



 , Jun Hu 1,2, Qing Feng 1,2, Jingying Sun 1,2, Xiaoyan Huang 1,2,*
, Jun Hu 1,2, Qing Feng 1,2, Jingying Sun 1,2, Xiaoyan Huang 1,2,* , Cuixiang Xu 1,2,*
, Cuixiang Xu 1,2,*1 Shaanxi Provincial Key Laboratory of Infection and Immune Diseases, Shaanxi Provincial People’s Hospital, 710068 Xi’an, Shaanxi, China
2 Research Center of Cell Immunological Engineering and Technology of Shaanxi Province, Shaanxi Provincial People’s Hospital, 710068 Xi’an, Shaanxi, China
Abstract
Activation of autoreactive lymphocytes leads to cellular and tissue damage, which results in the development of autoimmune diseases. External environmental changes, such as chronic microbial infections, can alter the immune homeostasis and disrupt the balance of autoreactive T and B cells. In this review, we first summarize immune tolerance mechanisms of T and B cells, and then describe the breakthroughs of immune tolerance in T and B cells, followed by related autoimmune diseases. Furthermore, we explore how microbial infections can induce the production of autoreactive antibodies via carrier effects when the balance of autoreactive T and B cells is disrupted. These kinds of antibodies can lead to autoimmune diseases through molecular mimicry mechanisms. Our perspective provides a theoretical framework and novel insights into the mechanism of autoreactive antibodies in the pathogenesis of autoimmune diseases associated with microbial infections. This analysis may offer novel directions for drug discovery of autoimmune diseases.
Keywords
- autoreactive antibody
- autoimmune disease
- immune tolerance
- molecular mimicry
- carrier effect
In 1960, Burnet proposed the theory of clonal selection and was awarded the Nobel Prize for his groundbreaking work in this area [1, 2]. This theory posits that during embryogenesis, specific lymphocyte subsets encountering their corresponding antigens are eliminated or inactivated, thereby leading to immune tolerance [3, 4]. These lymphocyte subsets, termed forbidden clones, retain the potential for immune responses to their constituent antigens [5, 6]. Based on clonal selection, Burnet argued that autoimmune diseases arise due to the persistence of autoreactive lymphocytes, which should normally be eliminated through immune tolerance mechanisms. Wardemann et al., [7] observed that 75% of antibody clones in healthy individuals deriving from early immature B cells exhibited autoreactivity. In contrast, the proportion of autoreactive antibody clones in later-stage immature B cells decreased to 43%. Furthermore, approximately 41% of B cells remained autoreactive when they first entered the peripheral blood. This percentage gradually declined over time. Concurrently, Bouneaud et al. [8] demonstrated that low-affinity autoreactive T cells persist in the periphery, while Danke et al. [9] experimentally confirmed the presence of autoantigen-specific T cells in the peripheral blood of healthy individuals. Specifically, these were CD4+ T cells that targeted autoantigens, such as glutamate decarboxylase 65, melanocyte differentiation antigen tyrosinase, and cancer/testis antigen NY-ESO-1. These subsets of autoreactive lymphocytes exist in the peripheral blood while maintaining tolerance to autoantigens. Under homeostatic conditions, these cell subsets exhibit non-reactivity to autoantigens. However, if the tolerance mechanism fails, these cells may mount an immune response against autoantigens, contributing to the development of autoimmune diseases [10, 11, 12].
Immune tolerance breakdown is significantly influenced by both intrinsic factors, such as genetics, and extrinsic factors, including pathogenic and commensal microorganisms. Currently, it is generally believed that chronic microbial infection can destroy immune tolerance through complex mechanisms, thus promoting the progression of autoimmune diseases [13]. A report described that myasthenia gravis (MG) occurred several months after West Nile virus infection. In terms of the mechanism, the infection may trigger the breakdown of self-tolerance, and autoimmunity development may require significant time to evolve in a pathogenic infection [14]. TMEV infection of the brain is a critical step in the initiation of T cell-mediated autoimmunity [15, 16]. Injection of TMEV into the central nervous system of mice, which lack natural killer dendritic cells, establishes a long-term persistent infection within the central nervous system and may trigger a chronic antiviral immune response targeting myelinated axons [17]. Meanwhile, the emergence of autoreactive antibodies and autoreactive T cells is a characteristic of autoimmune diseases associated with microbial infections.
The mechanism of auto-antibodies in the occurrence and development of autoimmune diseases related to microbial infection is complex and requires further elucidation [18, 19, 20]. For example, the mechanisms underlying the production of auto-antibodies and the pathogenesis of autoimmune diseases following microbial infection are still a mystery. Based on the explanations of the immune tolerance mechanisms in T cells and B cells, this review firstly discusses the breakdown of immune tolerance in these subsets of cells and the following autoimmune diseases. Then we elaborate on the potential mechanisms by which the generation of autoreactive antibodies is induced by microbial infections under conditions of breakthrough of immune tolerance. This serves as a complement to molecular mimicry during the development of autoimmune diseases related to microbial infection. We hope that our insights will provide a novel perspective on the pathogenesis of autoimmune diseases associated with microbial infections.
B cells originate in the bone marrow from hematopoietic stem cells and further develop into immature B cells [21]. Immature B cells express unique B cell receptors (BCRs) that are randomly assembled from the variable (V), diversity (D), and joining (J) segments of immunoglobulin genes. Due to the random rearrangement of V(D)J, these immature B cells may potentially recognize autoantigens [22, 23]. Approximately 75% of immature B cells express BCRs that bind to auto-antigens [7]. These autoreactive immature B cells are primarily eliminated in the bone marrow through central tolerance mechanisms.
