








1 Department of Child Healthcare, The Affiliated Children’s Hospital of Xi’an Jiaotong University (Xi’an Children’s Hospital), 710003 Xi’an, Shaanxi, China
2 Pediatric Orthopaedic Hospital, Honghui Hospital, Xi’an Jiaotong University, 710054 Xi’an, Shaanxi, China
†These authors contributed equally.
Abstract
Achondroplasia (ACH), the predominant inherited form of disproportionate short stature, results from specific genetic alterations in fibroblast growth factor receptor 3 (FGFR3). N6-methyladenosine (m6A) modification is reported to modulate mRNA stability and translation. The present investigation systematically explored the epigenetic regulatory function of METTL16, an m6A RNA methyltransferase, within the pathophysiological framework of ACH.
We generated an ACH mouse model via Fgfr3380R (Fgfr3ach) gene mutation. Primary chondrocytes were isolated from newborn mice and stimulated with IL-1β to induce cell death. Proximal tibia tissues were collected and analyzed with HE staining, toluidine blue staining, safranin O staining, and immunohistochemical (IHC) analysis. Bone structure was analyzed by measuring bone mineral density (BMD), ratio of bone volume to total tissue volume (BV/TV), trabecular number (TbN), and trabecular thickness (TbTh). Cell viability and proliferation were assessed using the Cell Counting Kit-8 (CCK-8) and colony formation assays. The levels of iron (Fe2+), malondialdehyde (MDA), and glutathione (GSH) were measured to assess ferroptosis. Protein and RNA levels were measured by western blotting and quantitative real-time PCR (qPCR) assay, respectively, while the m6A modification level was assessed by m6A mRNA immunoprecipitation (IP).
METTL16 improved bone chondrogenesis in the ACH mouse model, with METTL16 overexpression promoting the proliferation of primary chondrocytes. METTL16 decreased ferroptosis both in vitro and in vivo and increased glutathione peroxidase 4 (GPX4) expression. METTL16 enhanced m6A modification of GPX4 mRNA and suppressed its degradation. Depletion of GPX4 abolished the effects of METTL16 on ACH mice and chondrocytes.
Overexpression of METTL16 improved bone growth and alleviated ferroptosis of chondrocytes by increasing m6A modification of GPX4 mRNA and thus GPX4 expression in chondrocytes. The METTL16/GPX4 axis may be a promising therapeutic approach for ACH treatment.
Keywords
- achondroplasia
- N6-methyladenosine
- METTL16
- chondrocyte
- ferroptosis
- GPX4
Achondroplasia (ACH) is a condition resulting from a missense mutation in the fibroblast growth factor receptor 3 (FGFR3) gene. ACH is the leading cause of short stature in humans [1, 2]. It is passed down through an autosomal dominant inheritance pattern, leading to the presentation of ACH-related symptoms in carriers of the FGFR3 mutation [3]. Despite advances in research, many preventative, diagnostic, and therapeutic challenges remain with ACH [4].
Ferroptosis is a recently identified form of oxidative cell death marked by the iron-dependent build-up of lipid peroxides to toxic levels [5]. It has become an effective target for avoiding multiple types of cancers and degenerative conditions, such as Alzheimer’s disease, Parkinson’s disease, and kidney degeneration [6, 7, 8]. Several proteins have been recognized as essential regulators of ferroptosis, including glutathione peroxidase 4 (GPX4), which regulates the production of glutathione (GSH), and the cysteine/glutamate antiporter SLC7A11, which promotes the import of cysteine for GSH biosynthesis and antioxidant defense [9, 10]. Research has shown that maintaining a balance between anabolic and catabolic processes in cartilage, as well as the survival of chondrocytes, is essential for the health of articular cartilage and for preventing the progression of osteoarthritis. This underscores the importance of chondrocyte regulation for articular cartilage health.
Epigenetic regulatory processes, including DNA methylation and
N6-methyladenosine (m6A), are exciting new areas of investigation in the
field of tumor biology [11]. m6A modification can selectively modulate
polyadenylation and pre-mRNA splicing, thereby regulating mRNA stability and
translation [12, 13]. m6A modification is the most prevalent modification
after transcription, and is primarily mediated by m6A “writers”,
“erasers”, and “readers” (WER) [14]. The m6A methylation of RNA is
mediated by a multiprotein complex and initiated by methyltransferases such as
methyltransferase-like 3 (METTL3), METTL14, METTL16, and WTAP (WT1-associated
proteins). Moreover, it is modulated by “erasers” including AlkB congeners,
demethylase FTO (
In this study, we explored the role of METTL16 in ACH and investigated m6A-regulated ferroptosis during ACH development. Our findings reveal a novel and promising mechanism for the prevention and treatment of ACH.
