



 , Xuanhao Zhang 1,†, Hengzhou Zhu 1, Dong Niu 1, Xiaodan Zhu 1, Chunhui Jin 1,*
, Xuanhao Zhang 1,†, Hengzhou Zhu 1, Dong Niu 1, Xiaodan Zhu 1, Chunhui Jin 1,*
1 Department of Oncology, Wuxi Affiliated Hospital of Nanjing University of Chinese Medicine, 214071 Wuxi, Jiangsu, China
†These authors contributed equally.
Abstract
The dynamic evolution of signaling pathway remodeling and molecular regulation within the tumor microenvironment (TME) of hepatocellular carcinoma (HCC) play a critical role in the onset and progression of this malignancy. As chronic hepatitis progresses to cirrhosis and ultimately to HCC, the signaling pathways and TME show stage-specific characteristics that provide important insights into the therapeutic challenges and opportunities. In this review, we profiled the principal components of the HCC TME, along with pivotal signaling pathways, including the receptor tyrosine kinase (RTK) and extracellular signal-regulated kinase (ERK) pathways. Furthermore, we characterized the dynamic transformation of the TME from an inflammatory state in the hepatitis phase to a fibrotic state in the cirrhosis phase. Ultimately, we assessed the therapeutic potential of current HCC targets emphasizing emerging strategies for precision and personalized treatment.
Keywords
- hepatocellular carcinoma
- tumor microenvironment
- signal transduction
- disease progression
- molecular targeted therapy
- immunotherapy
Hepatocellular carcinoma (HCC), the most prevalent primary liver cancer globally, is notorious for an alarmingly high mortality rate. Its epidemiological profile resembles that of chronic liver diseases, with the main etiological factors including hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, alcohol consumption, and metabolic diseases. In developed countries, the incidence of HCC has kept rising over decades [1]. Additionally, HCC may also arise from non-viral factors, such as metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease [2]. These signals indicate the heavy load that HCC poses on the global public health.
Generally, hepatitis and liver cirrhosis develop sequentially prior to HCC. Firstly, in the hepatitis stage, HBV and HCV infections are significant inducers for HCC. In the case of HBV infection, HBV proteins, such as HBV X protein (HBx), can facilitate the development of HCC by modulating chromatin and dysregulating the expression of tumor suppressor genes [3]. HCV infection promotes carcinogenesis through arousing chronic inflammation and the interaction between viral proteins and cell cycle regulatory factors [4]. Subsequently, hepatic stellate cells (HSCs) become activated. These activated HSCs not only interact with immune cells by secreting cytokines and chemokines, but also provoke excessive extracellular matrix (ECM) deposition and liver fibrosis, thus transforming the liver microenvironment into immunosuppressive [5, 6]. Immune cells in the fibrotic liver, such as M2-type macrophages and regulatory T cells (Tregs), suppress anti-tumor immune responses, thereby facilitating tumor survival [7, 8]. After entering the cirrhosis stage, the immune and non-immune cells, HSCs, and hepatocytes in the liver continue to interact to promote the occurrence of tumors [9]. The infiltration of macrophages in the liver, especially M2-type macrophages, and the secretion of related factors keep continually enhanced, thus promoting tumor invasion and metastasis [10, 11]. Finally, the tumor microenvironment (TME) of HCC also undergoes metabolic reprogramming, which facilitates the continuous growth and survival of the tumor [12]. Overall, the dynamic interaction and evolution of various components within the TME drive the formation and progression of HCC. Instead of focusing on a static TME, this review depicted the evolution of the signaling pathways and molecular regulation in the dynamic TME. During this evolution, a “time window” may be utilized for precise treatment.
We systematically screened the existing literature by searching major databases, including PubMed, Web of Science, and Elsevier ScienceDirect. Studies dedicated to the TME of HCC over the past three years (2022–2025) were selected to guarantee the novelty of literature, mainly including original research studies, high-quality reviews, and key clinical trials published in peer-reviewed journals. Meanwhile, a small number of early foundational studies were also selected through methods such as manual screening of reference lists. In addition, studies irrelevant to the theme were excluded after reading their titles and abstracts, so as to ensure a high relevance of the literature to the present research theme.
The TME of HCC represents a highly intricate and dynamic ecosystem comprising tumor cells, various immune cells, stromal cells, ECM, and soluble factors, all of which interact intimately to influence the onset, progression, metastasis, and therapeutic responses of HCC. Notable immunosuppressive constituents include tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), Tregs, and cancer-associated fibroblasts (CAFs). Conversely, cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells serve as the primary fighters against tumor growth. Other significant components include dendritic cells (DCs), tumor-associated endothelial cells (TECs), and tumor-associated neutrophils (TANs) (Fig. 1, Ref. [13, 14]).
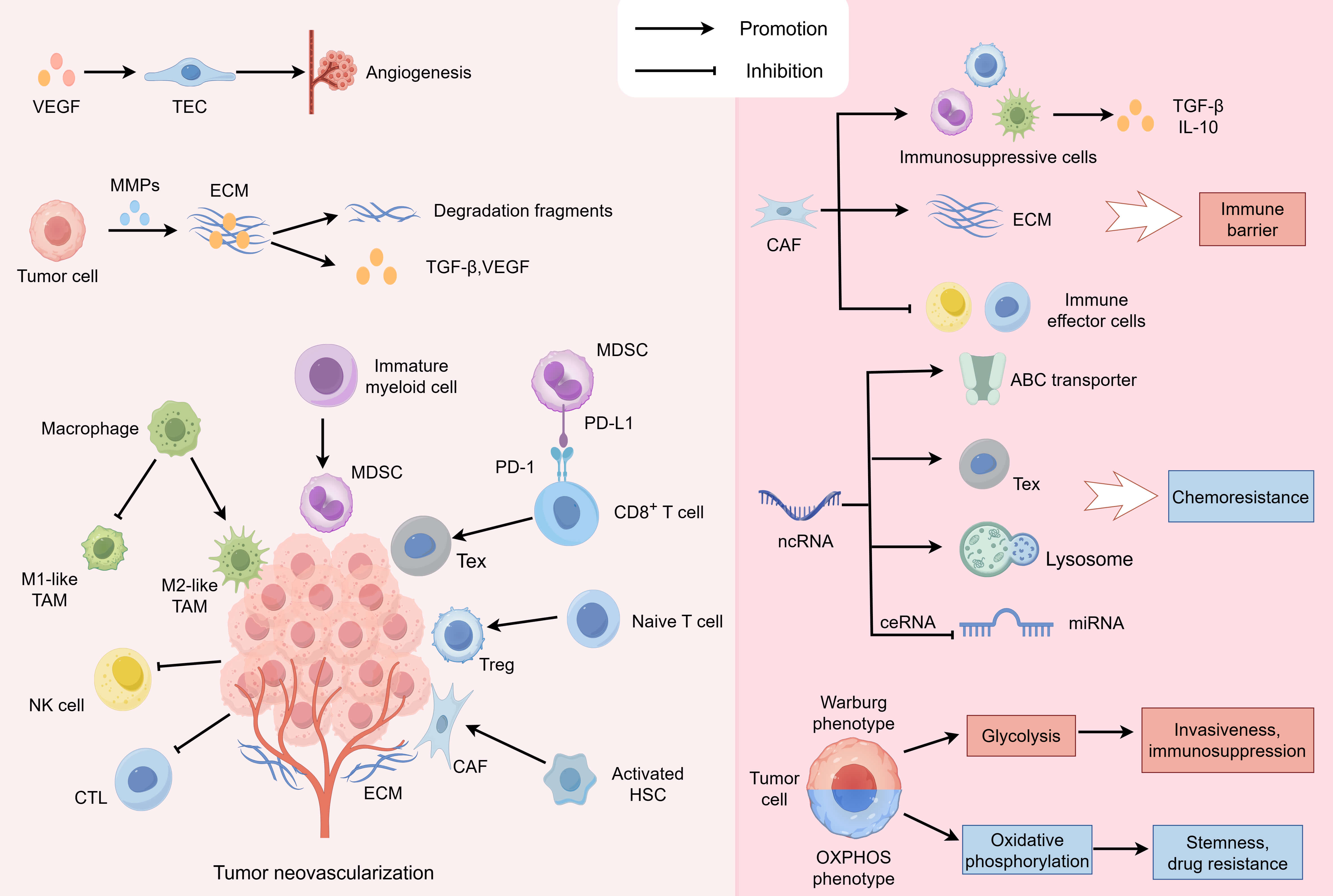 Fig. 1.