The central tolerance of B cells is achieved through clonal deletion, anergy induction, and receptor editing in response to autoantigens expressed in the bone marrow [24, 25, 26, 27]. (i) Clonal deletion: High concentrations of autoantigens in the bone marrow are recognized by immature B cells via their BCRs, resulting in the transmission of strong signals that trigger apoptosis in these B cells. (ii) Anergy: If the BCR on immature B cells encounters low levels of autoantigen or if BCR signaling is down-regulated, these cells exhibit low-affinity binding to autoantigens and become functionally unresponsive or anergic. (iii) Receptor editing: Some B cells that bind to autoantigens may re-express recombinase activating gene 1 (RAG1) and recombinase activating gene 2 (RAG2), initiating a new rearrangement of V(D)J segments. This process generates a new BCR that no longer reacts with autoantigens.
After undergoing central tolerance in the bone marrow, approximately 40% of immature B cells remain autoreactive [7]. This relatively high proportion of residual autoreactive immature B cells might result from the limited exposure of immature B cells to autoantigens, which are only present in the bone marrow microenvironment [28].
Immature B cells that have undergone central tolerance in the bone marrow enter the periphery and home to the spleen, where they transition into transitional B cells and subsequently mature into primary (follicular) B cells [29, 30, 31]. During this process, B cells are subjected to peripheral tolerance mechanisms. Peripheral tolerance of B cells encompasses both transitional B cells and primary B cells.
Transitional B cell tolerance: Transitional B cells serve as a critical link between immature bone marrow B cells and peripheral mature B cells during peripheral B cell maturation. Newly generated immature B cells that are located in the lymphoid sheath of the bone marrow and splenic red pulp are referred to as T1 B cells. After entering the splenic follicle, these cells acquire surface markers IgD, CD21, and CD23, as well as the ability to recirculate. However, they still retain immature markers and are termed T2 B cells [32, 33]. Both T1 and T2 B cells that bind to autoantigens were presented in the spleen via their BCRs. These cells undergo apoptosis due to proliferation failure or upregulation of the co-stimulatory molecule CD86, a process referred to as clonal deletion [34, 35]. The tolerance of early transitional B cells is a key component of peripheral B cell tolerance.
Primary B cell tolerance: In peripheral lymphoid tissues, primary B cells that recognize self-antigens in the absence of specific T helper cells become anergic or undergo apoptosis, thereby establishing immune tolerance [36, 37]. In the presence of T cells, an autoreactive B cell may survive if repeatedly stimulated by autoantigen due to higher levels of B cell activating factor (BAFF). However, these cells are rapidly eliminated through apoptosis during competition with normal B cells that are less dependent on BAFF. Additionally, apoptosis of this subset of B cells can also occur via the mitochondrial pathway in a BAFF-independent manner [38, 39]. There are also some B cells with low-affinity binding to autoantigens that are subject to various inhibitory receptors, which set a threshold for B cell activation and prevent responses to autoantigens [40]. Because the repertoire of autoantigens presented in the spleen differs from that in the bone marrow, the frequency of autoreactive B cells is further reduced in the periphery [41]. Ultimately, after undergoing peripheral immune tolerance mechanisms, the number of autoreactive B cells is significantly diminished.
Immune cytokines and molecules involved in the growth, differentiation, maturation, and activation of B cells play critical roles in maintaining immune tolerance [42, 43]. Dysregulation of these cytokines and molecules can lead to the breakdown of B-cell immune tolerance, resulting in autoimmune responses (Table 1, Ref. [44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63]). The roles of these checkpoints in the breakdown of immune tolerance are described below. Their relevance to specific autoimmune diseases is also discussed (Fig. 1).
| Checkpoint | Stimulation/regulation signaling | Autoimmune diseases | Mechanism | Reference |
| BAFF | Regulation signaling | MS, SLE | B cell activating | [44, 45, 46, 47] |
| IL-6 | Regulation signaling | SLE, RA | B cell growth; plasma cell differentiation | [48, 49, 62] |
| CD19 | Regulation signaling | SLE, SS | regulate B cell signaling thresholds | [50, 51, 52, 63] |
| CD22 | Regulation signaling | SLE, MG | regulating B cell responses | [53, 54] |
| TLR | Stimulation signaling | RA | activation of B cells | [50, 55] |
| CD40/CD40L | Stimulation signaling | SLE, RA, MS | co-stimulatory for B cell activation | [56, 57, 58, 59, 60, 61] |
BAFF, B cell activating factor; TLR, Toll-like receptors; MS, multiple sclerosis; RA, rheumatoid arthritis; SS, Sjögren’s syndrome; IL-6, interleukin-6; MG, myasthenia gravis; SLE, systemic lupus erythematosus.
