




 , Jianping Wang 1,*
, Jianping Wang 1,*
1 Department of Colorectal Surgery, Affiliated Jinhua Hospital, Zhejiang University School of Medicine, 321000 Jinhua, Zhejiang, China
2 Department of General Surgery, The Second Affiliated Hospital of Soochow University, 215100 Suzhou, Jiangsu, China
3 Department of Scientific Research, The Affiliated Women and Children’s Hospital of Ningbo University, 315100 Ningbo, Zhejiang, China
†These authors contributed equally.
Abstract
Colorectal cancer (CRC) is a globally prevalent malignancy with rising incidence and mortality rates over the past decades. N6-methyladenosine (m6A) is the most abundant internal RNA modification in eukaryotes, and plays a pivotal role in post-transcriptional regulation. m6A is dynamically modulated by three core components, namely methyltransferases (writers), demethylases (erasers), and binding proteins (readers), which together govern the transcription, processing, translation, decay, and stability of mRNA. There has been accumulating evidence for the association of dysregulated m6A modification with CRC pathogenesis, metastasis, and therapeutic resistance. This review summarizes the biogenesis of m6A modification and its regulatory mechanisms, and discusses the dysregulation of m6A-related factors in CRC and the functional impacts. Most importantly, the review highlights the key roles of m6A modification in mediating CRC resistance to chemotherapy, targeted therapy, and immunotherapy. These insights may facilitate the development of novel therapeutic strategies for CRC.
Keywords
- colorectal neoplasms
- N6-methyladenosine
- drug resistance
- RNA methyltransferases
- RNA demethylases
Colorectal cancer (CRC) is one of the most prevalent malignancies of the digestive system, ranking the third most common cancer only next to breast and lung cancers, which poses significant threats to public health [1]. Early-stage CRC is primarily characterized by non-specific gastrointestinal symptoms, such as abdominal distension and dyspepsia, which are easily overlooked in clinical practice, and therefore many patients miss the window of opportunity for early intervention. Furthermore, management of advanced CRC is associated with high healthcare costs and suboptimal therapeutic efficacy, which further exacerbates the burden on public health systems [2, 3, 4].
The pathogenesis, progression, metastasis, and drug resistance of CRC involve multilevel changes spanning genetics, epigenetics, and transcriptomics. Among these changes, epigenetic changes, particularly N6-methyladenosine (m6A) modification, which is the most prevalent internal transcript modification in eukaryotic mRNA, have attracted considerable research interests in the past decade. There has been emerging evidence indicating the pivotal regulatory role of m6A modification in CRC, demonstrating its dual impacts on malignant behaviors (such as proliferation, invasion, and metastasis) and therapeutic resistance to chemotherapeutic agents, targeted drugs, and immunotherapies [5]. Dysregulation of m6A modification can affect the processing, degradation, and translation of mRNA, thereby activating oncogenes while silencing tumor suppressors. These processes are intricately linked to malignant progression. In summary, the m6A regulatory network serves as an ‘epitranscriptomic switch’ governing CRC initiation and progression, and more importantly it represents a critical bottleneck in overcoming therapeutic resistance in CRC.
In this review, we comprehensively summarize the functions of m6A modification in regulating CRC and drug resistance, providing novel perspectives for clinical translation. In addition, we contend that while current research has sufficiently elucidated the mechanistic diversity, future efforts should prioritize clinical validation of druggable targets rather than remaining mired in repetitive mechanistic characterization.
m6A modification is recognized as the most prevalent internal modification of RNA in eukaryotes among the over 170 identified chemical modifications, and is extensively distributed across a variety of RNA species, including mRNA, lncRNA, and circRNA [6]. Since the first discovery in mammalian mRNA in 1974, m6A modification has been detected in over 7000 human genes and demonstrated to be evolutionarily conserved across viruses, bacteria, yeast, plants, and vertebrates [7]. m6A modification involves methylation at the nitrogen-6 position of adenosine, and predominantly occurs at RRACH motifs (where R = G/A and H = A/C/U) and is enriched in regions such as those near stop codons, within 3′ untranslated regions (3′UTRs), and in long internal exons [8].
Generally, the m6A regulatory system functions via three coordinated protein classes, namely methyltransferases (writers), demethylases (erasers), and binding proteins (readers). It is a multi-dimensional, dynamic, and precise process centered around the core “methylation writing-erasing-reading” pathway, and is simultaneously regulated by intracellular and extracellular signals. Ultimately, it achieves precise control over the post-transcriptional fate of RNA, thereby influencing gene expression and cellular functions. The mechanism for the action of m6A modification proteins is illustrated in Fig. 1.
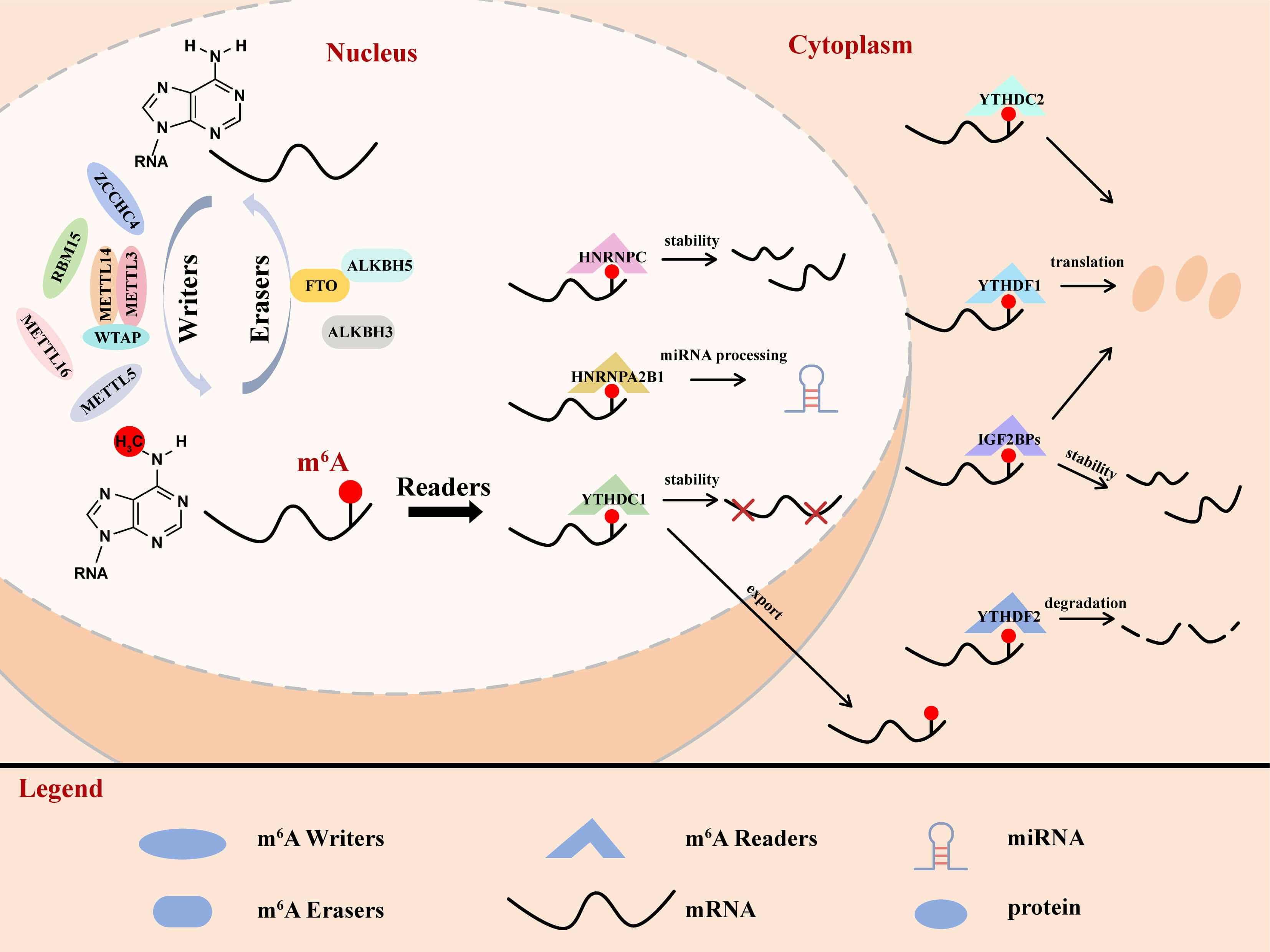 Fig. 1.
Fig. 1.
Roles of different m6A modification proteins. The m6A modification of mRNA is mainly catalyzed by the core methylase complex methyltransferase-like 3 (METTL3)- wilms tumor 1-associated protein (WTAP) - methyltransferase-like 14 (METTL14). The m6A modification is ‘erased’ by fat mass and obesity-associated protein (FTO), alkB homolog H5 (ALKBH5), and ALKBH3. The readers recognize m6A and affect various functions of the RNA, and mainly include members in the YT521-B homology (YTH) domain-containing family, the insulin-like growth factor 2 mRNA-binding protein (IGF2BP) family, and the heterogeneous nuclear ribonucleoproteins (hnRNPs) family. m6A, N6-methyladenosine; METTL3, methyltransferase-like 3; FTO, fat mass and obesity-associated protein; YTH, YT521-B homology; ALKBH, AlkB homolog H.