Transgenic mice with a C57BL/6 background and carrying the heterozygous Fgfr3𝑎𝑐ℎ transgene were obtained from Shanghai Model Organisms (China). They were housed in SPF conditions with free access to water and food. The mean bodyweight of mice at 8 weeks of age was approximately 20 g. Mouse genotypes were ascertained by qPCR. Wild-type C57BL/6 mice were used as controls. For all procedures involving tissue harvesting, mice were first anesthetized using isoflurane (2% for induction, 1.5% for maintenance) delivered via a precision vaporizer (2V22105103, RWD Life Science Co., Ltd., Shenzhen, Guangdong, China), ensuring a consistent and humane depth of anesthesia. Following anesthesia, mice were euthanized by cervical dislocation to minimize suffering and ensure ethical compliance. For treatment, lentivirus with METTL16 overexpression vectors (pCDH-CMV vectors; GenePharma, Shanghai, China) was intraperitoneally injected into newborn mice for 20 days. The tibiae from mice were collected and processed into paraffin-embedded samples or stored in liquid nitrogen for subsequent experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Animal Care and Use Committee of Pediatric Orthopaedic Hospital, Honghui Hospital, Xi’an Jiaotong University (No. KT2023-01-05-01) and conducted in accordance with the relevant guidelines and regulations.
Chondrocytes were prepared from the tibia of newborn mice [17] and cultured in
DMEM medium with 10% fetal bovine serum (FBS) in a 37 °C incubator.
Cell transfection was conducted using Lipofectamine 2000 (11668027,
Invitrogen™, Carlsbad, CA, USA) for 48 h. Chondrocytes were
stimulated with IL-1
All primary chondrocytes were freshly isolated and used at early passage. STR authentication is not applicable to primary cells.
Primary cells were observed daily under a phase-contrast microscope. The cells exhibited typical morphology consistent with human nucleus pulposus/endplate chondrocyte characteristics, including a polygonal or round shape, relatively large nuclei, and a pericellular lacuna-like structure. No fibroblast-like overgrowth or abnormal morphological changes were observed within passages P2–P4, which were used for subsequent experiments.
The micro-architecture parameters of the tibia from mice were analyzed by assessing the bone mineral density (BMD), bone volume to total tissue volume (BV/TV), trabecular number (TbN), and trabecular thickness (TbTh).
For histological examination, tibial tissues were preserved in 10% formalin, demineralized using 0.5 M EDTA, embedded in paraffin, and then cut into 5 µm sections. The histomorphology was observed by staining with hematoxylin and eosin (H&E; C0105S, Beyotime, Shanghai, China). The expression of METTL16 was assessed by immunohistochemical (IHC) staining using anti-METTL16 antibody (17676, Cell Signaling Technology, Danvers, MA, USA, 1:1000) and visualized by DAB (Beyotime, China) as the substrate.
The samples were stained with toluidine blue (89640, Sigma, St. Louis, MO, USA). This stains cartilage and osteoblasts a dark blue color against a light blue background.
Bone samples were stained with safranin O solution, Weigert hematoxylin solution, and solid green staining solution (Sigma, USA). This results in chondrocyte cytoplasm presenting as red, and chondrocyte nuclei presenting as gray black.
Cells induced by IL-1
Chondrocytes were digested into a single cell suspension and seeded into 6-well plates at a density of 10,000 cells per well. After incubation for 10 days, the colonies were stained with crystal violet (Sigma, USA) for 20 min, and the images were recorded with a camera.
The concentration of malondialdehyde (MDA), which is the final product of lipid peroxidation, was determined using a lipid peroxidation assay kit (Doojindo, Kumamoto, Japan). Additionally, total iron and Fe2+ levels were evaluated with an iron assay kit (ab83366, Abcam, Woburn, MA, USA), as per the instructions provided by the manufacturer.
Cell lysates were obtained using RIPA buffer (89900, Invitrogen™, Carlsbad, CA, USA). Following loading and separation on an SDS-PAGE gel, the proteins were transferred to PVDF membranes and blocked with 5% skim milk. The membranes were then incubated overnight at 4 °C with primary antibodies against METTL16 and GPX4 (1:1000, ab252420; ab125066, Abcam, USA). They were subsequently treated with HRP-conjugated anti-rabbit secondary antibodies (1:1000, ab6721, Abcam, USA), and protein bands were visualized using an ECL chemiluminescence substrate (WBKLS0500-2, Millipore, Burlington, MA, USA).
Tissues and chondrocytes were treated with Trizol reagent to isolate total RNA.