Fig. 1.
Cellular interactions within the HCC TME. In the TME, tumor
cells act as the core drivers, and immunosuppressive cells are enriched in their
surroundings. Different HCC subtypes utilize unique glucose metabolism patterns
(favoring either the Warburg effect or OXPHOS) to perform their distinct
functions. Macrophages tend to differentiate into the immunosuppressive M2
phenotype, rather than the pro-inflammatory M1 phenotype; immature myeloid cells
into MDSCs; naive T cells into Tregs; and activated HSCs into CAFs. And MDSCs can
induce CD8+ T cell exhaustion via the PD-L1/PD-1 pathway. Concurrently, the
functionality of immune effector cells such as NK cells and CTLs is suppressed.
Tumor cells are capable of secreting MMPs to degrade the ECM and liberate
cytokines like TGF-
HCC cells, the major player within the TME, actively modify their surrounding
environment to enhance their survival, proliferation, invasion, and metastasis.
HCC cells exhibit a substantial heterogeneity, as well as obvious intra-tumor and
inter-tumor variations. A single HCC tumor displays a spatial heterogeneity. Its
regions undergo distinct evolutions, resulting in multiple phenotypic subtypes
that facilitate local adaptation of the tumor [15]. Guo et al. [16]
delineated three subtypes of HCC tumor cells (metabolic, proliferative, and
pro-metastatic), and the activation of the transforming growth factor-
HCC cells engage in complex interactions with other cells within the TME, establishing an environment favorable for their growth. HCC cells can secrete various cytokines, growth factors and exosomes, to remodel the microenvironment. Tumor-derived alpha-fetoprotein (AFP) promotes macrophage polarization to an immunosuppressive phenotype and inhibits macrophage phagocytic function [19]. Besides, HCC cells secrete erythropoietin to activate the erythropoietin receptor signal in TAMs, promoting the construction of a tumor immunosuppressive microenvironment (TIME) [20]. HCC cells can transform normal HSCs into CAFs. Apart from secreting cytokines, tumor cells can also activate the protein kinase B (Akt) pathway in HSCs through exosomes, thereafter promoting the differentiation of HSCs into CAFs [21, 22].
The exosomes released by HCC contain various bioactive molecules. Once
internalized by other TME cells they deliver functional contents to target cells
and alter their functions. For example, exosomes derived from cancer cells have
been shown to interrupt the expression of neutrophil microRNA (miRNA) and
activate the NF-
Immunosuppressive cells inhibit immune effector cells by upregulating inhibitory immune checkpoint molecules and producing immunosuppressive factors and chemokines, till the setup of the TIME. TAMs tend to grow into the M2 phenotype, exerting a variety of pro-tumorigenic effects [25]. During the close signaling between M2 macrophages and HCC cells, HCC cells secrete cytokines to recruit M2 macrophages, and then M2 macrophages TAMs promote tumor metastasis by facilitating angiogenesis, EMT, and vascular permeability [26]. Using a Transwell co-culture model, Zhang et al. [27] revealed bi-directional interactions between TAMs and HBV-associated HCC cells, which promoted M2 polarization as well as stemness maintenance of cancer cells. Wang et al. [28] demonstrated that M2 macrophages induce sorafenib resistance by secreting CXCL1 and CXCL2, with the downstream extracellular signal-regulated kinase (ERK) signaling playing a crucial role.
MDSCs suppress the immunity to promote tumor invasion, so a high MDSC
infiltration is linked to a poor prognosis of HCC [29]. The main subtypes of
MDSCs are polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs).
PMN-MDSCs suppress T cell activity through the release of oxygenated lipids via
ferroptosis [30]. They can also enhance their immunosuppressive function by
upregulating the programmed death-ligand 1 (PD-L1) expression [31]. M-MDSCs exert
immunosuppressive functions by inducing adenosine accumulation, which inhibits
cluster of differentiation 8 (CD8)+ T cell activity [32]. M-MDSCs can also
highly express PD-L1 to induce CD8+ T cell exhaustion, which is associated
with liver transplant rejection [33]. Mutations in HCC induce the hyperactivity
of
Tregs, contributing to the immunosuppressive properties of the TME, present a
major hurdle for immunotherapy to function in solid tumors. Tregs secrete a lot
of immunosuppressive factors including TGF-
HCC cells recruit and activate CAFs to level up CAF infiltration, thus promoting
tumor proliferation and metastasis linked to a poor prognosis [44]. Within HCC,
many subgroups of CAFs display their own characteristics, and the heterogeneity
of CAFs deserves special attention. For example, Periostin-positive
CAFs, mainly located in the peripheral region of the tumor, can dominate ECM
remodeling and induce macrophage M2 polarization, thus contributing significantly
to the formation of an immune barrier [45]. Platelet-derived growth factor
receptor
CD8+ T cells and NK cells, two types of primary antitumor effector cells, can directly eliminate cancer cells. However, immune effector cells within the TME are often dysfunctional, limiting their efficacy in combating tumor growth. The accumulation of certain metabolites in HCC, such as glycerol-3-phosphate and bile acids, can diminish the cytotoxic capacity of CTLs [49, 50]. Some cells in the TME can also inhibit the function of CTLs. For example, a subset of TECs secretes CXCL12 to suppress the activation of CD8+ naive T cells [51].
In addition, chronic antigen exposure, such as persistent viral infections, activates the TOX and EOMES transcriptional programs, driving CD8+ T cells into an “exhausted” state [52]. It is worth noting that under the upregulation of co-inhibitory receptors like PD-1 and immunosuppressive molecules like CD39, the immunosuppressive effect is amplified, indicating that Texs (exhausted T cells) also play an immunosuppressive role within the TME [52, 53].
NK cells, integral to the innate immune system, act in anti-tumor immunity by
recognizing and eliminating cancer cells without prior sensitization. In the TME
of HCC, NK cells exhibit a low level, and their antitumor function is
significantly inhibited. In addition, a study found that one type of bacteria in
HCC can promote lipolysis into acetyl coenzyme A to inhibit NK cell ferroptosis
and enhance the antitumor function of NK cells [54]. Although NK cells have
antitumor effects, they can disrupt CD8+ T cell differentiation by blocking
retinoic acid receptor
In the HCC TME, the abnormal activation of multiple key signaling pathways drives the hepatocarcinogenesis, progression, metastasis, and therapeutic resistance. These pathways form an intricate regulatory network underpinning cell proliferation, apoptosis, angiogenesis, and immune evasion. The main signaling pathways, functions, molecules and targeted drugs are summarized in Table 1 (Ref. [57, 58, 59, 60, 61, 62, 63, 64]).
| Signaling pathways | Functions | Key molecules | Targeted drug | Mechanism | Clinical phase | Refs. |
| RTK | Angiogenesis, immunosuppression, stem cell maintenance, drug resistance, anti-apoptosis | VEGF, VEGFR, EGF, EGFR, HGF, c-Met, FGF, FGFR | TKI (lenvatinib) | Inhibits VEGFR and FGFR pathways to suppress angiogenesis | III | [57] |
| ERK | Cell proliferation, invasion and metastasis, EMT, PD-L1 expression, immunosuppression, drug resistance | Ras, Raf, MEK, ERK | MEK inhibitor (refametinib) | Directly inhibits MEK to block ERK signaling and suppress abnormal proliferation | II | [58] |
| Wnt/ |
Promote tumor growth and metastasis, EMT, immunosuppression | TBL1 inhibitor (tegavivint) | Interferes with |
I/II | [59] | |
| TGF- |
Immunosuppression, EMT, angiogenesis, maintenance of stem cell stemness, fibrosis, drug resistance | TGF- |
anti-ALK-1 monoclonal antibody (GT90001) | Blocks the TGF- |
Ib/II | [60] |
| PI3K/Akt/mTOR | Cell proliferation, metabolic regulation, immunosuppression, angiogenesis, drug resistance | PI3K, Akt, mTOR | mTOR inhibitor (everolimus) | Inhibits mTOR to block the cell cycle and suppress abnormal proliferation | II | [61] |
| Hedgehog | Cell proliferation, stem cell maintenance, drug resistance, immunosuppression, fibrosis | Hh, Smo, Gli | Smo inhibitor (vismodegib) | Inhibits Smo activation to cut off Hh pathway signaling | II | [62] |
| NF- |
Stem cell maintenance, immunosuppression, EMT, drug resistance | IKK, NF- |
IKK |
Binds to IKK |
III | [63] |
| HIF | Metabolic reprogramming, angiogenesis, stem cell maintenance, drug resistance, EMT, immunosuppression, promote metastasis | HIF-1 |
HIF-1 |
Directly inhibits HIF-1 |
Ib | [64] |
RTK, receptor tyrosine kinase; VEGF, vascular endothelial growth factor; EGF,
epidermal growth factor; HGF, hepatocyte growth factor; FGF, fibroblast growth
factor; VEGFR, vascular endothelial growth factor receptor; EGFR, epidermal
growth factor receptor; c-Met, hepatocyte growth factor receptor; FGFR,
fibroblast growth factor receptor; TKI, tyrosine kinase inhibitor; PD-L1,
programmed death-ligand 1; EMT, epithelial-mesenchymal transition; Ras, rat
sarcoma; Raf, rapidly accelerated fibrosarcoma; FZD, Frizzled; TCF/LEF, T-cell
factor/lymphoid enhancer factor; TBL1, transducin beta-like protein 1;
TGF-
Receptor tyrosine kinases (RTKs), a category of surface receptors with transmembrane structures, exert a pivotal regulatory effect on HCC development. When bound by ligands such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF), RTKs can activate downstream signaling cascades that collectively orchestrate cellular behaviors. The most common RTKs include vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), hepatocyte growth factor receptor (c-Met), and fibroblast growth factor receptor (FGFR). They are inhibited by tyrosine kinase inhibitors (TKIs) like lenvatinib [65].