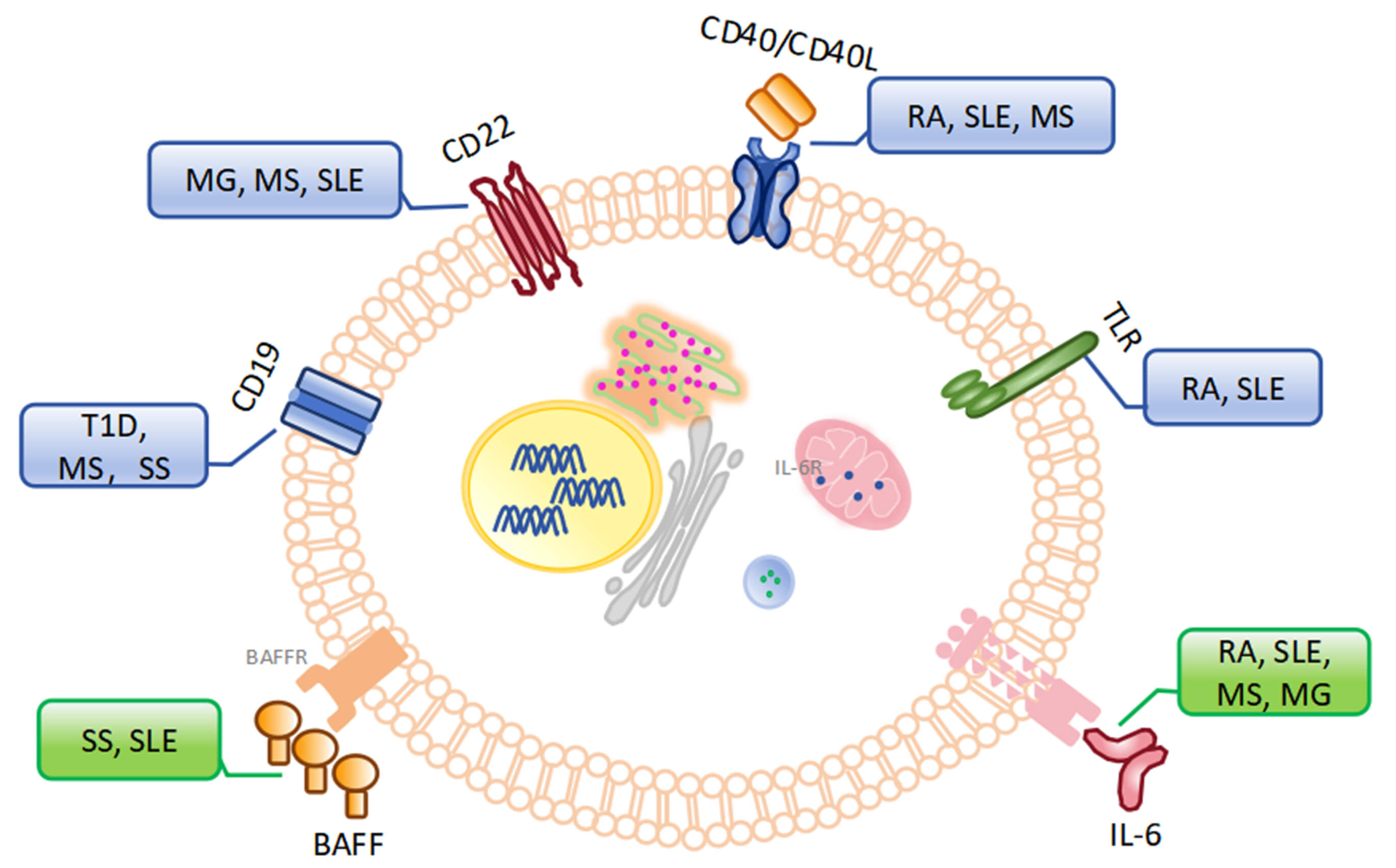 Fig. 1.
Fig. 1.
Checkpoints in the breakthrough of immune tolerance for B cells and related autoimmune diseases. (The blue box represents autoimmune diseases related to regulatory signals of B cells, the green box represents autoimmune diseases related to stimulatory signals of B cells). MG, myasthenia gravis; T1D, type 1 diabetes; MS, multiple sclerosis; SLE, systemic lupus erythematosus; BAFFR, B cell activating factor receptor; SS, Sjögren’s syndrome; BAFF, B cell activating factor; RA, rheumatoid arthritis; IL-6R, interleukin-6 receptor; TLR, Toll-like receptors.
Immune checkpoints in regulation signaling: ① BAFF, also known as TNFSF13B, is essential for B cell survival. During the establishment of immune tolerance, BAFF promotes the survival of B cells that weakly bind to autoantigens and contributes to the breakdown of immune tolerance [44]. Elevated BAFF expression has been observed and is associated with the loss of B-cell tolerance and production of auto-antibodies in Sjögren’s syndrome (SS) [45]. Additionally, serum BAFF concentrations are positively correlated with anti-double-stranded DNA (dsDNA) antibody titers in patients with systemic lupus erythematosus (SLE) [46]. Belimumab, a human monoclonal antibody that targets BAFF, has demonstrated therapeutic efficacy in patients with autoantibody-positive SLE [47]. ② Interleukin-6 (IL-6): IL-6 was originally identified as a B cell growth factor and a plasma cell differentiation factor. In RA, elevated serum levels of IL-6 are associated with joint injury. And blocking IL-6 receptor with Tocilizumab alleviates symptoms in patients with RA [48]. Research by Arkatkar et al. [49] has shown that IL-6 plays a pivotal role in B-cell-driven autoimmunity through the formation of autoimmune germinal centers and follicular helper T cell differentiation in SLE. Administration of anti-IL-6 antibodies to myasthenic rats suppressed experimental autoimmune myasthenia gravis (EAMG), accompanied by a reduced number of B cells [64]. These data identify IL-6 as an important target for modulation of autoimmune responses. Lv et al. [65] reported that chlorzoxazone could ameliorate experimental autoimmune encephalomyelitis (EAE) pathogenesis via inhibiting IL-6 production by dendritic cells. ③ CD19: CD19 is a central regulator of B cell signaling thresholds. High CD19 expression lowers the B cell signaling threshold and potentially increases susceptibility to autoimmune development [50]. The role of autoantigen-binding B cells and CD19+ plasma cells is key antigen-presenting cells in “T cell-mediated” autoimmune disorders such as type 1 diabetes (T1D) [66]. In tight-skin (TSK/+) mice, a genetic model of human SS, deficiency in CD19 expression significantly reduced skin fibrosis and autoantibody production [51]. It has been reported that the anti-human CD19 antibody LY3541860 demonstrates efficacy in various B-cell-dependent autoimmune disease models such as nonobese diabetic (NOD) mice and EAE mice [52]. ④ CD22: CD22 is a transmembrane receptor molecule associated with BCR signaling. CD22 plays an important role in regulating B cell responses to autoantigens. CD22 contains three tyrosine-based cytoplasmic immunoreceptor inhibitory motifs. Phosphorylation of these motifs can promote the activation of autoreactive B cells [67, 68]. In addition, studies in a number of mouse models have shown that B cells exhibit an “over-activated” phenotype in the absence of functional CD22 [69]. Single-cell RNA-seq analysis revealed that plasma soluble CD22 levels were correlated with myasthenia gravis (MG) severity and B cell frequency [70]. CD22 blockade aggravated EAE in mice [53]. Furthermore, Epratuzumab, a monoclonal antibody targeting CD22, has been evaluated in clinical trials for the treatment of SLE [54].