This was converted into cDNA using the PrimeScript RT reagent kit (RR037A,
Takara, Kyoto, Japan). Real-time qPCR was performed with the SYBR Green qPCR
Master Mix (Thermo, Waltham, MA, USA). Gene expression levels were measured
relative to the
RNAs were isolated from cells and incubated with protein G beads (70024, CST, Danvers, MA, USA) that had previously been reacted with anti-m6A monoclonal antibody at 4 °C for 12 h in rotation. After reaction at 4 °C for 6 h, the beads were eluted and washed, and RNA was extracted using Trizol. The level of METTL16 was measured by qPCR assay.
Statistical analyses were conducted using GraphPad Prism 7.0 software (GraphPad
Software, Inc,San Diego, CA, USA). The paired Student’s t-test was
utilized to compare two groups. For analyzing differences among multiple groups,
either a one-way Analysis of Variance with Tukey’s post hoc test or a
Mann–Whitney U test was employed. The criterion for statistical significance was
defined as p
We first examined the in vivo effects of METTL16 on ACH using transgenic mice. The expression of METTL16 was suppressed in the proximal tibia of ACH mice compared with the control, but was recovered by treatment for METTL16 overexpression (Fig. 1A). Further analysis of the proximal tibia revealed an increased number of chondrocytes and cartilage, as well as an organized tissue structure after METTL16 overexpression compared with ACH mice (Fig. 1A). Treatment with METTL16 overexpression vectors also increased the BMD, BV/TV, TbN and TbTh compared to ACH (Fig. 1B). Notably, the