






 , Young-Wook Seo 1,†, Yae-Ji Kim 1, Hui-Ju Lee 1, Jin-Li Ryu 1, Hyun-Tae Kim 2, Geum-Lan Hong 1, Ju-Young Jung 1,*
, Young-Wook Seo 1,†, Yae-Ji Kim 1, Hui-Ju Lee 1, Jin-Li Ryu 1, Hyun-Tae Kim 2, Geum-Lan Hong 1, Ju-Young Jung 1,*
1 Department of Veterinary Medicine & Institute of Veterinary Science, Chungnam National University, 34134 Daejeon, Republic of Korea
2 Aging Research Unit, Honam Regional Center, KBSI Institute of Regional Hub, Korea Basic Science Institute(KBSI), 61751 Gwangju, Republic of Korea
†These authors contributed equally.
Abstract
Aging is frequently accompanied by chronic, low-grade inflammation, often referred to as “inflammaging”, which contributes to functional decline of multiple organs including the liver. The NLRP3 inflammasome has emerged as a key mediator of age-related inflammation; however, its pharmacological inhibition in the context of hepatic aging remains insufficiently explored. In this study, we investigated the effects of the selective NLRP3 inflammasome inhibitor MCC950 on inflammatory responses in the liver of aged mice.
Aged C57BL/6 mice (18 months old) were administered MCC950 intraperitoneally for four weeks, and liver tissues were analyzed for inflammatory and stress-related markers.
MCC950 treatment significantly reduced hepatic expression of NLRP3, caspase-1 activation, and IL-1β production, accompanied by a decrease in proinflammatory cytokines such as p-STAT3. Histological analysis demonstrated attenuation of age-associated hepatic inflammatory infiltration and improved tissue architecture. Furthermore, MCC950 administration restored autophagy-related proteins (LC3B, p62) indicating broader protective effects on liver homeostasis.
These findings suggest that NLRP3 inflammasome inhibition with MCC950 alleviates age-associated hepatic inflammation and may represent a potential therapeutic strategy for mitigating inflammaging and preserving liver function in the elderly.
Keywords
- inflammasomes
- liver
- aging
- NLRP3 inflammasome inhibitor
- autophagy
Aging is accompanied by progressive structural and functional deterioration in various organs, leading to increased susceptibility to chronic diseases and reduced physiological resilience. Among these age-related changes, chronic, low-grade systemic inflammation—commonly termed “inflammaging”—has emerged as a major contributor to the onset and progression of aging-associated pathologies [1]. The liver, a central organ in metabolism, detoxification, and immune regulation, is particularly vulnerable to age-driven inflammatory and oxidative insults. Accumulating evidence suggests that age-related hepatic inflammation accelerates fibrosis, metabolic dysregulation, and diminished regenerative capacity, ultimately impairing liver homeostasis and functional reserve [2, 3].
A key molecular driver of inflammaging is the nucleotide-binding oligomerization
domain (NOD)-like receptor family pyrin domain-containing protein 3 (NLRP3)
inflammasome [4]. The NLRP3 inflammasome is a cytosolic multiprotein complex that
detects cellular stress signals and activates caspase-1, resulting in the
maturation and release of proinflammatory cytokines such as
interleukin-1
Despite its pathogenic significance, pharmacological interventions targeting NLRP3 in the context of hepatic aging remain limited. MCC950 is a selective and potent small-molecule inhibitor of the NLRP3 inflammasome that prevents inflammasome assembly and caspase-1 activation without affecting other inflammasome complexes [7]. Preclinical studies have demonstrated the therapeutic benefit of MCC950 in metabolic, autoimmune, and neuroinflammatory disorders [5]; however, its effects on age-associated liver inflammation and tissue homeostasis have not been fully elucidated.
Therefore, the present study aimed to evaluate whether NLRP3 inhibition by MCC950 can attenuate inflammaging-related changes in the liver of aged mice. We assessed inflammatory cytokine expression, inflammasome activation, and autophagy-related markers to determine the therapeutic potential of MCC950 in restoring hepatic function during aging. Our findings provide evidence that MCC950 alleviates age-related hepatic inflammation and may serve as a promising strategy to preserve liver health in the elderly population.
MCC950 was purchased from AppexBio (Houston, TX, USA). The following primary
antibodies were used in this study: NLRP3 (NLR family pyrin domain containing 3,
68102-1-1g, Proteintech, Rosemont, IL, USA); ASC (apoptosis-associated speck-like
protein containing a CARD, A1170); VPS34 (vacuolar protein sorting 34, A12483),
and LC3B (microtubule-associated protein 1A/1B-light chain 3B, A19665, Abclonal,
Woburn, MA, USA); caspase-1 (sc-392736) and IL (interleukin)-1
The animal experimental protocols were approved by the Institutional Animal Ethics Committee of Chungnam National University (approval number: 202307A-CNU-136). Two-months old C57BL/6J male mice and 18-month-old C57BL/6J mice were obtained from the Animal Facility of Aging Science, Korea Basic Science Institute Gwangju Center (Gwangju, Korea). The mice were acclimated for 1 week and fed a pelleted diet and tap water ad libitum under specific pathogen-free conditions. The mice were randomly divided into three groups (n = 6/group) as follows: 2-months old C57BL/6J male mice (Young mice), 18-months old C57BL/6J male mice (Aged mice), and 18-months old C57BL/6J male mice intraperitoneally injected 20 mg/kg of MCC950 every other day (once every 48 hours) for 4 weeks (Aged mice + MCC). The mice were fasted prior to autopsy. Mice were euthanized using CO2, with compressed CO2 introduced at a rate of 30–70% of the chamber volume per minute (gradual-fill method) using a calibrated flow meter, and cardiac blood was subsequently collected.