Immune checkpoints in stimulation signaling: ① Toll-like receptors (TLRs): TLRs contribute to the activation of autoreactive B cells. The dual recognition of auto-DNA by TLR9 and BCR has been reported to activate autoreactive B cells, such as rheumatoid factor-specific B cells [55]. Other studies have found that single-stranded RNA or RNA-binding proteins (e.g., small nuclear ribonucleoproteins, Sm, Ro, and La) can stimulate the activation of rheumatoid factor-specific B cells in the presence of both TLR7 and BCR in patients with RA [50]. TLR7 drives the extrafollicular B cell response and the germinal centre reaction that are involved in autoantibody production and disease pathogenesis in SLE [71]. ② CD40/CD40L: CD40/CD40L is a co-stimulatory molecule that mediates B cell activation. The interaction between CD40 and CD40L is necessary to support the autoantibody response of autoreactive B cells [72]. In lupus-prone MRL/lpr mice, macrophages regulate autoreactive B cells by secreting CD40L [56]. It has also been reported that CD40L is upregulated in T cells from various autoimmune diseases (including SLE, RA, and MS), and that soluble CD40L levels correlate with autoantibody titers and disease activity [57, 58, 59, 60, 61]. Frexalimab is a second-generation anti-CD40L monoclonal antibody being evaluated for the treatment of MS. Inhibition of CD40L with frexalimab slows new brain lesions in MS [73, 74]. Dazodalibep, a novel anti-CD40L nonantibody fusion protein, appears to be a potential new therapy for SS, and its efficacy implies an important role for the CD40/CD40L pathway in its pathogenesis [75].
T cells originate from bone marrow-derived pluripotent stem cells, which commit
to the T lineage and subsequently migrate to the thymus for differentiation and
maturation. In the thymus, T cells undergo random V(D)J recombination of the T
cell receptor
In the early stages of T cell development in the thymic cortex, progenitor T cells develop into immature CD4+CD8+ double-positive (DP) thymocytes during TCR gene rearrangement and upregulation of CD4 and CD8 [78]. After interaction with Class I or Class II MHC-auto-peptide complexes, these DP thymocytes are presented on cortical epithelial cells and differentiate into single-positive (SP) CD4+ or CD8+ thymocytes. This process is known as positive selection [79, 80]. Following positive selection, CCR7 expression on SP thymocytes is upregulated, enabling migration of SP thymocytes to the cortico-medullary junction of the thymus [81]. At this location, medullary thymic epithelial cells (mTECs) and/or dendritic cells present diverse MHC-auto-peptide complexes to SP thymocytes. SP thymocytes that bind to these complexes with relatively high affinity are induced to undergo apoptosis, which is called negative selection [82, 83]. Negative selection is mediated by chemokines such as CCL19 and CCL21, which facilitate the interaction between thymocytes and antigen-presenting cells [84, 85]. Central tolerance, primarily achieved through negative selection, ensures the clearance of most autoreactive T cells within the thymus [86]. Only 3–5% of all thymocytes survive both positive and negative selection. These cells then emerge as mature SP CD4+ or CD8+ T cells. Additionally, mTECs play a critical role in promoting the differentiation of autoreactive CD4+ T cells into regulatory T cells (Tregs), a type of T cell characterized by the expression of the transcription factor FoxP3 [87]. Tregs migrate into peripheral tissues and contribute to the suppression of autoreactive T cells that may have escaped central tolerance mechanisms [88].
Although T cells suffer central tolerance in the thymus, Bouneaud et al. [8] reported that 25%-40% of autoreactive T cells escape clonal deletion and enter the periphery. Consequently, the initial peripheral T cell repertoire contains a considerable number of autoreactive T cells [78, 79]. Peripheral autoantigens can reduce the pool of autoreactive T cells through various mechanisms [89, 90, 91, 92] or subject their responses to strict regulation, such as that of regulatory T cells [93].
Peripheral tolerance mechanisms of T cells: (1) Clonal clearance: T cells recognize autoantigens in the periphery via TCR and then induce apoptosis of activated autoreactive T cells through Fas-mediated “death receptor” signaling and Bcl-2-regulated apoptosis pathways. This elimination of autoreactive T cells is called peripheral clonal clearance [94, 95]. (2) Immune incompetence: T cells become non-responsive when they either lack co-stimulatory signals or receive inhibitory signals upon encountering autoantigens in the periphery. Programmed cell death protein 1 (PD-1/PD-L1) and Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) are key co-inhibitory molecules involved in immune incompetence and tolerance [96, 97]. (3) Immune neglect: Autoantigen expression is either too low or anatomically inaccessible to be recognized by TCR. In this case, T cells cannot be stimulated sufficiently to elicit a response to autoantigen [98, 99]. (4) Immune silence: T cells remaining in the G0 phase of the cell cycle exhibit minimal metabolic activity and size. These T cell subsets do not respond to autoantigens [100, 101]. Immune silence can be modulated to either allow initial T cells to respond to low-affinity antigens or to more strictly limit immune responses [102]. (5) Regulatory T cells: Regulatory T cells suppress autoreactive T cells through direct contact or cytokine secretion [103].
Although autoreactive T cells are cleared during a series of tolerance mechanisms in thymus development and peripheral circulation, some checkpoints can break through the immune tolerance of T cells at different stages (Table 2, Ref. [104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119]). We will discuss these breakthroughs during the derivation, maturation, and activation of T cells, followed by autoimmune diseases (Fig. 2).
| Checkpoint | Stimulated/inhibitory signaling | Autoimmune diseases | Mechanism | Reference |
| AIRE | stimulated signaling | APS-1 | upregulate tissue restricted antigens | [104, 105, 106] |
| Fas/FasL | stimulated signaling | ALPS | Fas-mediated apoptosis | [107] |
| Bcl-2 | stimulated signaling | SLE, T1DM | mitochondrial apoptosis | [108, 109, 110] |
| CTLA-4 | inhibitory signaling | MS, EAE | mediates T-cell non-responsiveness; | [112, 119] |
| altered chronic antigen stimulation | ||||
| PD-1/PD-L | inhibitory signaling | T1DM | inhibit T cell activation | [111, 113, 114, 115] |
| TIGIT | inhibitory signaling | SLE | inhibit T and B cell activation | [116, 117, 118] |
TIGIT, T cell immunoglobulin and immunoreceptor tyrosine inhibitory motif domain protein; CTLA-4, Cytotoxic T lymphocyte-associated protein 4; APS-1, autoimmune polyglandular syndrome Type I; AIRE, autoimmune regulatory transcription factor; EAE, experimental autoimmune encephalomyelitis; PD-1, programmed death 1; T1D, type 1 diabetes; ALPS, autoimmune lymphoproliferative syndrome.