The core function of writers is to transfer the methyl group (-CH3) from S-adenosylmethionine (SAM, the methyl donor) to the N6 position of adenine (A) in RNA molecules, a process executed by a multi-subunit complex [9], where the methyltransferase-like 3/14 (METTL3/14) heterodimer serves as the core catalyst. Both METTL3 and METTL14 contain SAM-binding domains, though only METTL3 possesses direct methyltransferase activity. Although METTL14 is lack of intrinsic catalytic activity, it can facilitate substrate recognition by assisting METTL3 to identify specific RNA sequence motifs [10]. Wilms’ tumor 1 associated protein (WTAP) does not directly participate in catalytic reactions; instead, it primarily functions to recruit the METTL3/14 heterodimer to the transcriptional sites in cell nucleus (such as the spliceosome-associated regions) and regulate their subcellular localization [11]. Moreover, VIRMA recruits the METTL3/METTL14/WTAP complex to preferentially modify 3′UTRs and regions near the stop codons. Some other regulatory proteins including vir-like m6A methyltransferase associate (KIAA1429) and RNA-binding motif protein15 (RBM15) contribute to complex assembly and functional regulation, thereby ensuring precise m6A deposition. Notably, METTL5 and ZC3H13 govern the m6A modification of 18S and 28S ribosomal RNAs, while METTL16 regulates m6A on non-coding RNAs [12].
The dynamic reversibility of m6A is mediated by fat mass and obesity-associated
protein (FTO) and alkB homolog H5 (ALKBH5), two
m6A-modified RNAs are decoded by specialized “reader” proteins that trigger downstream biological responses, and the key families include YT521-B homology (YTH) domain proteins, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), and heterogeneous nuclear ribonucleoproteins (HNRNPs). YTH m6A RNA-binding protein (YTHDF)1/2/3 are mainly located in the cytoplasm, targeting the 3′UTR region of mRNA at the m6A site. YTHDF1 recruits ribosomes or translation initiation factors (such as eukaryotic initiation factors 3 (eIF3)) to enhance the mRNA translation efficiency. In contrast, YTHDF2 facilitates mRNA decay by recruiting the CCR4-NOT deadenylase complex and other regulatory proteins. YT521-B homology domain-containing family 3 (YTHDF3) exerts dual functions: it synergizes with YTHDF1 to promote mRNA translation, and also interacts with YTHDF2 to enhance YTHDF2-mediated RNA degradation. YTHDC1/2 are mainly located in the cytoplasm, and YTHDC1 regulates pre-mRNA splicing and nuclear export [15]. The IGF2BP family proteins (IGF2BP1–3) recognize m6A modification sites through their unique domains. After binding to mRNA, they can enhance its stability and promote its translation process [16]. Heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1) regulates pre-miRNA processing, whereas HNRNPC and HNRNPG mediate alternative splicing of m6A-containing transcripts [17]. In addition, HUR, eIF3, and Prrc2a further expand the functional repertoire of m6A recognition, linking RNA methylation to diverse cellular processes from translation control to epigenetic memory. The roles of different regulatory factors in m6A modification are summarized in Table 1.
| Type | Regulator | Mechanism |
| Methyltransferase (Writers) | METTL3 | Transfers methyl from SAM to adenine bases |
| METTL14 | Recognizes the m6A-specific sequence | |
| WTAP | Facilitates m6A modification by guiding METTL3/14 heterodimer localization to nuclear spots | |
| RBM15 | Binds to u-rich region and guide METTL3/METTL14/WTAP complex to m6A-specific motifs | |
| ZC3H13 | Combines with WTAP and enhances m6A modification | |
| ZCCHC4 | Participates in translation biology | |
| METTL16 | Targets U6 small nuclear RNA and recognizes the splice site during pre-mRNA splicing | |
| METTL5 | Catalyzes m6A installation for 18s rRNA to increase metabolic stability | |
| Demethylases (Erasers) | FTO | Mediates the demethylation of m6A in RNA |
| ALKBH5 | Preferentially recognizes the m6A for demethylation | |
| ALKBH3 | Mediates the demethylation of m6A in tRNA | |
| RNA-binding Proteins (Readers) | YTHDC1 | Regulates mRNA splicing and export of m6A-containing mRNA from the nucleus to the cytoplasm |
| YTHDC2 | Enhances translation efficiency | |
| YTHDF1 | Mediates translation promotion via the interaction with translation initiation complex | |
| YTHDF2 | Induces the instability and accelerates the degradation of m6A-methylated mRNA | |
| YTHDF3 | Facilitates translation and affects the decay of m6A-containing mRNA in synergy with YTHDF1 and YTHDF2, respectively | |
| HNRNPA2B1 | Binds m6A-bearing RNAs to elicit alternative splicing effects and promotes primary miRNA processing | |
| HNRNPC | Affects the abundance and alternative splicing of target RNAs | |
| IGF2BP1/2/3 | Promotes mRNA stability and translation | |
| eIF3 | Initiates translation in a cap-independent manner |
Abbreviations: METTL, methyltransferase-like; WTAP, wilms’ tumor 1 associated protein; ALKBH, AlkB homolog H; FTO, fat mass and obesity-associated protein; ZC3H, zinc finger CCCH-type containing; IGF2BP, insulin-like growth factor 2 mRNA-binding proteins; HNRNP, heterogeneous nuclear ribonucleoprotein; YTHDF, YTH m6A RNA-binding protein; HNRNPA2B1, heterogeneous nuclear ribonucleoprotein A2B1; RBM15, RNA-binding motif protein15; ZCCHC4, zinc finger CCHC-type containing 4; eIF3, eukaryotic initiation factors 3; YTHDF3, YT521-B homology domain-containing family 3.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) and colorimetric assays are commonly employed for global m6A quantification. LC-MS/MS involves the enzymatic digestion of RNA, followed by chromatographic separation and mass spectrometric analysis to accurately determine the ratio of m6A to total adenosine [18]. Colorimetric assays, which are based on ELISA-like principles, utilize m6A antibodies and color development for semi-quantitative detection [19]. Both approaches are straightforward and appropriate for assessing the overall m6A level. However, they cannot provide transcript- or site-specific information, which limits their utility in mechanistic studies.
MeRIP-seq (m6A-seq) remains the most widely used method for m6A detection, which combines m6A-specific antibody-based immunoprecipitation of methylated RNA fragments with high-throughput sequencing for transcriptome-wide mapping of m6A modifications [20]. This approach benefits from a well-established workflow and commercially available reagents, but suffers from several limitations, including low resolutions (typically spanning 100–200 nucleotides), inability to pinpoint the modified adenosines at single-base resolution, low reproducibility, and requirement for a substantial amount of input RNA. To overcome the resolution constraint, a m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) method was developed [21], which employs UV cross-linking to covalently link antibodies to m6A sites, introducing mutations or truncations during reverse transcription to allow base-resolution mapping. However, the miCLIP method is still highly dependent on antibody quality and cross-linking efficiency, and struggles in resolving closely adjacent m6A sites.
Advancements in epitranscriptomics have led to the emergence of several novel m6A detection technologies. In 2019, Meyer [22] developed DART-seq, which employs a fusion protein of the YTH domain and APOBEC1 enzyme to identify m6A sites in an antibody-free manner. In the same year, researchers introduced MAZTER-seq and m6A-REF-seq, both leveraging the m6A-sensitive MazF ribonuclease for site-specific cleavage within defined motifs [23]. Subsequent innovations include m6A-SEAL-seq, which employs the demethylase FTO for selective chemical labeling [24], and m6A-SAC-seq based on selective allyl chemical labeling [25]. Beyond these next-generation sequencing-based methods, nanopore direct RNA sequencing represents a third-generation strategy that enables the detection of modifications in native RNA, and does not require reverse transcription, antibodies, or enzymatic steps [26]. This method provides single-molecule resolution and can simultaneously identify a broad spectrum of RNA modifications beyond m6A.
Despite these advancements, m6A detection is still faced with multiple challenges, including limited resolution and specificity, antibody dependency, sample throughput restrictions, lack of data standardization, difficulties in detecting isoform-specific modifications, and high sequencing costs [27]. Looking forward, the field is moving toward the integration of multiple technologies, single-cell multi-omics analyses, and clinical translation. In parallel, the expansion of public datasets and development in deep learning are expected to yield more accurate and efficient algorithms for m6A identification and quantification.
The m6A modification has been reported to participate in nearly all aspects of RNA metabolic regulation, including mRNA polyadenylation, pre-mRNA splicing, nuclear export of mRNA, as well as mRNA stability and translation under normal physiological conditions [28]. m6A modification plays a pivotal role in regulating critical biological processes such as cell proliferation, differentiation, development, and senescence [29]. Dysregulation of m6A modification is closely linked to the pathogenesis and progression of multiple diseases, particularly in cancers. During tumorigenesis, aberrant expression or functional perturbation of m6A regulatory enzymes, including “writers” (such as METTL3 and METTL14), “erasers” (such as FTO and ALKBH5), and their associated regulators, disrupts global and transcript-specific m6A methylation landscapes. This dysregulation subsequently modulates a broad spectrum of malignant phenotypes, encompassing cancer initiation, progression, metastasis, metabolic reprogramming (such as glycolysis), therapy resistance, immune evasion, cancer stem cell self-renewal, and remodeling of the tumor microenvironment. These findings collectively underscore the significant therapeutic potential of targeting the dysregulated m6A machinery. The role of m6A modification in other types of cancer has been reviewed in detail by other studies [30, 31].
Aberrant m6A modification frequently occurs under CRC, and is closely associated with tumor initiation, progression, metastasis, and drug resistance. Therefore, comprehensive insights into the mechanistic roles of m6A modification in CRC hold significant theoretical and clinical importance in elucidating the disease pathogenesis and identifying novel therapeutic targets.
Numerous studies have demonstrated aberrant expression of m6A modification proteins in both CRC tissues and cell lines, and their dysregulation is mechanistically linked to CRC and disease progression. It has been shown that most m6A-related genes such as METTL3, WATP, and YTHDF1 are significantly up-regulated in tumor tissues, while METTL14, YTHDF3, and ALKBH5 were downregulated in CRC as determined with multiple databases (The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), Human Protein Atlas (HPA) and TMA) [32, 33]. An analysis of MeRIP-seq data revealed the presence of m6A peaks in most mRNAs in CRC patients, and CRC samples had 1343 out-of-balance m6A peaks (625 significantly up-regulated and 718 significantly down-regulated) [34]. Wang et al. [35] further demonstrated that elevated m6A modification levels and expression of METTL3, METTL16, and WTAP are significantly associated with poor clinical outcomes in CRC patients, suggesting their potential as prognostic biomarkers. The dysregulation of m6A modification proteins confer their oncogenic or anti-oncogenic functions in CRC pathogenesis and progression.