expression of METTL16 and GPX4 were suppressed in the tibia tissues of ACH mice, while METTL16 overexpression recovered the levels of both (Fig. 1C). This observation suggests a potential correlation between the two genes and participation in ferroptosis during ACH. We next investigated the level of ferroptosis in the tibia of mice. The elevated levels of total iron and Fe2+ in the ACH group compared with the control group indicated an increase in cell ferroptosis. This was suppressed upon METTL16 overexpression (Fig. 1D). Perls’ Prussian blue staining demonstrated minimal iron deposition in control tibiae, whereas ACH mice exhibited prominent deposits concentrated in the growth plate and trabecular regions. Quantitative image analysis confirmed a significantly larger Perls’-positive area in ACH mice compared with controls, which was significantly reduced by METTL16 overexpression (Fig. 1E). In line with this, IHC for 4-Hydroxynonenal (4-HNE) showed markedly elevated lipid peroxidation in ACH tibiae, with intense cytoplasmic and extracellular 4-HNE staining. Overexpression of METTL16 substantially decreased this 4-HNE positivity (Fig. 1F). These histological findings, together with increased tissue iron and Fe2+ levels and decreased GPX4 expression in ACH, indicate enhanced ferroptosis in ACH that can be alleviated by restoring METTL16 expression. To further delineate the therapeutic effects of METTL16 on bone microstructure and cartilage morphology, we performed micro-computed tomography (CT) reconstruction of the proximal tibiae (Fig. 1G). CT