Liver tissues were fixed in 10% neutral-buffered formalin, embedded in
paraffin, and sectioned at 4 µm thickness. After serial gradient hydration,
sections were stained with hematoxylin and eosin (H&E) and Sirius Red using
standard protocols. For immunohistochemistry (IHC), antigen retrieval was
performed by heating the sections in citrate buffer (pH 6.0), followed by
incubation with an anti-NLRP3 antibody (Proteintech, 1:200 dilution) and
anti-F4/80 (Invitrogen, 1:400 dilution) overnight at 4 °C. Signal was
detected using a avidin-biotin complex kit and DAB chromogen (VectorLabs,
Burlingame, CA, USA). Each stained section (ten randomly selected fields) was
photographed at 200
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were determined using commercial assay kits (ALT: AM103-K; AST: AM102; Assan Pharmaceutical, Seoul, Republic of Korea) according to the manufacturer’s instructions. Briefly, the substrate reagent was pre-incubated at 37 °C, serum was added, and the reaction was allowed to proceed at 37 °C (ALT: 30 min; AST: 60 min). After color development with 2,4-dinitrophenylhydrazine followed by alkalinization, absorbance was measured at 505 nm (acceptable range: 490–530 nm) using a microplate reader, and enzyme activities were calculated from the manufacturer-provided standard curve and expressed as units per liter (U/L). All samples were assayed in triplicate to ensure analytical reproducibility.
Liver tissues were homogenized in radio-immunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, Danvers, MA, USA) containing protease and phosphatase inhibitors and clarified by centrifugation. Protein concentrations were determined prior to analysis. Equal amounts of protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred using a wet transfer system (Bio-Rad, Hercules, CA, USA) onto polyvinylidene fluoride membranes (Millipore, Boston, MA, USA). Following transfer, membranes were blocked to minimize nonspecific binding and subsequently incubated with the appropriate primary antibodies under controlled conditions. After washing, membranes were incubated with the corresponding horseradish peroxidase–conjugated secondary antibodies. Immunoreactive signals were developed using a chemiluminescence-based detection reagent and captured with an image acquisition system (ATTO, Tokyo, Japan). Band intensities were quantified by densitometric analysis, and protein expression levels were normalized to the respective loading controls.
All experiments were conducted in a double-blind manner. Results were randomly
selected and expressed as the mean
To investigate whether hepatic NLRP3 expression changes with aging, we first
analyzed RNA-seq data from human liver samples in the GTEx cohort. NLRP3
transcript levels showed no significant correlation with chronological age
(Spearman’s
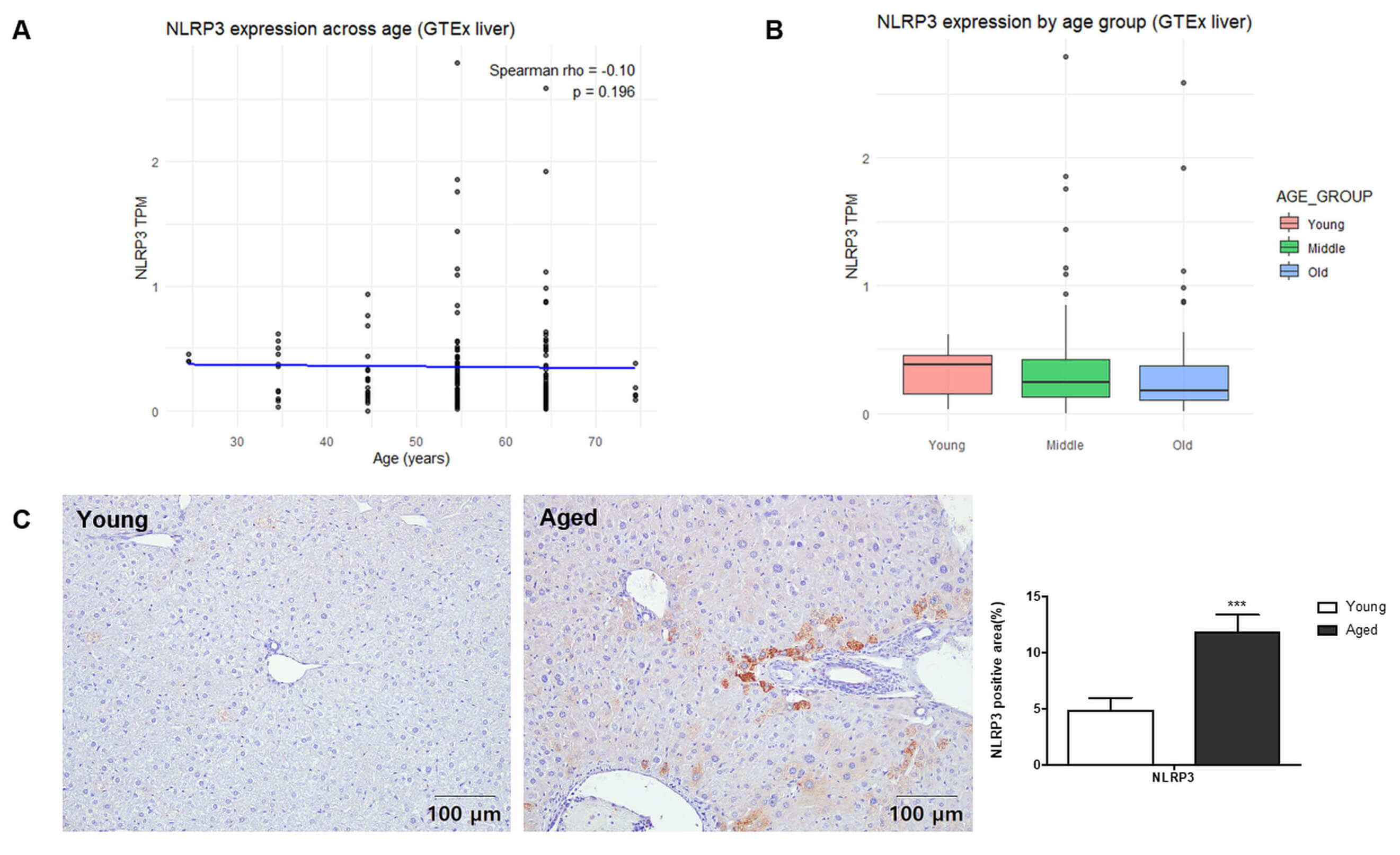 Fig. 1.