Consistently, it has been demonstrated that METTL3 and WTAP are overexpressed in CRC tissues and strongly correlated with poor prognosis in CRC patients. From a functional perspective, these m6A writers act as oncogene by promoting CRC cell invasion, migration, tumor progression, cancer stem cell maintenance, and drug resistance [36, 37, 38]. In contrast to METTL3, METTL14 shows opposite expression patterns and functions in CRC. METTL14 is downregulated and regarded as an independent risk factor in CRC. It has been shown that KDM5C-mediated demethylation of H3K4me3 in the METTL14 promoter reduces METTL14 transcription. Moreover, METTL14 increases the m6A methylation of SOX4 mRNA in a YTHDF2-dependent manner, thereby inhibiting SOX4-mediated EMT process and the PI3K/AKT pathway, ultimately suppressing CRC tumorigenesis [39].
FTO is significantly upregulated in CRC tissues and cells, where it promotes tumor cell proliferation while suppressing apoptosis [40, 41]. In contrast, the role of another demethylase ALKBH5 in CRC remains controversial: one study proposed that ALKBH5 downregulation is significantly associated with poor prognosis in CRC patients [42], while another study demonstrated that elevated ALKBH5 expression represents unfavorable prognosis in CRC [43].
m6A modification plays an important role in the occurrence and development of
CRC by modifying the mRNA of some key CRC genes, affecting their stability,
translation efficiency, splicing, and other processes, thereby regulating their
expression levels (Table 2, Ref. [41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57]). METTL3 affects the m6A
modification level of Myc proto-oncogene protein (MYC), influences the stability
of MYC mRNA in an IGF2BP1-dependent manner, and promotes CRC cell proliferation,
migration, and invasion [44]. Moreover, it can increase the transcripts of
Sry-related HMG box (SOX)2 and maintain the mRNA stability of SOX2 together with
IGF2BP2, thereby affecting the expression of downstream target genes MYC,
CCND1, and pOU5F1, and further influencing tumorigenesis and tumor
metastasis [45]. METTL3 further drives CRC progression by regulating
proliferation-related genes. Overexpression of METTL3 was found to enhance m6A
modification in the 5′UTR region of cyclin E1 (CCNE1) mRNA, thereby
increasing its stability and ultimately promoting the proliferation and colony
formation capabilities of CRC cells [46]. METTL3 also promotes m6A modification
on lncRNA hepatocyte nuclear factor 1-
| Gene | Expression | Target genes | Reader | Function | Sample source | Detection method | Ref. |
| METTL3 | Up | MYC | IGF2BP1 | Promotes MYC and cell proliferation, migration, and invasion | 20 pairs of CRC patient tissues; the TCGA database (434 cases) | IHC, flow cytometry, RIP | [44] |
| METTL3 | Up | SOX2 | IGF2BP2 | Promotes SOX2 and CRC cell stemness and metastasis | 432 CRC specimens (primary, lymph node, liver metastases); TCGA database | IHC, MeRIP-seq, RNA-seq, RNA pull-down, RIP | [45] |
| METTL3 | Up | CCNE1 | - | Stabilizes CCNE1 and facilitates CRC progression | Human CRC tissues (32 pairs) | MeRIP; Luciferase | [46] |
| METTL3 | Up | YPEL5 | YTHDF2 | Represses YPEL5 to accelerate CRC formation and metastasis | Human CRC tissues | MeRIP; RIP; Luciferase | [47] |
| METTL3 | Up | STAG3 | IGF2BP2 | Promotes STAG3 and promotes cell proliferation and migration while inhibiting apoptosis | CRC tissues (30 cases) and adjacent normal tissues (30 cases) | MeRIP, flow cytometry, RIP, pull-down assays | [48] |
| METTL14 | - | KLF4 | IGF2BP2 | MeCP2 binds to METTL14 to co-regulate KLF4 and promote metastasis in CRC cells | 216 paired human CRC and normal tissues; TCGA data | IHC; Co-IP; RNA-seq; MeRIP; Dual-luciferase reporter assay | [49] |
| WTAP | Up | SOD2 | IGF2BP3 | Elevates SOD2 and promotes the CRC tumorigenesis | 30 paired human CRC and normal tissues | MeRIP-qPCR, RIP, Bioinformatics | [50] |
| KIAA1429 | Up | WEE1 | - | Inhibits WEE1 and promotes CRC cell proliferation | 43 paired human CRC tissues; 111 CRC tissues | IHC, RNA-seq, RIP-seq, MeRIP, Luciferase Reporter Assay | [51] |
| KIAA1429 | Up | SIRT1 | - | Increases SIRT1 and promotes tumor progression | Human CRC and paired normal tissues, TCGA/GEPIA database | IHC, qRT-PCR, RIP, m6A-RIP | [52] |
| RBM15 | Down | E2F2 | - | RBM15 stabilizes E2F2 and promotes malignant cellular processes in CRC cells. | Human CRC tissues (n = 57 pairs) | m6A RNA Methylation Quantification, RIP | [53] |
| FTO | Up | MZF1 | - | GSK3 |
Human CRC tissues (n = 57) | MeRIP, PAR-CLIP | [41] |
| FTO | - | KCTD15 | YTHDF2 | Prevents KCTD15 mRNA degradation, inhibits CRC cell growth and triggers apoptosis | Human CRC tissues (n = 125) | RIP, Me-RIP, Dual-luciferase reporter assay | [57] |
| ALKBH5 | Down | PHF20 | - | Decreases PHF20 to suppress CRC progression | Human CRC tissues (n = 57) | MeRIP, PAR-CLIP | [42] |
| ALKBH5 | - | AXIN2 | IGF2BP1 | ALKBH5 inhibits AXIN2 to foster an immunosuppressive tumor microenvironment. ALKBH5 knockdown enhances the efficacy of anti-PD-1 therapy | - | MeRIP-qPCR, MeRIP-seq, RIP | [43] |
| ALKBH5 | Up | RAB5A | YTHDF2 | Promotes RAB5A and the proliferation, migration and invasion abilities of CRC cells | Human CRC tissues (n = 35) | MeRIP-seq, MeRIP-qPCR, Luciferase reporter assay, RIP | [54] |
| YTHDF1 | UP | SH3TC2 | Enhances SH3TC2 and promotes proliferation and tumor growth | 12 paired tumour and adjacent normal tissues | MeRIP-qPCR, RIP | [55] | |
| IGF2BP3 | Up | CCND1 | Enhances CCND1 to promote proliferation and cell cycle progression | Colon cancer specimens and paired non-tumor bowel tissues | MeRIP-qPCR, RIP | [56] |
CRC, Colorectal cancer; CCNE1, Cyclin E1; CCND1, cyclin D1; SH3TC2, SH3 domain
and tetratricopeptide repeats 2; AXIN2, axis inhibition protein 2; KCTD15, potassium channel tetramerization
domain containing 15; PHF20, plant homeodomain finger protein 20; SIRT1, Sirtuin 1; WEE1, Wee1-like protein
kinase 1; YTHDF2, YTH domain family 2; RIP, RNA Immunoprecipitation; PAR-CLIP,
Photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation;
Co-IP, Co-Immunoprecipitation; AMPK
Zhou et al. [50] showed that silencing of WTAP potently restrained the
CRC tumorigenesis in virto and in vivo. Mechanically, WTAP
regulates SOD2 m6A modification in an IGF2BP3-dependent manner to maintain its
mRNA stability. Notably, SOD2 overexpression could rescue the tumor-suppressive
effects induced by WTAP knockdown. The m6A methyltransferase KIAA1429 acts as an
oncogenic factor in CRC. It has been suggested that KIAA1429 promotes the
proliferation, colony formation, and growth of CRC cells by regulating the level
of m6A and expression of Wee1-like protein kinase 1 (WEE1), SIRT1, and
NF
Numerous studies have demonstrated that FTO plays pivotal roles in regulating cell cycle progression and apoptosis in CRC. Mechanistically, FTO reduces the m6A modification level of the key oncogenes MYC and myeloid zinc finger 1 (MZF1), thereby increasing the proportion of S-phase CRC cells [41]. Downregulation of ALKBH5 is significantly associated with poor prognosis in CRC patients, and ALKBH5 has been revealed to suppress the proliferation, migration, and invasion capacities of LOVO and RKO cells. In terms of mechanism, ALKBH5 may destabilize PHF20 mRNA by removing m6A modification in the 3′UTR region [42]. In contrast, another study demonstrated that elevated ALKBH5 expression generally represents unfavorable prognosis in CRC [43]. Shen et al. [54] found that ALKBH5 promotes the proliferation, migration, and invasion abilities of CRC cells in vitro and enhances subcutaneous tumor growth in vivo. Mechanistically, ALKBH5 can perform post-transcriptional activation of RAB5A by m6A demethylation through a YTHDF2-mediated pathway.
Reader proteins regulate RNA metabolic processes through m6A and are involved in the regulation of CRC. YTHDF1 directly binds to m6A-modified SH3TC2 mRNA and enhances its expression, thereby accelerating cell cycle progression and promoting tumor growth in CRC [55]. Yang et al. [56] demonstrated that IGF2BP3 promotes the cell cycle progression and proliferation of CRC cells by recognizing m6A modification in CCND1 and enhancing its mRNA expression.
