




 , Baiwei Zhang 1, Zuodong Yuan 1, Chansachak Chea 1, Fei Ye 1,*
, Baiwei Zhang 1, Zuodong Yuan 1, Chansachak Chea 1, Fei Ye 1,*
1 Department of Neurosurgery, Tongji Hospital, Tongji Medical School, Huazhong University of Sciences and Technology, 430030 Wuhan, Hubei, China
Abstract
Malignant gliomas remain largely refractory to current therapies, in part because abnormal tumor vasculature and a disrupted blood-brain barrier limit intratumoral drug delivery and cause severe tumor-associated edema. The oncolytic adenovirus KD01 is a conditionally replicating adenovirus type 5 with a 27-bp deletion in Early Region 1A (E1A) and a truncated BH3-Interacting Domain Death Agonist (tBID) expression cassette inserted into the E3 region. This study investigated whether bevacizumab-induced vascular normalization would enhance the delivery and efficacy of KD01 in gliomas.
The oncolytic activity and mitochondrial effects of KD01 were evaluated in human glioma cell lines using cell viability assays, JC-1 staining, quantitative real-time-polymerase chain reaction (qRT-PCR), and western blotting for BID/tBID. An orthotopic LN229 nude mouse model was used to assess a sequential bevacizumab→KD01 regimen. Mice were randomized to receive PBS, bevacizumab, KD01, or combination treatment. Body weight and survival were recorded. Tumor cell proliferation (Ki-67), tumor vasculature (CD31), brain water content (ΔWater%), serum biochemistry, coagulation parameters, and organ weights were analyzed to evaluate antitumor activity, edema, and systemic safety.
KD01 induced robust dose- and time-dependent cytotoxicity in glioma cells and caused marked mitochondrial depolarization, accompanied by increased BID mRNA expression, loss of full-length BID, and accumulation of tBID. In the orthotopic LN229 model, bevacizumab administered 48 h before KD01 significantly improved overall outcomes compared with either monotherapy. The bevacizumab→KD01 group showed improved preservation of body weight, pronounced prolongation of survival, and the lowest Ki-67 labeling index. This group also exhibited reduced brain water content (ΔWater%), consistent with sparser CD31-positive vessels resulting from vascular normalization and oncolysis, indicating effective attenuation of tumor-associated edema. Serum liver and kidney function tests, platelet counts, coagulation indices, and major organ weights were comparable across treatment groups, suggesting no additional systemic toxicity associated with combination treatment.
KD01 exerts potent tBID-mediated mitochondrial oncolytic activity against glioma cells. When used as a priming strategy, transient vascular normalization induced by bevacizumab enhanced the intratumoral efficacy of KD01 in an orthotopic glioma model while maintaining a favorable safety profile. These findings support a simple, sequence-dependent combination approach integrating anti-VEGF therapy with oncolytic virotherapy for the treatment of malignant gliomas.
Graphical Abstract
Keywords
- glioma
- adenovirus
- oncolytic virotherapy
- KD01
- bevacizumab
- vascular normalization
Malignant gliomas, particularly glioblastomas, are among the most aggressive primary brain tumors in adults [1]. Even with current standard-of-care regimens combining maximal safe resection with radiotherapy and temozolomide, median overall survival rarely exceeds 15–20 months [1]. Treatment failure reflects not only marked genetic and phenotypic heterogeneity but also profoundly abnormal tumor vasculature and disruption of the blood-brain barrier. These vascular abnormalities severely restrict intratumoral drug delivery and distribution and contribute to pronounced tumor-associated brain edema [2]. Together, these features highlight the need for therapeutic strategies that can efficiently kill tumor cells while overcoming vascular barriers within the brain.
Bevacizumab, a recombinant humanized monoclonal antibody targeting vascular endothelial growth factor (VEGF), has been incorporated into glioblastoma management as an antiangiogenic agent [3]. Clinical trials have shown that bevacizumab can rapidly reduce contrast enhancement and peritumoral edema and improve progression-free survival; however, its impact on overall survival is modest, and resistance is nearly universal [4]. The concept of “vascular normalization” provides a plausible explanation for these paradoxical findings. Short-term, appropriately dosed VEGF blockade can transiently remodel chaotic, leaky tumor vessels into a more structurally organized and homogeneously perfused network, thereby improving drug penetration during a limited time window [5]. However, in malignant glioma, it remains unclear how best to exploit this normalization window, particularly in combination with biological agents such as oncolytic viruses, and how treatment sequence and timing influence therapeutic efficacy.
Oncolytic virotherapy offers a complementary approach for malignant gliomas by combining selective tumor cell lysis with the potential to activate antitumor immune responses [6]. Among available platforms, adenovirus type 5–based vectors are particularly attractive because of their well-characterized biology and amenability to genetic engineering [7]. KD01 is a recombinant oncolytic adenovirus derived from adenovirus type 5, engineered with a 27-bp deletion in a conserved region of Early Region 1A (E1A) to confer preferential replication in tumor cells. In addition, the E3 region is modified by deletion of the adenovirus death protein (ADP) and insertion of a truncated BH3-Interacting Domain Death Agonist (tBID) expression cassette [8]. tBID is a potent pro-apoptotic molecule that translocates to mitochondria and promotes mitochondrial outer membrane permeabilization, thereby amplifying intrinsic apoptotic signaling [9]. Previous studies in non–central nervous system tumor models have shown that KD01 and related tBID-armed vectors induce strong oncolytic effects, mitochondrial apoptosis, and immunogenic cell death with favorable safety profiles. However, their activity in gliomas and interactions with abnormal cerebral vasculature have not been systematically characterized [10].