Fig. 1.
Age-dependent hepatic NLRP3 expression in human and murine
samples. (A) Correlation analysis of NLRP3 mRNA expression with chronological
age in human liver samples using GTEx RNA-seq data. (B) Group-wise comparison of
NLRP3 transcript levels among human donors classified into Young (
Given the increased hepatic NLRP3 protein expression in aged mice, we next examined the effects of MCC950 treatment on age-associated inflammatory and pathological changed in the liver. Aging was associated with pronounced inflammatory changes in the liver. H&E staining revealed substantial infiltration of inflammatory cells in aged mice, particularly around periportal and perisinusoidal regions, whereas young mice showed minimal inflammatory cell presence. Quantitative analysis confirmed a significant increase in inflammatory cell numbers in aged livers. Importantly, MCC950 treatment markedly reduced inflammatory cell infiltration, restoring liver architecture toward that of young controls (Fig. 2A). To assess age-associated fibrotic remodeling, Sirius Red staining was performed. Aged mice showed robust collagen deposition, with increased perivascular and periportal fibrosis. Sirius Red–positive area was significantly higher in aged livers, while MCC950 administration significantly reduced collagen accumulation, indicating attenuation of aging-related hepatic fibrosis (Fig. 2B). We next evaluated whether these histological improvements were accompanied by functional restoration. Serum ALT and AST levels were markedly elevated in aged mice, reflecting hepatocellular injury. MCC950 treatment significantly decreased both ALT and AST, although levels remained slightly above those of young mice (Fig. 3). These findings demonstrate that MCC950 mitigates age-related hepatic injury, improving both structural and biochemical indicators of liver damage.
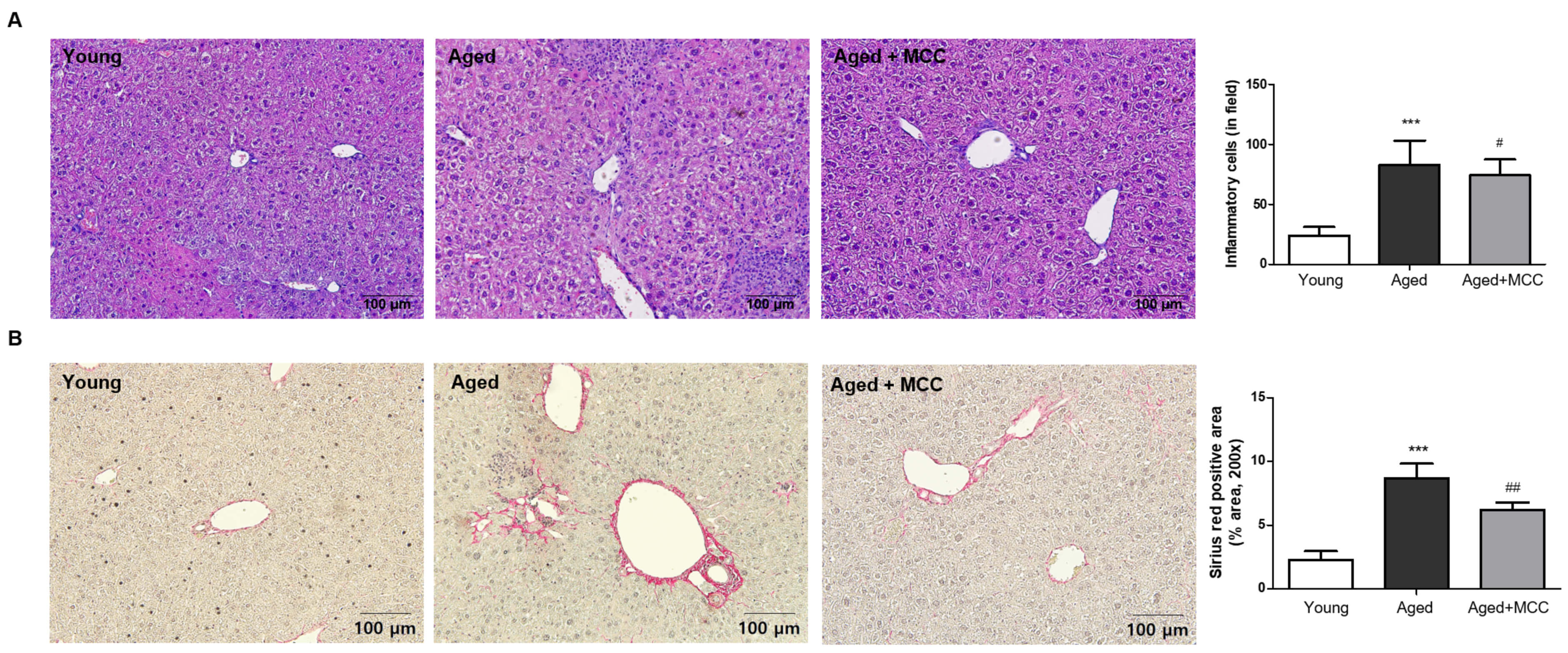 Fig. 2.