Extensive research has demonstrated that m6A modification regulates CRC
progression through multiple signaling pathways (Table 3, Ref. [59, 60, 61, 62, 63, 64, 65, 66, 67, 68]). METTL3
modulates the m6A modification level of crumbs protein homolog 3 (CRB3)
mRNA in CRC cells. The m6A-YTHDF2 axis suppresses CRB3 protein translation
efficiency, ultimately reducing the phosphorylation levels of key Hippo pathway
components (MST1, LATS1, MOB1, and YAP) [59]. METTL3 upregulates plasminogen
activator (PLAU) mRNA in an m6A-dependent manner, and then participates in the
MAPK/ Extracellular signal-regulated kinase (ERK) pathway to promote angiogenesis
and metastasis in CRC [60]. METTL3 also regulates the m6A modification of
Membrane-bound erythropoietin-producing hepatocellular receptor tyrosine kinase
class A2 (EphA2) and vascular endothelial growth factor A (VEGFA), and stabilizes
their transcripts through IGF2BP2/3 to prevent their degradation, thereby
activating both the PI3K/AKT and ERK1/2 signaling pathways and promoting
vasculogenic mimicry formation in CRC [61]. Notably, METTL3 upregulates JAK1 and
Signal transducer and activator of transcription 3 (STAT3) expression through
both m6A-dependent and -independent mechanisms, and cooperates with
NF-
| Gene | Expression | Target genes | Reader | Function | Sample source | Detection method | Ref. |
| METTL3 | Up | CRB3 | YTHDF2 | Inhibits CRB3 expression and Hippo pathway, promotes the proliferation, migration, and invasion of CRC cells | Human CRC, adenoma, and normal tissues | m6A epitranscriptomic microarray, MeRIP-qPCR, RIP, Luciferase reporter | [59] |
| METTL3 | Up | PLAU | - | Promotes PLAU and MAPK/ERK pathway, promotes angiogenesis and metastasis | CRC tissues (32 paired) and adjacent normal tissues | RNA-seq, MeRIP-seq, MeRIP-PCR, luciferase reporter assay | [60] |
| METTL3 | Up | EphA2 VEGFA | IGF2BP2/3 | Promotes EphA2 and VEGFA vasculogenic mimicry via PI3K/AKT and ERK1/2 signaling | Human CRC and paired adjacent tissues | RNA-seq, MeRIP-qPCR, Luciferase reporter | [61] |
| METTL3 | Up | JAK1 | YTHDF1 | Promotes JAK1 and interacts with NF- |
Human CRC and paired normal tissue | MeRIP-seq, ChIP, RIP, Luciferase reporter | [62] |
| METTL3 | - | p38 and ERK | - | Inhibits p38/ERK pathways and proliferation, migration and invasion in CRC cells | CRC tissues (tissue microarray, 181 patients) | [63] | |
| YTHDF1 | Up | GMEB2 | Promotes GMEB2, thereby activating NF- |
Clinical samples. The expression data of GMEB2, ADRM1 and YTHDF1 were downloaded from the TCGA and GEO | MeRIP-qPCR, RIP, SRAMP website predict the m6A modification sites | [64] | |
| YTHDF1 | - | TCF7L2, TCF4 | Promotes TCF7L2/TCF4, thereby activating Wnt/ |
- | MeRIP-qPCR, m6A-seq, RIP-qPCR, Ribo-seq, Luciferase reporter assay | [65] | |
| YTHDF1 | Up | ARHGEF2 | Enhances ARHGEF2, thereby activating RhoA signaling to promote cell growth and lung and liver metastasis | Human CRC tissues (n = 151), 208 CRC TMA | MeRIP-qPCR, RIP-qPCR, MeRIP-seq, RIP-seq, Ribo-seq | [66] | |
| YTHDF2 | Up | GSK3 |
MiR-6125 downregulates YTHDF2, thereby increasing GSK3 |
Human CRC tissues (n = 150) | MeRIP-qPCR, RIP-qPCR, Luciferase reporter assay | [67] | |
| YTHDF2 | - | STEAP3 | STEAP3-AS1 interacts with YTHDF2 to increase STEAP3, subsequently activating Wnt signaling to support CRC progression | - | Me-RIP, RIP, RNA pulldown SRAMP website predict the m6A modification sites | [68] |
GSK3
YTHDF1 is widely involved in the regulation of signaling pathways. It can
promote the expression of ARHGEF2, TCF7L2, T-cell factor 4 (TCF4), and
Glucocorticoid modulatory element-binding proteins 2 (GMEB2) in an m6A-dependent
manner, thereby activating multiple oncogenic signaling pathways including RhoA,
Wnt/
There have been emerging studies revealing the dual substrate specificity of m6A modification in CRC, as it mediates both protein-coding transcripts and non-coding RNAs to orchestrate oncogenic programs (Table 4, Ref. [58, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82]). It has been revealed that porcine retina-derived POU domain factor 1 (POU6F2)-AS1 may function as a novel oncogenic lncRNA in CRC with potential diagnostic and therapeutic value. In vitro studies have demonstrated that POU6F2-AS1 promotes the growth of both CRC cells and patient-derived organoids (PDOs). METTL3 induces the modification of m6A and stability of POU6F2-AS1 through IGF2BP2, and upregulation of POU6F2-AS1 could tether YBX1 to the FASN promoter to induce its transcriptional activation, thus facilitating the growth and lipogenesis of CRC cells [69]. Similarly, the methyltransferase KIAA1429 regulates POU6F2-AS1 to modulate CRC cell survival, migration, and metastasis, underscoring the diversity of m6A-mediated lncRNA regulation in cancer [70]. METTL3-mediated m6A modification can induce abnormal LINC02418 expression in CRC, where LINC02418 regulates the proliferation and migration of CRC cells by interacting with YBX1 to activate CTNNB1 transcription, and promoting the interaction between IGF2BP1 and CTNNB1 mRNA to enhance CTNNB1 stability [71]. It has also been confirmed that METTL3 is critical for maintaining the RNA stability of alpha/beta hydrolase domain-containing protein 11-antisense RNA 1 (ABHD11-AS1) and Family with sequence similarity 83 member H (FAM83H)-AS1 through IGF2BP2/3, where ABHD11-AS1 enhances FOXM1 stability and FAM83H-AS1 specifically binds to PTBP1 to regulate its phosphorylation and splicing function in CRC cell proliferation and migration and suppress ferroptosis [72, 73].
| Gene | Expression | Target genes | Reader | Function | Sample source | Detection method | Ref. |
| METTL3 | Up | lncRNA HNF1A-AS1 | IGF2BP3 | Upregulates HNF1A-AS1and promotes proliferation and cell cycle progression | Human CRC tissues (52 pairs) | RIP; RNA Pull-down; Luciferase; MeRIP; FISH | [58] |
| METTL3 | Up | lncRNA | IGF2BP2 | Upregulates POU6F2 to promote CRC cell lipogenesis and growth | CRC tissues (84 paired fresh frozen, 60 paired paraffin-embedded for TMA) | RNA-seq, RNA pull-down, RIP, immunofluorescence, luciferase reporter assay, ChIP, MeRIP-qPCR | [69] |
| POU6F2 | |||||||
| METTL3 | - | LINC02418/CTNNB1 | IGF2BP1 | METTL3 induces LINC02418-mediated CTNNB1 transcription, while LINC02418 enhances CTNNB1 stability via IGF2BP1, collectively promoting CRC cell proliferation and metastasis | 10 pairs of human CRC/adjacent tissues; TCGA/GEO datasets | RNA-FISH; RNA pull-down; RIP; ChIP; MeRIP-qPCR; Dual-luciferase | [71] |
| METTL3 | - | lncRNA | IGF2BP2/IGFBP3 | Promotes FAM83H-AS1 to promotes CRC progression | Human CRC tissues | RNA-seq; RIP; MeRIP; MeRIP-qPCR | [72] |
| FAM83H-AS1 | |||||||
| METTL3 | - | lncRNA | IGF2BP2 | Upregulates ABHD11-AS1 to promote colorectal cancer progression and inhibit ferroptosis | Human CRC tissues | RNA-seq; RIP; Ubiquitination assay; Ferroptosis assays | [73] |
| ABHD11-AS1 | |||||||
| METTL14 | Down | lncRNA XIST | YTHDF2 | Downregulates XIST to suppress proliferation and metastasis | 37 paired CRC/normal tissues | RNA-seq; Me-RIP; RIP | [74] |
| KIAA1429 | Up | lncRNA POU6F2-AS1 | - | Promotes POU6F2-AS1 to facilitate CRC cell malignancy | 32 paired human CRC and normal tissue | MeRIP, Bioinformatics Analysis | [70] |
| ZCCHC4 | Up | LncGHRLOS | - | Downregulates lncGHRLOS to promote CRC cell survival, migration, and metastasis | 243 CRC tissues | RNA-seq; MeRIP-seq; RIP; RNA pull-down | [75] |
| ALKBH5 | - | LncRNA | - | Promotes NEAT1 to enhance proliferation and migration while inhibiting apoptosis | Human CRC tissues (n = 70) | Me-RIP, RIP | [76] |
| NEAT1 | |||||||
| ALKBH5 | - | lncRNA CARMN | YTHDF2/YTHDF3 | Enhances CARMN to suppress CRC cell proliferation, invasion, and migration potential | - | MeRIP, RIP, SRAMP website predict the m6A modification sites | [77] |
| YTHDF2 | - | circ0003215 | YTHDF2 decreases circ 0003215 to promote proliferation, invasion and migration | 100 pairs tumor and paracancerous tissues | MeRIP, RIP, RNA pulldown SRAMP website predict the m6A modification sites | [78] | |
| YTHDF3 | - | circ-YAP | YTHDF3 and eIF4G2 enhance | Normal cases, CRC cases, CRC with liver metastasis, cases with lung metastasis and cases with brain metastasis | Me-RIP, RIP, RNA pulldown | [81] | |
| circ-YAP to promote migration, invasion and liver metastasis | |||||||
| YTHDC1 | - | circFNDC3B | Facilitates cytoplasmic translocation of circFNDC3B to inhibit CRC stemness and metastasis | Human CRC tissues (n = 58) | MeRIP-qPCR, RIP, RNA pulldown | [79] | |
| IGF2BP2 | - | circEZH2/CREB1 | circEZH2 interacts with IGF2BP2 to block its degradation and sponges miR-133b to upregulate IGF2BP2. The circEZH2/IGF2BP2 axis stabilizes CREB1 mRNA, aggravating CRC progression | Human CRC tissues (n = 124) | MeRIP-seq, RIP, RNA pulldown | [80] | |
| PRRC2A | Up | CSNK1E | Promotes CSNK1E to enhance CRC progression | Human CRC tissues (n = 89) | MeRIP-seq, MeRIP-qPCR, RIP-seq, RIP-qPCR, Dual-Luciferase Reporter Assays, RNA pulldown | [82] |
CSNK1E, encoding CK1
It has been demonstrated that METTL14 significantly suppresses CRC cell proliferation and migration potential through YTHDF2-mediated inhibition of the oncogenic lncRNA XIST [74]. ZCCHC4, a newly identified m6A methyltransferase, has been functionally validated in CRC, where it downregulates the lncRNA GhrelinOS (GHRLOS) to modulate KDM5D expression, thereby promoting CRC cell survival and migration [75].