Based on these considerations, we hypothesized that transient vascular normalization induced by bevacizumab could enhance intratumoral delivery and antitumor efficacy of KD01 in gliomas. Specifically, we postulated that administering bevacizumab within an appropriate time window before local KD01 injection would reduce vascular leakiness and tumor-associated brain edema, improve perfusion and viral distribution, and thereby potentiate tBID-mediated tumor cell killing without increasing systemic toxicity. To test this hypothesis, we evaluated the oncolytic activity of KD01 and its effects on mitochondrial function in human glioma cell lines in vitro. We further used an orthotopic LN229 nude mouse model to systematically assess the effects of a sequential bevacizumab–KD01 regimen on survival, tumor cell proliferation, brain water content, and systemic safety. By combining a tBID-armed oncolytic adenovirus with a clinically available anti-VEGF therapy, we aimed to provide preclinical evidence for a simple, sequence-dependent combination strategy to improve oncolytic virotherapy for malignant gliomas.
Human glioma cell lines A172 and LN229 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and authenticated by short tandem repeat profiling within 6 months before use. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Thermo Fisher Scientific, Grand Island, NY, USA, catalog number 11965092) supplemented with 10% fetal bovine serum (FBS; Gibco, 16140071, Grand Island, NY, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2. Cells were routinely tested and confirmed to be mycoplasma-free.
KD01 is a recombinant oncolytic adenovirus type 5 carrying a 27-bp deletion in the conserved E1A region and an engineered E3 region in which ADP is deleted and replaced by a tBID expression cassette. A replication-competent adenovirus with an ADP deletion but without tBID insertion (M20) was used as the control virus. Viral stocks were propagated in HEK293 cells, purified by double cesium chloride gradient ultracentrifugation, and dialyzed against storage buffer (10 mM Tris–HCl, 2 mM MgCl2, 5% glycerol, pH 8.0). Viral particle (vp) concentrations were determined spectrophotometrically at 260 nm, and infectious titers PFU (Plaque-Forming Units) were determined using plaque assays in HEK293 cells. Viruses were stored at –80 °C until use.
To assess oncolytic activity in vitro, A172 and LN229 cells were seeded
into 96-well plates at a density of 5
Mitochondrial membrane potential (
Total RNA was extracted from LN229 cells using TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA), and 1 µg of RNA was reverse-transcribed using a PrimeScript RT kit (Takara Bio Inc., Kusatsu, Shiga, Japan, catalog number RR037A). Quantitative real-time PCR (qRT-PCR) was performed using TB Green Premix (Takara) on a QuantStudio 6 System (Applied Biosystems, Thermo Fisher Scientific, Carlsbad, CA, USA). Primers for human BID and GAPDH (internal control) were designed using Primer-BLAST. Relative BID mRNA expression was calculated using the 2-ΔΔCt method. The detailed gene sequences are listed in Table 1.
| Gene | Primer type | Primer sequence |
| BID | Forward | 5′-CCAGGCTGTTTGAGGACCTC-3′ |
| Reverse | 5′-TGTGGGCTGCTTGTCTCTG-3′ | |
| GAPDH | Forward | 5′-GGAGCGAGATCCCTCCAAAAT-3′ |
| Reverse | 5′-GGCTGTTGTCATACTTCTCATGG-3′ |
qRT-PCR, Quantitative real-time PCR; BID, BH3-interacting domain death agonist; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
Protein expression of BID and tBID was examined by western blotting. LN229 cells
were infected with KD01 at different MOIs (0, 1, and 10) for 24 h and lysed in
RIPA buffer supplemented with protease inhibitors (Beyotime). Equal amounts of
protein were separated by SDS-polyacrylamide gel electrophoresis and transferred
onto PVDF membranes (Merck Millipore, Burlington, MA, USA). Membranes were
blocked with 5% nonfat milk and incubated overnight at 4 °C with
primary antibodies against BID (ABclonal Technology, Wuhan, China, catalog number
A23234) and
All animal procedures were approved by the Animal Experimentation Ethics Committee of Tongji Medical College (protocol code 4704) and were conducted in accordance with institutional guidelines.
Male BALB/c nude mice (4–6 weeks old; Jicui Biotechnology, Guangdong, China)
were anesthetized with isoflurane and positioned in a stereotactic frame. LN229
cells (1
Tumor-bearing mice were randomly assigned to four groups (n = 11 per group): PBS
control, bevacizumab alone, KD01 alone, and bevacizumab followed by KD01
(Bev
At the experimental endpoint, mice were humanely euthanized by intraperitoneal (i.p.) injection of an overdose of sodium pentobarbital (200 mg/kg body weight). This method induces rapid, painless anesthesia, followed by the cessation of vital functions, ensuring minimal distress to the animals. Successful euthanasia was confirmed by the absence of corneal reflexes and respiratory and cardiac activity.