Fig. 2.
MCC950 treatment attenuates age-associated hepatic inflammation
and fibrosis. (A) Representative hematoxylin and eosin (H&E)-stained liver
sections at 200
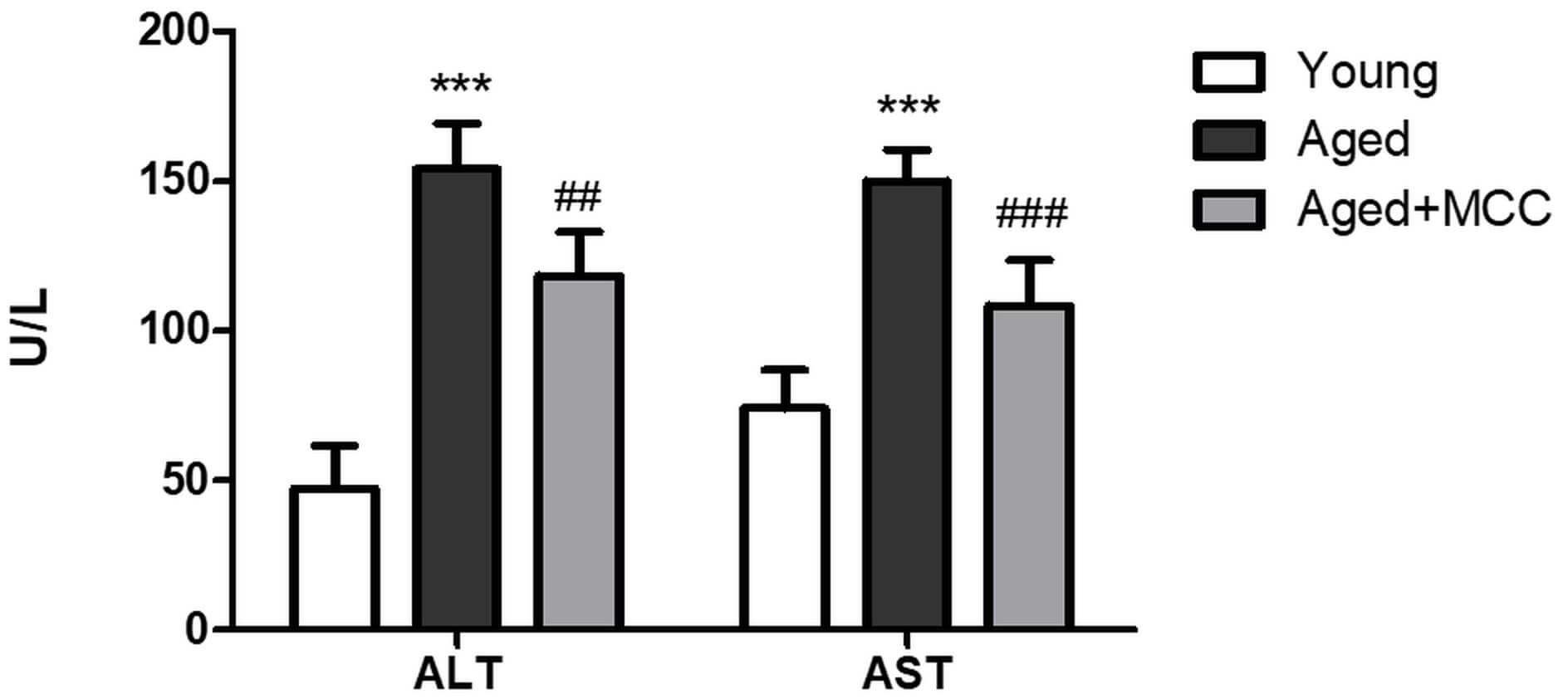 Fig. 3.
Fig. 3.
Effects of MCC950 on serum liver injury markers in aged mice.