Guo et al. [76] has confirmed that ALKBH5 increases the expression level of lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) by lowering its m6A enrichment to enhance cell proliferation and tumor growth while inhibit apoptosis in CRC cells. Inversely, another study reported that ALKBH5-mediated demethylation of m6A on lncRNA Cardiac mesoderm enhancer-associated non-coding RNA (CARMN) can enhance its stability through YTHDF2 recognition, thereby sustaining the expression level of CARMN. Functionally, the CARMN-miR-5683-FGF2 axis suppresses CRC cell proliferation, invasion, and migration potential [77].
Circular RNAs (circRNAs) have emerged as pivotal players in deciphering cancer pathogenesis due to their exceptional stability and resistance to RNase R digestion. Circ ubiquitin-like with PHD and ring finger domains 2 (UHRF2), a novel circRNA, plays a critical oncogenic role in CRC progression by facilitating the binding of IGF2BP1 to DDX27 mRNA, thereby enhancing its stability and expression.
Circ_0003215 is significantly downregulated in CRC and demonstrates strong negative correlations with tumor size, TNM stage, and lymph node metastasis. Mechanistically, YTHDF2 binds to circ_0003215 and promotes its RNA degradation. Functionally, circ_0003215 acts as a tumor suppressor by sponging miR-663b to upregulate DLG4 expression and G6PD ubiquitination [78]. Zeng et al. [79] demonstrated that YTHDC1 facilitates cytoplasmic translocation of m6A-modified circ fibronectin type III domain containing 3B (FNDC3B), thereby enhancing the stability of RNF41 mRNA to promote ASB6 degradation, and ultimately inhibiting CRC stemness and metastasis. CircEZH2 interacts with IGF2BP2, impeding its ubiquitination-dependent degradation, and also acts as a miR-133b sponge to upregulate IGF2BP2 expression. Taken together, the circEZH2-IGF2BP2 axis stabilizes cyclic-AMP response element-binding protein 1 (CREB1) mRNA and exacerbates CRC cell proliferation and migration [80].
Dysregulation of YAP has significant implications for the pathological biology
and progression of CRC. The lncRNA pituitary tumor-transforming 3, pseudogene
(PTTG3P) undergoes m6A methylation mediated by METTL3 and IGF2BP2 to enhance its
stability, which promotes glycolytic CRC reprogramming and proliferation in
cooperation with YAP1 [38]. YTHDF3 has been identified as a novel target of YAP,
and their mRNA and protein levels have significant positive correlations. The circ-YAP
transcript encodes a novel 220-amino acid truncated protein (YAP-220aa) that
functions as a competitive inhibitor of LATS1, which promotes YAP
dephosphorylation and nuclear translocation, thereby activating a pro-metastatic
transcriptional program. Notably, YTHDF3 can cooperate with eIF4G to enhance the
translation of circ-YAP, while the YAP/TEAD transcription factor complex
simultaneously upregulates circ-YAP expression, establishing a self-reinforcing
positive feedback loop to promote liver metastasis of CRC [81]. It has also been
found that the m6A reader protein PRRC2A directly targets CSNK1E (encoding
CK1
The regulation of lncRNAs and circRNAs by m6A modification offers novel perspectives for CRC diagnosis. However, its clinical application is faced with two major challenges: the stability of non-coding RNAs and insufficient biomarker specificity due to tumor heterogeneity. Therefore, integration of m6A sequencing with liquid biopsy techniques (such as exosomal RNA analysis) may enhance the sensitivity of early detection in future studies. Notably, although many lncRNAs and circRNAs demonstrate promising potential in mechanistic research, their validity as independent prognostic indicators still requires large-scale validation with collaboration to mitigate sampling bias.
Cancer cells mainly rely on glycolysis for energy supply, which enables cancer cells to tolerate hypoxic environments, while also increases the lactate level in the microenvironment of cancer cells, thereby stimulating tumor growth and development (Table 5, Ref. [38, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93]) [94]. There has been accumulating evidence demonstrating that m6A modification regulates tumor glycolysis through multiple pathways, thereby exerting critical impacts on cancer proliferation, metastasis, and therapeutic response. In CRC cells, METTL3 directly targets lncRNA PTTG3P, HK2, glucose transport protein type 1 (GLUT1) and PNN genes, enhancing their mRNA stability by m6A modification through IGF2BP2/3 or YTHDF1, ultimately promoting glycolysis and proliferation in CRC cells [38, 83, 84]. The methyltransferase KIAA1429 also enhances HK2 mRNA methylation in an m6A-dependent manner, leading to increased transcription level and stability of HK2. This metabolic reprogramming accelerates aerobic glycolysis in CRC cells, thereby promoting tumor aggressiveness [85]. METTL14 inhibits glycolysis, and wild-type p53 activates its transcription, which promotes the biogenesis of pri-miR-6769b and pri-miR-499a. Ultimately, the miR-6769b-3p-GLUT3 and miR-499a-3p-PGAM1 axes suppress aerobic glycolysis and malignant phenotypes in p53 WT CRC cells [86]. Wu et al. [87] proposed that ALKBH5/IGF2BPs bind to mjC domain-containing protein 8 (JMJD8) and modulate its stability in an m6A-dependent manner, increasing glycolysis and accelerating the development of CRC by enhancing the enzymatic activity of PKM2. Interestingly, the synthesized ALKBH5 mRNA-loaded folic acid-modified exosome–liposome hybrid nanoparticles were found to significantly inhibit the progression of CRC.
| Gene | Expression | Target genes | Reader | Function | Sample source | Detection method | Ref. |
| METTL3 | - | lncRNA PTTG3P | IGF2BP2 | Increases PTTG3P to facilitate CRC glycolysis and proliferation | Human CRC tissues (n = 120) | flow cytometry, MeRIP-qPCR | [38] |
| METTL3 | - | HK2/GLUT1 | IGF2BP2/3 | Stabilizes HK2/GLUT1 to induce CRC tumorigenesis depends on cell glycolysis | CRC patient tissues (n = 47) | MeRIP-seq, MeRIP-qPCR, RIP | [83] |
| METTL3 | Up | Pinin | YTHDF1 | Enhances PNN to induce glycolysis, proliferation and metastasis | Human COAD tissues | RIP, m6A assay, IHC | [84] |
| METTL3 | Up | SNAIL | YTHDF1 | Promotes SNAIL to promote colorectal cancer pulmonary metastasis | Clinical CRC and lung metastasis specimens | ChIP, RNA-seq, MeRIP-qPCR, IHC, metabolite assays | [88] |
| METTL3 | - | HMGA1 | IGF2BP2 | Elevates HMGA1 to promote CRC proliferation and metastasis | 498 paired human CRC and adjacent normal tissues | RNA pull-down, RIP, MeRIP | [89] |
| METTL14 | Down | pri‐miR‐6769b/pri‐miR‐499a | YTHDF2 | Promotes pri-miR-6769b/499a to suppress aerobic glycolysis and malignant phenotypes in p53 WT colorectal cancer cells | Human p53-wild-type and p53-mutant colorectal cancer tissues and cell lines | ChIP-qPCR, RNA-seq, miRNA microarray, MeRIP-qPCR, Co-IP, IHC, ISH | [86] |
| WTAP | Up | VEGFA | YTHDC1 | Upregulates VEGF to exacerbate cell proliferation, migration, invasion, and angiogenesis | Human CRC tissues | RNA-seq, MeRIP-seq, RIP | [92] |
| KIAA1429 | Up | HK2 | Increases HK2 to accelerate the aerobic glycolysis and malignant phenotype of CRC cells | 48 paired human CRC and adjacent normal tissues | RIP, MeRIP | [85] | |
| RBM15 | - | HUNK | - | LncSLERT binds to RBM15 to decrease HUNK and enhance liver metastasis in CRC | Human colorectal cancer tissues | RNA-FISH, qRT-PCR, RNA-seq, Western Blot, RIP-qPCR, Luciferase reporter assay | [87] |
| FTO | - | ATF4 | YTHDF2 | Promotes ATF4 to inhibit tumor cells autophagy while promoting cell survival | - | RIP, ChIP | [93] |
| ALKBH5 | Down | JMJD8 | IGF2BPs | Inhibits JMJD8 to suppress glycolysis and accelerate the development of CRC | 8 pairs of CRC tissues and adjacent normal tissues; 192 pairs of CRC tissues and matched nontumor tissues | MeRIP-seq, MeRIP-qPCR, RNA pull-down, Luciferase reporter assay | [87] |
| 1078 CRC tumor and matched normal tissues | |||||||
| IGF2BP2 | - | HMGA2 | circNSUN2 and IGF2BP2 stabilize HMGA2 to promote hepatic metastasis in CRC | - | Me-RIP, RIP, RNA-EMSA assay | [90] | |
| IGF2BP2 | Up | LncRNA BACE1-AS | Stabilizes BACE1-AS to facilitate CRC liver metastasis | Human CRC tissues (n = 4) | RIP, S1m-tagged immunoprecipitation | [91] |
HUNK, Hormonally up-regulated neu-associated kinase; HMGA2, High mobility group
A2; JMJD8, mjC domain-containing protein 8; BACE1,
In CRC patients, cancer metastasis is the primary cause of deaths, with the
liver and lungs being the most common target organs for metastasis. A previous
study has shown that METTL3 facilitates epithelial-mesenchymal transition of
pulmonary metastasis by targeting the m6A-Snail- CXC motif chemokine ligand
(CXCL)2 axis to recruit M2-type immunosuppressive macrophages [88]. LINC00460
promotes EMT, CRC cell proliferation, migration, invasion in vitro, and
tumor growth and metastasis in vivo. Mechanistically, METTL3 elevates
high mobility group A1 (HMGA1) m6A methylation, while LINC00460 directly
interacts with IGF2BP2 and DHX9 to bind HMGA1 mRNA, thereby enhancing its
stability [89]. High mobility group A2 (HMGA2) (a member of the HMGA1 protein
family) has been confirmed to play an oncogenic role in CRC liver metastasis.