Tumor-associated brain edema was assessed by measuring brain water content. On Day 5 after KD01 treatment (day 14 after tumor implantation), three mice from each group were randomly selected and subjected to deep anesthesia followed by decapitation. The brain was rapidly removed, and the tumor-bearing and contralateral hemispheres were dissected and weighed to obtain wet weights. Samples were then dried at 100 °C for 24 h and reweighed to obtain dry weights. Brain water content (%) was calculated as:
Water content = (wet weight – dry weight)/wet weight
The difference in water content between the tumor-bearing and contralateral
hemispheres (
For Ki-67 and CD31 staining, mice were transcardially perfused with PBS,
followed by 4% paraformaldehyde. Brains were harvested, fixed overnight,
paraffin-embedded, and sectioned at 4 µm. Sections were deparaffinized,
rehydrated, subjected to antigen retrieval (citrate buffer, pH 6.0), and blocked
with 5% bovine serum albumin. Sections were incubated overnight at 4 °C
with primary antibodies against Ki-67 (Abcam, Waltham, MA, USA, catalog number
ab15580) or CD31 (Abcam), followed by incubation with HRP-conjugated secondary
antibodies. Immunoreactivity was visualized with DAB substrate and counterstained
with hematoxylin. The Ki-67 labeling index was calculated as the percentage of
Ki-67-positive nuclei counted in
To evaluate systemic toxicity, blood samples were collected from the retro-orbital sinus at the experimental endpoint. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and creatinine (CREA) were measured using an automated biochemical analyzer (Cobas 8000 Modular Analyzer; Roche Diagnostics, Switzerland). Platelet counts (PLT) and prothrombin time (PT) were measured using hematology and coagulation analyzers, respectively. Major organs, including the brain, liver, and spleen, were excised and weighed.