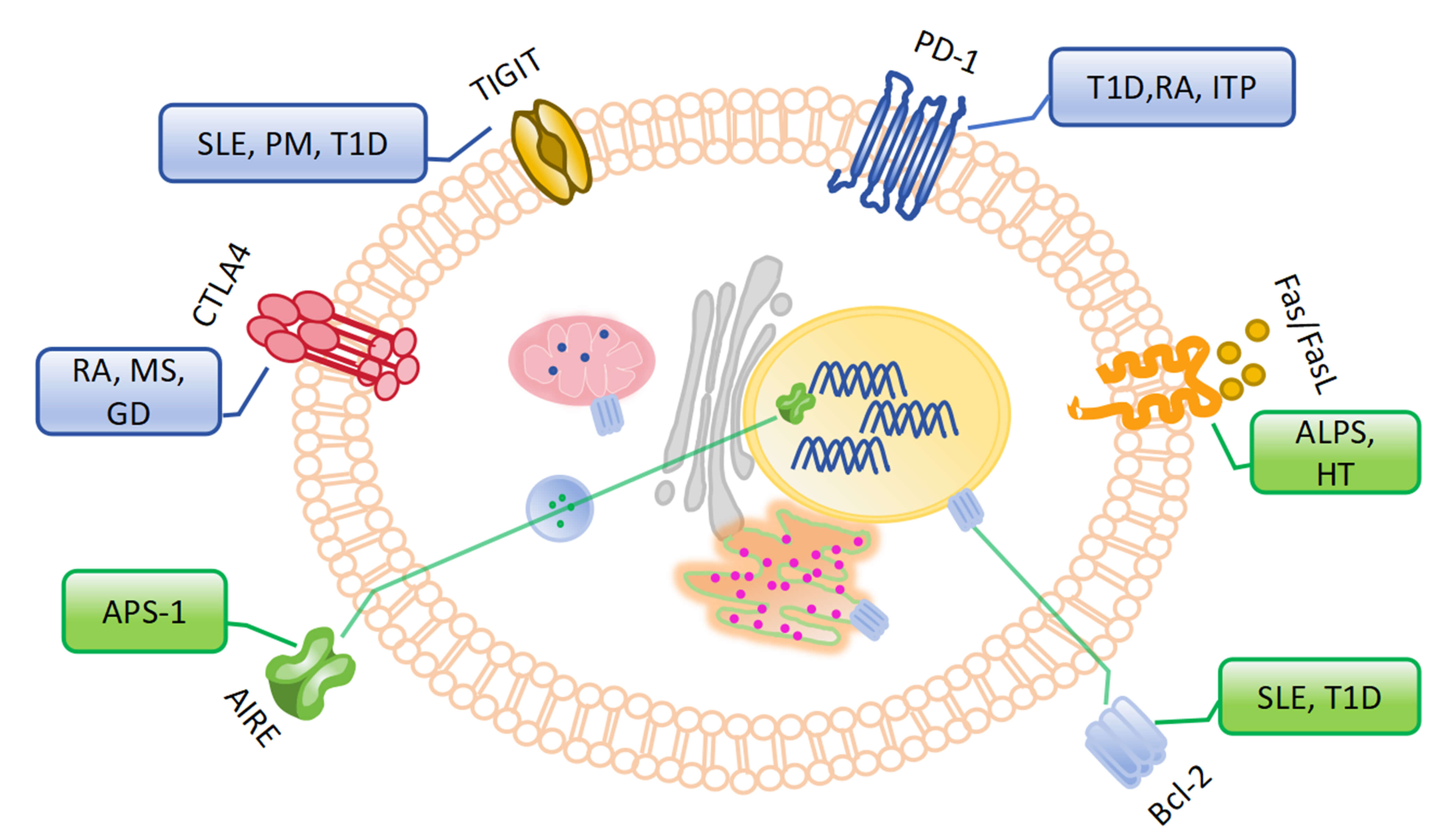 Fig. 2.
Fig. 2.
Checkpoints in the breakthrough of immune tolerance for T cells and related autoimmune diseases. (The blue box represents autoimmune diseases related to stimulatory signals of B cells, the green box represents autoimmune diseases related to inhibitory signals of B cells). TIGIT, T cell immunoglobulin and immunoreceptor tyrosine inhibitory motif domain protein; CTLA-4, Cytotoxic T lymphocyte-associated protein 4; APS-1, autoimmune polyglandular syndrome Type I; AIRE, autoimmune regulatory transcription factor; PD-1, programmed death 1; T1D, type 1 diabetes; ALPS, autoimmune lymphoproliferative syndrome; HT, Hashimoto’s thyroiditis; GD, Graves’ disease; PM, polymyositis.
