliva and tears, such as proteomic, metabolomic, and exosomal miRNA signatures, have the capacity to detect glandular inflammation and immune dysregulation before the emergence of overt histopathological changes, thereby facilitating substantially earlier case identification and therapeutic intervention [44, 226, 228].
Machine learning models integrated with fluid biomarkers address the inherent heterogeneity of pSS by simultaneously processing complex, multi-omic data to identify disease-specific diagnostic patterns [61, 229]. These algorithms capture dynamic molecular changes and immune responses that vary significantly across individuals and disease subtypes, improving diagnostic sensitivity and specificity in diverse patient populations [61, 226]. When analyzed using machine learning approaches such as random forests and elastic-net regression, salivary and tear proteomic/metabolomic panels achieve high diagnostic accuracy (AUC 0.82–0.88, sensitivity 80–100%, specificity 84–100%) for pSS, including in early-stage and anti-SSA-negative cases where conventional biopsy interpretation remains challenging [42, 44, 61, 172, 180, 226].
Beyond diagnostic accuracy, non-invasive biomarker collection represents a distinct clinical advantage. Saliva and tear sampling is safe, painless, and readily repeatable, facilitating longitudinal disease monitoring and reducing patient burden compared to invasive tissue biopsy. These fluids directly reflect the pathophysiology of affected salivary and lacrimal glands, providing organ-specific molecular insights without requiring invasive procedures [226, 228, 230]. Identified diagnostic markers include immune-metabolites (kynurenine), phospholipids, and immunologically-active proteins [lipocalin 2 (LCN2), signal regulatory protein alpha (SIRPA)], alongside exosomal RNAs [44, 61, 228, 231]. The combination of fluid biomarkers with objective, automated machine learning analysis replaces operator-dependent biopsy interpretation with standardized, reproducible diagnostic models [42, 61].
Despite these advantages, broader validation remains necessary before clinical translation. Large, multicenter studies are required to confirm biomarker robustness and reproducibility across geographically diverse populations and disease phenotypes [39, 42]. Standardization of sample collection protocols, analytical processing procedures, and rigorous machine learning model validation frameworks is essential for enabling clinical adoption [44, 232, 233]. Ultimately, integrating non-invasive fluid biomarkers with machine learning can overcome tissue biopsy limitations by enabling early, repeatable, and individualized detection of subclinical pSS, even in heterogeneous populations, though broad validation and standardization are prerequisites before these approaches can supplement or replace biopsy in routine clinical practice [233].
Salivary gland ultrasonography (SGUS) has become an essential non-invasive,
non-irradiating imaging technique for evaluating parotid and submandibular gland
involvement in both primary and secondary SS [158, 234]. Over the past decade,
SGUS has evolved from a supplementary imaging tool into a standardized diagnostic
component of SS assessment [235]. Before 2020, SGUS interpretation was largely
subjective, demonstrating limited reliability (
From 2023 to 2025, technological advances have transformed SGUS into a more
objective and reproducible diagnostic method through the integration of
AI-assisted ultrasonography [238]. AI-assisted tools have addressed
inter-observer variability in assessing glandular inflammation in early pSS by
enhancing reproducibility and diagnostic precision through deep learning and
radiomics-based approaches [239, 240]. Advanced architectures such as multilayer
perceptron, residual network (ResNet), fully convolutional DenseNet (FCN-DenseNet), and U-shaped network (U-Net), trained on large annotated SGUS
datasets, provide automated scoring and segmentation that match or exceed human
expert performance [239, 241, 242, 243]. These models achieve high reliability, with
the best-performing algorithms reporting Cohen’s kappa values around 0.7, which
surpass the average inter-observer agreement of
As a result, AI-driven SGUS scoring has markedly improved inter-rater
reliability (ICC = 0.9328) [248, 249], particularly benefiting anti-SSA-negative
patients (negative predictive value = 92%) [249]. Recent studies also show
strong correlations between SGUS findings and disease activity (ESSDAI: r =
–0.88) as well as inflammatory biomarkers (IL-6: r = 0.735;
Concurrently, molecular and omics-based approaches add complementary diagnostic insights. Gene-level analyses employing machine learning algorithms such as least absolute shrinkage and selection operator (LASSO) and random forest have identified transcriptomic biomarkers (IFI27, HES4) validated through reverse transcription quantitative polymerase chain reaction [18, 252]. Salivary exosome studies targeting interferon-stimulated genes (ISG15) provide non-invasive diagnostic opportunities, while proteomic and metabolomic profiling using LC-MS/MS and ultra-high-performance liquid chromatography high-resolution mass spectrometry differentiates SS patients based on unique protein and metabolic signatures [47, 229, 253]. Single-cell RNA sequencing of immune cells further elucidates disease-specific cellular heterogeneity and immune dysregulation [254, 255].