Data are presented as mean
To generate a conditionally replicating oncolytic adenovirus, KD01 was
engineered with a small deletion in the conserved region of E1A
(
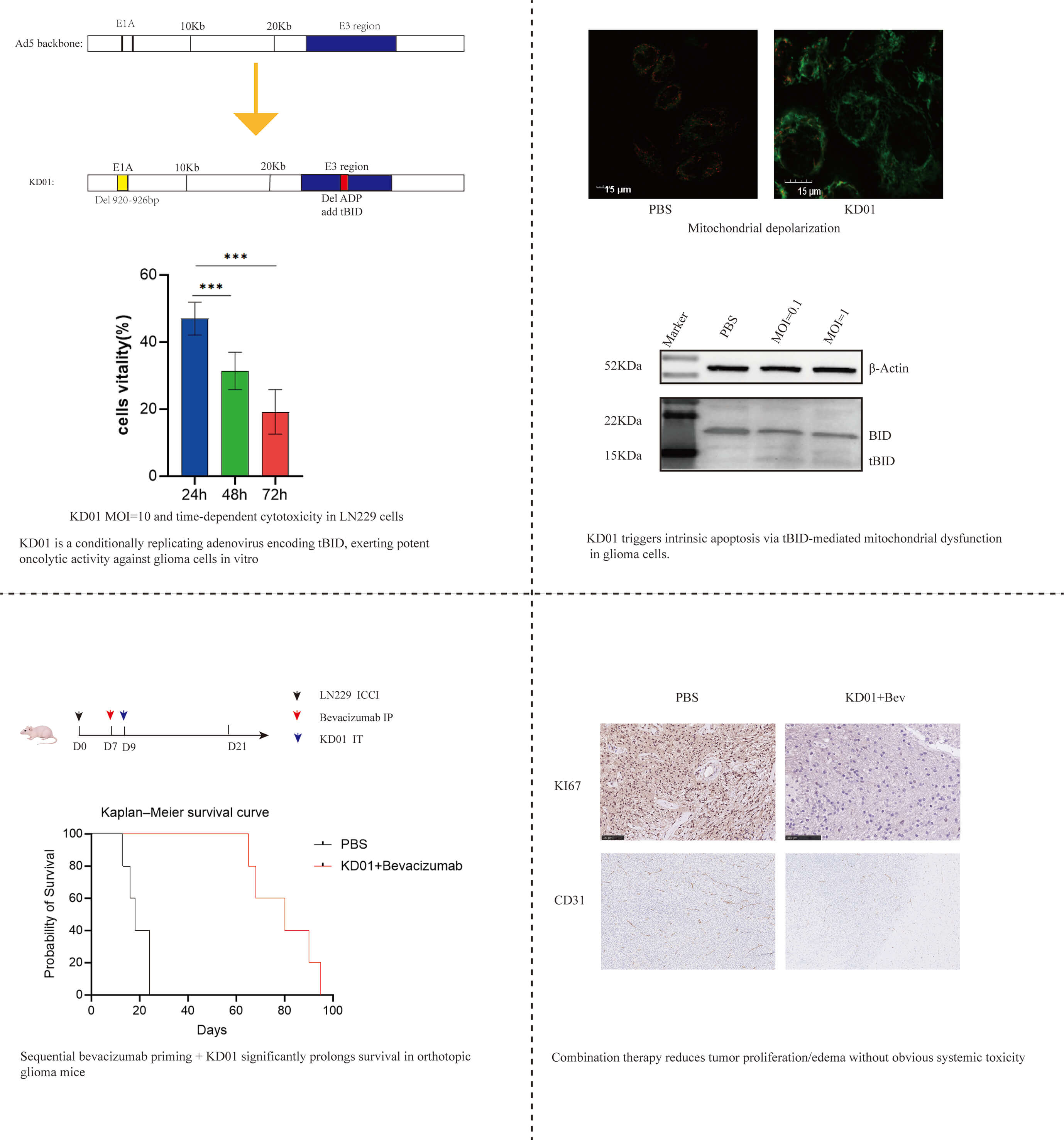
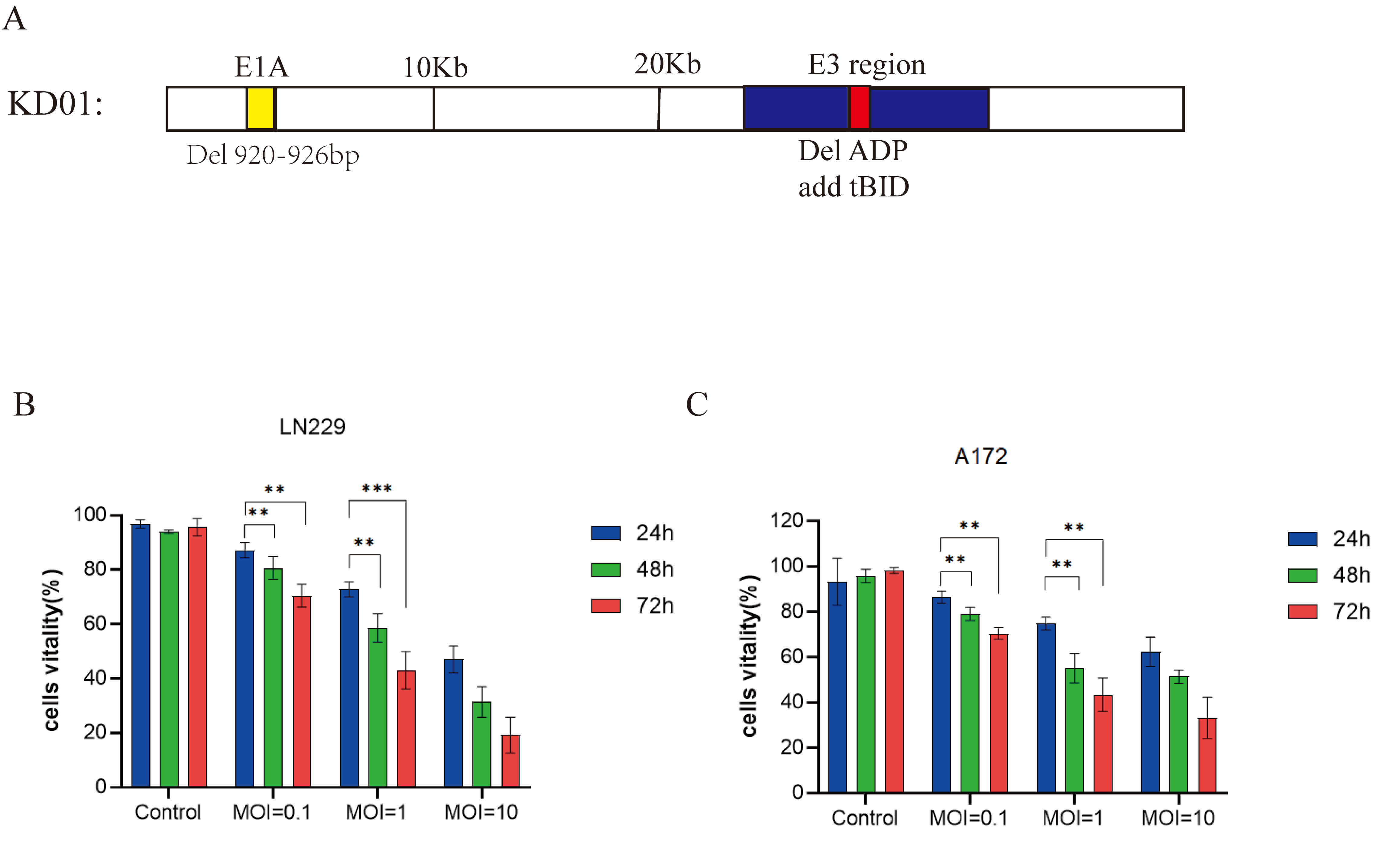 Fig. 1.
Fig. 1.
Structure of KD01 and its oncolytic activity in human glioma
cells. (A) Schematic representation of KD01. A 27-bp deletion was introduced
into the conserved region of E1A (
We first examined the intrinsic susceptibility of human glioma cells to KD01.
Infection of LN229 cells with increasing MOIs of KD01 (0.1, 1, and 10) resulted
in a clear dose- and time-dependent reduction in cell viability (Fig. 1B). Even
at an MOI of 0.1, KD01 significantly reduced cell viability compared with
controls at 48 and 72 h. At an MOI of 10, viability decreased to a small fraction
of control levels by 72 h (p
To investigate whether KD01 exerts cytotoxicity through mitochondrial apoptosis, we first assessed mitochondrial membrane potential using JC-1 staining. In LN229 cells treated with PBS, strong red JC-1 aggregates and weak green fluorescence were observed, indicating an intact mitochondrial membrane potential (Fig. 2A). Infection with the control virus M20 caused a moderate increase in green fluorescence and a reduction in red aggregates, whereas KD01 infection led to a marked loss of red fluorescence with predominantly green staining, consistent with mitochondrial depolarization. Quantification of the JC-1 aggregate/monomer ratio confirmed that KD01 induced significantly greater disruption of the mitochondrial membrane potential than M20 (Fig. 2B).