Immune checkpoints of stimulated signaling: ① Autoimmune regulatory transcription factor (AIRE): AIRE, a transcription factor specific to medullary thymic epithelial cells (mTECs), plays a pivotal role in central tolerance [104]. Thymic mTECs express tissue-restricted antigens that are specific to various tissues throughout the body. AIRE upregulates the expression of these tissue-restricted antigens to facilitate effective negative selection of autoreactive T cells [120]. Disruption of central tolerance due to mutations in AIRE can lead to the development of multi-system autoimmune diseases, such as autoimmune polyendocrine syndrome type 1 (APS-1, also known as APECED) [105, 106]. It is reported that mice with AIRE mutations develop pathological autoimmune features similar to those of APECED and characterized by multi-organ lymphocyte infiltration and autoantibody production [105, 106]. ② Fas/FasL: Autoreactive T cells that recognize self-antigen are eliminated through Fas death receptor signaling [121, 122]. Fas and FasL play an extremely important role in the pathogenesis of autoimmune lymphoproliferative syndrome (ALPS). And ALPS arises from mutations in Fas genes that impair T cell apoptosis regulation, which clinically manifests as autoimmune and lymphoproliferative diseases [107]. And autonomous interaction between thyrocyte Fas and FasL has been proposed as a major mechanism of thyrocyte depletion in Hashimoto’s thyroiditis (HT) [123]. Gene polymorphism of Fas and G allele frequency may play a role in the regulation of apoptosis in thyroid autoimmune disorders [124]. ③ Bcl-2: Bcl-2 regulates cell death via the mitochondrial apoptotic signaling pathway. This is essential for establishing and maintaining peripheral T cell tolerance [125]. In a NOD mouse model, failure to upregulate Bim in T cells stimulated by highly active autoantigens results in reduced apoptosis of autoreactive T cells [108]. And treatment of NOD mice with Bcl-2 inhibitors eliminated senescent beta cells and prevented diabetes [126]. Similarly, loss of Bim or overexpression of Bcl-2 in mice leads to a SLE-like disease and premature death [109, 110].
Immune checkpoints of inhibitory signaling: ① CTLA-4: CTLA-4 is a T
cell-dependent suppressor receptor that mediates T cell non-responsiveness or
altered chronic antigen stimulation through its interaction with B7
co-stimulatory factors [127]. The CTLA-4 gene has previously been shown to be
associated with RA, GD, and MG [111]. The G allele of the rs231775A
Regulatory T lymphocytes: A small number of autoreactive T cells in the thymus
differentiate into Foxp3+ regulatory T cell (Treg) lineages and migrate to the
periphery. These peripheral Foxp3+ Tregs inhibit autoreactive T and B cells and
help maintain peripheral immune tolerance [139, 140]. Animal studies have shown
that depletion/reduction of CD4+CD25+ Tregs can mitigate the suppression of
autoreactive T cell activation. This triggers an autoimmune response to certain
autoantigens [141, 142]. Additionally, it has been reported that CD4+CD25+ Tregs
and transforming growth factor-beta 1 (TGF-
It is widely accepted that both genetic predisposition and external risk factors, such as chronic microbial infections, physical factors, medications, and diet, contribute to the disruption of the autoimmune balance following the occurrence of autoimmune diseases [146]. The relationship between microbial infections and autoimmune diseases has been investigated for over a century. Molecular mimicry is a mechanism underlying the pathogenesis of microbe-induced autoimmune diseases. Molecular mimicry occurs when a microbe shares epitopes that are identical or similar to those of the host. In such cases, immune response products, autoreactive antibodies, may cross-react with autoantigens, resulting in autoimmunity and autoimmune diseases. For instance, Amin et al. [147] and Boettler et al. [148] identified sequence homology between Coxsackie B virus protein 2C and the pancreatic autoantigen glutamate decarboxylase (GAD) in T1D. Moreover, GAD autoreactive antibodies derived from lymphocytes of patients with T1D exhibit cross-reactivity with the Coxsackie B virus protein 2C. SLE is a classic autoimmune disease closely associated with microbial infections, such as Epstein-Barr virus (EBV) infection. Anti-Ro antibodies are frequently observed in patients with SLE [149], and it has been reported that antibodies against the Ro 60kDa peptide (TKYKQRNGWSHK) bind to the restriction region (GGSGSGPRHDGVRR) of the EBV nuclear antigen 1protein [150, 151].
An increasing number of studies have reported that autoreactive antibodies in both organ-specific and systemic autoimmune diseases exhibit cross-reactivity with microbial antigens or epitopes [152]. Autoreactive antibodies induced by microbes that recognize autoantigens are critical factors in the onset of autoimmune diseases associated with microbial infections [153, 154]. It should be noted that the presence of cross-antigens between host tissues and microorganisms serves as the foundation for the generation of autoreactive antibodies. Nevertheless, the underlying mechanisms governing the generation of autoreactive antibodies and their role in microbe-associated autoimmune diseases remain largely unknown and warrant further investigation.
In 1936, Landsteiner and van der Scheer proposed that an antigen consists of two parts [155, 156]. One part of the antigen is responsible for antibody generation, and the other part influences antibody generation. Several decades later, Ovary and Benacerraf [157] and Rajewsky et al. [158] demonstrated that antibody production requires the cooperation of two cell types. Each type of cell recognizes different epitopes of the antigen. After generations of exploration, the mechanism of antibody production has been clearly elucidated. A complete antigen comprises two components: carrier epitopes and antigenic epitopes. During antibody production, the receptor on the surface of B cells recognizes antigenic epitopes, while the receptor on the surface of T helper cells recognizes carrier epitopes. B-cell activation occurs when both the first signal (antigen recognition) and the second signal (provided by activated T helper cells) are present. Consequently, activated B cells differentiate into plasma cells that secrete antibodies. As is well known, T helper cells are essential for B cell activation.
Cross-antigens are present in both microorganisms (foreign antigens) and the host (autoantigens). As mentioned above, cross-antigens also consist of carrier epitopes and antigenic epitopes. The critical point is that although foreign antigens and autoantigens share similar antigenic epitopes, the carrier epitopes in foreign antigens differ from those in autoantigens (Fig. 3A). Carrier epitopes and similar antigenic epitopes of autoantigens are rendered tolerant through the central and peripheral tolerance mechanisms of T/B cells. However, when the body encounters microbial infections, either alone or in combination with autoimmune susceptibility genes or other risk factors, the autoimmune balance will be disrupted. Consequently, the immune tolerance of autoreactive T/B cells may be broken down through immune checkpoints. In such cases, when autoreactive B cells encounter microbial antigens, they will be activated.
