







 , Zengliang Gao 1,3,†, Yanqiu Zhao 2, Xiaoting Ni 1,4, Wenlei Zhang 1,3, Longyu Li 1,3, Shiao Ren 1,3, Qi Li 2, Dan Guo 2, Lijuan Yue 1,3, Yutong Liu 1,3, Liwang Lin 1,3, Shengjin Fan 2, Xin Hai 1,3,*
, Zengliang Gao 1,3,†, Yanqiu Zhao 2, Xiaoting Ni 1,4, Wenlei Zhang 1,3, Longyu Li 1,3, Shiao Ren 1,3, Qi Li 2, Dan Guo 2, Lijuan Yue 1,3, Yutong Liu 1,3, Liwang Lin 1,3, Shengjin Fan 2, Xin Hai 1,3,*
1 Department of Pharmacy, First Affiliated Hospital of Harbin Medical University, 150001 Harbin, Heilongjiang, China
2 Department of Hematology, First Affiliated Hospital of Harbin Medical University, 150001 Harbin, Heilongjiang, China
3 NHC Key Laboratory of Cell Transplantation, 150001 Harbin, Heilongjiang, China
4 Department of Clinical Pharmacy, Qiqihar Medical University, 161006 Qiqihar, Heilongjiang, China
†These authors contributed equally.
Abstract
Arsenic trioxide (ATO) is a cornerstone of acute promyelocytic leukemia (APL) therapy but induces severe gut microbiota dysbiosis, limiting its efficacy and safety. This study investigated whether adjunctive Bifidobacterium pseudolongum (BP) could mitigate these adverse effects and enhance therapeutic outcomes.
16S rRNA gene sequencing data of gut microbiota were obtained from a cohort of 22 APL patients treated with ATO-based regimens (20 of 22 data were obtained and analysis further), accessible under BioProject ID PRJNA935705. To evaluate the within-sample microbial community richness and evenness, alpha and beta diversity indices were calculated. Using a murine APL model, we compared ATO monotherapy with ATO+BP co-treatment. Analyses included fecal metagenomic sequencing, single-cell RNA sequencing (sc-RNA-seq), flow cytometric immune profiling, and assessment of intestinal tight junction proteins (claudin-1, occludin, and ZO-1) via immunofluorescence.
ATO treatment significantly reduced gut microbial diversity and depleted beneficial taxa. Sc-RNA-seq data showed that ATO could orchestrate the APL immune microenvironment mainly through functional activation of CD8+ T cells and monocytes. BP supplementation restored microbial homeostasis and synergistically enhanced ATO’s antileukemic effect, reducing the leukemic burden in peripheral blood by 72% and in bone marrow by 64% compared to ATO alone. Mechanistically, BP preserved intestinal barrier integrity by upregulating tight junction protein expression and modulated anti-tumor immunity, notably increasing bone marrow CD8+ T cells by 2.21-fold.
BP is an effective adjunct to ATO therapy, counteracting gut dysbiosis, intestinal damage, and the immune microenvironment while synergistically improving antileukemic efficacy. Targeting the gut–leukemia axis with BP represents a promising strategy for improving the precision and safety of APL treatment.
Keywords
- arsenic trioxide
- acute promyelocytic leukemia
- intestinal homeostasis
- bone marrow microenvironment
- Bifidobacterium pseudolongum
Arsenic trioxide (ATO), a derivative of arsenic, has revolutionized the
treatment of acute promyelocytic leukemia (APL), a subtype of acute myeloid
leukemia characterized by the promyelocytic leukemia–retinoic acid receptor
The gut microbiota plays a pivotal role in maintaining immune homeostasis and
metabolic health [9, 10]. A growing body of evidence indicates that the gut
microbiota dysbiosis impairs therapeutic efficacy in various hematological
malignancies, including lymphoma [11] and leukemia [10, 12], and even influences
outcomes following hematopoietic stem cell transplantation [13]. Chemotherapy and
targeted therapies, including ATO, disrupt microbial diversity, leading to
dysbiosis marked by reduced Bifidobacterium and Lactobacillus
abundance and increased pathogenic Enterococcus and Escherichia
species [8]. This imbalance compromises intestinal barrier integrity, increases
systemic inflammation, and promotes leukemic cell survival through dysregulated
cytokine production (e.g., TNF-
For APL, emerging resistance mechanisms include PML/RAR
This study identifies Bifidobacterium pseudolongum (BP), a gut commensal with immunomodulatory properties, as a key protector for ATO related intestinal barrier integrity impairment and enhancer for therapeutic efficacy. In our investigation, BP attenuated ATO-induced intestinal barrier disruption by upregulating tight junction proteins (e.g., Claudin-1, Occludin, ZO-1). Murine model data demonstrate that BP co-administration with ATO reduced the APL cell burden. This dual therapeutic action positions BP as a novel adjunct to ATO therapy, addressing both efficacy and safety limitations in APL treatment. By elucidating BP’s role in remodeling the gut–leukemia axis, this work bridges a critical gap in APL therapeutics. The integration of microbial with leukemia omics represents a paradigm shift, highlighting microbiota-targeted interventions as essential components of precision oncology.
To validate the impact of ATO on the human gut microbiota, a comprehensive bioinformatic re-analysis was performed using publicly available data from the National Center for Biotechnology Information (NCBI) Sequence Read Archive. Raw 16S rRNA gene sequencing data of gut microbiota were obtained from BioProject ID PRJNA935705, which included 22 APL patients receiving ATO-based regimens. 20 of 22 fecal samples data were obtained and further analyzed, including pre-treatment (APL group, n = 5) and post-ATO treatment (ATO group, n = 15). Raw sequencing data were processed using the QIIME 2 pipeline (version 2023.2, https://qiime2.org/). After quality control, denoising, and chimera removal via the DADA2 plugin, amplicon sequence variants (ASVs) were generated. Taxonomic classification was performed using the SILVA database (version 138.1, https://www.arb-silva.de/).