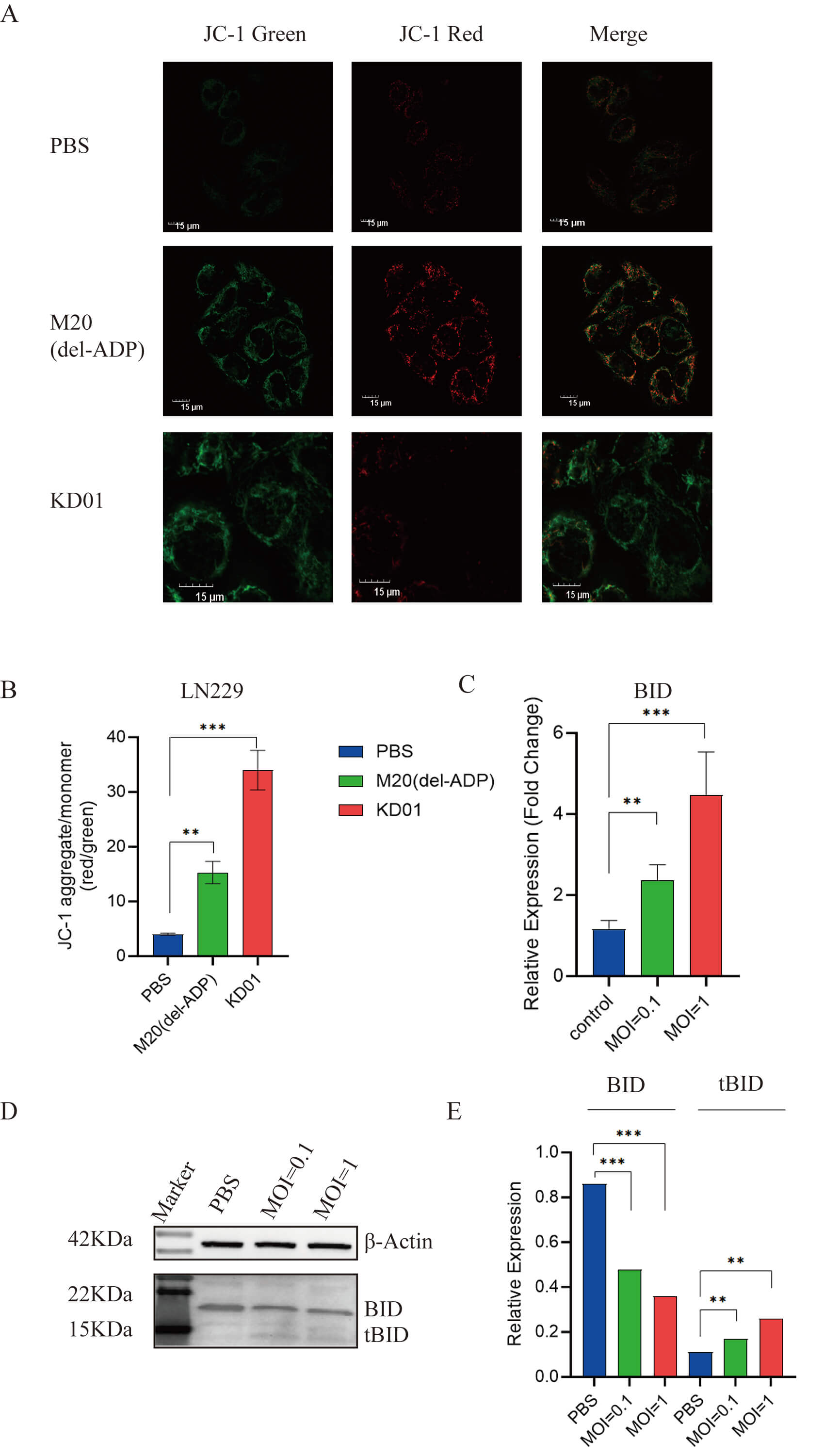 Fig. 2.
Fig. 2.
KD01 induces mitochondrial dysfunction via activation of the
BID–tBID pathway in glioma cells. (A) Representative JC-1 staining of LN229
cells treated with PBS, control virus M20 (Del-ADP), or KD01. Red fluorescence
indicates JC-1 aggregates in mitochondria with intact membrane potential, and
green fluorescence indicates JC-1 monomers in depolarized mitochondria (scale
bar, 15 µm). (B) Quantification of mitochondrial membrane potential
expressed as the ratio of JC-1 aggregates to monomers (red/green) in LN229 cells
after the indicated treatments. (C) Relative BID mRNA expression in LN229 cells
infected with KD01 at MOI 0.1 or 1 compared with PBS control, determined by
qRT-PCR. (D) Representative Western blot of full-length BID and tBID protein in
LN229 cells infected with KD01 at the indicated MOIs;
Given that KD01 encodes tBID, we next examined BID/tBID expression. KD01 infection increased BID transcript levels in a dose-dependent manner (Fig. 2C). At the protein level, full-length BID was progressively reduced, whereas tBID accumulated with increasing MOIs of KD01 (Fig. 2D,E), indicating efficient expression and processing of the tBID cassette. Taken together, these data demonstrate that KD01 potently activates the BID–tBID pathway and induces profound mitochondrial dysfunction in glioma cells, consistent with engagement of the intrinsic apoptotic pathway.
As shown in Fig. 3A, we evaluated the effects of bevacizumab combined with KD01 using a sequential treatment schedule in a nude mouse LN229 intracranial xenograft model. On day 0 (D0), LN229 cells were inoculated into the brain by stereotactic injection; on day 7 (D7), mice received an intraperitoneal injection of bevacizumab or PBS; and on day 9 (D9), KD01 or PBS was administered via stereotactic intratumoral injection. This design generated four treatment groups: PBS, KD01, bevacizumab, and KD01 + bevacizumab, and animals were monitored until death.
 Fig. 3.
Fig. 3.
Bevacizumab priming enhances antitumor efficacy of KD01 in an
orthotopic LN229 glioma model. (A) Schematic of the treatment schedule. LN229
cells were implanted intracranially on day 0 (D0). Mice received intraperitoneal
bevacizumab or PBS on day 7 (D7) and stereotactic intratumoral injection of KD01
or PBS on day 9 (D9), generating four groups: PBS, KD01, bevacizumab, and KD01 +
bevacizumab. (B) Body weight changes in each group at the indicated time points.