analysis revealed that ACH mice exhibited severe trabecular bone deterioration, characterized by fragmented, disconnected trabeculae with reduced spatial continuity. However, METTL16 overexpression substantially restored the integrity of the trabecular network.
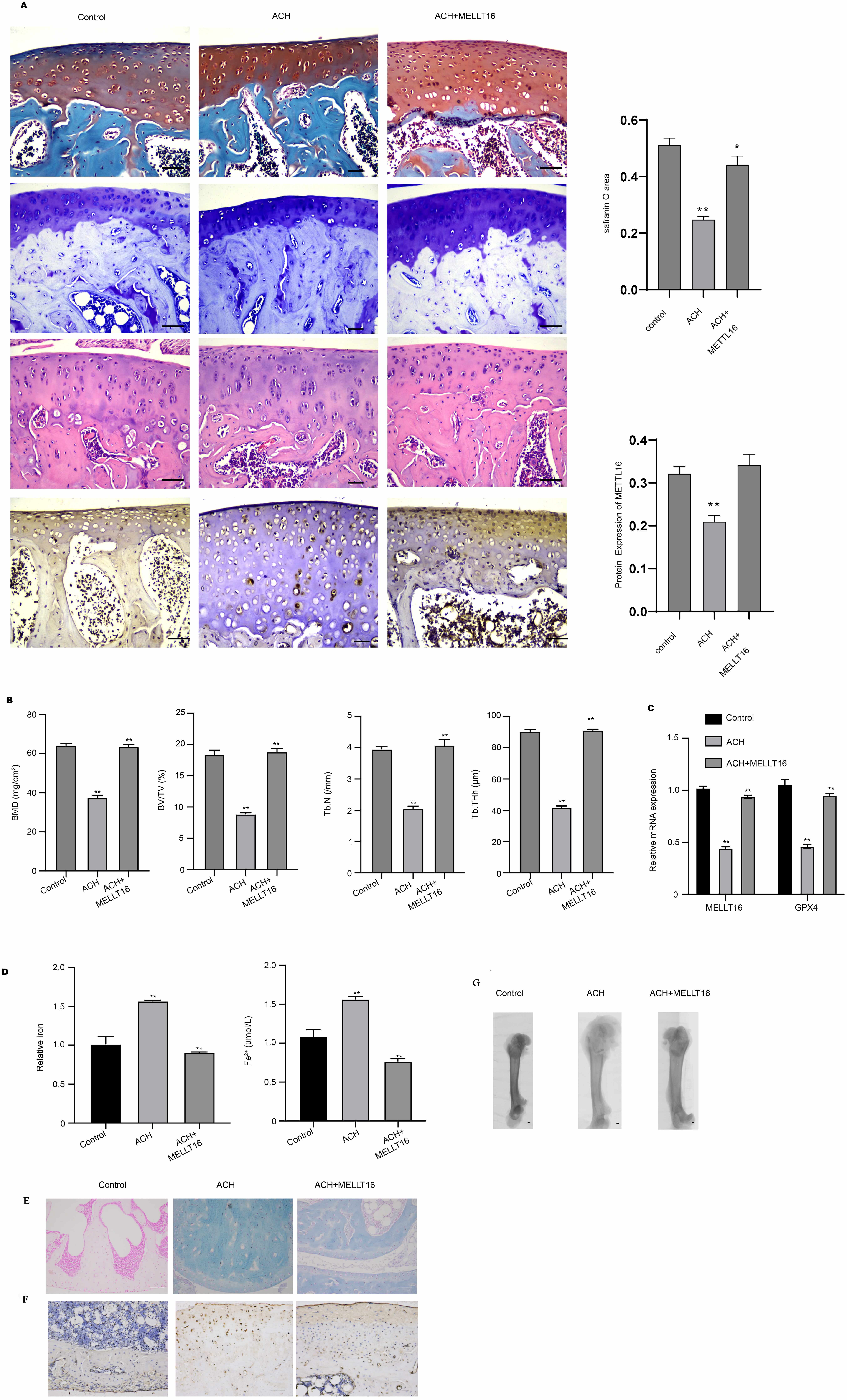 Fig. 1.
Fig. 1.
METTL16 promotes bone chondrogenesis and suppresses ferroptosis
in the ACH mouse model. Fgfr3𝑎𝑐ℎ transgenic mice were treated with
METTL16 lentivirus, and the proximal tibia tissues were collected for analysis.
(A) The morphology of cartilage, chondrocyte, and tissues was examined by
toluidine blue, safranin O, and H&E staining, while the expression of METTL16
was examined by IHC. Scale bar = 100 µm. (B) Results for bone mineral
density (BMD), bone structure via the bone volume to total tissue volume ratio
(BV/TV), trabecular number (TbN), and trabecular thickness (TbTh). (C) RNA levels
of METTL16 and GPX4, as measured by qPCR assay. (D) The levels of total iron and
Fe2+ were measured to assess ferroptosis. (E) Representative Perls’ Prussian
blue staining of tibial sections showing iron deposition (blue). Quantitative
analysis of Perls-positive area. Scale bar = 100 µm. (F) Representative IHC
staining for 4-HNE showing the levels of lipid peroxidation in tibiae.
Quantitative analysis of 4-HNE–positive signal intensity. Scale bar = 100
µm. (G) CT reconstruction for observing the microstructure of bone
cartilage in ACH mice. Scale bar = 100 µm. **p
To further examine whether ferroptosis is a major form of regulated cell death