Mechanistic studies have revealed that circNSUN2 forms a complex with IGF2BP2 to
directly bind and stabilize HMGA2 mRNA, thereby driving liver metastasis in CRC
[90]. IGF2BP2 also binds to two m6A motifs on LncRNA
Tumor-associated macrophages (TAMs) can reduce the antitumor activity of T cells.
Angiogenesis plays an important role in the occurrence and development of CRC. Ye et al. [92] found that WTAP overexpression exacerbates CRC cell proliferation, migration, invasion, and angiogenesis. WTAP upregulates VEGF expression in a YTHDC1-dependent manner, which subsequently activates the MAPK signaling pathway to drive angiogenesis.
Upregulation of glutaminolysis facilitates energy generation and biomass accumulation, thereby providing energy for CRC cell proliferation. Han et al. [93] demonstrated that FTO promotes ATF4 expression through m6A-dependent stabilization of its mRNA, which can counteract YTHDF2-mediated decay under glutaminolysis inhibition. Functionally, ATF4 activates DDIT4 transcription to inhibit mTOR signaling, thereby triggering pro-survival autophagy in CRC cells. Fig. 2 shows the schematic diagram illustrating the role of m6A modification in CRC.
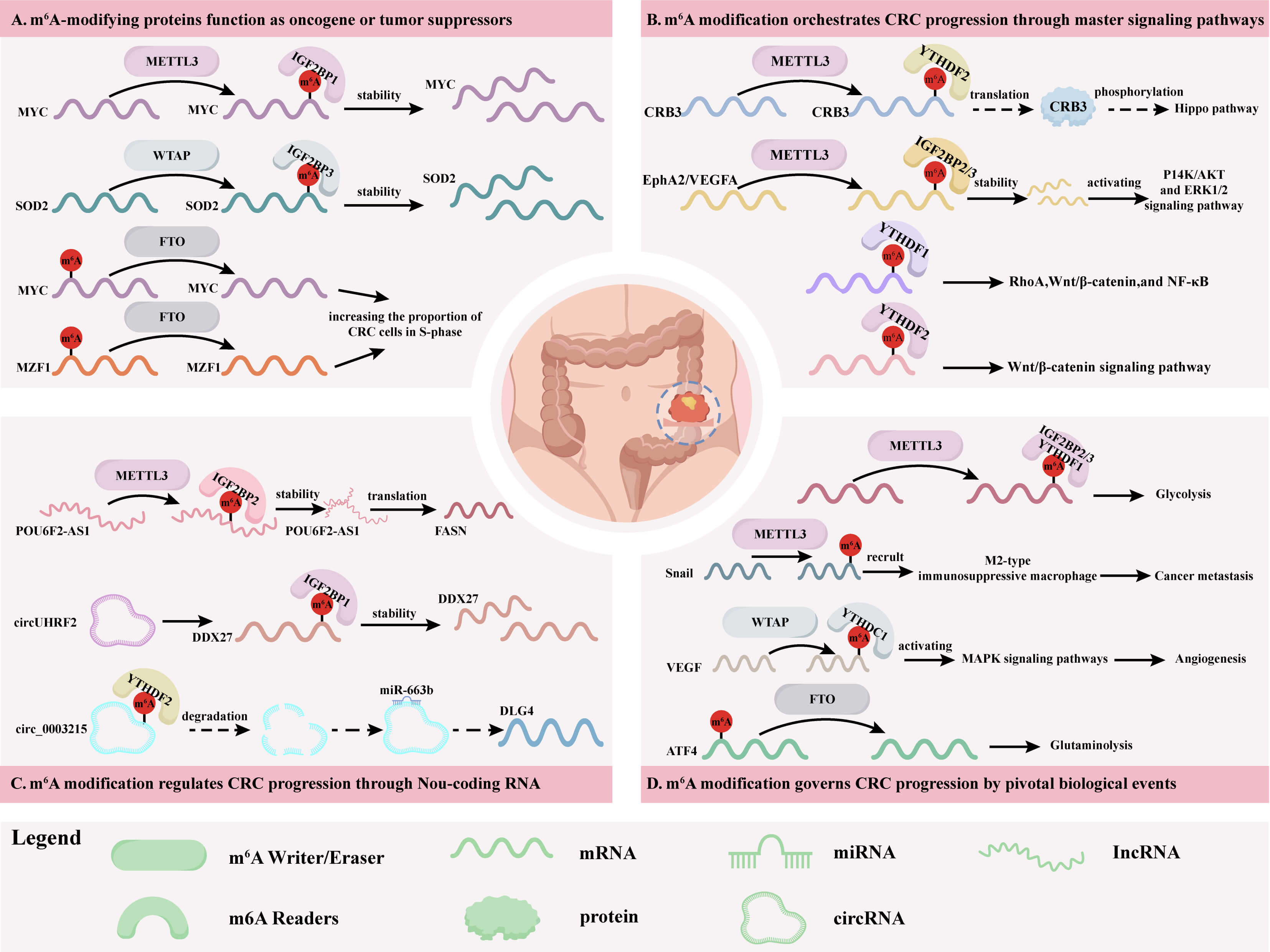 Fig. 2.
Fig. 2.
Roles of m6A modification in CRC progression. (A) m6A modification proteins function as oncogene or tumor suppressors. (B) m6A modification orchestrates CRC progression through master signaling pathways. (C) m6A modification regulates CRC progression through non-coding RNA. (D) m6A modification governs CRC progression by pivotal biological events.
METTL3 and METTL14 are core components of methyltransferases, and can form a heterodimer to exert consistent regulatory functions to control other biological events. However, they play opposite roles in CRC: METTL3 generally acts as a promoter, while METTL14 mostly functions as a suppressor, which may be attributed to the differences in their specific upstream and downstream mechanisms within cells. Methyl-CpG binding protein 2 (MeCP2) is a methylated DNA-binding protein, and has been reported to have oncogenic functions in gastric cancer, CRC, hepatocellular carcinoma, and osteosarcoma. It was identified that MeCP2 can bind to METTL14 to occupy the interaction interface between METTL3 and METTL14, indicating a competitive relationship between MeCP2 and METTL3 for METTL14. Furthermore, MeCP2 acts as an oncogene in CRC progression by interacting with METTL14 and reducing m6A modification [49]. Some studies have shown that METTL3 can promote glycolysis by affecting the downstream target genes such as lncRNA PTTG3P, HK2, GLUT1, and pNN, thereby facilitating the proliferation of CRC cells [38, 83, 84]. In contrast, METTL14 inhibits glycolysis via pri-miR-6769b and pri-miR-499a, thus suppressing the progression of CRC [86]. Therefore, the contrasting roles of METTL3 and METTL14 in CRC may be ascribed to their distinct upstream binding factors and downstream regulatory pathways.
In addition, the demethylases FTO and ALKBH5 exhibit paradoxical roles in CRC. Many studies have demonstrated that FTO plays a promoting role in CRC progression. However, another study showed that FTO may act as anti-tumor factor in CRC progression. FTO expression was found to prevent YTHDF2-mediated degradation of the anti-proliferative and pro-apoptotic factor potassium channel tetramerization domain containing 15 (KCTD15) mRNA. Furthermore, KCTD15 overexpression downregulates HDAC1 expression and enhances p53 acetylation, thereby establishing a tumor-suppressive signaling cascade [57]. We hypothesize that these paradoxical roles may be attributed to two factors. On the one hand, it may be caused by differences in experimental samples; on the other hand, it may be associated with the microenvironment for the CRC cells. Specifically, in tumors with abundant immune cell infiltration, FTO tends to regulate apoptosis-associated genes (such as KCTD15).
ALKBH5 plays even more complex bidirectional roles in CRC. First, different studies have reported inconsistent expression patterns of ALKBH5 in CRC, with some showing its upregulation while others displaying its downregulation. Second, numerous studies have documented both pro-tumorigenic and anti-tumorigenic effects of ALKBH5 in CRC. It remains challenging to elucidate the mechanism underlying such paradoxical roles. We suspect that one possible reason may be the variations in clinical stages across different studies. For instance, in early-stage CRC, ALKBH5 appears to suppress cell proliferation by inhibiting PHF20 [42]; in contrast, in advanced metastatic lesions, it seems to promote tumor invasion by activating RAB5A [54]. This stage-dependent effect suggests that the clinical utility of ALKBH5 as a therapeutic target must be evaluated in the context of tumor progression.
The main treatment for CRC is usually surgery combined with radiotherapy,
chemotherapy, targeted therapy, and immunotherapy [96]. Chemotherapy is key for
advanced/metastatic CRC, and the first-line drugs such as 5-fluorouracil (5-FU)
and oxaliplatin (OX) form the FOLFOX regimen (OX inhibits DNA replication) [97].