The VEGFR signaling pathway mainly promotes tumor growth and spread by easing angiogenesis and increasing tumor blood supply. Abnormal vascular networks and angiogenesis in HCC are essential for growth, progression, invasion, and metastasis [66]. In addition, VEGFR, particularly VEGFR2, participates in TIME formation by facilitating Treg expansion, disrupting effector T cell and DC activities [67].
EGFR, integral to HCC proliferation and metastasis, also plays a role in establishing the TIME. Liver Kupffer cells depend on EGFR to produce IL-6, which stimulates compensatory proliferation and correlates with unfavorable survival outcomes in HCC patients [68]. Zhang et al. [69] demonstrated that EGFR participates in the formation of invasive pseudopodia of HCC cells, driven by increased stiffness of the ECM, and enhances the malignant traits of cancer cells. The activation of EGFR in HCC not only maintains the stemness and tumorigenicity of HCC cells, but also increases resistance to TKIs, such as lenvatinib and sorafenib, while targeted inhibition on EGFR can reverse this resistance [70, 71, 72]. Furthermore, this signaling pathway can interact with downstream signaling pathways to promote cancer progression. For instance, it stimulates the PI3K-Akt pathway to promote the Warburg effect and activate glycolysis to support the rapid proliferation of cancer cells, and also stimulate the downstream ERK1/2 pathway to encourage Treg differentiation and drug resistance [73, 74, 75].
As an oncogene, c-Met is overexpressed to promote the formation and proliferation of HCC [76]. HGF is the ligand for c-Met, and their binding activates downstream ERK1/2, rat sarcoma (Ras)/MAPK, and PI3K/Akt pathways to modulate HCC cell proliferation and migration [77]. In addition, the HGF-Met axis is a significant contributor to drug resistance and apoptosis resistance [78, 79].
Fibroblast growth factor (FGF) and its high-affinity receptor FGFR can benefit tumor survival. On the one hand, it can induce the proliferation of hepatic endothelial cells and angiogenesis to support tumor metastasis [80]. On the other hand, it can also induce TAMs to cause tumor phenotypic changes, helping to form a TIME [81]. In a cohort study of 870 samples, FGFR4 is the most frequently expressed among HCC patients, implying it as a carcinogenic driver of HCC [82, 83].
ERK is a subfamily of MAPKs that mediate various cellular responses, including inflammation, cell proliferation, cell differentiation, and apoptosis [84]. The classical MAPK pathway transmit signals through a highly conserved three-tiered cascade, in which the MAP kinase kinase kinase (MAP3K) phosphorylates the MAP kinase kinase (MAP2K) and then phosphorylates the MAPK to amplify signals for precise regulation [85]. Once activated, MAPKs target specific serine and threonine residues on downstream protein kinases or transcription factors, thus fulfilling a role in gene transcription [86]. Different MAPK subfamilies have their specific upstream kinases. Having been activated by rapidly accelerated fibrosarcoma (Raf) protein kinase, kinase MEK1 and MEK2 activate ERK1/2 [87]. Clinical studies have been conducted aiming to block the ERK pathway via MEK inhibitors [58].
The ERK signaling pathway modulates the immune response and the secretion of
cytokines, including chemokines, TGF-
In the context of HCC, the ERK pathway poses a profound effect on the myeloid
TME. Activation of MEK-ERK1/2 drives cancer cells to secrete
granulocyte-macrophage CSF, and monocyte-derived cells to develop with
immunosuppressive and pro-inflammatory features [89]. Moreover, the glycolytic
switch upregulates CD93 expression on monocytes through the ERK pathway, which
enhances PD-L1 expression and also induces monocytes to produce the ECM
component, thereby refraining CD8+ T cells from migrating into the tumor
[90]. High expression of AlkB homolog 5 (ALKBH5) in
HCC can activate the ERK pathway through upregulating MAP3K8, which in turn
increases the chemokine IL-8 expression to recruit PD-L1+ macrophages [91].
ERK/NF-
The ERK pathway can transform tumor-associated cells immunosuppressive, and enhance their pro-tumorigenic effects. For example, the Raf/ERK pathway can be activated by lactate in-flow, which then induces macrophage M2 polarization and suppresses the immunity in HCC [93]. In addition, HBV can induce high IL8 production through triggering the MEK-ERK signaling, which not only enhances endothelial permeability to promote vascular invasion, but also switches Tregs polarization for easier immune escape [94].
The ERK pathway drives tumor growth, invasion, metastasis, and drug resistance.
Deficient in p90 ribosomal S6 kinase 2, human HCC cells rely on the Ras/MAPK
signaling pathway for proliferation, and this proliferation can be effectively
curbed by MEK inhibitors [95]. The ERK pathway mediates TGF-
The Wnt/
The Wnt/
The TGF-
In response to chronic liver injury, TGF-
The PI3K/Akt pathway is notably upregulated in patients with HCC [119]. The mTOR complex 1 (mTORC1), a vital downstream effector of the PI3K/Akt pathway, plays a key role in autophagy [120]. In HCC, autophagy occurs to promote tumor progression and drug resistance [121]. Within the TME, the activation of the PI3K/Akt pathway in TAMs promotes M2 polarization, allowing tumor cells to evade the immune system [122]. HBV-expressed hepatitis B surface antigen (HBsAg) and HBx can activate this pathway to induce cellular transformation, thus propeling the development of HBV-related HCC [123]. As an mTOR-targeted drug, everolimus remains under intensive investigation, despite its unsatisfactory results in previous trials [61].
The aberration of the Hedgehog (Hh) pathway is closely linked to oncogenesis. Hh ligands bind to inhibitory receptors Patched (Ptch) to disrupt the inhibitory effect to Smoothened (Smo), phosphorylating Glioma-associated oncogene homolog (Gli) transcription factors that move into the nucleus to initiate the transcription of target genes [124]. The Hh pathway can inhibit cell cycle arrest, promote HCC cell proliferation, and permit the self-renewal of liver CSCs, thus inducing an acquired drug resistance [125, 126, 127]. Previous research has shown that a key enzyme in cholesterol synthesis can activate the Hh signal to promote HCC regeneration and metastasis [128]. Hh signals can also stimulate various immune cells to foster an immunosuppressive vibe. Tumor cells secrete Hh ligands that drive PD-L1 expression in TAMs, which subsequently inhibits the CD8+ T cell function [129].
Chronic inflammation is an important process involved in the development of HCC.
The NF-
Owing to the outgrowth of tumor cells, oxygen within the tumor is overconsumed
to create a hypoxic microenvironment. The hypoxia-inducible factor (HIF) pathway
is dominant in the hypoxic response. Abnormal activation of the HIF pathway is
involved in the occurrence, progression, metastasis, and treatment resistance of
HCC. HIF regulates VEGF transcription and promotes the angiogenesis in HCC [139].
The activation of the HIF-1
Signaling pathways within the HCC TME do not function independently; rather, they form a complex regulatory network through intricate crosstalk, feedback loops, and compensatory mechanisms. As shown in (Fig. 2).