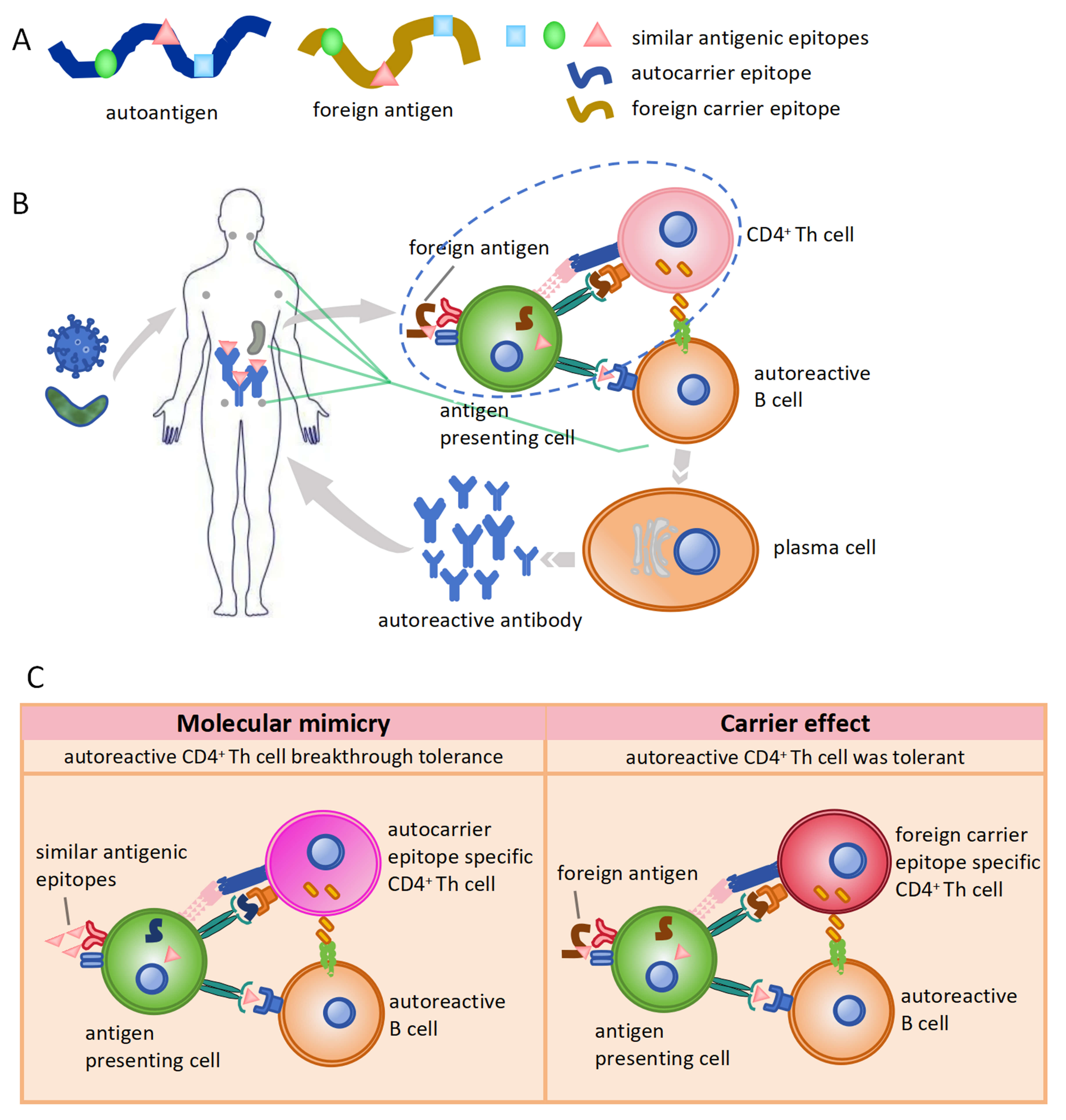 Fig. 3.
Fig. 3.
Carrier effect in the generation of autoreactive antibodies. (A) Different carrier epitope in auto-antigen or foreign antigen with the similar antigenic epitopes. The blue square, green circle, and pink triangle represent antigenic epitopes. The brown curve represents the foreign carrier epitope. The blue curve represents the autocarrier epitope. (B) Generation of autoreactive antibody induced by microbial infection in case of immune tolerance breakthrough. (C) Differences between molecular mimicry and carrier effect during autoreactive antibody generation induced by microbial infection.
The activation of autoreactive B cells depends on the carrier epitopes on foreign antigens derived from microbes due to the carrier effect, while the immune tolerance of autoreactive T cells remains intact. The detailed mechanism for the generation of autoreactive antibodies regarding immune tolerance is as follows: (1) Immune tolerance of autoreactive B cells remains intact. In this case, regardless of the breakdown of tolerance of autoreactive T cells, autoreactive B cells cannot be activated by either auto-antigens or foreign antigens. Consequently, there is no immune response against cross-antigens. (2) Both the immune tolerance of autoreactive T and B cells is broken down. In this scenario, autoreactive T cells are activated and then provide a second signal to autoreactive B cells in response to autoantigens. Thereby, autoreactive B cells were activated to secrete autoreactive antibodies, which results in autoimmune diseases (Fig. 3B). As is well known, this mechanism of autoreactive antibody production is generally explained by molecular mimicry (Fig. 3C). (3) The immune tolerance of autoreactive B cells is broken, but autoreactive T cells remain intact. In such cases, the carrier epitopes of foreign antigens activate T cells and provide the second signal required for the activation of autoreactive B cells. Autoreactive antibodies against cross-antigens are generated via the carrier effect due to differences in carrier epitopes in foreign antigens (Fig. 3C, Table 3). These autoreactive antibodies can recognize the corresponding autoantigens, which leads to tissue and organ damage and ultimately causes autoimmune diseases.