in chondrocytes and to confirm the protective role of METTL16, we employed
specific inhibitors of ferroptosis (Ferrostatin-1) and apoptosis (Z-VAD-FMK) in a
model of IL-1
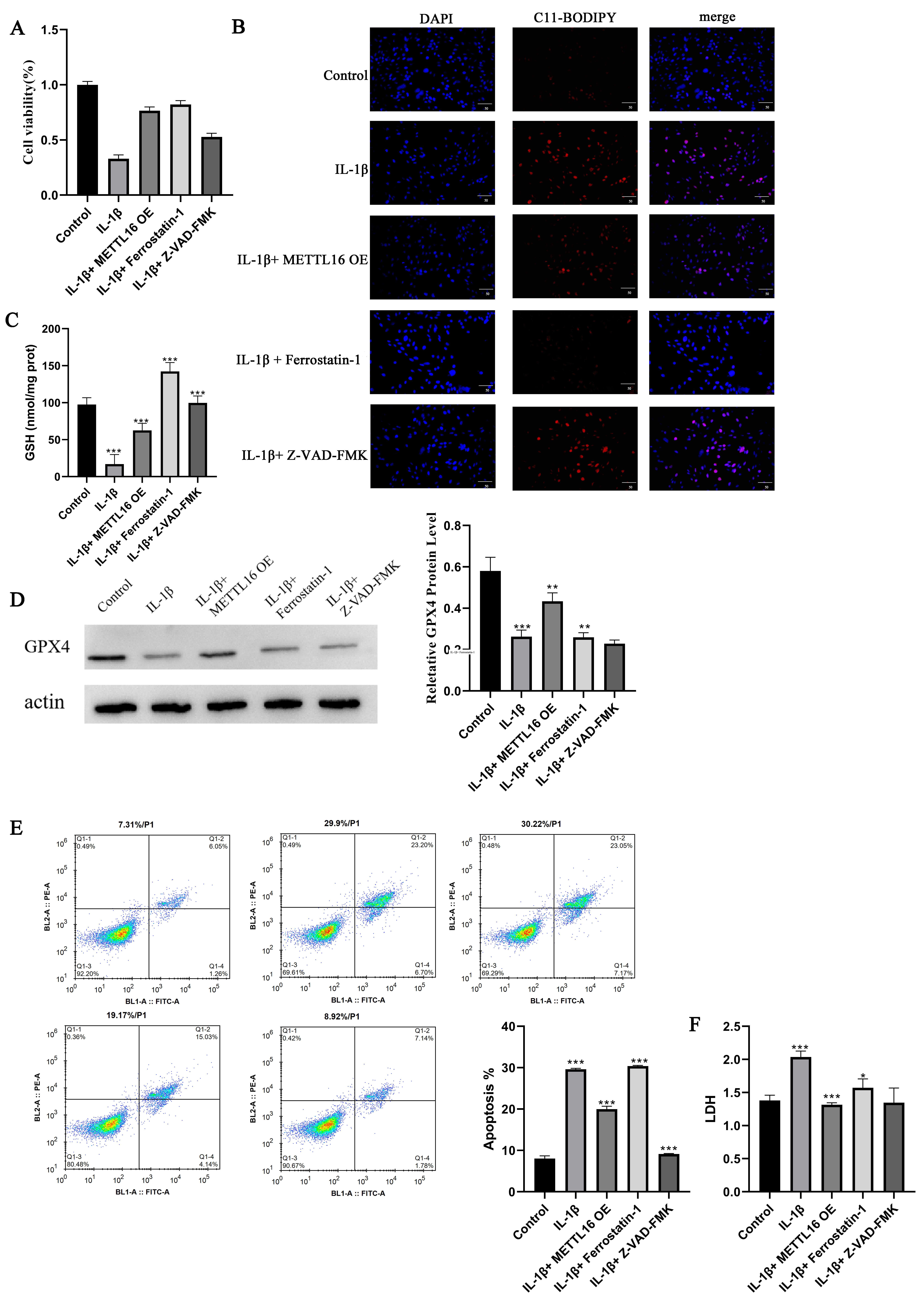 Fig. 2.
Fig. 2.
METTL16 protects chondrocytes from IL-1β-induced injury primarily by inhibiting ferroptosis. (A) Cell viability assessed by CCK-8 assay in control,
IL-1
Subsequently, we investigated the effects of METTL16 on chondrocytes. Cells from
primary culture were treated with IL-1
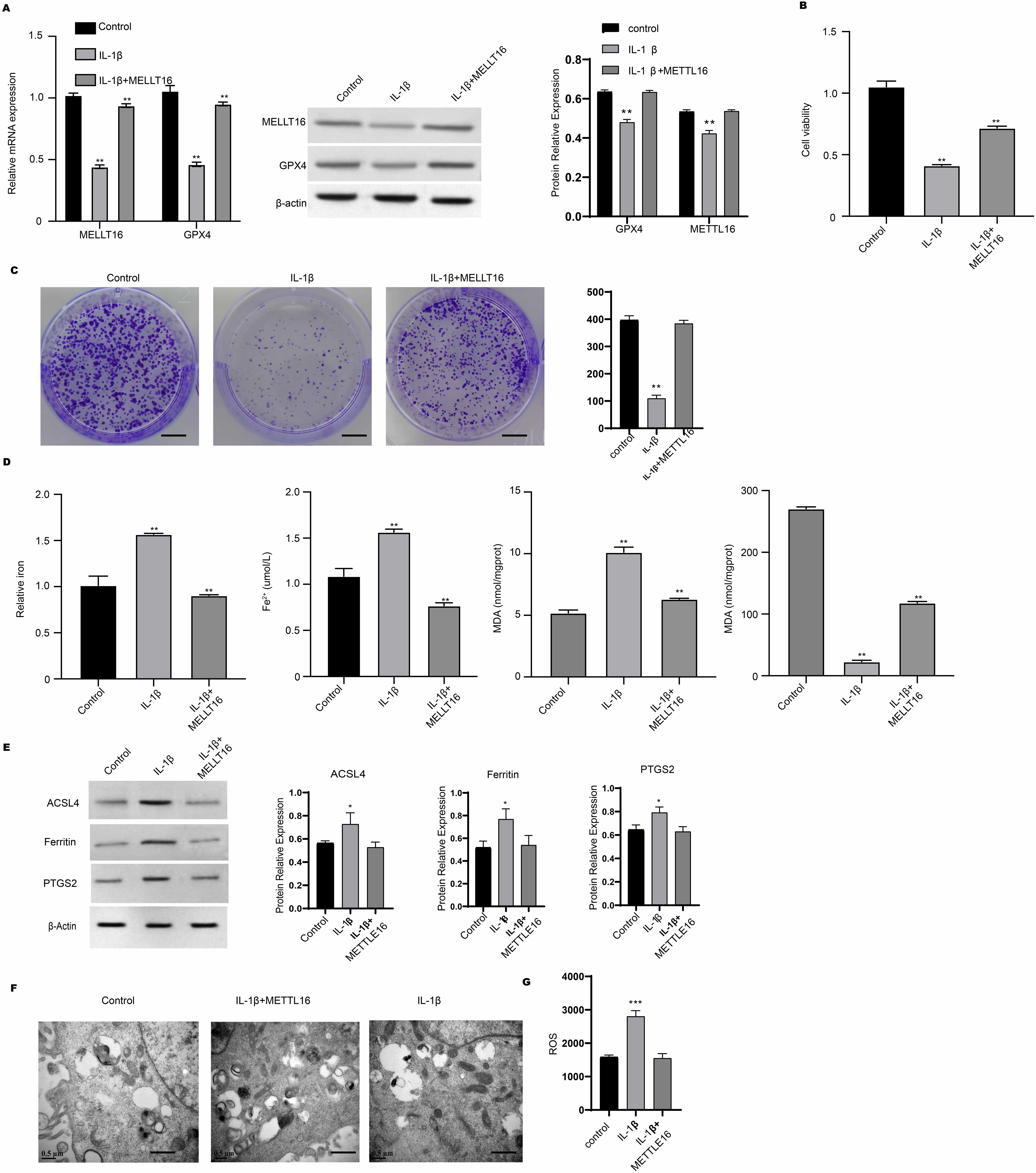 Fig. 3.
Fig. 3.
METTL16 enhances chondrocyte proliferation and suppresses cell
ferroptosis. Primary chondrocytes were stimulated with IL-1
To investigate how METTL16 influences GPX4 expression, we assessed the m6A methylation levels of GPX4 mRNA. The reduction of METTL16 levels using siRNAs was found to markedly decrease m6A modification of GPX4 mRNA (Fig. 4A). Moreover, depletion of METTL16 decreased the protein level of GPX4 (Fig. 4B). We also utilized CHX to block protein synthesis. The protein level of GPX4 was notably decreased 6 h after CHX treatment, and depletion of METTL16 induced faster degradation of GPX4 (Fig. 4C). These results indicate that knockdown of METTL16 can reduce the stability of GPX4 mRNA.
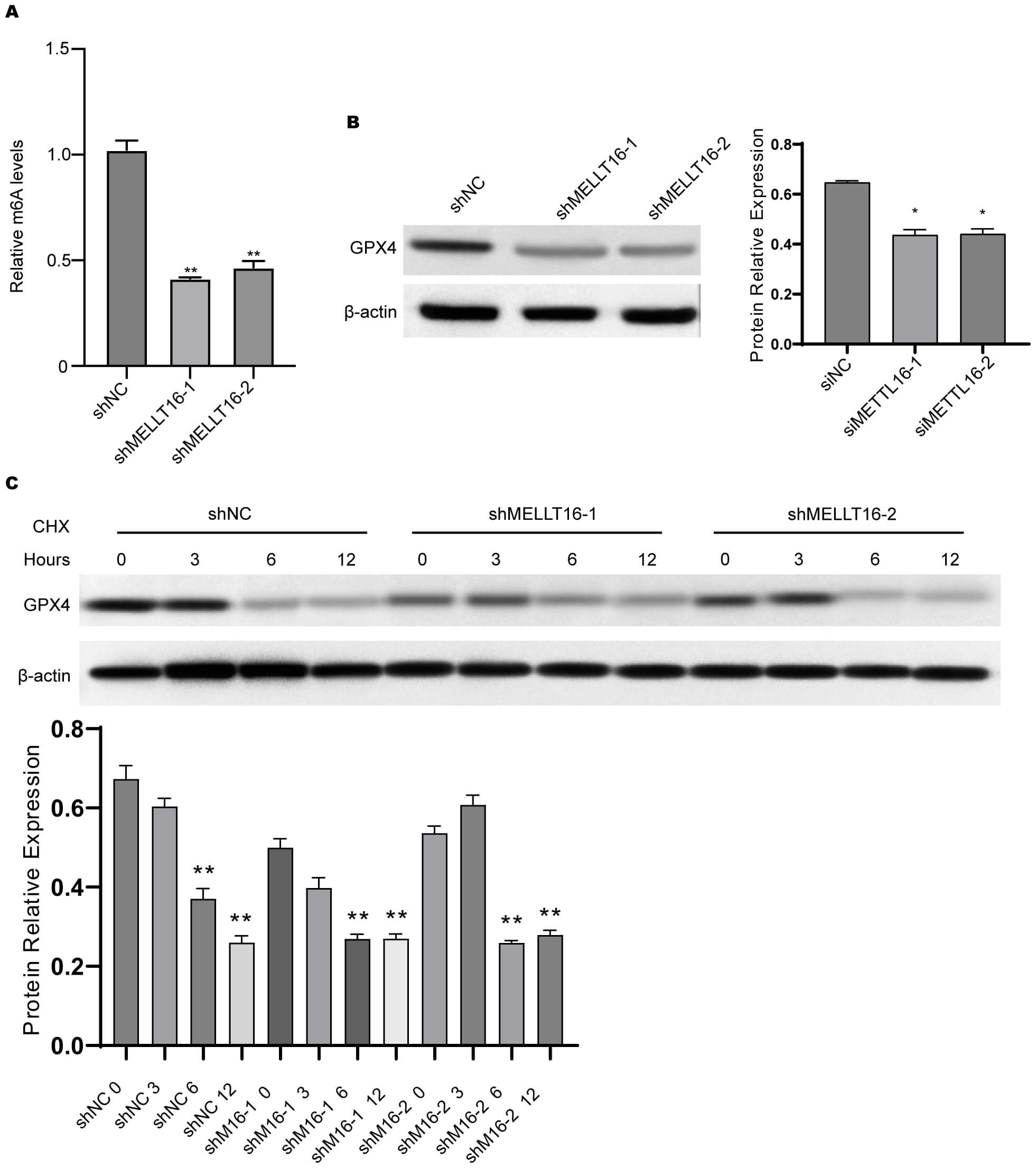 Fig. 4.
Fig. 4.
METTL16 regulates m6A modification and degradation of GPX4
mRNA. (A) The level of m6A modification of GPX4 mRNA was measured by the
m6A mRNA IP assay. (B) The protein level of GPX4 in chondrocytes was
measured by Western blotting. (C) Chondrocytes were first transfected with