Despite these considerable achievements, several validation and implementation challenges remain. Broader multicenter and multiethnic studies are required to assess AI model generalizability across populations and imaging systems [246, 247, 256]. Establishing international standardization of SGUS acquisition, scoring, and AI integration is essential for consistent performance and regulatory approval [256, 257]. Furthermore, clinical integration demands explainable, user-friendly AI interfaces, while prospective and longitudinal studies should clarify their impact on disease monitoring and outcomes [247, 256, 257]. Finally, compliance with medical device regulations and ethical standards remains pivotal to achieving safe, equitable, and effective clinical deployment [247, 256].
AI and machine learning methods are reshaping the diagnostic and prognostic landscape of pSS by augmenting histopathology, imaging, and multi-omics analysis. Together, these approaches aim to reduce observer variability, quantify tissue injury with greater precision, and enable endotype-guided clinical decision-making.
Advanced neural network models for automated analysis of labial salivary gland
biopsies achieve high AUC values for both focus score classification and disease
diagnosis on digitized hematoxylin and eosin-stained slides, with external
validation across six European expert centers confirming diagnostic accuracy
while identifying novel histological disease subtypes [258]. Notably, YOLOv8
models enhanced with multi-dimensional attention modules and custom S-MPDIoU loss
functions have been specifically optimized for precise lymphocyte detection in
biopsy images, addressing the challenge of small cell size and distinguishability
[241]. The improved YOLOv8 model achieved a 9.1% increase in recall and 3.2%
improvement in mAP.5 compared to baseline YOLOv8, with an average of
Beyond histopathology classification, machine learning algorithms effectively predict disease severity and complications through multiple complementary approaches. Random forest algorithms achieve an AUC of 0.854 for differentiating rheumatoid arthritis-Sjögren’s overlap from uncomplicated SS through LASSO-based feature selection combined with Shapley Additive exPlanations (SHAP)-based model interpretability analysis, establishing age, anti-Ro/SSA antibodies, and systemic inflammatory markers (C-reactive protein, RF) as key discriminators with quantifiable feature importance values [259]. In parallel, XGBoost models integrate molecular genetic data (including N7-methylguanosine-related genes and their expression profiles) to predict pSS risk stratification with high accuracy and specificity, providing gene-level disease prediction [260].
Recent advances in deep learning have shown promising potential in overcoming the limitations of manual operator-dependent scoring systems in SGUS by allowing objective image interpretation, thereby addressing the significant inter-rater disagreement [De Vita score, Outcome Measures in Rheumatology (OMERACT) score] that has limited the clinical application of SGUS despite its value as a non-invasive diagnostic tool for pSS [234, 261]. For instance, convolutional neural networks based on the ResNet-50 architecture achieve a specificity of 86.9% for pSS detection, compared to 80.1% with conventional operator-based scoring, while maintaining a comparable sensitivity of approximately 59.4% versus 61.4% [245]. A more complex model employing five convolutional layers has demonstrated diagnostic accuracy of 99%, with corresponding sensitivity and specificity nearing 98% and 99%, respectively, in test datasets [247]. In addition to classification, deep learning-based segmentation algorithms such as fully convolutional networks, deep neural networks, DenseNet, U-Net, and LinkNet enable high-fidelity delineation of salivary gland boundaries, matching the precision of expert radiologist annotations [242, 262]. These technologies facilitate advanced radiomic feature extraction and texture analysis, significantly reducing evaluation time and minimizing inter-reader subjectivity [242, 262]. Collectively, these developments highlight the role of AI in standardizing SGUS interpretation and advancing its integration as a reproducible, operator-independent diagnostic tool in routine pSS care [234]. However, despite these encouraging results, AI-assisted SGUS still faces several challenges in SS diagnosis. Conventional SGUS shows reduced diagnostic sensitivity in anti-Ro/SSA–negative patients, suggesting that algorithms trained mainly on anti-Ro/SSA–positive cases may underperform in this subgroup and require targeted validation [249]. Generalizability is also limited by variations across imaging devices and acquisition protocols [263]. Furthermore, inter-rater variability persists despite OMERACT standardization, although consensus atlases and structured training can improve consistency [235, 264]. Finally, the inherently low signal-to-noise of ultrasound further complicates the automated feature extraction [262]. To overcome these obstacles, future models should be trained on multi-center and multi-device datasets, validated externally, and supported by standardized imaging protocols and bias-mitigation strategies to ensure reproducibility and broad clinical adoption.