Serum alanine aminotransferase (ALT) and Serum aspartate aminotransferase (AST)
levels. Experimental groups were Young (2 months, PBS, IP), Aged (18 months, PBS,
IP), and Aged + MCC950 (18 months, 20 mg/kg MCC950, IP). Data are presented as
mean
We next investigated whether MCC950 effectively suppresses NLRP3 inflammasome signaling, the primary pharmacological target of this compound. Aged livers showed strong upregulation of NLRP3, ASC, and cleaved caspase-1, indicating increased inflammasome activation during aging. Quantification revealed significant increases in all three proteins in aged mice, whereas MCC950 markedly reduced NLRP3 and ASC levels and strongly suppressed caspase-1 activation (Fig. 4). These results showed that MCC950 effectively inhibits NLRP3 inflammasome activity in aged liver tissue.
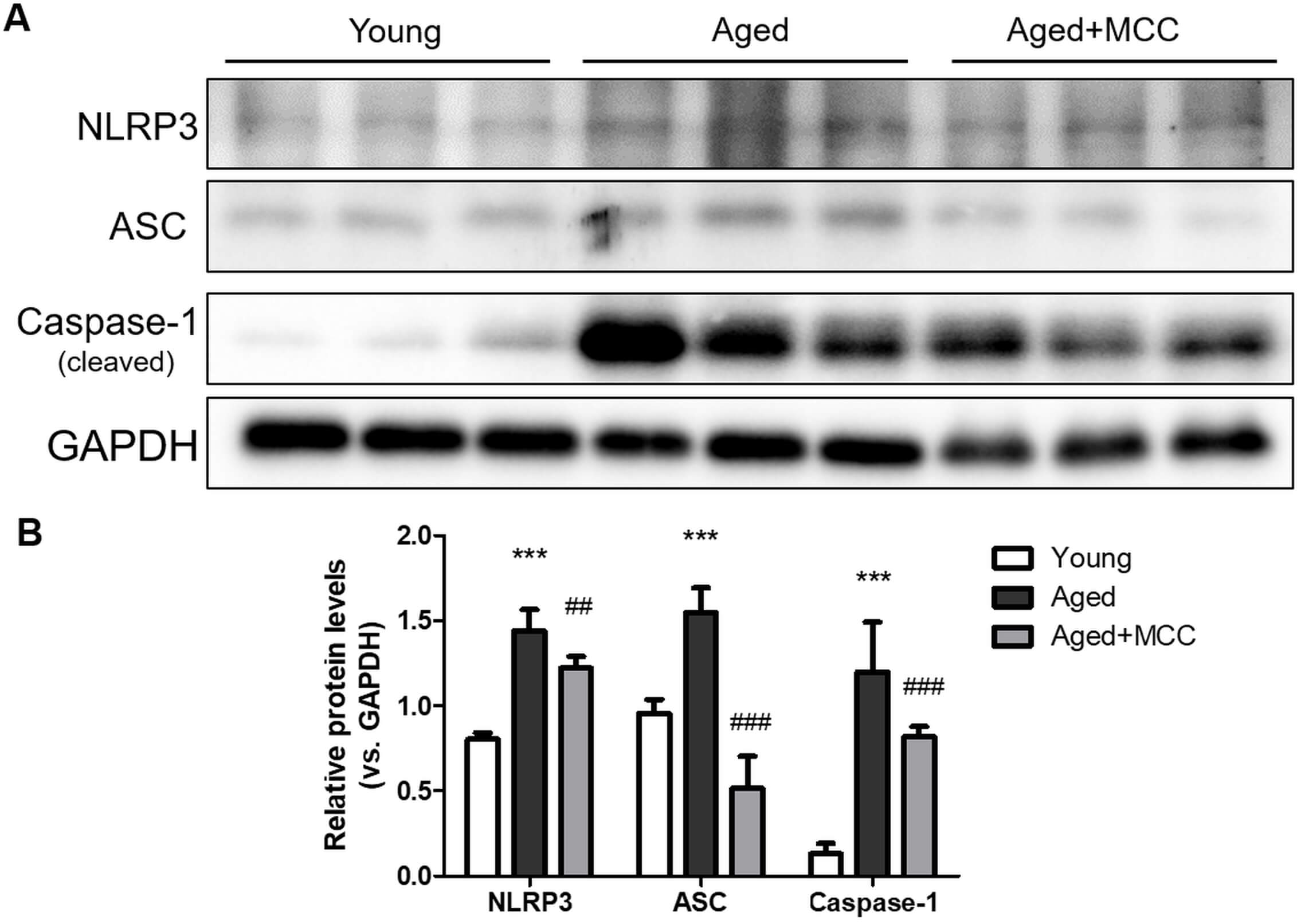 Fig. 4.
Fig. 4.
NLRP3 inflammasome activation in aged liver and its suppression
by MCC950. (A) Representative Western blot images showing hepatic protein
expression of NLRP3, ASC, and cleaved caspase-1, with GAPDH as a loading control.