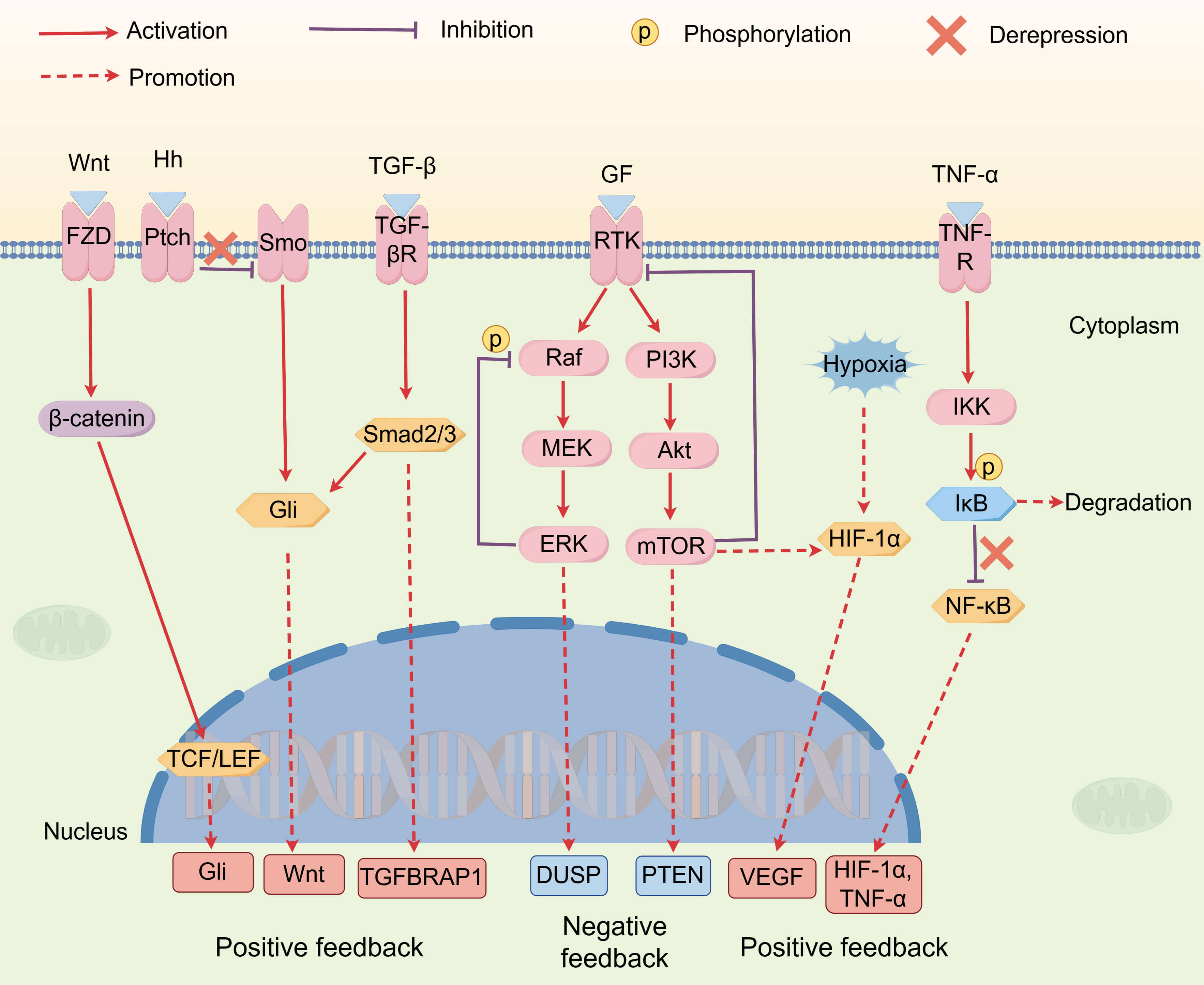 Fig. 2.
Fig. 2.
Intricate network of signaling crosstalk. Crosstalk among
various cellular signaling pathways forms a dynamic regulatory network. This
network contains negative feedback mechanisms, such as the ERK and PI3K/Akt/mTOR
pathways, which prevent pathway hyperactivation by inhibiting upstream signals
and promoting the production of negative regulators. Simultaneously, the network
also includes multiple positive feedback mechanisms that collectively drive tumor
growth and drug resistance. For example, mTOR promotes the synthesis of
HIF-1
RTKs, upstream of the signaling cascade, can activate various downstream
pathways, including the ERK and PI3K/Akt/mTOR pathways to promote tumor survival
and proliferation [145, 146]. Furthermore, RTKs can re-activate the ERK and Akt
pathways, conferring HCC with a resistance to targeted therapies [147]. The
PI3K/Akt pathway can then promote the translation of HIF-1
Negative feedback is a common mechanism for maintaining biological homeostasis.
The negative regulation of the pathway by growth factor-activated ERK involves a
feedback loop, where activated ERK not only inhibits Raf activity via feedback
phosphorylation but also promotes the expression of the downstream negative
regulator, dual-specificity phosphatase (DUSP) [158, 159]. Similarly, the
activation of the PI3K/Akt/mTOR pathway not only increases the expression of its
negative regulator, phosphatase and tensin homolog (PTEN), but also negatively
regulates the pathway itself by inhibiting the expression of RTKs [160, 161].
These negative feedback loops serve as a compensatory mechanism against
single-target inhibitors, thus conferring drug resistance.Conversely, positive
feedback amplifies pro-cancer signals, and the TGF-
Targeting a single pathway with an inhibitor can easily trigger a compensatory
effect from other pathways, suggesting that multi-target combination therapies
may be more effective. For example, VEGF signaling can be selectively silenced to
suppress tumor growth, but his suppression can also lead to hypoxia, resulting in
HIF-1
PD-1, an immune checkpoint molecule predominantly expressed on T cells,
interacts with PD-L1, which is mainly located on tumor cells, macrophages, and
DCs. This interaction facilitates tumor cell proliferation and dissemination by
suppressing immune responses [165]. The expression of PD-L1 is intricately linked
to immune evasion, and its interaction with PD-1 induces T cell dysfunction and
exhaustion [166]. A study on HCC demonstrated that a high PD-L1 expression
correlates with aggressive tumor characteristics and poor clinical outcomes
[167]. Additionally, PD-L1 overexpression may facilitate tumor progression via
evoking the Akt/
Beyond PD-1/PD-L1, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), a classic immune checkpoint molecule, significantly contributes to the progression of HCC. High CTLA-4 expression contributes to the construction of the TIME, which involves various mechanisms that impair the ability of the immune system for tumor surveillance and clearance, including the secretion of inhibitory cytokines and metabolites [171]. CTLA-4 maintains immune tolerance primarily by inhibiting T cell activation, thus fostering tumor proliferation and immune evasion [172].
T cell immunoglobulin and mucin-domain containing-3 (TIM-3), a newly found immune checkpoint molecule, is broadly expressed on various immune cells, and commit multiple roles in the TME. Tan et al. [173] demonstrated that TIM-3 promotes HCC progression by impairing the function of NK cells: specifically, TIM-3 is upregulated in tumor-infiltrating NK cells and inhibits their cytokine-secreting and cytotoxic abilities. Thus, blocking TIM-3 can restore NK cell function and suppress HCC growth. Additionally, TIM-3 inhibition can enhance the ability of DCs to regulate innate and adaptive immunity and promote the M2-to-M1 polarization of macrophages, thereby countering tumor progression [174].
Another immune checkpoint molecule is lymphocyte-activation gene 3 (LAG-3), and its expression is also closely linked to the transformation into TIME. A study found that the co-expression of LAG-3 and PD-1 may further suppress T cell function, suggesting that the blockade of both LAG-3 and PD-1 may open a new therapeutic avenue [175].
Cytokines in the HCC TME not only regulate immune responses, but also tumor
growth, invasion, and metastasis through multiple mechanisms. TAMs secrete
cytokines, such as TGF-
Furthermore, chemokines, a specialized subset of cytokines, precisely guide immune cells to migrate to the TME during tumor progression. Specifically, they interact with their receptors to regulate the accumulation of immune cells, thereby transforming the TME, so the expression and activity of chemokines and their receptors are inevitably closely related to cancer prognosis [178, 179]. For example, CXCL12 can support tumor growth and metastasis by abnormalizing the biological properties of endothelial cells [51]. Liu et al. [180] found that CCL15 promotes immune evasion in HCC by recruiting CCR1+ monocytes. Chemokines, such as CXCL5, not only function in tumor progression, but also may serve as diagnostic and prognostic biomarkers [181].