Alpha diversity indices, including Chao1 (species richness) and Shannon (richness and evenness), were calculated to assess within-sample diversity. Group differences were evaluated using the Mann–Whitney U test. Beta diversity was visualized using principal coordinate analysis (PCoA) based on weighted UniFrac distances. Differentially abundant bacterial genera between groups were identified using R package “DESeq2” (v1.38.3, https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html). Spearman’s rank correlation analysis was conducted among the top 10 most abundant genera, and a correlation heatmap was generated to explore interactions, especially involving key beneficial taxa such as Bifidobacterium.
The human APL cell line NB4 (ATCC® CRL-2047™) was cultured in RPMI-1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 1% penicillin/streptomycin (Gibco), and 1% L-glutamine (Gibco) at 37 °C in a 5% CO humidified incubator (Thermo Fisher Scientific, Waltham, MA, USA). ATO (GBW08666, purchased from the National Institute of Metrology, Beijing, China) was dissolved in sterile phosphate-buffered saline (PBS) to a final concentration of 200 µg/mL for in vivo assays. All cell lines were validated by STR profiling and tested negative for mycoplasma.
Six-week-old female C57BL/6J mice were maintained under specific pathogen-free
(SPF) conditions (22
Fecal samples were collected from mice in the ATO and ATO+BP groups (n = 8 per group). Genomic DNA was extracted using the DP328 Fecal Genomic DNA Extraction Kit (Centrifugal Column Type, TIANGEN, Beijing, China), and DNA quality was assessed based on 260/280 nm and 260/230 nm absorbance ratios.
Library preparation was performed using the Rapid Plus DNA Library Prep Kit for Illumina (RK20208, ABclonal, Wuhan, China). DNA fragmentation was carried out with a Covaris LE220R-plus Ultrasonicator (Covaris, Woburn, MA, USA), and sequencing was conducted on an Illumina NovaSeq X Plus System (PE150, Illumina, San Diego, CA, USA).
Following ATO treatment, alpha diversity (Chao1, Shannon, and Simpson) and beta diversity (Bray–Curtis dissimilarity) indices were calculated to assess microbial community richness, evenness, and structural variation.
Bone marrow aspirates were collected from tibial bone marrow cavities using
1
CD11b immune cells were isolated using CD45 microbeads and MACS LS columns
(Miltenyi Biotec, Bergisch Gladbach, Germany, 130-042-301) following the
manufacturer’s protocol. Single-cell libraries were generated using the
10
Raw reads were processed using Cell Ranger v2.1.0 (10
The normalized data (NormalizeData function in Seurat package) was performed for extracting a subset of variable genes. Variable genes were identified while controlling for the strong relationship between variability and average expression. Next, we integrated data from different samples after identifying “anchors” between datasets using FindIntegrationAnchors and IntegrateData in the Seurat package. The clusters were visualized by Uniform Manifold Approximation and Projection (UMAP). In order to understand the BMME in APL, HALLMARK analysis was used to evaluate immune cells in samples [17]. The difference in the abundance of immune cell infiltration was analyzed.
Spleens and distal colons were fixed in 10% neutral-buffered formalin (Leica Biosystems, Nussloch, Germany) for 24–48 h, processed routinely, embedded in paraffin, and sectioned at 5 µm. Sections were stained with hematoxylin and eosin (H&E) for histopathological evaluation or subjected to immunohistochemistry (IHC) for Ki67 detection (DAKO, Glostrup, Denmark).
For immunofluorescence (IF), paraffin-embedded sections were deparaffinized, rehydrated, and subjected to antigen retrieval (citrate buffer, pH 6.0, 95 °C for 20 min). After blocking, sections were incubated with primary and fluorescent-conjugated secondary antibodies, followed by DAPI counterstaining. Fluorescence microscopy images were acquired using a Zeiss AXIO Imager M2 system (ZEISS, Oberkochen, Denmark), while bright-field (white light) images were captured using an Olympus CKX53 microscope (OLYMPUS, Tokyo, Japan). Antibody panels including anti-Ki67 antibody (DAKO, Glostrup, Denmark, 1:200) and, for immunofluorescence, Claudin-1 (Servicebio, Wuhan, China, GB11032, 1:500), Occludin (Abcam, Cambridge, UK, ab53032, 1:500), and ZO-1 (Servicebio, Wuhan, China, GB111402, 1:500) were employed. Secondary antibodies (ZSGB-BIO, Beijing, China) and all reagents were prepared following the manufacturers’ instructions.
Bone marrow cells were isolated by flushing the bilateral tibiae and femurs with PBS supplemented with 2% FBS, followed by filtration through a 70 µm nylon mesh for FCM analysis. Bone marrow and peripheral blood cells were treated with red blood cell (RBC) lysis buffer and then filtered through 70 µm nylon strainers (BD Falcon, Franklin Lakes, NJ, USA). Antibodies for FCM were purchased from Thermo Fisher Scientific, MA, USA. The panels included (1) APL cells: anti-hCD11b (11-0118-42) and hCD33 (12-0339-42); (2) monocytes: anti-mCD11b (12-0112-81) and Ly6C (53-5932-82); (3) T cells: anti-mCD3 (11-0032-80), CD8 (17-0081-82), and CD4 (12-0041-81). Each panel included the live/dead stain eBioscience™ Fixable Viability Dye eFluor™ 780 (65-0865-14). Data were acquired on a BD FACSCanto™ II (Becton, Dickinson and Company, NJ, USA) and analyzed using FlowJo software (version 10.8.1, https://www.flowjo.com) .
Data are presented as mean
Building upon previous reports [8], we analyzed 16S rRNA sequencing data from 20
APL patients treated with ATO based regimens (BioProject ID: PRJNA935705).