(B) Densitometric quantification of the corresponding protein levels normalized
to GAPDH. Experimental groups were Young (2 months, PBS, IP), Aged (18 months,
PBS, IP), and Aged + MCC950 (18 months, 20 mg/kg MCC950, IP). Data are presented
as mean
To further elucidate the mechanisms underlying the protective effects of MCC950
in aged liver, we examined downstream inflammatory and fibrogenic signaling
pathways. Aged mice exhibited pronounced activation of inflammatory mediators,
characterized by elevated IL-1
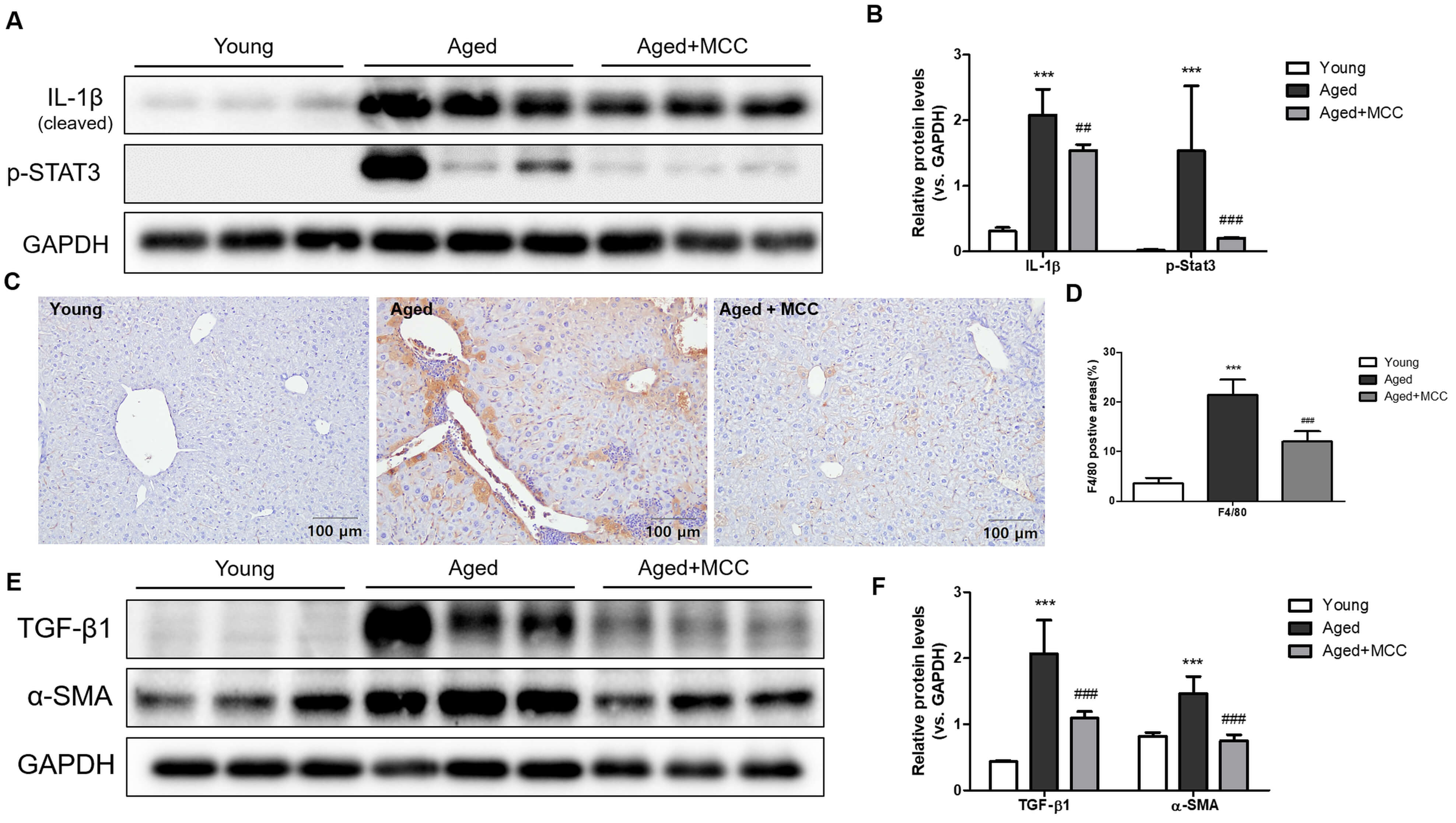 Fig. 5.
Fig. 5.
MCC950 suppresses inflammatory and fibrogenic signaling in aged
liver. (A) Representative Western blot images showing hepatic protein expression
of the inflammatory markers IL-1
Given the known interplay between inflammasome activation and autophagy, we next evaluated whether MCC950 influences autophagy status in aged liver. Aged mice displayed profound impairment of autophagy, characterized by decreased expression of Vps34 and Atg14L, key components of the autophagosome initiation machinery. Additionally, aged liver exhibited a reduced LC3-II/LC3-I ratio and marked accumulation of p62, indicating suppressed autophagic flux. MCC950 treatment effectively reversed these autophagy defects, restoring Vps34 and Atg14L expression, increasing LC3-II/LC3-I ratio, and reducing p62 accumulation (Fig. 6). These findings suggest that suppression of NLRP3 inflammasome activity not only dampens inflammation and fibrosis but also reactivates autophagy pathways compromised during aging, contributing to improved hepatic homeostasis.