Computed tomography (CT)-based AI achieves performance comparable to expert readers in pSS. In a head-to-head study with 50 test patients, an AI system reached 96% accuracy with 100% sensitivity and 92% specificity for pSS detection, matching experienced radiologists and clearly outperforming inexperienced readers [265]. Radiomics pipelines that extract first-order, shape, and texture features from segmented parotid glands further improve discrimination and maintain strong performance on temporal validation with an AUC of 0.96 [266]. These approaches enable objective quantification of CT signs of gland injury, including parenchymal atrophy, ductal changes, and fat deposition, thereby improving reproducibility and standardization [266].
Beyond single-modality imaging, multi-omics integration with machine learning
has identified distinct pSS endotypes characterized by differential immune
dysregulation. A landmark European study (Soret et al., 2021 [74])
identified four molecular clusters using comprehensive multi-omics data from 304
pSS patients: an IFN-
In summary, AI-enabled histopathology, ultrasound, CT, and multi-omics analysis collectively advance objective assessment of glandular damage and systemic risk in pSS while laying the groundwork for precision stratification. Real-world deployment now depends on robust external validation, standardized acquisition and analysis protocols, and regulatory-grade evidence that demonstrates reproducibility, clinical utility, and economic value across diverse care settings.
Future research in pSS should focus on validating multi-omics biomarkers and integrating novel autoantibodies, epigenetic markers, and non-invasive diagnostics into clinical practice. The development of DNA assembly strategies in both basic research and biomedical applications could open up opportunities for the exploration of novel biomarkers [274]. Moreover, crucial biomarkers of TJ dysfunction in saliva should be considered for both non-invasive diagnostic tests and the management of pSS [275]. Stratification based on immune endotypes may enable personalized therapy, while AI-assisted imaging and machine learning can further refine diagnosis and monitoring. Elucidation of the links between IFN signaling, B-cell activation, and metabolic dysfunction should provide a scientific basis for the development of targeted, mechanism-based treatments.
Conventional autoantibodies and newly developed biomarkers are now being combined to allow earlier and more precise diagnosis of pSS, as well as improved risk stratification, even in seronegative cases. Advances in salivary and tear-based multi-analyte panels, AI-driven imaging, and multi-omics profiling have contributed to more precise disease monitoring and facilitated the development of targeted, pathway-specific therapies. While these innovations hold substantial promise for personalized management, their implementation into routine clinical practice will require the resolution of challenges relating to test standardization, different patient demographics, and clinical adoption.
pSS, primary Sjögren’s syndrome; ILD, interstitial lung disease; ACR/EULAR, American College of Rheumatology/European League Against Rheumatism; SSA, Sjögren’s-syndrome-related antigen A (Ro); SSB, Sjögren’s-syndrome-related antigen B (La); RF, rheumatoid factor; FLS, focal lymphocytic sialadenitis; SP-1, salivary protein 1; PSP, parotid secretory protein; CA6, carbonic anhydrase VI; DTD2, D-aminoacyl-tRNA deacylase 2; RESF1, retroelements silencing factor-1; IgG, immunoglobulin G; FNBP4, formin-binding protein 4; SNRPC, small nuclear ribonucleoprotein polypeptide C; CCL, C-C motif chemokine ligand; SUMO2, small ubiquitin-like modifier 2; OAS3, 2′-5′-oligoadenylate synthetase 3; IFI44L, interferon-induced protein 44-like; HES4, hes family bHLH transcription factor 4; OTOF, otoferlin; TTC21A, tetratricopeptide repeat domain 21A; ZCCHC2, zinc finger CCHC-type containing 2; SGECs, salivary gland epithelial cells; TLRs, toll-like receptors; TJs, tight junctions; M3R, M3 muscarinic acetylcholine receptor; APCs, antigen-presenting cells; DCs, dendritic cells; NK, natural killer; IL, interleukin; TNF-
TTD and THN conceived and designed the review. VTN, KTTD, HGT, DNTL, and QMML were responsible for manuscript writing. KTTD, HGT, DNTL, and QMML created the tables and contributed to the critical interpretation of existing data. The graphic figures were created and edited by TNT, VTN, KTTD, and THN. TTD, VTN, and THN contributed to preparing the draft and editorial revisions. TTD and THN served as supervisors and administrators. All authors reviewed critically for important intellectual content and contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We sincerely acknowledge Can Tho University of Medicine and Pharmacy for their dedicated time and commitment to this study. We are also deeply grateful to our mentor, Professor Yoshihiro Kubo, MD & PhD, from the Division of Biophysics and Neurobiology, National Institute for Physiological Sciences, 38 Nishigonaka Myodaiji, Okazaki, Aichi, 444-8585, Japan, for his invaluable guidance and support throughout our research.
This research received no external funding.
The authors declare no conflict of interest.
During the preparation of this manuscript, the authors used ChatGPT (OpenAI) and Grammarly as language assistance tools for checking English spelling and grammar. The authors have reviewed and edited the output and take full responsibility for the integrity of this publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.