Exosomes, as a mediator on intercellular communication, also exert multiple biological functions in the HCC TME. First, exosomes derived from HCC cells can deliver biomolecules to disrupt the function of immune cells, promoting the formation of TIME. For example, non-coding RNAs (ncRNAs) in exosomes can promote the expansion of Tregs to interfere with the immune system and thereby promote tumor proliferation and metastasis [182]. Second, exosomes are integral to the processes of metastasis and invasion in HCC. For example, exosomal miR-92a-3p derived from cancer cells with a high metastatic potential enhances the EMT and metastasis of low-metastatic cancer cells by modulating the PTEN/Akt pathway [183]. Additionally, exosomes released by HCC cells can disturb the action of drugs by delivering specific RNA molecules and proteins, leading to drug resistance [184]. Finally, exosomes have shown potential values in diagnosing and treating HCC: either as biomarkers for early diagnosis and prognosis, or carriers for targeted drug delivery [185, 186].
The mechanisms by which ncRNAs act in the TME have slipped into the research
hotspot in recent years. ncRNAs influence tumor immune evasion and immune
responses by regulating immune cell infiltration and activation. It can shape the
TIME by regulating the polarization of TAMs, thereby influencing HCC progression
[187]. Besides, ncRNAs are implicated in the proliferation, invasion, and
metastasis of tumor cells. Xue et al. [188] found that certain long
non-coding RNAs (lncRNAs) tune the proliferation and migration of HCC cells
through interactions with proteins. Additionally, ncRNAs regulate the expression
of pivotal genes through a competing endogenous RNA (ceRNA) mechanism during the
occurrence and development of HCC [189]. Multiple ncRNAs can upregulate
ATP-binding cassette (ABC) transporters, which promotes the efflux of
chemotherapy drugs [190]. By promoting chemotherapy drug efflux and immune
evasion, ncRNAs can mediate cancer cell drug resistance, which positions them as
promising biomarkers and therapeutic targets [191]. For example, cancer
cell-derived lncRNA HDAC2-AS2 promotes CD8+ T cell exhaustion and inhibits
anti-tumor immunity [192]. LINC00680 can activate Akt3 to decrease the
chemosensitivity of HCC to 5-fluorouracil [193]. LINC-ROR enhances resistance to
adriamycin by activating the Wnt/
Although they have the potential to serve as biomarkers, their clinical translation still faces significant challenges. While chemokines can predict therapeutic responses, their clinical translation is hampered by a lack of large-scale, prospective studies [198]. Similarly, although exosomes show favorable predictive sensitivity and specificity, their extraction and isolation are technically demanding, and their functional mechanisms require in-depth investigations [199].
Metabolic reprogramming, a significant hallmark of HCC, is engaged in the proliferation, survival and immune evasion of tumor cells. In HCC, dysregulation of glucose, fatty acid, and amino acid metabolism represents a key component in metabolic reprogramming, and significantly distorts the functions of TAMs and other immune cells [200]. Reprogramming of glucose metabolism is one of the most significant metabolic changes in HCC, with HCC cells tending to acquire energy via glycolysis rather than oxidative phosphorylation (OXPHOS)—a phenomenon known as the “Warburg effect” [201]. Under the Warburg effect, HCC cells preferentially produce energy through glycolysis, even under aerobic conditions, manifesting increases in glucose uptake and lactate production. Cancer cells exhibit metabolic plasticity, where OXPHOS is not abolished but co-regulates energy provision with glycolysis [202]. Some HCC subtype cells with reactivated OXPHOS tend to generate energy through the oxidative phosphorylation pathway, resulting in oxygen overconsumption [203]. And OXPHOS is also associated with cancer stemness maintenance and drug resistance, driving the development of OXPHOS inhibitors [204]. The shift in metabolic pathways not only satisfies the demands of cancer cells for rapid proliferation, but also provides a microenvironment conducive for tumor advancement [205].
Fatty acid metabolism is also reprogrammed in HCC. Obesity and metabolic dysfunction-associated steatohepatitis (MASH) are recognized as risk factors for HCC; under these conditions, fatty acid metabolic reprogramming provides tumor cells with abundant exogenous fatty acids, thereby promoting tumor progression [206]. This reprogramming not only drives the growth of tumor cells, but also promotes them to release metabolites to interact with other non-cancerous cells, leading to the acidification of microenvironmental and suppression on immune cell activity [207].
Reprogramming of amino acid metabolism, particularly that of glutamine, also plays a vital role in the occurrence and development of HCC. Glutamine metabolism provides carbon and nitrogen sources for HCC cells, which are used for the synthesis of glutathione and nucleotides, promoting cell proliferation [208]. In addition, glutamine metabolism is also associated with the activation of the mTOR signaling pathway, which hinders immunotherapy [209]. In summary, metabolic reprogramming in HCC reshapes the TME through multiple pathways to promote tumor progression and immune evasion.
The metabolites derived from the gut microbiota exert a dual effect on HCC through the gut-liver axis. Short-chain fatty acids from a balanced microbiota can suppress HCC. For example, butyrate can stimulate the production of chemokines, thereby favoring the infiltration of NK cells [210]. Isobutyrate suppresses tumor growth by augmenting CD8+ T cells and muting the JAK/STAT3 pathway [211]. Conversely, dysbiotic gut microbiota can accelerate the intrahepatic metastasis of HCC [212]. A reduction in vitamin B6 synthesis by dysbiotic microbiota can disrupt the amino acid metabolism, and impair the function of immune cells in HCC [213, 214]. Moreover, the dysregulation of secondary bile acids (BAs) metabolism is linked to carcinogenesis. Owing to the scarcity of gut bacteria rich in bile salt hydrolase, obesity induces increases in carcinogenic deoxycholic acids and reductions in protective secondary BAs, jointly promote tumor growth [215, 216]. Additionally, due to the impairment of intestinal barrier in pathological conditions, excessive lipopolysaccharide (LPS) enters the liver through intestinal leakage, binds to TLR4 on hepatocytes, and enhances the invasive capacity of HCC [217].
Before tumorigenesis, the hepatic microenvironment is remodeled profoundly and dynamically, creating a niche favoring tumor initiation and progression (Fig. 3, Ref. [124, 218, 219, 220, 221, 222]).
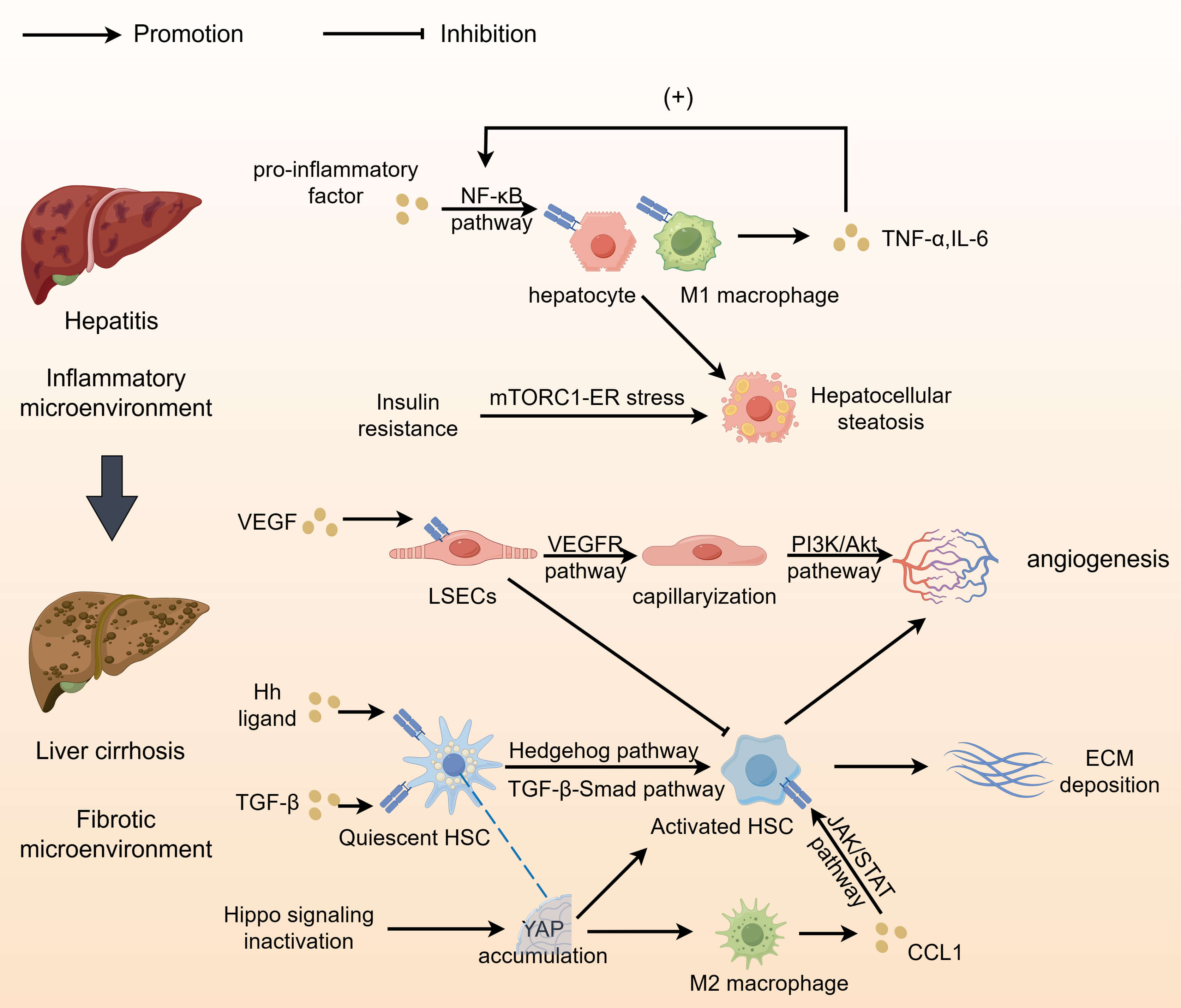 Fig. 3.