Immunotherapy, especially the use of immune checkpoint inhibitors (ICIs) such as
PD-1/PD-L1 inhibitors (approved by the FDA in 2014), shows promise for metastatic
CRC by activating T cells in the tumor microenvironment [98]. Although
immunotherapy has advanced significantly in CRC treatment, its clinical use is
limited by low response rate, unique toxicity, and acquired resistance. Thus,
targeted therapy has become the optimal option for personalized and comprehensive
CRC treatment [99]. It targets dysregulated pathways (such as EGFR and
Wnt/
Chemoresistance to OX and 5-FU agents in CRC cells represents a major
therapeutic challenge in advanced CRC, and is the primary cause of treatment
failure and poor patient prognosis. Lan et al. [103] demonstrated that
OX-resistant CRC tissues exhibit elevated total m6A RNA levels and increased
expression of METTL3. Mechanistically, M2-polarized TAMs enhance the OX
resistance via elevating METTL3-mediated m6A modification of tumor necrosis
factor receptor associated factor 5 (TRAF5) in cells [103]. Zhang et al.
[104] and Li et al. [105] observed upregulation of both METTL3
expression and global m6A level in 5-FU-resistant CRC cell lines. METTL3
increases lactate dehydrogenase A (LDHA) transcription by stabilizing
HIF-1
Wnt/
The TAMs, which consist of extracellular matrix, myofibroblasts, cytokines,
fibroblasts, neuroendocrine cells, adipocytes, immune-related cells, and blood
vessels, induce phenotypic changes in cancer cells and immune cells through
complex molecular mechanisms to promote immune escape [112]. In CRC cells,
deletion of METTL3/METTL14 causes dysfunction of CD8+ T cells, thereby
restricting the anti-tumor T cell response in the tumor immune microenvironment.
Wang et al. [113] found that deficiency of METTL3/METTL14 in CRC
significantly increases the infiltration of CD8+ T cells and promotes the
secretion of cytokines such as IFN-
Zhai et al. [43] reported that elevated ALKBH5 expression represents
unfavorable prognosis in CRC. ALKBH5 demethylates the m6A of axis inhibition
protein 2 (AXIN2) mRNA, leading to its dissociation from IGF2BP1 and subsequent
degradation, which upregulates the downstream Wnt/
In terms of targeted therapy drugs and resistance to them, lncRNA MIR100HG
sustains cetuximab resistance and facilitates invasion and migration of CRC cells
both in vitro and in vivo. HNRNPA2B1 interacts with MIR100HG to
maintain the mRNA stability of TCF7L2, which activates Wnt/
Notably, drug-resistant cancer cells, particularly those in a mesenchymal state with high metastatic potential, generally exhibit marked vulnerability to ferroptosis. This unique vulnerability makes pharmacological induction of iron oxidation a promising therapeutic strategy against refractory malignancies [118]. Curdione can induce ferroptosis to trigger necrotic death of CRC cells. Mechanistically, it promotes the expression of METTL14 and YTHDF2, which in turn elevates m6A methylation and upregulates the expression of light chain subunit solute carrier family 7 member 11 (SLC7A11) and Human class I homeobox A13 (HOXA13) genes [119].
Zhang et al. [120] found that targeting AKT could significantly induce GPX4-dependent ferroptosis and suppress CRC growth both in vitro and in vivo. AKT inhibition downregulates FTO expression, increasing the m6A modification of GPX4 mRNA. The m6A-marked GPX4 transcripts are subsequently recognized and degraded by YTHDF2, which subsequently suppresses CRC progression. The demethylase ALKBH5 also exhibits pro-ferroptotic effects on CRC cells. ALKBH5 is downregulated in CRC and its overexpression reduces SLC7A11 transcription by erasing m6A modification, thus promoting ROS release and ferroptosis [121]. The schematic diagram for the role of m6A modification in CRC drug resistance is shown in Table 6 (Ref. [103, 104, 105, 106, 107, 108, 109, 110, 111, 113, 114, 115, 116, 117, 119, 120, 121]) and Fig. 3.
| Gene | Expression | Target genes | Reader | Function | Sample source | Detection method | Ref. |
| METTL3 | - | TRAF5 | - | M2-polarized TAMs enable the OX resistance via elevating METTL3-mediated TRAF5 | 20 human CRC tissues (OX-sensitive vs. OX-resistant | Flow cytometry; Dot blot; MeRIP | [103] |
| METTL3 | - | LDHA | YTHDF1 | Triggers LDHA to promote glycolysis and inducing resistance to 5-FU | Using colorectal cancer cell lines and their drug-resistant variants, public database data | Metabolic profiles | [104] |
| METTL3 | Up | RAD51AP1 | - | Promotes RAD51AP1 to enhance 5-FU resistance in CRC cells by attenuating DNA damage accumulation and cell apoptosis | Human CRC cell lines (HCT-8, 5-FU-resistant HCT-8R) | Flow Cytometry, Immunofluorescence, Comet Assay | [105] |
| METTL3 | - | Sec62 | IGF2BP1 | Upregulates Sec62 to promote the stemness and chemoresistance of CRC by enhancing Wnt signaling | 102 CRC patient tissue | Co-IP, GST Pull-down, IHC, Immunofluorescence, m6A-MeRIP, Flow Cytometry | [108] |
| METTL3/14 | - | Stat1 and Irf1 | YTHDF2 | Deficiency of METTL3/METTL14 promotes IFN‐ |
Human pMMR-MSI-L CRC tissues (n = 59). | RNA-seq, MeRIP-seq, flow cytometry | [113] |
| METTL3 | - | CircQSOX1 | IGF2BP2 | Improves CircQSOX1 to facilitate immune escape of CRC, which reduces the therapeutic effect of anti-CTLA-4 | Human CRC tissues (n = 60) | RNA pull-down, MeRIP, flow cytometry, glycolysis assays | [114] |
| METTL14 | - | SLC7A11/HOXA13 | YTHDF2 | Curdione induces ferroptosis in CRC by upregulating METTL14 | - | Flow Cytometry, Dot Blot (global m6A), MeRIP-qPCR | [119] |
| METTL16 | Down | PD-L1 | - | Decrease PD-L1 to enhance the tumor suppressive effect of anti-PD-1 antibody | Human CRC tissues (n = 20) | IHC, RIP, MeRIP-qPCR, flow cytometry | [115] |
| FTO | Up | NUPR1 | YTHDF2 | Prevents degradation of NUPR1 to facilitate CRC chemoresistance | - | RNA m6A dot blot assay, MeRIP-qPCR, RIP | [106] |
| FTO | - | G6PD/PARP1 | YTFDF2 | Promotes G6PD/PARP1 to enhance CRC progression and chemotherapy resistance | Human CRC tissues | RNA m6A dot blot assay, Me-RIP, MeRIP-qPCR | [107] |
| FTO | - | GPX4 | YTHDF2 | AKT inhibition downregulates FTO, decreasing GPX4 to activate ferroptosis and suppress CRC progression | - | RNA m6A dot blot assay, MeRIP-qPCR | [120] |
| ALKBH5 | Down | SLC7A11 | - | Reduces SLC7A11 to promote ferroptosis of CRC | Human CRC tissues (n = 82) | m6A RNA methylation quantification, MeRIP-qPCR | [121] |
| YTHDF1 | Up | GLS1 | Promotes GLS1, which finally decreases the cisplatin sensitivity of CRC through the GLS1-glutamine metabolism | - | RIP, RNA pulldown, Luciferase reporter assay | [109] | |
| YTHDF3 | - | ATP7A, DYRK1B, ERCC1 | Promotes ATP7A, DYRK1B and ERCC1 via eIF3A and eIF2AK2 to promote oxaliplatin-resistant | - | Me-RIP, LC-MS/MS, RNA pulldown assay, Co-IP | [110] | |
| IGF2BP3 | - | ABCB1 | Enhances ABCB1 mRNA, which leads to DOX resistance in CRC cells | - | RIP-qPCR, m6A RIP-qPCR | [111] | |
| IGF2BP3 | Up | EGFR | Enhances EGFR to increase CRC tumorigenesis, progression and drug resistance to cetuximab | - | RNA m6A dot blot assay, MeRIP-qPCR, RNA-EMSA assay | [117] | |
| hnRNPA2B1 | Up | TCF7L2 | HNRNPA2B1 interacts with MIR100HG to maintain TCF7L2, sustains cetuximab resistance and facilitates invasion and metastasis | 12 paired tumor specimens and 14 paired specimens of primary CRC tissues | MeRIP-seq, SRAMP website predict, S1m aptamer-based pull-down assay, RIP | [116] |
TRAF5, Tumor necrosis factor receptor associated factor 5; DYRK1B,
Dual-specificity tyrosine-regulated kinase 1B; ABCB1, ATP-binding cassette
sub-family B member 1; eIF2AK2, EIF2
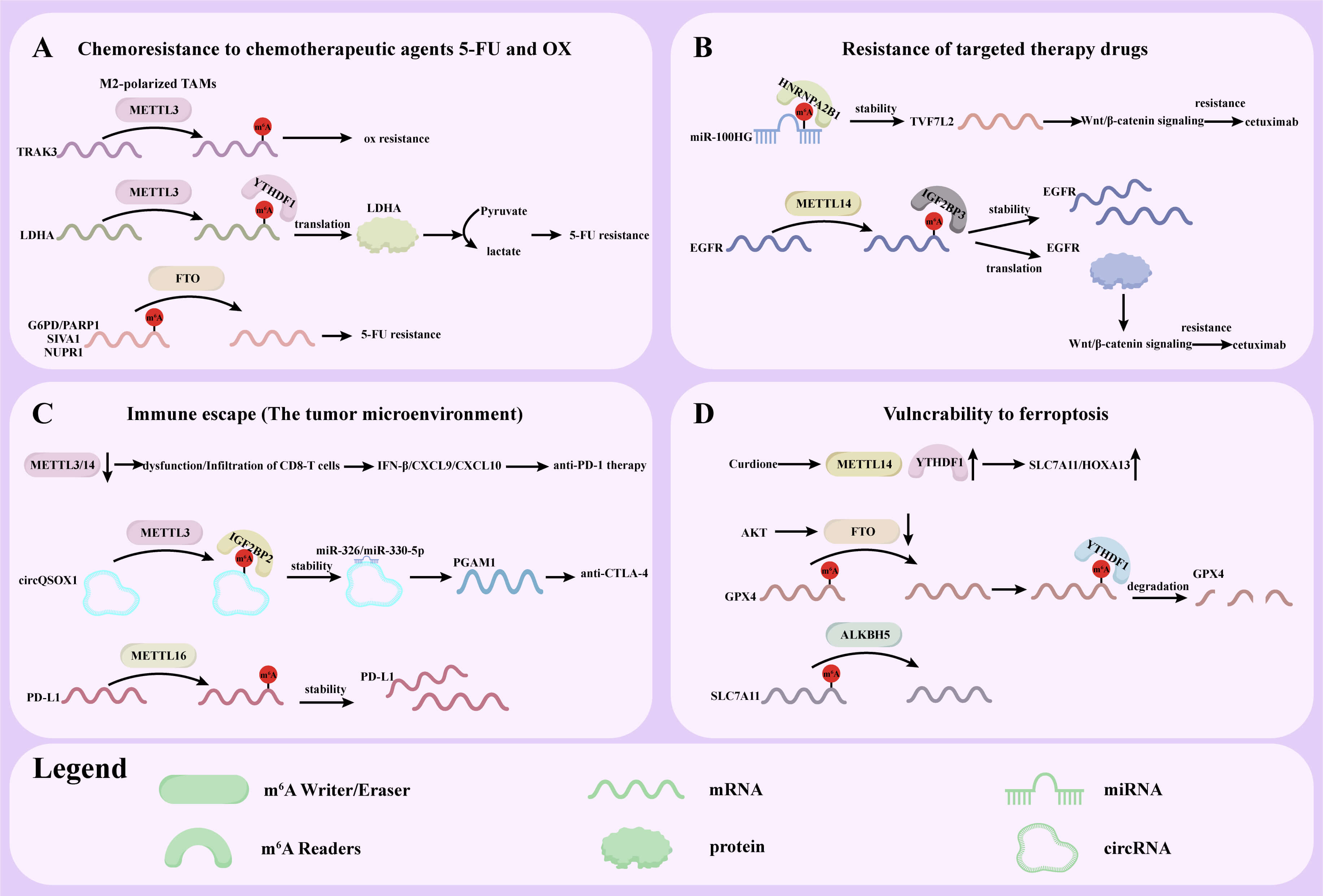 Fig. 3.