Our analysis revealed that ATO therapy induced significant alterations in gut
microbiota composition, including reduced microbial diversity and depletion of
beneficial taxa such as Bifidobacterium. A significant difference in
Shannon diversity was observed between ATO-based treatment patients (ATO group)
and the non-treated group (APL group), which demonstrated a significantly lower
diversity in the feces of ATO group (Fig. 1A). Principal coordinate analysis
(PCoA) demonstrated a significant difference in
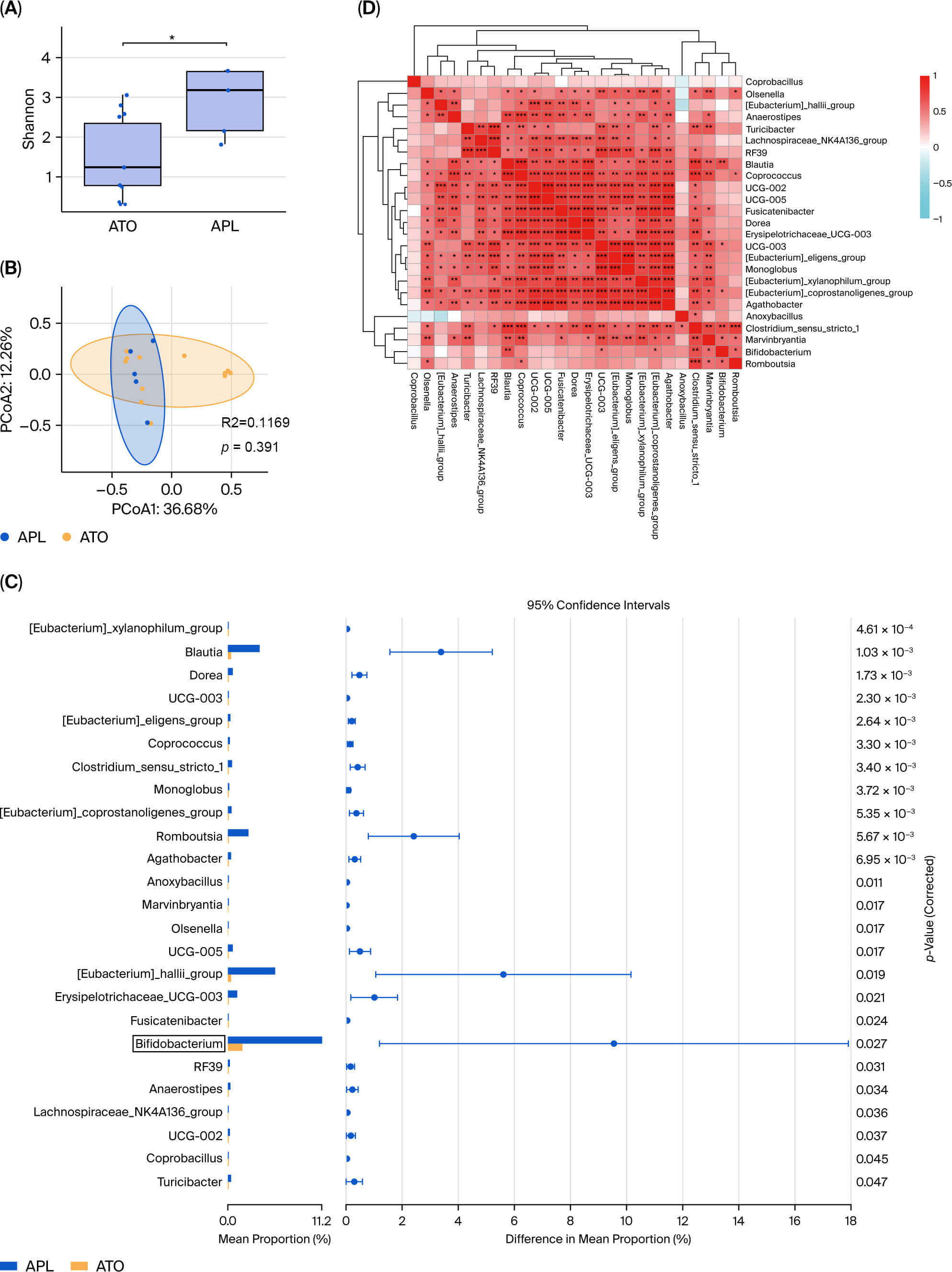 Fig. 1.
Fig. 1.
The diversity and composition of the gut microbiota are
significantly altered in APL patients after treatment with ATO. Data of 16S rRNA
genes sequenced from APL patients after or before ATO treatment. (A) The
diversity and richness of the gut microbiota in APL patients before (APL n = 5)
and after ATO treatment (ATO n = 15). (B) PCoA of a weighted UniFrac distance
analysis (n = 20), R2 = 0.1169, p = 0.0391. (C) Relative taxon
abundance comparison among the APL and ATO groups, with Bifidobacterium
showing a significant change (n = 17). (D) Spearman correlations between the
intestinal content of the 10 genera in the APL and ATO groups (red: positive
correlation; blue: negative correlation). *: p
To investigate whether ATO affects the gut microbiota in murine models, we established an APL mouse model and collected fecal samples from both ATO-treated (ATO group) and control (APL group) mice before euthanasia (Fig. 2A). A total of 16 fecal samples (8 per group) were subjected to metagenomic sequencing. Consistent with the human data, murine APL models recapitulated similar dysbiotic patterns. ATO-treated mice showed a relative reduction in OTUs compared to controls (Fig. 2B–D). Principal component analysis (PCA) of UniFrac distances confirmed microbiota shifts (Fig. 2E), and the heatmap showed a decrease in Bifidobacterium abundance, especially BP (Fig. 2F).