siMETTL16-1 or siMETTL16-2 and then treated with CHX, an inhibitor of protein
synthesis. The protein level of GPX4 at 0, 3, 6, and 12 h after CHX treatment was
assessed by Western blot assay. **p
To further validate the involvement of ferroptosis in ACH pathology and clarify
the therapeutic potential of METTL16, ACH mice were treated with METTL16
overexpression (OE) vectors, the ferroptosis inhibitor Ferrostatin-1 (Fer-1, 1
mg/kg i.p.), or Caspase inhibitor/Necrostatin-1 (Casp/Nec-1). Normal mice served
as controls. Proximal tibiae and vertebrae were analyzed histologically and
biochemically to assess ferroptosis and inflammation. H&E staining revealed that
ACH mice exhibited a disorganized growth plate, reduced cartilage thickness, and
decreased chondrocyte density compared with control animals. These pathological
alterations were markedly attenuated by METTL16 OE and Fer-1 treatment, whereas
Casp/Nec-1 intervention resulted in minimal improvement. Perls’ Prussian blue
staining revealed extensive iron accumulation in the trabecular and growth plate
regions of ACH tibiae, which was significantly reduced in METTL16 OE- and
Fer-1-treated mice (Fig. 5A). IHC for 4-HNE showed pronounced lipid peroxidation
in ACH mice, reflected by intense cytoplasmic and extracellular staining, which
was substantially alleviated by METTL16 OE and Fer-1 (Fig. 5B). Biochemical
assays further supported these observations, with MDA and Fe2+/total iron
levels being significantly elevated, while GSH content was reduced in ACH mice.