Fig. 3.
Microenvironmental changes during different stages of chronic
liver diseases. During the phase of hepatitis, an inflammatory microenvironment
predominates. Pro-inflammatory factors trigger the NF-
As the initial stage of HCC, hepatitis is a vital contributor to the development
of HCC. Its inflammatory microenvironment provides the basis for subsequent
lesions to build up. Many signaling pathway start to show abnormalities during
this stage. Persistent inflammation in the liver can cause hepatic injury and
fibrosis, thus elevating the risk of cancer. During the chronic inflammatory
phase, pro-inflammatory cytokines like TNF-
Viral infection stands as the principal cause of hepatitis, especially HBV, and
chronic hepatitis B constitutes a high-risk factor for HCC. In the context of HBV
infection, the HBx continuously activates the NF-
Moreover, the dysbiosis of the gut microbiota is intimately connected with
chronic liver disease. LPS from gut Gram-negative bacteria enter the bloodstream
and are transported to the liver, where LPS binds to TLR4 on Kupffer cells and
other cells, activating the LPS/TLR4 signaling pathway, as well as the MAPK and
NF-
The incidence of MASLD/MASH has kept rising steadily in recent years. In
MASLD/MASH, Hh signaling is consistently involved, as liver injury increases the
secretion of Hh ligands to activate the Hh pathway, thus facilitating the
deterioration from MASLD to HCC [124]. The Hh pathway not only promotes MASH
progression, but also regulates HSC-mediated angiogenesis in liver fibrosis and
the malignant conversion of the TME in HCC [124]. Hu et al. [230] found
that ductal reaction originating from HPCs can activate the Hh pathway and
promote liver fibrosis. Highly sensitive to energy metabolism, the mTORC1
signaling promotes lipid synthesis, increases lipid storage, impairs insulin
signaling, and triggers inflammation in MASLD, thereby resulting in insulin
resistance [231]. High-level fructose can activate the mTOR-autophagy-endoplasmic
reticulum (ER) stress signaling pathway to inhibit hepatic autophagy, increase ER
stress, enhance fatty acid synthesis, form fatty liver; then with activation of
inflammatory pathways, fibrosis is induced, thus accelerating the advancement of
chronic liver disease [232]. The AMP-activated protein kinase (AMPK) pathway,
closely implicated in lipid metabolism, inhibits lipid formation and promots
fatty acid oxidation [233]. Some natural compounds, such as acacetin and
Buddleoside, have been found to target and inhibit mTORC1, enhance the AMPK
signaling pathway, activate autophagy mediated by the transcription factor EB,
reduce fat accumulation, and ultimately alleviate MASLD [234, 235]. Smad is a
downstream signaling molecule of the TGF-
Cirrhosis, a progressive disease, is characterized by liver fibrosis, nodule formation, and functional impairment, and its progression parallels with the changes in the liver microenvironment. This microenvironment serves as a physical support for hepatocytes, and also a complex ecosystem undergoing cell-cell interactions, ECM remodeling, and crosstalk among diverse signaling molecules.
Liver fibrosis, either as the consequence of persistent inflammation and or the
preliminary stage of cirrhosis, is mainly related to the activation of HSCs.
Liver fibrosis is mainly reflected as the changes in the ECM. Activated quiescent
HSCs differentiate into myofibroblastic HSCs to promote the deposition of ECM,
thus increasing the stiffness of the ECM, which in turn activates of HSCs and
worsens liver fibrosis [237]. THe TGF-
Macrophages also takes on a critical profile in the microenvironment of cirrhosis. They secrete various cytokines and chemokines that act on HSC activation and inflammatory responses. Macrophages can promote or inhibit hepatic fibrosis during cirrhosis. Macrophage-secreted CCL1 can target and activate the CCR8 on HSCs, thereby igniting the JAK/STAT signaling pathway and accelerating the progression of liver fibrosis [221]. Silencing the JAK/STAT pathway in HSCs can impede EMT to alleviate liver fibrosis [243, 244]. However, the monocytes can also pool to differentiate into reparative macrophages, which primarily express MMPs and induce the regression of fibrosis [245]. Given their high heterogeneity and plasticity, macrophages reprogramming therapy may be feasible for liver diseases. In addition, in the decompensated stage of cirrhosis, the abundance of follicular helper T cells decreases and the intensity of IL-2 signaling increases, collaboratively triggering cirrhosis-related immune dysfunction [246].
Moreover, the capillarization of liver sinusoidal endothelial cells (LSECs) is observed over the progression of cirrhosis. Healthy LSECs, characterized by transcellular pores and the absence of basement membranes, interact with surrounding cells through angiocrine signaling to maintain liver homeostasis [247]. In the context of liver injury, LSECs lose their membrane fenestrae and function to regulate blood flow, manifesting abnormal liver hemodynamics and hypoxia; meanwhile, they lose their ability to maintain HSCs quiescent, further promoting fibrosis [247]. VEGFR and other angiogenesis-related signaling pathways undertakes a central role in the phenotypic changes of LSECs. The VEGFR2-dependent PI3K/Akt pathway in LSECs promotes angiogenesis, which in turn induces liver fibrosis [222].
The progression from hepatitis to HCC is a complex process characterized by dynamic changes in cellular signaling pathways. Each stage of this disease is dominated by distinct signaling characteristics. Therefore, a targeted approach to inhibit these stage-specific pathways may offer a promising strategy to slow down disease progression. Further clinical studies are summarized in Table 2.
| Disease stage | Primary signaling pathways | Biomarkers | Drugs | Target Disease | Mechanism | Clinical Trial |
| Hepatitis | NF- |
IL-1 | Canakinumab | Alcoholic hepatitis | Inhibition of inflammation by suppressing the key mediator IL-1 |
NCT03775109 (Completed) |
| TLR7 mRNA | RO7020531 | Chronic hepatitis B | Enhancement of adaptive immunity via TLR7 activation | NCT02956850 (Completed) | ||
| HFF | Cotadutide | MASH | Improvement of hepatic steatosis through GLP-1R/GCGR co-agonism | NCT05364931 (Completed) | ||
| Adiponectin concentration, PRO-C3 | BMS-986036 | MASH | Regulation of lipid and glucose metabolism as an FGF21 analog | NCT03486912 (Completed) | ||
| Liver fibrosis/liver cirrhosis | TGF- |
p-Smad3 | Pirfenidone | Liver fibrosis | Suppresses the phosphorylation of signaling proteins in the TGF- |
NCT05542615 (Unknown status) |
| Smad7, TGF |
Hydronidone | Liver fibrosis associated with chronic hepatitis B | Promotion of TGF |
NCT05115942 (Completed) | ||
| LOXL2 | Simtuzumab | Compensated cirrhosis | An anti-LOXL2 monoclonal antibody that blocks a key step in liver fibrosis | NCT01672879 (Terminated) | ||
| HCC | RTKs, PD-1/PD-L1, CTLA-4 | AFP, Anti-PD-1 autoantibody | Bevacizumab+atezolizumab | Locally advanced or metastatic HCC | Simultaneous inhibition of VEGF and PD-L1 to block angiogenesis and immune evasion | NCT03434379 (Completed) |
| FGFR4 and Treg infiltration | Lenvatinib+pembrolizumab | Advanced HCC | Targets VEGFR, FGFR, and PD-1 to counter angiogenesis and immune suppression | NCT03713593 (Completed) | ||
| NLR | Durvalumab+tremelimumab | Advanced HCC | Synergistic activation of anti-tumor immunity via PD-L1 and CTLA-4 inhibition | NCT03298451 (Completed) |
GLP-1R, glucagon-like peptide-1 receptor; GCGR, glucagon receptor; FGF,
fibroblast growth factor; FGFR, fibroblast growth factor receptor; HFF, hepatic
fat fraction; MASH, metabolic dysfunction-associated steatohepatitis; PRO-C3,
N-terminal type III collagen propeptide; TGF-
The TLR and NF-
Despite some breakthroughs in the treatment of hepatitis and liver cirrhosis, the field faces major challenges due to the complex interplay of multiple signaling pathways and the irreversibility of late-stage fibrosis. As a result, many patients still progress from cirrhosis to HCC, which has made HCC a central focus for drug development in liver diseases.