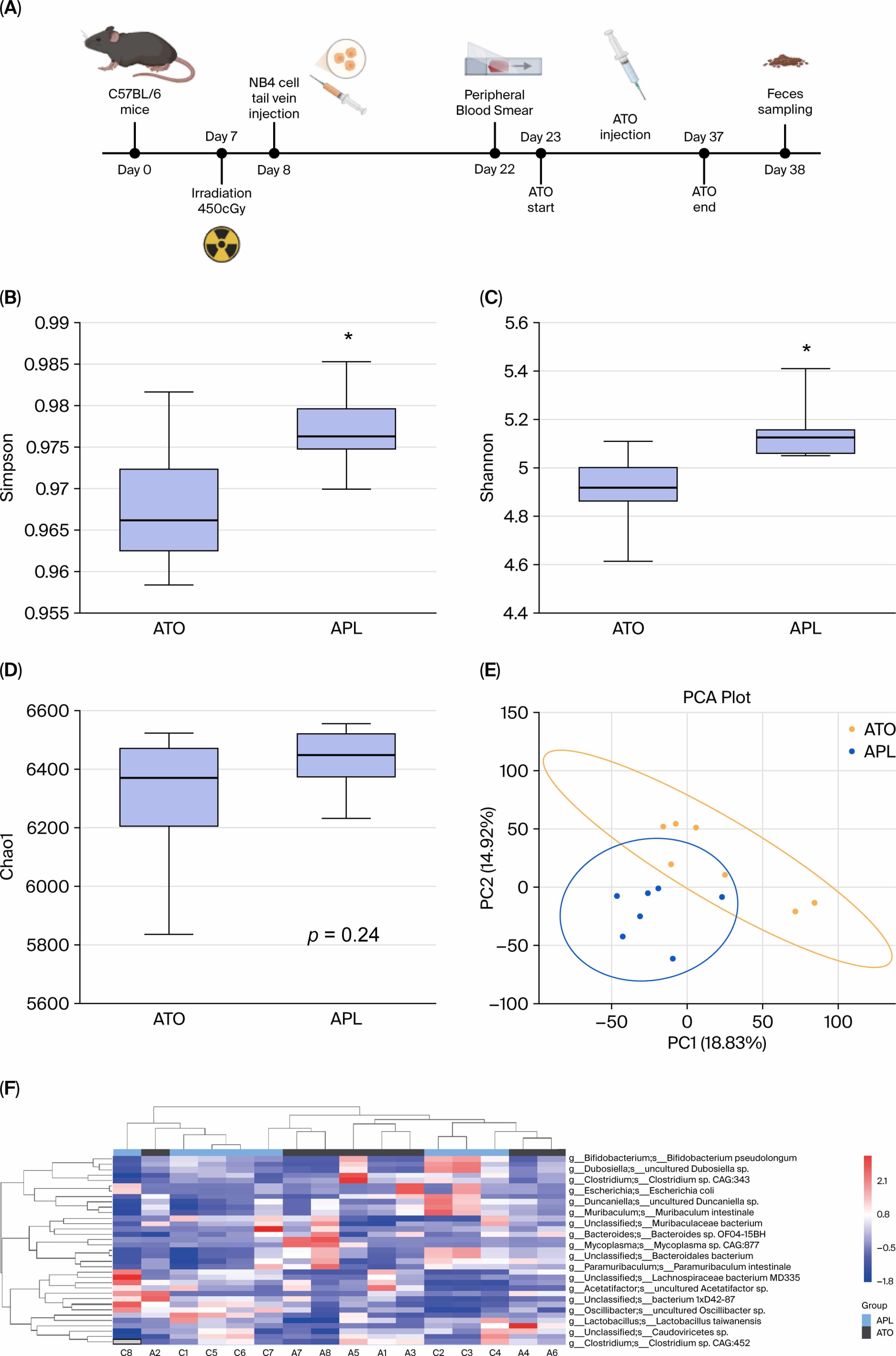 Fig. 2.
Fig. 2.
The diversity and composition of the gut microbiota are
significantly altered in APL mouse models after treatment with ATO. (A)
Schematic diagram of the mouse experimental process. (B–D) Alpha and beta
diversity based on data of bacterial metagenomics from APL murine models after or
before ATO treatment (n = 8, respectively), *p
Although LEfSe analysis did not identify significant abnormalities based on our
metagenomic sequencing data, we observed abundance differences of BP after ATO
treatment, consistent with evidence from the previous literature suggest that ATO
treatment markedly reduces Bifidobacterium abundance [8]. To investigate
the functional role of BP bacteria during ATO therapy, we supplemented ATO
administration with BP. After 14 days of treatment, bone marrow and peripheral
blood samples were collected for FCM analysis (Fig. 3A). Notably, the combination
of BP supplementation and ATO (BP+ATO) significantly reduced APL cell burden.
Specifically, in ATO+BP-treated mice, peripheral blood leukemic blasts were
reduced by 72% (4.57%
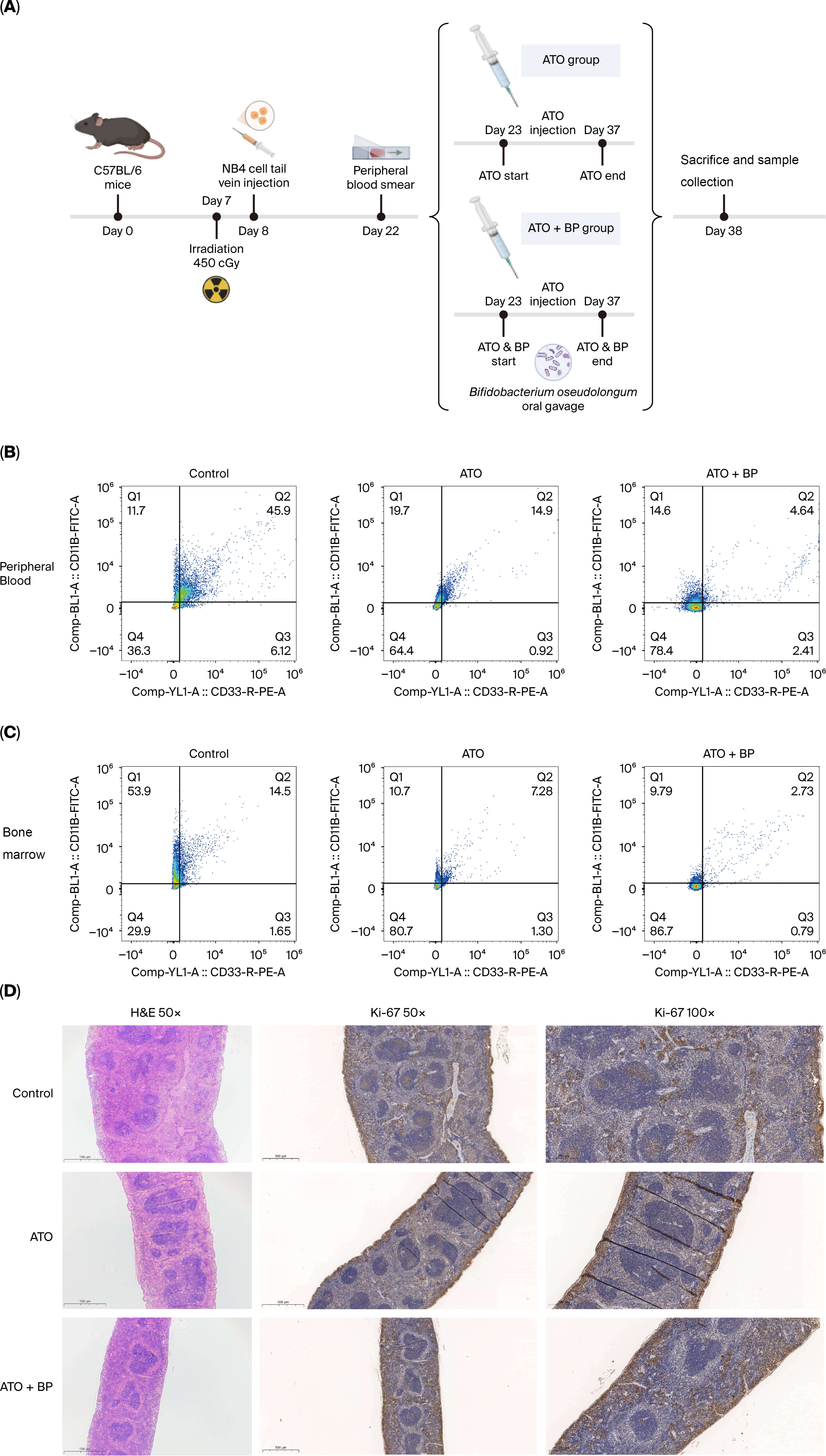 Fig. 3.
Fig. 3.
BP alleviated APL cell burden in mouse models. (A) Schematic diagram of the mouse experimental process. (B)Leukemia cells (CD11b+/CD33+ cells) in peripheral blood from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.043. (C) Leukemia