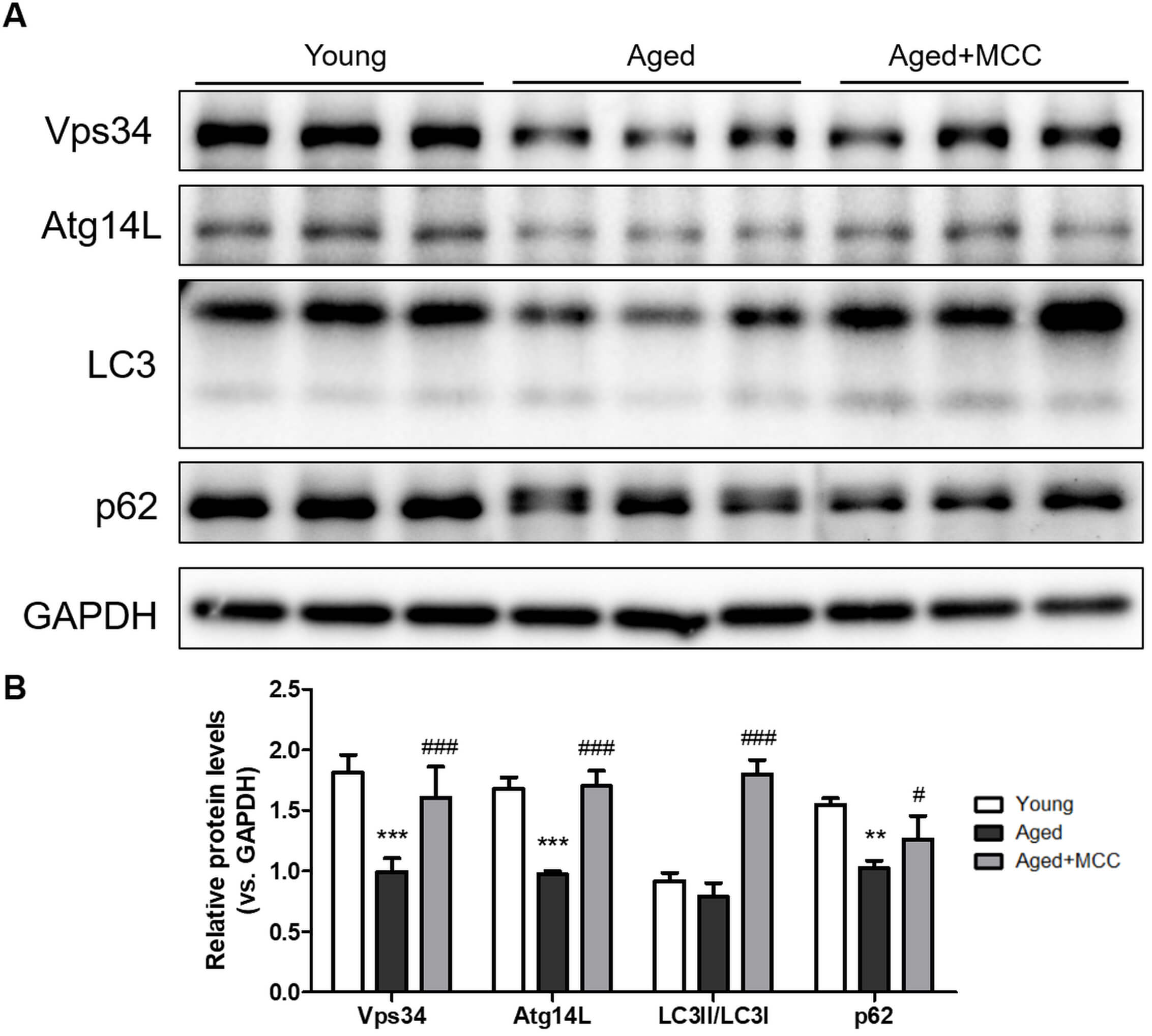 Fig. 6.
Fig. 6.
MCC950 partially restores autophagy-related proteins in aged
liver. (A) Representative Western blot images showing hepatic expression of
autophagy-related proteins Vps34, Atg14L, LC3B, and p62, with GAPDH used as a
loading control. (B) Densitometric quantification of the corresponding protein
levels normalized to GAPDH. Experimental groups were Young (2 months, PBS, IP),
Aged (18 months, PBS, IP), and Aged + MCC950 (18 months, 20 mg/kg MCC950, IP).
Data are presented as mean
In this study we demonstrate that aging is associated with increased NLRP3
inflammasome activation in the liver, which contributes to chronic low-grade
inflammation and fibrotic susceptibility. We observed that aged mice exhibit
higher NLRP3 expression, IL-1
Aging fosters a pro-inflammatory milieu in the liver partly through the NLRP3 inflammasome [5]. Consistent with this, our findings show that aged livers exhibit elevated NLRP3 activation and inflammatory cytokine expression relative to young adults, which likely contributes to their increased susceptibility to liver injury and fibrosis. Prior studies similarly report that older mice develop more severe and persistent fibrosis than young mice following hepatic insults, coinciding with heightened NLRP3 levels, greater immune cell infiltration, and amplified fibrogenic activation [8]. Chronic metabolic conditions common in the elderly (such as NAFLD) may further exacerbate this pathway, as they lead to excessive NLRP3 activation and inflammatory tissue damage [5]. Taken together, the NLRP3 inflammasome emerges as a central mediator of age-related liver pathophysiology by driving ongoing inflammation and priming the liver for fibrotic remodeling.