Fig. 3.
Role of m6A modification in CRC drug resistance. (A) Chemoresistance to chemotherapeutic agents 5-FU and OX. (B) Resistance to targeted therapy drugs. (C) Immune escape (tumor microenvironment). (D) Vulnerability to ferroptosis.
While the role of the m6A regulatory axis in drug resistance has been well-established, its clinical translation potential remains not fully explored. To date, inhibitors targeting m6A modification have been extensively reported [122]. It has been shown that STM2457 has great impacts on cell growth suppression and apoptosis of CRC cells in vitro and subcutaneous xenograft growth in vivo mediated by the METTL3/ASNS axis [123]. In 2019, a new, specific, and cell active FTO inhibitor compound FB23/FB23-2 was reported. Compared with FB23, FB23-2 has a slightly lower inhibitory activity but a significantly higher cell uptake rate [124]. Lin et al. [40] demonstrated that FB23-2 could increase the sensitivity of CRC cells to 5-FU both in vitro and in vivo by inhibiting FTO. Therefore, targeting m6A regulators with small molecules or modulating their expression represents a promising therapeutic strategy for cancer treatment and is worth of significant scientific interests.
In the treatment of certain cancers (such as oral squamous cell carcinoma), STM2457 has shown promising outcomes. The combination of STM2457 and anlotinib (targeting EGFR) can exert a multifaceted antitumor effect [125]. Despite the promising prospects of m6A-targeted drugs, their clinical translation is still confronted with significant challenges in terms of toxicity, bioavailability, and patient stratification.
The primary concern is “on-target/off-tumor” toxicity. Small-molecule inhibitors targeting m6A modification proteins may exert certain off-target effects on the enzymes with similar structures. Moreover, since m6A modification plays a fundamental role in normal physiology, such inhibitors may trigger systemic toxicity. For example, inhibition of METTL3 can impair postnatal liver regeneration in mice [126], while knockout of YTHDF2 causes hematopoietic system failure [127], indicating the risk of non-target tissue damage. Future studies should prioritize the development of tissue-specific delivery systems to mitigate such risks. In addition, utilization of well-established drugs with existing safety certifications can bypass the “de novo” development of small molecules, which is expected to accelerate clinical application.
In terms of bioavailability, although small-molecule inhibitors (such as FB23-2) exhibit robust activity in vitro, their in vivo efficacy is often compromised by poor solubility, low metabolic stability, and insufficient tumor penetration. Encouragingly, Huang et al. [128] recently proposed a novel strategy to circumvent this bottleneck. They found that homoharringtonine, a clinically approved drug, does not directly inhibit FTO enzymatic activity; instead, it promotes FTO proteasomal degradation, thereby upregulating the global m6A level in acute monocytic leukemia.
Precise patient stratification remains a critical bottleneck. The functional roles of m6A regulators are highly context-dependent, showing significant variations across different cancer types and even subtypes [129]. Currently, there is still a lack of validated biomarkers for prediction of the treatment response or drug resistance, making it difficult to identify patients who would truly benefit from m6A-targeted therapies. The treatment of CRC is developing toward greater precision and individualization, and biomarker-driven trial design has become a focus of current research. Molecular biomarkers such as MSI/dMMR, BRAF, KRAS, and HER-2 have been widely used to guide treatment decisions, while the development of liquid biopsy technologies such as ctDNA has provided new tools for CRC management [130]. Therefore, combining m6A expression profiles with these biomarkers may facilitate the clinical application of m6A modification.
Finally, in current CRC treatment strategies, single therapeutic approaches usually have certain limitations, and the optimization of combined therapies will become a future hotspot. Multiple strategies, including immunotherapy combinations, chemotherapy combined with targeted therapy, and novel ADC drugs, have shown significant efficacies [131]. For example, regarding microsatellite instability-high (MSI-H) and microsatellite stable (MSS) subtypes, a key research direction is to determine whether further combining PD-1 antibodies with chemotherapy plus bevacizumab can improve the efficacy. The complexity of the m6A regulatory network, its functional redundancy, and the dynamic evolution of the epitranscriptome under therapeutic pressure all make it necessary to develop combination therapies. For instance, YTHDF1-mediated chemoresistance is closely associated with glutamine metabolism, suggesting the potential of combining its inhibitors with GLS1 blockers. Similarly, the immunosuppressive microenvironment induced by METTL14 deficiency may be reversed through combination with PD-1 inhibitors. Nevertheless, combination regimens may also introduce new complexities regarding optimal dosage determination and additive toxicity [132].
In our view, the field of m6A research in CRC is at a critical juncture. While preclinical data are robust, clinical translation largely lags behind due to insufficient understanding of the dose-dependent effects and long-term safety. To successfully integrate m6A-targeted strategies into the therapeutic arsenal for CRC, these challenges must be addressed by developing more specific inhibitors, advanced drug delivery systems, and reliable predictive biomarkers, as well as by better understanding the biological mechanisms through which m6A regulates CRC.
Through the coordinated actions of methyltransferases, demethylases, and reader proteins, m6A modification dynamically regulates the metabolic processes of RNA. This process maintains the molecular equilibrium within cells under normal physiological conditions. The abnormal expression of m6A modification-related proteins is closely associated with the proliferation, migration, invasion, apoptosis, and drug resistance of CRC. In this review, we summarize the roles of several key m6A modification proteins in CRC. Their mechanisms involve the regulation of multiple key genes, signaling pathways, and specific biological processes of non-coding RNAs. It can be concluded that certain methyltransferases, demethylases, and reader proteins can serve as potential biomarkers for the diagnosis of CRC and crucial targets for CRC treatment.
Despite the significant progress achieved in the research on m6A modification in CRC, numerous gaps still exist. In the future, in-depth research on the mechanism of m6A modification in CRC, especially its interactions with other epigenetic modifications and roles in the tumor microenvironment, holds great theoretical significance. Moreover, it is of great practical value to develop drugs targeting m6A modification-related proteins, establish m6A modification-based diagnostic and prognostic assessment models for CRC, and explore m6A modification-based immunotherapeutic strategies.
Notably, while existing evidence strongly indicates that m6A modification is closely associated with CRC drug resistance, there are still several challenges for the clinical translation. Future research must go beyond the single-gene approach and instead establish a clinically actionable integrated framework, specifically by combining m6A expression profiles with well-established biomarkers and developing real-time clinical m6A monitoring systems. Furthermore, although combinatorial strategies may overcome chemoresistance, they require rigorous safety assessment. These efforts will provide new theoretical basis and treatment approaches for the prevention and treatment of CRC, offering greater opportunities to improve the prognosis and enhance the life quality of CRC patients.
MY, ZD, YH, YS, JW carried out the design, the acquisition, analysis and interpretation of the data; MY wrote the draft manuscript; YS, JW critically reviewed the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the [Jinhua Municipal Science and Technology Projects] under grants [number 2023-3-103, 2023-3-101, and 2021-3-039], [Special Research Fund for Basic Research of Jinhua Central Hospital] under grants [number JY2022-6-09].
The authors declare no conflict of interest.
During the preparation of this work, we used ChatGpt to check spelling and grammar. After using this tool, we reviewed and edited the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.