cells (CD11b+/CD33+ cells) in bone marrow from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.005. (D) H&E histopathology sections and Ki-67 IHC of a representative spleen: control, ATO group, and ATO+BP group. All microscopic analyses were performed (Scale bar = 500 µm, left and middle; scale bar = 200 µm, right). BP, Bifidobacterium pseudolongum; H&E, hematoxylin and eosin.
Here, we conducted paired scRNA-seq and scTCR/BCR-seq profiling on 6 bone marrow (BM) aspirates (3 with and 3 without ATO) (Fig. 4A). As the mice had undergone irradiation prior to leukemia cell inoculation, both groups exhibited markedly low hematopoietic stem cell (HSC) proportions compared to clinical APL patients. ATO treatment induced profound remodeling of the immune microenvironment. Unsupervised immune cell clustering and scoring revealed widespread transcriptional alterations across major immune populations (Fig. 4B,C). Notably, while CD8+ T cells showed reduced abundance, post-ATO treatment. This depletion might reflect exhaustion following APL remission, suggesting that strategies to preserve or expand CD8+ T cells could amplify ATO’s immunotherapeutic efficacy. In contrast, B cells and basophils remained stable. HALLMARK analysis demonstrated commonly enhanced inflammatory responses especially in neutrophilsm monocytes and CD8+ T cells (Fig. 4D).
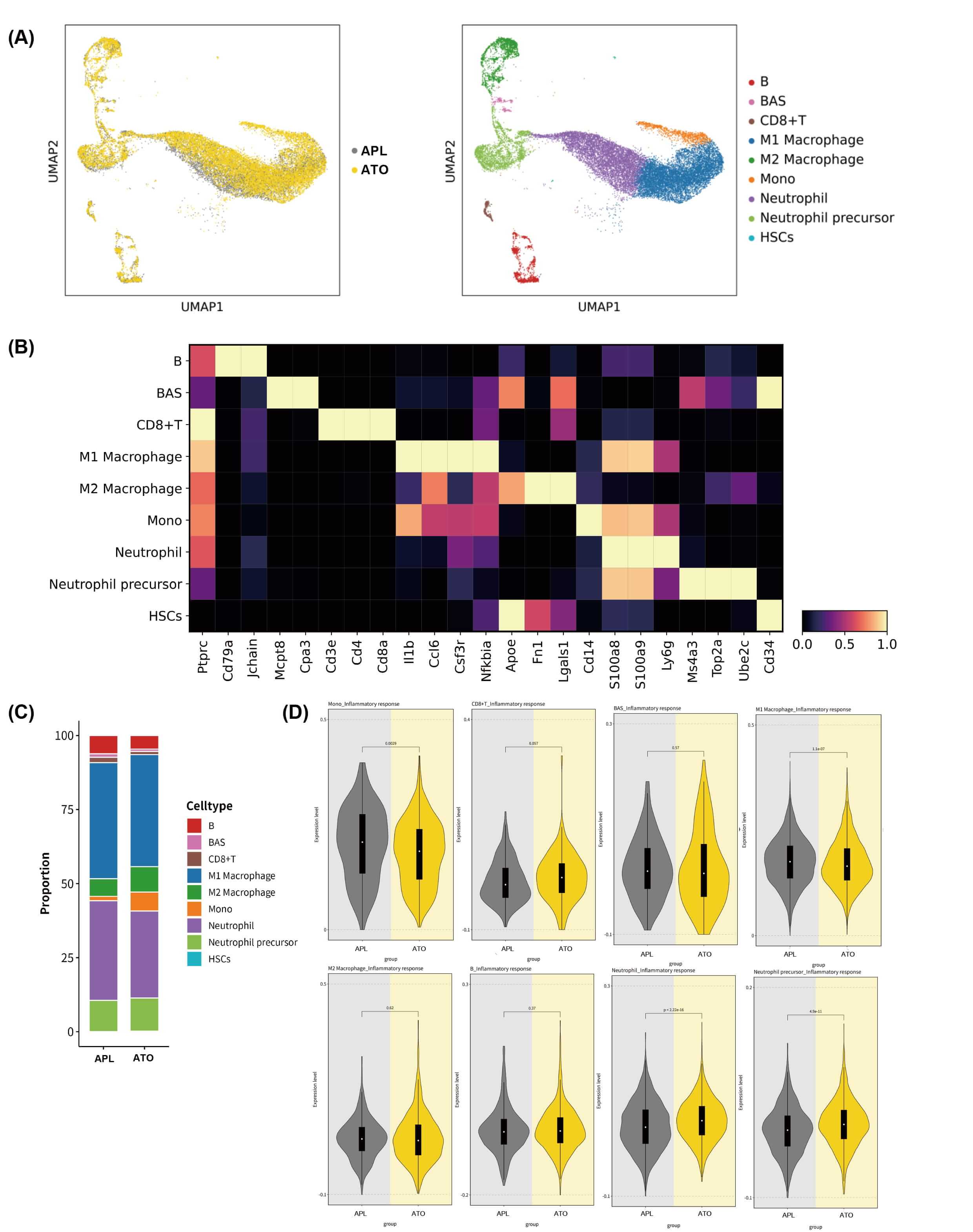 Fig. 4.