Interestingly, our findings also highlight a divergence between human and murine aging with respect to NLRP3 expression. Specifically, analysis of the GTEx transcriptome data revealed no significant age-related increase in hepatic NLRP3 expression in healthy humans, consistent with previous reports indicating that sterile inflammation in human aging is often subtle and context-dependent [1, 9]. However, it should be noted that GTEx-derived human transcriptomic data represent a heterogeneous population and may be influenced by multiple confounding factors, including underlying comorbidities, medication use, lifestyle differences, and postmortem interval. Accordingly, the absence of a significant age-dependent change in hepatic NLRP3 mRNA levels in GTEx data should be interpreted with caution. This discrepancy may be attributable, at least in part, to regulatory processes beyond steady-state mRNA abundance (e.g., translational and protein turnover mechanisms) and to inherent differences between human donor-based transcriptomic datasets and controlled murine aging models. In the present study, GTEx analysis was used to provide contextual insight into human liver aging at the transcriptomic level, rather than to establish definitive causal relationships between aging and inflammasome activation. In contrast, our murine model exhibited a robust age-associated increase in hepatic NLRP3 protein expression, indicating more pronounced inflammasome activation during mouse liver aging, even in the absence of overt metabolic stress. These species-specific differences may reflect variations in immune surveillance, lifespan, or subclinical inflammatory burden, and underscore the inherent limitations of large-scale human postmortem datasets. Together, these findings highlight the value of complementary experimental models, such as spontaneously aged mice, for mechanistic investigation of age-related hepatic inflammation.
A major pathological consequence of NLRP3 inflammasome hyperactivation in the
aging liver is the promotion of fibrosis. Persistent inflammasome signaling
drives chronic release of IL-1
Importantly, although pharmacological inhibition of NLRP3 with MCC950 has been reported to ameliorate liver inflammation and fibrosis in previous studies, most of these investigations were conducted in acute injury– or metabolically stressed models, such as toxin-induced or diet-induced liver disease [14]. In contrast, the present study specifically employed spontaneously aged mice, which develop hepatic inflammation and fibrotic susceptibility in the absence of overt exogenous insults. This distinction is critical, as spontaneous aging models more closely reflect the physiological and progressive nature of hepatic inflammaging, rather than secondary inflammatory responses driven by experimental injury. Therefore, our findings extend prior work by demonstrating that NLRP3 inflammasome activation is not merely a consequence of liver damage, but a primary driver of age-associated hepatic inflammation during natural aging.
Given its central involvement in age-related liver pathology, NLRP3 represents
an attractive therapeutic target. MCC950, a highly selective small-molecule
inhibitor of NLRP3, suppresses inflammasome assembly, thereby preventing
caspase-1 activation and subsequent IL-1
Restoring autophagy represents a complementary therapeutic axis to inflammasome
blockade because autophagic flux declines with aging, allowing damaged organelles
and protein aggregates to accumulate and provide endogenous danger signals that
can prime and activate the NLRP3 inflammasome. Conversely, autophagy acts as a
negative regulator of NLRP3 signaling by removing inflammasome activators (e.g.,
damaged mitochondria) and, in some contexts, limiting inflammasome components and
cytokine output [17]. Notably, rather than reflecting a secondary consequence of
reduced inflammatory burden, our data indicate that MCC950 treatment
preferentially restores components of the early autophagy initiation machinery.
Consistent with this bidirectional crosstalk, our data show that MCC950-mediated
suppression of IL-1
While our study provides important insights, several limitations should be
acknowledged. First, murine findings may not directly translate to humans, as
GTEx-based analyses suggest that age-related inflammasome changes in human liver
are relatively subtle, warranting validation in elderly human cohorts. Second,
the present study was conducted exclusively in male mice, which may limit the
generalizability of our findings. Given well-documented sex-dependent differences
in hepatic metabolism and inflammaging, future studies will incorporate female
mice and apply appropriate experimental controls, such as estrous cycle staging
or hormonal profiling, to comprehensively evaluate sex-specific effects. Third,
MCC950 was administered for a relatively short duration in aged mice; therefore,
the long-term efficacy, optimal treatment window, and safety of sustained NLRP3
inhibition remain to be determined. Finally, our analyses were largely performed
at the whole-tissue level, and future work using cell-type–resolved approaches
(e.g., single-cell profiling or conditional knockouts) will be required to define
the principal hepatic cell populations driving NLRP3 activation and IL-1
Taken together, our results indicate that targeting the NLRP3 inflammasome while restoring autophagy is an effective strategy to counter age-associated hepatic inflammation and fibrosis. By simultaneously reducing inflammasome-driven cytokine output and reactivating cellular quality-control pathways, this approach disrupts the self-perpetuating cycle of hepatic inflammaging. Although additional studies are required to establish translational applicability, our findings provide clear proof-of-concept that coordinated modulation of NLRP3 signaling and autophagy can improve liver homeostasis in aging.
The datasets generated and analyzed during the current study are available from the corresponding authors upon request.
KHK: Writing-original draft, Animal modeling, Investigation, Review & Editing. YWS: Animal modeling, Investigation. YJK: Animal modeling, Investigation. HJL: Animal modeling, Investigation. JLR: Investigation. HTK: Animal modeling. GLH: Investigation. JYJ: Conceptualization, Funding acquisition, Supervision, Project Administration. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experimental protocols were approved by the Institutional Animal Ethics Committee of Chungnam National University (approval number: 202307A-CNU-136). This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals of National Institutes of Health.
Not applicable.
This research was supported by the 2025 Daejeon RISE Project (DJR2025-12), funded by the Ministry of Education and Daejeon Metropolitan City, in the Republic of Korea. This work was supported by research fund of Chungnam National University.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.