Data are shown as mean
Body weight changes were compared among groups as an indirect measure of
systemic toxicity and overall condition. Throughout the observation period, mice
in the PBS and KD01 groups showed a gradual decline in body weight, whereas body
weight in the bevacizumab group remained largely stable. Notably, in the
combination group, body weight did not decrease and was consistently higher than
that in the PBS and KD01 groups from D10 onward (Fig. 3B). On D14, D21, and D28,
body weight in the KD01 + bevacizumab group was significantly higher than that in
the KD01 monotherapy group (p
Kaplan–Meier survival analysis further confirmed that KD01 combined with bevacizumab significantly prolonged survival in tumor-bearing mice (Fig. 3C). Animals in the PBS group developed neurological deterioration shortly after implantation and died sequentially. Bevacizumab or KD01 monotherapy delayed death to some extent, but survival curves declined rapidly. In contrast, the survival curve of the KD01 + bevacizumab group shifted markedly to the right, with most mice surviving longer than those in the three control groups. Log-rank testing revealed statistically significant differences between the combination group and the KD01, bevacizumab, and PBS groups, indicating that sequential bevacizumab priming markedly enhanced the in vivo antitumor efficacy of KD01.
Tumor cell proliferation was next assessed using Ki-67 immunohistochemistry. In the PBS group, strong brown nuclear Ki-67 staining was diffusely distributed throughout the tumor, resulting in the highest Ki-67 labeling index. The proportion of Ki-67–positive cells was clearly reduced in both the KD01 and bevacizumab groups, although numerous positive cells remained. In the KD01 + bevacizumab group, Ki-67–positive cells were markedly decreased, with only scattered weakly positive nuclei and much lower overall staining intensity. Semi-quantitative analysis showed that the Ki-67 labeling index was lowest in the combination group and was significantly lower than that in the PBS and monotherapy groups (Fig. 3D).
To evaluate the effect of combination therapy on tumor-associated brain edema,
we measured the difference in water content between the tumor-bearing and
contralateral hemispheres (
Tumor vascular density was evaluated using CD31 (an endothelial cell marker) staining (Fig. 3F,G). The PBS group exhibited dense, disorganized CD31-positive vessels. KD01 monotherapy mildly reduced vascular density, whereas bevacizumab alone induced a more pronounced decrease. Notably, the KD01 + bevacizumab combination group displayed the sparsest CD31 staining, with only scattered vascular structures, consistent with synergistic vascular remodeling by bevacizumab-mediated normalization and KD01-induced oncolytic disruption.
To further evaluate systemic safety of sequential KD01 and bevacizumab treatment, serum biochemical and coagulation parameters were measured at the study endpoint, and major organ weights were compared among groups. Serum AST, ALP, and CREA levels remained within physiological ranges in all groups, with no statistically significant differences between the KD01, bevacizumab, or KD01 + bevacizumab groups and the PBS control group (Fig. 4A–C). ALT levels were slightly higher in the KD01 and bevacizumab monotherapy groups than in the PBS group; however, the increase was modest, and ALT levels in the KD01 + bevacizumab group were comparable to PBS (Fig. 4D). These findings suggest that either virus or anti-VEGF monotherapy may cause mild, tolerable fluctuations in liver function that are not exacerbated by the combination regimen.
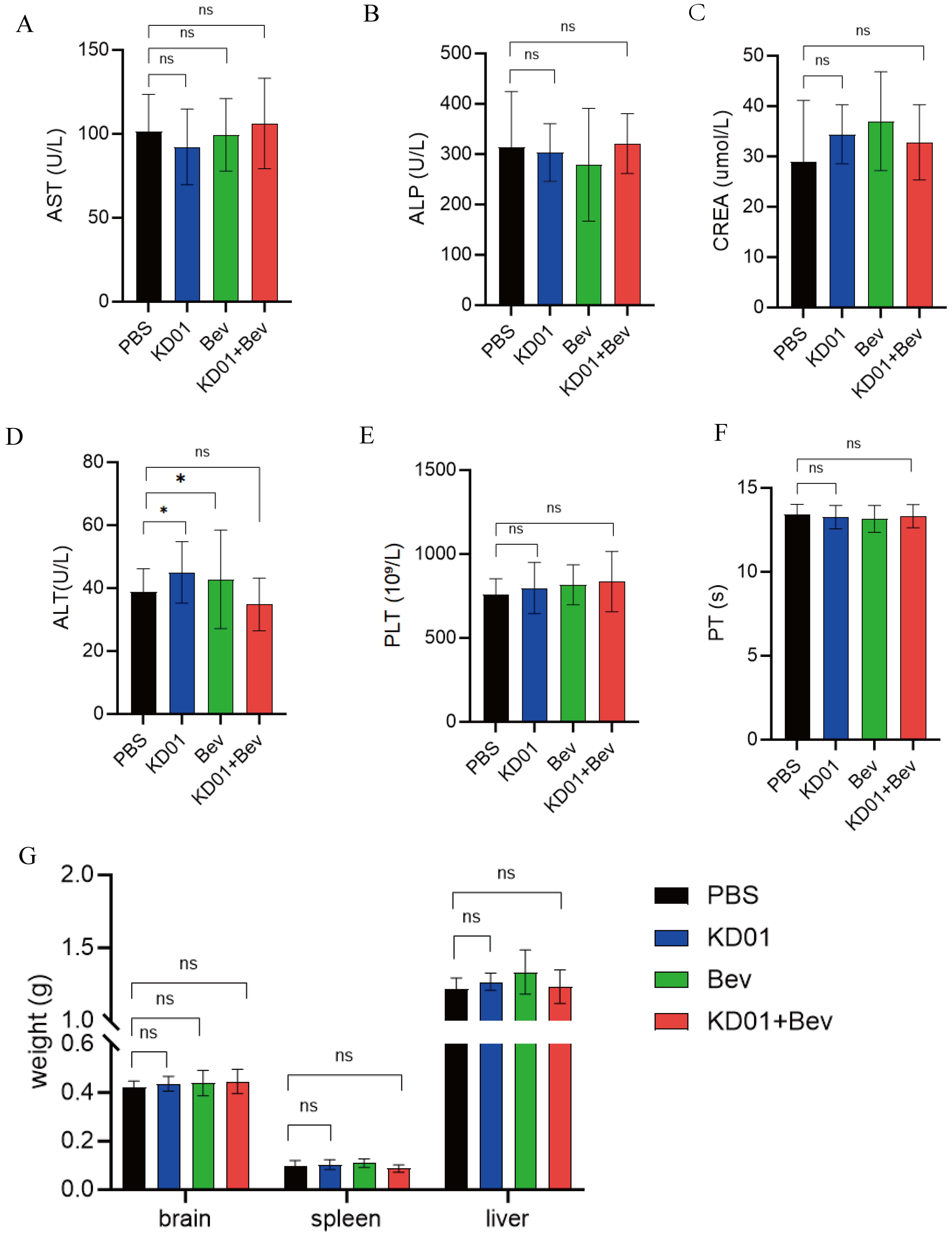 Fig. 4.