Fig. 4.
Landscape of Microenvironment in APL murine models
with ATO treatment by single-cell RNA sequencing and the functional states of APL
subtypes. (A) UMAP plot of BM samples from murine models with or without ATO.
(B) Heatmap showing the scaled expression levels of genes in specific subtypes.
(C) Proportion comparison of cell fraction between APL and ATO group. (D)
Comparative immunophenotypic analysis of ATO-treated versus untreated mice
revealed distinct immune modulation patterns. ATO administration induced
significant immunomodulatory effects across multiple immune cell subsets:
monocytes: p = 0.0029, CD8+ T cells: p = 0.057, BAS: p
= 0.57, M1 macrophages: p = 1.1
Previous studies have demonstrated that probiotics can mitigate intestinal
barrier dysfunction caused by pathological conditions or pharmacological
interventions [8, 10, 18]. In this study, we investigated whether BP
supplementation could counteract ATO-induced intestinal barrier impairment. Using
immunofluorescence analysis, we quantified the expression of tight junction
proteins (Claudin-1, Occludin, and ZO-1) in colonic epithelial cells. ATO
treatment alone compromised intestinal barrier function, as evidenced by the
decreased expression of tight junction proteins Claudin-1, Occludin, and ZO-1
(Fig. 5A, Supplementary Fig. 1A–C). Strikingly, BP supplementation in
ATO-treated mice restored tight junction integrity, as shown in Fig. 5A.
Interestingly, previous studies have demonstrated that BP bacteria modulate the
tumor microenvironment, particularly by increasing CD8+ T cell proportions and
enhancing antitumor immunity [19]. Consistently, our flow cytometry analysis of
peripheral blood and bone marrow from ATO
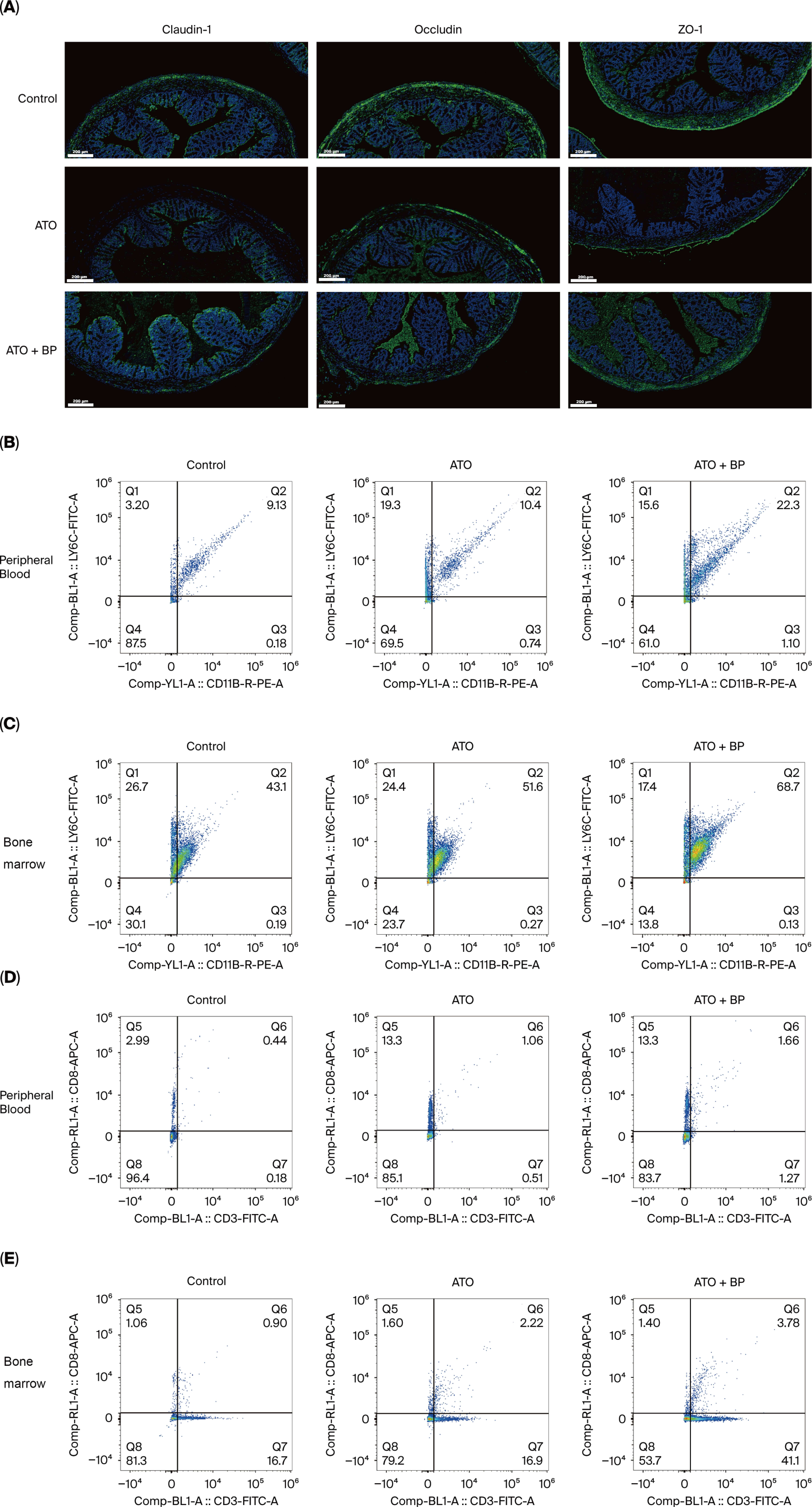 Fig. 5.