As suggested by the ESMO guidelines, a range of treatments, such as liver resection, thermal tumour ablation, liver transplantation, radiotherapy and transarterial therapies, can be adopted to fight early HCC [255]. But HCC is usually not suitable for surgery due to its high aggressiveness, as it has usually already spread or invaded important blood vessels at diagnosis. Consequently, targeted therapy and immunotherapy may bring with encouraging results in HCC treatment [256]. Several targeted drugs and immunotherapeutic agents, which have been evaluated in clinical trials in recent years, are listed in Table 3.
| Treatment | Clinical trial number | Target | Phase | Enrollment | Primary endpoint | Overall status | |
| Targeted therapy | Tivozanib | NCT01835223 | VEGFR | Ib/II | 33 | PFS | Completed |
| Apatinib | NCT02329860 | VEGFR-2 | III | 400 | OS | Completed | |
| Anlotinib | NCT02809534 | VEGFR, FGFR, PDGFR, c-Kit | II | 50 | PFR | Completed | |
| Fruquintinib | NCT06446154 | VEGFR | II | 36 | ORR | Recruiting | |
| Chiauranib | NCT03245190 | VEGFR/Aurora B/CSF-1R | Ib | 27 | PFR | Completed | |
| Tepotinib | NCT01988493 | c-Met | Ib/II | 117 | TTP | Completed | |
| Cabozantinib | NCT04767906 | VEGFR, c-Met, AXL | II | 16 | TT | Completed | |
| Cabozantinib+sapanisertib | NCT06811116 | VEGFR, c-Met, AXL, mTOR | I/II | 92 | DLTs, AEs, PFS | Recruiting | |
| Trametinib+sorafenib | NCT02292173 | MEK, VEGFR, Raf, PDGFR | Ia/Ib | 17 | MTD | Completed | |
| Temsirolimus+sorafenib | NCT01687673 | mTOR, VEGFR, Raf, PDGFR | II | 29 | TTP | Completed | |
| Immunotherapy | Pembrolizumab | NCT03062358 | PD-1 | III | 453 | OS | Completed |
| Camrelizumab | NCT02989922 | PD-1 | II | 220 | ORR, OS rate | Completed | |
| Tislelizumab | NCT03412773 | PD-1 | III | 674 | OS | Completed | |
| Nivolumab | NCT02576509 | PD-1 | III | 743 | OS | Completed | |
| Nivolumab+ipilimumab | NCT04039607 | PD-1, CTLA-4 | III | 732 | OS | Active, not recruiting | |
| HBV-TCR-T | NCT04677088 | HBV antigens | I | 7 | AEs, SAEs | Completed | |
| GPC3 CAR-T (CBG166) | NCT06461624 | GPC3 | I | 15 | DLTs, AEs, MTD | Recruiting | |
| CAR-NK (SN301A) | NCT06652243 | GPC3 | I | 12 | DLTs, AEs, SAEs | Recruiting | |
| Autologous NK | NCT06044506 | / | I | 3 | OTD | Recruiting | |
| TIL therapy (BST02) | NCT06526832 | / | I | 9 | AEs, SAEs, DLTs, MTD | Recruiting | |
| Immunotherapy combined with targeted therapy | TACE+atezolizumab+bevacizumab | NCT04712643 | PD-L1, VEGF | III | 342 | PFS, OS | Active, not recruiting |
| Anlotinib+penpulimab | NCT04344158 | PD-1, VEGFR, FGFR, PDGFR, c-Kit | III | 649 | OS | Active, not recruiting | |
| Sintilimab+bevacizumab biosimilar (IBI305) | NCT03794440 | PD-1, VEGF | II-III | 595 | PFS, OS | Completed | |
| Camrelizumab+rivoceranib | NCT03764293 | PD-1, VEGFR2 | III | 543 | PFS, OS | Completed | |
| Toripalimab+bevacizumab | NCT04723004 | PD-1, VEGF | III | 326 | PFS, OS | Completed | |
| TACE+durvalumab+bevacizumab | NCT03778957 | PD-L1, VEGF | III | 724 | PFS | Active, not recruiting | |
| KN046+lenvatinib | NCT04542837 | PD-L1, CTLA-4, VEGFR, FGFR | II | 55 | ORR | Completed | |
HCC, hepatocellular carcinoma; VEGFR, vascular endothelial growth factor
receptor; PDGFR, platelet-derived growth factor receptor; c-Kit, cellular
proto-oncogene tyrosine-protein kinase Kit; FGFR, fibroblast growth factor
receptor; Aurora B, aurora kinase B; CSF-1R, colony-stimulating factor 1
receptor; AXL, axl receptor tyrosine kinase; HIF1
Angiogenesis is a prerequisite for the rapid growth and metastasis of HCC. Targeting receptors such as VEGFR can inhibit tumor angiogenesis. As multi-targeted TKIs, sorafenib and lenvatinib have been approved as the first line for the treatment of advanced HCC, but adverse reactions and drug resistance limit their application [65, 257]. As the cornerstone in the second line, regorafenib and cabozantinib are multi-kinase inhibitors that can prolong OS in patients with advanced HCC who show progression after sorafenib treatment [258, 259, 260]. Ramucirumab, a monoclonal antibody that specifically targets VEGFR-2, is specifically selected to treat HCC patients with elevated AFP [261]. Apart from above drugs recommended in the guidelines, apatinib and donafenib have also received approval in China for HCC [262, 263, 264]. Other new drugs, such as anlotinib, have shown therapeutic potential and controllable safety in both first-line and second-line treatments [265]. Tivozanib and fruquintinib have been approved for other cancers, but their efficacy for HCC should be further explored [266].
Galunisertib, as a TGF-
Refametinib, an MEK inhibitor, has been found to exhibit synergistic antitumor
effects for RAS-mutated HCC when used in combination with sorafenib [58]. The
mTOR inhibitor sapanisertib is currently assessed in the clinical trial.
Moreover, early clinical trials have employed mRNA antagonists to inhibit
HIF1
The immunosuppression in the HCC TME is the primary obstacle that the immunotherapy aims to resolve. Such suppression can be relaxed to reactivate the host’s intrinsic anti-tumor immune response. Therefore, the immunotherapy is often combined with targeted therapy.
ICIs, the cornerstone of HCC immunotherapy, can restore T lymphocyte activity by blocking immune checkpoint molecules to strengthen cancer cell recognition and killing. ICIs are often used in combination with anti-angiogenic agents. The combination of atezolizumab (a PD-L1 inhibitor) and bevacizumab (an anti-VEGF monoclonal antibody) serves as the first-line defense against advanced HCC. Atezolizumab blocks the binding of PD-L1 to PD-1/CD80 to unleash T-cell suppression and restore anti-tumor immunity, while bevacizumab inhibits tumor angiogenesis (blocking nutrient supply) and repels VEGF-mediated immunosuppression in the TME to enhance T-cell infiltration. Their combination exerts a dual effect of “relieving immunosuppression + improving immune microenvironment”. Phase III IMbrave150 trial results confirmed its significant advantages: in treatment-naive patients with unresectable HCC, the combination reduced death risk by 42%, with a 12-month OS rate of 67.2% (vs. 54.6% in the sorafenib group) [267]. This regimen is the first to verify the OS benefit of the “immuno + anti-angiogenesis” strategy in first-line settings. As the new standard treatment for Child-Pugh Class A patients, this combination proves effective for high-risk groups. Marking an era advancing from “targeted monotherapy” to “immune combination”, this combination reshapes the current first-line treatment, and lays the groundwork for future immune treatment optimization. In addition, the phase III clinical trials of toripalimab combined with bevacizumab and durvalumab combined with bevacizumab have also harvested encouraging outcomes [268]. These trials provide multiple options for the clinical treatment of HCC, as well as inspirations for future research.