Fig. 4.
Systemic safety of sequential KD01 and bevacizumab treatment in
tumor-bearing mice. (A‒D) Serum biochemical parameters at the end of the study:
AST (A), ALP (B), CREA (C), and ALT (D) in mice treated with PBS, KD01, Bev, or
KD01 + Bev. AST, ALP, and CREA remained within the physiological range with no
significant intergroup differences; ALT showed a mild increase in the KD01 and
Bev monotherapy groups but returned to control levels in the combination group.
(E,F) PLT (E), and PT (F) among the four groups, showing no significant effects
of KD01, Bev, or their combination on coagulation function or platelet
production. (G) Relative weights of major organs (brain, spleen, and liver) at
sacrifice. No significant differences were observed among groups, and no obvious
organ atrophy or enlargement was detected. Data are presented as mean
PLT and PT values were also similar across all four groups (Fig. 4E,F), indicating that neither KD01 nor bevacizumab, alone or in combination, significantly affected coagulation function or platelet production. Likewise, relative weights of major organs, including the brain, spleen, and liver, did not differ significantly among the PBS, KD01, bevacizumab, and KD01 + bevacizumab groups (Fig. 4G), and no obvious organ atrophy or enlargement was observed.
Taken together, these results indicate that sequential bevacizumab priming combined with KD01 oncolytic therapy did not induce hepatic or renal injury, coagulation abnormalities, or major organ toxicity at the doses and schedules used in this study, supporting a favorable systemic safety profile for this combination strategy.
In this study, we systematically investigated the application of the tBID-armed, conditionally replicating adenovirus KD01 in gliomas and evaluated the feasibility of its sequential combination with bevacizumab. We demonstrated that KD01 exerts robust, dose- and time-dependent oncolytic activity against the human glioma cell lines LN229 and A172, indicating that glioma cells with distinct molecular backgrounds are highly susceptible to this vector. Mechanistically, KD01 infection led to a marked loss of mitochondrial membrane potential, accompanied by a decrease in full-length BID and pronounced accumulation of tBID, indicating activation of the BID–tBID axis and engagement of the intrinsic mitochondrial apoptotic pathway. These findings not only confirm effective expression and functional activity of tBID within the KD01 backbone but also provide a molecular basis for the strong oncolytic effects of this virus.
We therefore focused on the in vivo effects of combining anti-VEGF therapy with KD01. Compared with either KD01 or bevacizumab alone, sequential bevacizumab plus KD01 treatment produced a more pronounced survival benefit in an orthotopic LN229 nude mouse model. Mice in the combination group exhibited a smaller decline in body weight, and Kaplan–Meier analysis revealed a significant prolongation of median survival, indicating that bevacizumab priming can markedly enhance the overall therapeutic efficacy of KD01. At the histopathological level, Ki-67 staining was weakest in the combination group, with the lowest proportion of Ki-67–positive tumor cells among all treatment arms, further supporting the superior antiproliferative effect of the combination regimen.
A major cause of treatment failure in gliomas is the combination of severely disorganized tumor vasculature and blood-brain barrier disruption, which results in poor drug delivery and pronounced cerebral edema [11, 12]. Previous clinical trials have shown that bevacizumab can rapidly reduce radiographic enhancement and brain edema; however, its impact on overall survival remains limited, suggesting that VEGF blockade alone is insufficient to fundamentally alter disease progression [13]. The vascular normalization hypothesis proposes that short-term, moderate inhibition of VEGF can remodel abnormal tumor vessels into a more orderly and less permeable network, thereby improving perfusion and drug delivery [14]. In the present study, sequential administration of bevacizumab and KD01 significantly reduced water content in the tumor-bearing hemisphere, with marked alleviation of peritumoral interstitial edema and exudation. These changes were consistent with improvements in body weight and survival and indirectly support the existence of a bevacizumab-induced “vascular normalization window” that is conducive to viral delivery in this model [15]. Administration of KD01 within this window likely allows more uniform and deeper viral penetration into the tumor core and infiltrative margins, thereby amplifying its oncolytic and pro-apoptotic effects.