Fig. 5.
BP remodeled the immune microenvironment and repaired the intestinal barrier in murine models. (A) Representative IF images showing Claudin-1, Occludin, and ZO-1 (all in green) localization with DAPI nuclear counterstain (blue) (n = 4 per group), quantification data were shown in Supplementary Fig. 1. Scale bar: 200 µm. (B) Monocytes (CD11b+Ly6c+ cells) in peripheral blood from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.006. (C) Monocytes (CD11b+Ly6c+ cells) in bone marrow from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.068. (D) CD8+ T cells (CD3+/CD8+ cells) in peripheral blood from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.012. (E) CD8+ T cells (CD3+/CD8+ cells) in bone marrow from ATO mice (n = 4) and ATO+BP mice (n = 4). p = 0.009. IF, immunofluorescence; DAPI, 4′,6-diamidino-2-phenylindole.
In this study, we systematically investigated the synergistic effects of ATO and BP in treating APL, with a focus on mitigating ATO-induced intestinal dysbiosis and enhancing therapeutic efficacy. Using integrated in vitro, in vivo, and multi-omics approaches, we demonstrated that BP administration restored gut barrier integrity, modulated the immune microenvironment, and amplified ATO-induced leukemic cell apoptosis. Key findings included (1) ATO-induced microbiota dysbiosis characterized by Bifidobacterium depletion and pathogenic taxa expansion; (2) ATO orchestrates the APL immune microenvironment mainly by activating CD8+ T cells and monocytes; (3) BP-mediated restoration of tight junction protein (Claudin-1, Occludin, and ZO-1) production; and (4) BP increased the amount of CD8+ T cells and monocytes to remodel microenvironment (Fig. 6). These results position BP as a novel adjunct to ATO therapy, addressing both efficacy and toxicity challenges in APL treatment.
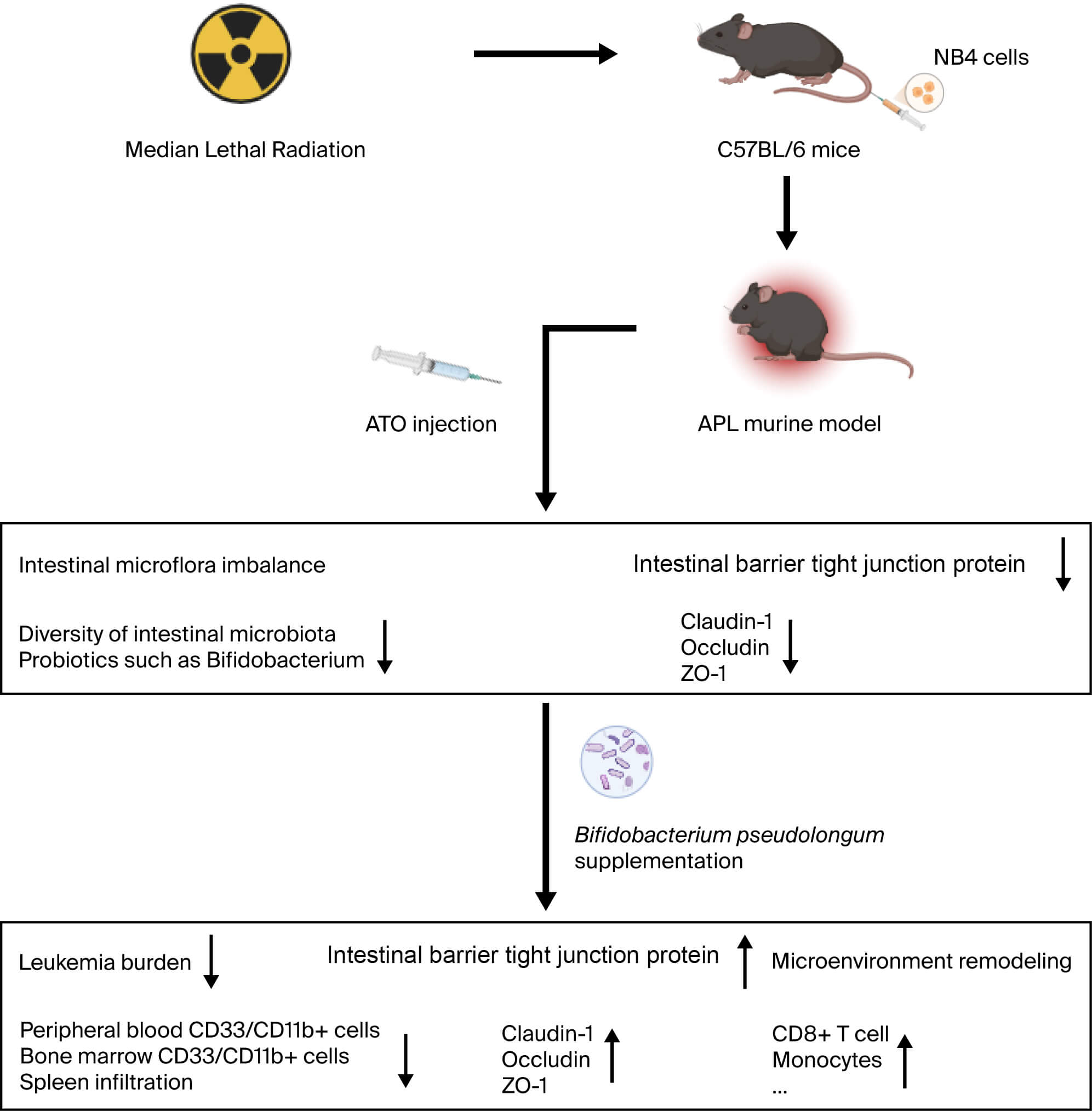 Fig. 6.