Sintilimab combined with bevacizumab biosimilar (IBI305), versus sorafenib, has exhibited marked benefits to progression-free survival (PFS) and OS with a good safety, and the combination of sintilimab with apatinib plus capecitabine also exhibits favorable anti-tumor activity [269, 270]. Pembrolizumab has proven effective in post-sorafenib patients [271]. Moreover, the effect of pembrolizumab as a combination agent cannot be ignored. Studies have shown that transarterial chemoembolization (TACE) combined with lenvatinib and pembrolizumab can extend the PFS [272]. Additionally, a multicenter trial found that compared with sorafenib, anlotinib plus pembrolizumab significantly prolongs the median OS [273]. Camrelizumab-based regimens, including triple therapies with lenvatinib+raloxifene-based hepatic arterial infusion chemotherapy (HAIC), shows a significant anti-tumor activity and a well-tolerated profile [274]. In a phase II study, the combined treatment of camrelizumab, apatinib, and HAIC achieves a good efficacy in specific HCC patients [275]. Furthermore, in a multicenter study, camrelizumab combined with rivoceranib displays an evident efficacy [276]. Tislelizumab, another immunotherapeutic agent, does not show a significant effect, but a higher safety profile than sorafenib [277]. In addition, nivolumab and ipilimumab have also been written into guidelines. In a phase III trial, the combination of nivolumab and ipilimumab significantly improves the OS, compared with the control group [278].
In addition to ICIs, cellular immunotherapy has emerged to counter HCC. T cell receptor-engineered T cells (TCR-T) therapy can be designed by engineering the patient’s own T cells. Relevant studies have shown that HBV-TCR-T therapy is safe and tolerant, and some patients have experienced long-term disease-free survival [279]. In patients with HBV-related HCC recurrence after liver transplantation, multiple infusions of mRNA-electroporated HBV-specific TCR-T cells are well-tolerated [280]. Chimeric antigen receptor T cell (CAR-T) therapy has been introduced into the treatment of gastric cancer, but the evidence about HCC is still under exploration. Moreover, CAR-NK therapy has been set up. Glypican-3 (GPC3)-specific NK cells exhibit a significant toxicity against GPC3-positive HCC in vitro, with a stable activity under hypoxic conditions and a remarkable anti-tumor effect in xenograft models [281]. Furthermore, tumor-infiltrating lymphocyte (TIL) therapy is designed through isolating and expanding tumor-reactive T cells from the patient’s tumor tissue and then infusing them back into the patient. This therapy, such as drug BST02 injection, overcomes the TME barrier, but its application is still in the early explorational stage.
Bispecific therapy refers to a class of therapeutics specially designed to simultaneously target two distinct biomolecules. Currently, bispecific antibodies represent its primary form, among which KN046 is a relatively well-developed candidate. As an anti-PD-L1/CTLA-4 bispecific antibody, KN046 in combination with lenvatinib has achieved an objective response rate (ORR) of 45.5%, thereby providing a promising first-line treatment option for patients with advanced HCC [282].
Although the aforementioned therapies have transformed the HCC treatment landscape, their efficacy remains limited in patients presenting a heterogeneity, or having developed primary or acquired resistance. Therefore, predictive biomarkers should be explored to personalize treatments. The immune cell infiltration in the TME is directly related to the therapeutic efficacy. Based on the degree of functional immune cell infiltration, particularly CD8+ T cells, tumors can be classified as “cold” or “hot”, with hot tumors more responsive to immunotherapy [283]. A high intratumoral immune infiltration predicts a better response to immunotherapy and a favorable prognosis [284].
Moreover, the activation of
Although significant progress has been made in HCC diagnostics and therapy, patient outcomes remain poor. This is primarily attributed to primary resistance caused by intratumor heterogeneity and acquired resistance that arises from the tumor’s adaptation to therapeutic pressure [290]. Further investigation into drug resistance mechanisms provides key insights into the factors driving treatment failure in HCC patients and offers guidance for developing more effective combination therapies. The major mechanisms of drug resistance in HCC are summarized in Table 4.
| Category | Mechanism | Related pathways/cells | Description |
| Cellular intrinsic mechanisms | Pathway compensation and reactivation | ERK, Akt, c-Met, EGFR, TGF- |
Compensatory activation of alternative pathways in response to single-pathway targeted therapy |
| Drug efflux | ABC transporter | Drug efflux mediated by ABC transporters, leading to a reduction in intracellular drug concentration | |
| Metabolic reprogramming | OXPHOS | A metabolic shift towards OXPHOS-based survival, rendering certain HCC subtypes insensitive to glycolysis inhibitors | |
| TME-mediated mechanisms | Immune evasion | PD-L1, Tregs, M2-TAMs, MDSCs, Texs | Immunosuppression leading to severe T cell exhaustion and impaired function, resulting in immunotherapy failure |
| Hypoxia | HIF | Anti-angiogenesis therapy induced hypoxia activates the HIF pathway, leading to enhanced tolerance to an anoxic microenvironment |
HCC, hepatocellular carcinoma; ERK, extracellular signal-regulated kinase; Akt,
protein kinase B; c-Met, hepatocyte growth factor receptor; EGFR, epidermal
growth factor receptor; TGF-
In a majority of cases, HCC develops with the long-term progression of chronic
liver disease. The liver microenvironment demonstrates dynamic changes as chronic
hepatitis aggravates to cirrhosis, including inflammatory cell infiltration,
activation of HSCs, deposition of extracellular matrix, and gradual accumulation
of immunosuppressive cells, collectively laying the soil for the growth of HCC.
Some signaling pathways may dominate in one stage, but also continue to function
in other stages. The inflammatory pathway NF-
Targeting TME components and their aberrantly activated signaling pathways has become a crucial strategy for current HCC treatment. The progression from multikinase inhibitors targeting RTKs to ICIs highlights the effectiveness of intervening in the HCC TME. But these treatments are also confronted with the high heterogeneity of tumors, the complexity of the TME, and the acquired drug resistance. Sensitive biomarkers will help guide the creation of individualized therapy. Extensive crosstalk and compensatory activation among signaling pathways often lead to resistance against single-pathway targeted therapies. Thus, combination therapies will remain the mainstream to combat drug resistance. Furthermore, cell therapies, RNA interference therapeutics, and nanodrug delivery systems will offer new treatment options. Future research should dig deeper into the TME complexity for deciphering the molecules capable of preventing resistance and immune evasion, as well as reversing the pro-tumorigenic microenvironment.
New analytical tools can be utilized to gain a clearer insight into the evolution of the HCC TME. Through spatial transcriptomic analysis, gene expression can be integrated with spatial information to construct the maps of functional genes within tissues [291]. The combination of spatial transcriptomics and single-cell sequencing enables a deeper elucidation of the mechanisms underlying the interactions between cancer cells and other cell types in tumor tissues [21]. Multi-omics integration analysis allows to synthesize multidimensional biological processes to investigate the mechanisms of cancer initiation, and facilitate the early screening, exploration of therapeutic targets, and discovery of biomarkers [292]. Additionally, patient-derived organoid models are capable of mimicking the structure and function of tumors in vivo, thereby offering sparks for designing new personalized therapy and precision medicine [293]. Finally, liquid biopsy biomarkers, such as methylated cell-free DNA and miRNAs in exosomes, have promise a high value in the diagnosis and dynamic monitoring of HCC, thus enabling real-time identification of the therapeutic window [294, 295]. In conclusion, these emerging tools are expected to provide more information about the dynamics in HCC TME, which may be exploited to realize the early detection and precise treatment of HCC.
LX and XZhang designed, wrote and revised the manuscript. LX and XZhu prepared figures of the manuscript. XZhang and DN contributed to making table of the manuscript. LX and XZhang participated in collecting data of the manuscript. HZ and CJ participated in the conception and design of the review, undertook significant revisions to optimize the academic content, and completed thorough proofreading of the manuscript. CJ also provided support for the publication of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Figures were generated using Figdraw.com (https://www.figdraw.com).
This work was funded by the National Natural Science Foundation of China (No: 82274269, 82304936) and the Natural Science Foundation of Nanjing University of Chinese Medicine (No: XZR2023024).
The authors declare no conflict of interest.
During the preparation of this work the authors used DeepSeek in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.