| Molecular mimicry | Carrier effect | |
| Antigenic epitope | similar antigenic epitopes from a foreign antigen | similar antigenic epitopes from a foreign antigen |
| Carrier epitope | autocarrier epitope | foreign carrier epitope |
| T cell | autocarrier epitope-specific CD4+ Th cell | foreign carrier epitope-specific CD4+ Th cell |
| B cell | similar antigenic epitopes, specific B cell | similar antigenic epitopes, specific B cell |
Foreign antigens from microorganisms share similar antigenic epitopes but possess different carrier epitopes compared to those in autoantigens. Following microbial infection and the subsequent alteration of the autoimmune balance, the T cell receptor recognizes the carrier epitopes in foreign antigens, while the B cell receptor on autoreactive B cells recognizes similar antigenic epitopes. Subsequently, activated T cells provide the second signal necessary for the activation of autoreactive B cells, which then differentiate into plasma cells to secrete autoreactive antibodies. Such autoreactive antibodies may serve as a basis for the occurrence of autoimmune diseases through molecular mimicry mechanisms mediated by the carrier effect. Our perspective can offer a complementary explanation for molecular mimicry in elucidating the roles of autoreactive antibodies in autoimmune diseases associated with microbial infections. Although this is a narrative review with its own advantages and is capable of achieving conceptual integration, it lacks systematic experimental evidence. Future systematic reviews combined with meta-analysis may be able to verify the universality of carrier effect-mediated autoreactive antibody production in autoimmune diseases related to microbial infections.
We proposed experimental techniques to test the carrier effect hypothesis. The SLE animal model induced by phytanic acid does not produce anti-Ro60kD antibodies and can be used to verify the carrier effect. The SLE study induced by Campylobacter jejuni (CJ-S131) in Balb/c mice has certain advantages in the investigation of immune response and can also be used to verify the carrier effect. The experimental protocol is to stimulate SLE model animals with the restriction region (GGSGSGPRHDGVRR) of the EBV nuclear antigen 1 or EBV nuclear antigen 1, respectively. The examination of the binding of serum antibodies from the two groups of animals with the complexes of small RNAs and 60 kD proteins could verify whether the absence of carrier epitopes but only the presence of antigenic epitopes could lead to the production of corresponding antibodies.
Microbial infection is one of the critical factors contributing to autoimmune diseases [159]. Anti-citrullinated peptide antibodies (ACPAs) derived from EBV nuclear antigens have been shown to cross-react with human citrullinated fibrin in RA [160]. ACPA is widely regarded as one of the most important diagnostic biomarkers of early-stage RA [161]. A case report showed that arthritis and ACPAs were resolved by antibiotic treatment in a patient with Actinomyces actinomycetemcomitans endocarditis [162]. Meanwhile, the microbial imbalance of oral microbiomes in patients with RA shows partial normalization following treatment with methotrexate, a commonly prescribed disease-modifying antirheumatic drug [163]. Rheumatic fever is triggered by group A streptococcus and is accompanied by rheumatic heart disease [164]. Zhang et al. [165] reported that both rheumatic valvular heart disease and glomerulonephritis are dramatically decreased in incidence after adoption of therapeutic and preventive measures for Streptococcal infection in the US. These results demonstrate that microbial infection is deeply implicated in autoimmune diseases.
In this review, we first elucidate the immune tolerance mechanisms of T/B lymphocytes and their incomplete tolerance due to immune checkpoints, as well as autoimmune disorders. Subsequently, we discuss the mechanism underlying the generation of autoreactive antibodies induced by foreign microbial antigens under conditions of autoimmune imbalance. We propose that the carrier effect represents one of the mechanisms for the activation of autoreactive B cells following changes in autoimmune balance after microbial infection. This mechanism of autoreactive antibody production through the carrier effect complements the molecular mimicry theory for understanding microbial infection-related autoimmune disease. We provide a novel perspective for understanding the role of autoreactive antibodies and microbial infections in the pathogenesis of autoimmune diseases. We hope that our insights will provide new avenues for exploring the mechanisms of autoimmune diseases and offer innovative strategies for drug discovery in this field.
BCR, B cell receptor; TCR, T cell receptor; MHC, major histocompatibility complex; RAG1/2, recombinase activating gene 1/2; BAFF, B cell activating factor; BAFFR, B cell activating factor receptor; SLE, systemic lupus erythematosus; MS, multiple sclerosis; RA, rheumatoid arthritis; TLR, Toll-like receptors; CCR7, chemokine receptor 7; mTECs, thymus medullae epithelial cell; Tregs, regulatory T cells; AIRE, autoimmune regulatory transcription factor; APS-1, autoimmune polyglandular syndrome Type I; ALPS, autoimmune lymphoproliferative syndrome; CTLA-4, Cytotoxic T lymphocyte-associated protein 4; PD-1, programmed death 1; PD-L1, programmed death-ligand 1; EAE, experimental autoimmune encephalomyelitis; TIGIT, T cell immunoglobulin and immunoreceptor tyrosine inhibitory motif domain protein; GAD, glutamate decarboxylase; NOD, nonobese diabetic; T1D, type 1 diabetes; ACPA, anti-citrullinated peptide antibodies; EAMG, experimental autoimmune myasthenia gravis; MG, myasthenia gravis; SSc, systemic sclerosis; HT, Hashimoto’s thyroiditis; GD, Graves’ disease; EBV, Epstein-Barr virus; MG, myasthenia gravis; SS, Sjögren’s syndrome; IL-6R, interleukin-6 receptor; PM, polymyositis; ITP, idiopathic thrombocytopenic purpura.
LTY, XYH and CXX designed the research. JH developed the overall concept. LTY and CXX integrated and refined the key highlights. QF, JYS conducted the literature search. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by the Natural Science Basic Research Program of Shaanxi (2023-JC-QN-0855), the Shaanxi Provincial People’s Hospital Science and Technology Talent Support Program Project (2021JY-17).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.