Fig. 6.
Impact of ATO on intestinal microbiota and tissue damage in
mice, with BP supplementation alleviating toxicity and potentiating drug
efficacy.
While ATO combined with ATRA achieves
BP is an anaerobic, Gram-positive probiotic bacterium within the Bifidobacterium genus that ferments sugars to produce lactic acid and acetate, while synthesizing various metabolites including L-arginine and short-chain fatty acids, synergistic exerting multiple anti-inflammatory and tumor effects [19, 26]. Intestinal barrier impairment exacerbates inflammation through inflammatory cytokines that might promote tumor progression [27]. Our scRNA-seq and flow cytometry analyses demonstrated that ATO activates cytotoxic CD8+ T cells and monocytes, while BP further expands CD8+ T cell frequencies in bone marrow (2.21-fold increase). These results directly support BP’s role in modulating anti-leukemic immunity. As crucial components of the immune system, CD8+ T cells influence the therapeutic efficacy of AML treatment [28, 29, 30], which serve as pivotal effectors in adaptive anti-tumor immunity [31, 32, 33]. Tumor-infiltrating CD8+ T cell density correlates positively with improved prognosis across multiple cancers [34, 35, 36]. Supplementing BP further enhances CD8+ T cell proliferation to exert anti-tumor effects. Previous studies propose that CD8+ T cells facilitate monocyte infiltration [37]. In our study, BP cooperates with ATO to enhance CD8+ T cell effector function. The reciprocal interaction between these immune cells presents a fascinating area worthy of in-depth investigation [38].
This study has several limitations that should be acknowledged. First, the murine cohort and limited human microbiota data may restrict the generalizability of findings, with the confounding factors could not be ruled out yet, necessitating validation in larger, more diverse cohorts. Comparing the roles of BP and other Bifidobacteria in the treatment of APL would be also an interesting topic for future research. And the contributions of direct microbial anti-leukemic effects and ATO pharmacokinetics cannot be ruled out and warrant further investigation. Second, the irradiation (450 cGy) used in our murine model likely contributed to transient lymphopenia [39], a limitation inherent to preclinical APL studies. Moreover, the use of lethally irradiated mice to establish APL models, while critical for leukemia engraftment, introduces confounding factors such as radiation-induced immunosuppression and other lesions, which may not fully recapitulate human APL microenvironment. Future studies will further investigate the effects of ATO and BP on the immune microenvironment using immunocompetent animal models. Third, direct metabolomic validation in intestinal epithelial cells or leukemia blasts remains unexplored, leaving mechanistic gaps in how BP modulates host–microbe interactions. Current studies confirm BP repairs the intestinal barrier via immunofluorescence (Claudin-1, Occludin, and ZO-1). These functional validations—independent of metabolomics—demonstrate BP indirectly modulates host-microbe interactions by restoring barrier integrity. While metabolomics uncovers molecular details, it is complementary: key findings (e.g., immune reprogramming) are already validated via flow cytometry (CD8+ T cell expansion) and microbiome analysis. We will further conduct immune cell depletion studies to strengthen causal inference. Fourth, the therapeutic window and long-term safety of BP supplementation in APL patients, particularly its impact on gut barrier integrity during ATO-induced mucositis, require further investigation. We also need to evaluate the survival benefit and safety of probiotics in immunocompromised hosts. Finally, the absence of immune checkpoint inhibitor co-therapy in our model limits insights into BP’s role in synergizing with modern immunotherapies, highlighting a critical avenue for future research.
This work provides a foundation for optimizing ATO-based APL therapy through microbiota modulation. Mechanistically, targeting the gut–leukemia axis offers a dual benefit: enhancing leukemia cell apoptosis while preserving intestinal homeostasis. Future trials should evaluate BP as an adjunct to ATO regimens, particularly in high-risk APL patients with baseline dysbiosis. Additionally, integrating BP with fecal microbiota transplantation or engineered probiotics could further personalize therapy. By addressing both efficacy and toxicity, this strategy aligns with the evolving paradigm of “precision oncology” , where microbial interventions complement conventional therapies.
This study reveals that ATO therapy disrupts gut microbiota balance in APL by depleting Bifidobacterium, exacerbating intestinal permeability and restoring microenvironment. Co-administration of BP restores microbial diversity, enhances gut barrier integrity, and synergizes with ATO in rewiring microenvironment to reduce leukemic burden.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Conceptualization, ZGuo, ZGao (equal), and XH; methodology, XN; software, XN and WZ; validation, LL, SR; formal analysis, ZGuo; investigation, QL and SF; resources, DG; data curation, LY and DG; writing—original draft preparation, ZGuo, DG and SF; writing—review and editing, ZGao and XH; visualization, LL and YL; supervision, SF; project administration, YZ and XH; funding acquisition, ZGuo, YZ, and XH. All authors have read and agreed to the published version of the manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethical approval was granted by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University (IACUC No.2025032), and this study was conducted in strict accordance with this institution’s ethical guidelines and relevant national/institutional regulations for animal experimentation.
Not applicable.
This study was supported by the National Natural Science Foundation of China (No.82274028), the Heilongjiang Key R&D Program (No.2022ZX02C09), the Fundamental Research Funds for the Provincial Universities in Heilongjiang Province (2025, Zhibo Guo), Natural Science Foundation of Heilongjiang Province (No.JJ2025PL0189), the Innovation Fund of the First Affiliated Hospital of Harbin Medical University (2024M10 and 2024M25), and 2025 Central Government Fiscal Subsidy Fund for Medical Care Compliance and Capacity Enhancement (Traditional Chinese Medicine Undertakings and Inheritance and Development Component) (230000253533210000086).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL48584.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.