Importantly, the enhanced antitumor activity achieved with the combination regimen was not accompanied by increased systemic toxicity. Serum biomarkers of liver and kidney function (AST, ALT, ALP, CREA) did not differ significantly among groups, and coagulation parameters and platelet counts were unaffected. Consistent with these findings, no obvious changes were observed in the weights of major organs, including the brain, spleen, and liver. Together, these results suggest that, at the doses and schedules used in this study, bevacizumab priming does not exacerbate the systemic toxicity of KD01, supporting further development of this combination strategy from a safety perspective.
Our findings are also consistent with accumulating evidence supporting the combination of oncolytic viruses with antiangiogenic or immune-based therapies [16]. Previous studies have shown that appropriately tuned vascular normalization can facilitate intratumoral viral spread and enhance immune cell infiltration into the tumor microenvironment [17]. KD01 is characterized by tumor-selective replication, potent activation of mitochondrial apoptosis, and the potential induction of immunogenic cell death through tBID expression. In principle, this mode of tumor cell death, together with improved perfusion and reduced interstitial pressure during vascular normalization, may elicit a stronger antitumor immune response [18]. Although immune cell infiltration and functional antitumor immunity were not directly assessed in this study, these potential synergistic mechanisms warrant further investigation in immunocompetent models.
Some limitations of this study should be acknowledged. First, the in vivo efficacy of KD01 and bevacizumab was evaluated using a single human glioma cell line–derived orthotopic nude mouse model. Validation in additional models, including patient-derived organoids and glioma stem-like cells, is necessary to improve the generalizability of these findings [19]. Second, assessment of vascular normalization and edema was based primarily on brain water content measurements and conventional histology. Advanced imaging modalities, such as dynamic contrast-enhanced MRI, perfusion imaging, or spatiotemporal mapping of viral distribution, were not employed. Consequently, the precise onset and duration of the vascular normalization window could not be defined or quantitatively linked to viral delivery [20]. Third, the doses and timing of bevacizumab and KD01 administration were empirically selected rather than systematically optimized. Future dose–response and schedule–optimization studies may further enhance therapeutic efficacy or permit dose reductions [21]. Finally, immune effects are an integral component of oncolytic virotherapy [22]; however, our in vivo experiments were conducted predominantly in immunodeficient mice, limiting our ability to fully capture the impact of KD01 and bevacizumab on the tumor immune microenvironment. Addressing these issues is important in future studies.
It is also important to consider potential clinical challenges that may limit the translational applicability of this regimen. Pre-existing neutralizing antibodies (NAbs) against adenovirus type 5 are highly prevalent in human populations and could substantially reduce intratumoral delivery and replication of KD01 by sequestering viral particles before they reach glioma tissue. Strategies such as capsid engineering or transient immunosuppression may be required to mitigate this effect. Additionally, although bevacizumab can normalize tumor vasculature in preclinical models, its penetration across the intact blood-brain barrier in human gliomas is limited, particularly in infiltrative tumor regions with relatively preserved barrier integrity. This limitation may restrict the induction of a functional vascular normalization window in clinically relevant compartments, necessitating adjunctive approaches, such as blood-brain barrier-disrupting techniques or convection-enhanced delivery, to improve bevacizumab distribution within the brain. These challenges highlight the need for tailored strategies to bridge preclinical efficacy and clinical application.
In summary, this study demonstrates that the tBID-armed, conditionally replicating adenovirus KD01 exerts potent oncolytic activity against glioma cells in vitro and in vivo, and that bevacizumab-induced vascular normalization can significantly enhance its antitumor efficacy in an orthotopic glioma model while reducing brain edema without increasing systemic toxicity. These findings provide experimental support for a sequential “anti-VEGF priming plus oncolytic adenovirus” strategy for malignant glioma. This approach merits further evaluation in advanced preclinical models and early-phase clinical trials, including its integration with radiochemotherapy and immune checkpoint blockade as part of multimodal glioma treatment.
In conclusion, this study demonstrates that the tBID-armed, conditionally replicating adenovirus KD01 exerts potent mitochondria-dependent oncolytic activity against human glioma cells. In an orthotopic LN229 model, transient bevacizumab priming significantly enhanced the therapeutic efficacy of KD01, as evidenced by improved body weight maintenance, prolonged survival, reduced Ki-67 proliferation, and attenuation of tumor-associated brain edema, without introducing appreciable systemic toxicity. These findings support a simple, sequence-dependent combination strategy in which anti-VEGF-mediated vascular normalization is exploited to improve intratumoral delivery and antitumor activity of oncolytic adenoviruses in malignant gliomas. Further preclinical and early-phase clinical studies are warranted to optimize dosing and timing and to explore integration of this approach into multimodal treatment regimens.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
WG collected and analyzed whole-genome data, collected experimental data, drafted the initial manuscript, and prepared the figures. BZ collected experimental data. ZY collected mouse blood and organ/tissue samples and collated the experimental data. CC analyzed the experimental data. FY contributed to conception and supervision of the study, critical review and important revision of the manuscript, obtaining funding for the present study. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were approved by the Animal Experimentation Ethics Committee of Tongji Medical College (Approval No. 4704) on April 6, 2025, and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
We would like to thank the Animal Experiment Center of Tongji Medical College, Huazhong University of Science and Technology, for providing the experimental platform.
This research was funded by the National Key R&D Program of China (Nos.2022YFC2704204).
The authors declare no conflicts of interest.
During the preparation of this work, the authors used ChatGPT (OpenAI, San Francisco, CA, USA) to assist with translation and to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.