Both METTL16 OE and Fer-1 normalized these parameters, whereas Casp had little
effect (Fig. 5C). Western blot analysis revealed marked downregulation of GPX4
and SLC7A11, and upregulation of the pro-ferroptotic proteins ACSL4 and
transferrin receptor (TfR1) in ACH tissues. METTL16 OE and Fer-1 effectively
restored GPX4, SLC7A11 and ferritin (FTH1) expression, while reducing ACSL4 and
TfR1 levels (Fig. 5D). Furthermore, serum and tissue cytokine analyses showed
that TNF-
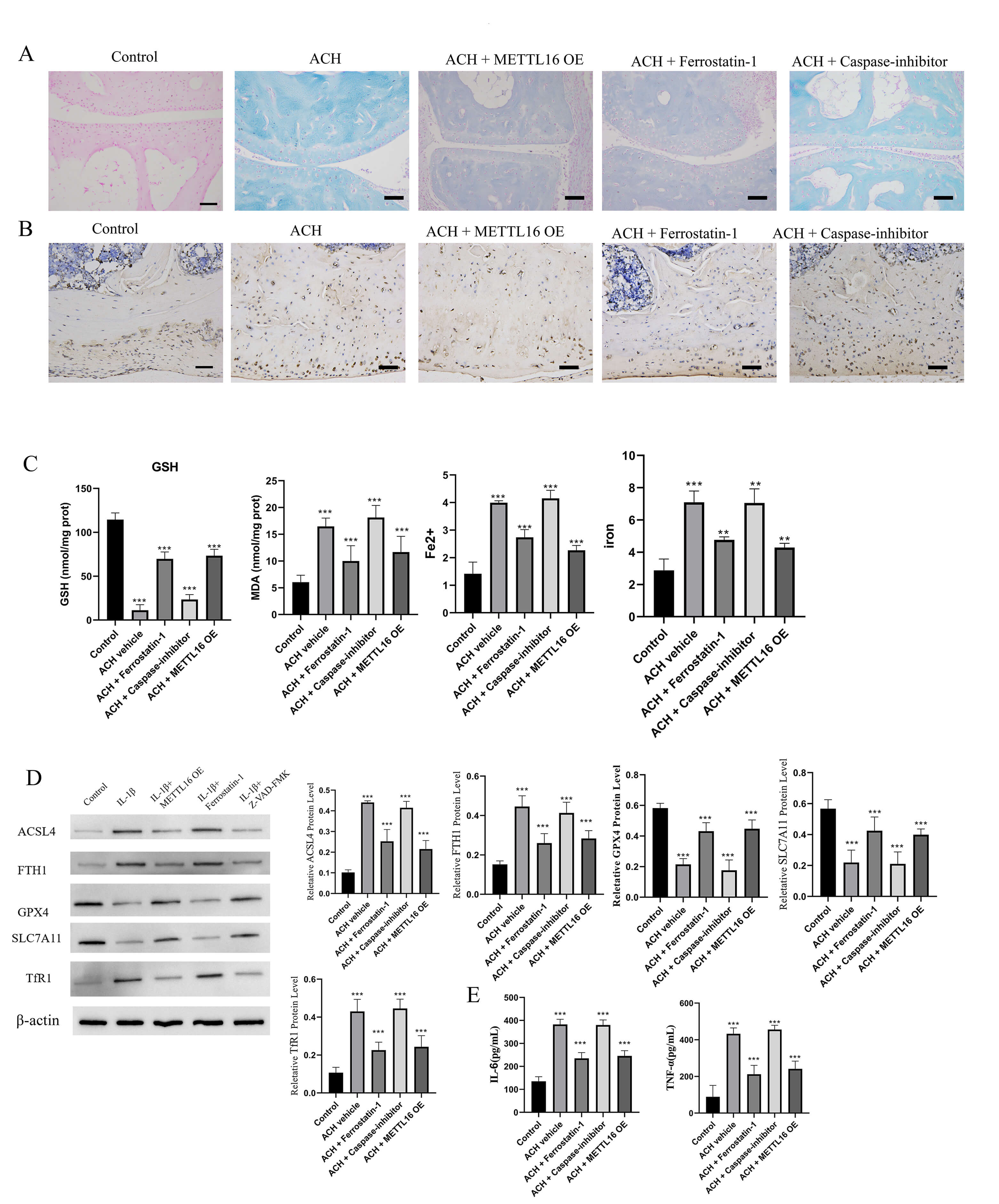 Fig. 5.
Fig. 5.
METTL16 overexpression and ferroptosis inhibition alleviate
ferroptosis and cartilage degeneration in ACH mice. (A) Perls’ Prussian blue
staining of proximal tibia sections showing iron accumulation in the trabecular
and growth plate regions of ACH mice. Iron deposition was significantly reduced
in groups with METTL16 overexpression (OE) and Ferrostatin-1 (Fer-1) treatment,
whereas Caspase inhibitor (Casp) and Necrostatin-1 (Nec-1) treatment had minimal
effects. Scale bar = 100 µm. (B) Immunohistochemistry for 4-HNE showed
lipid peroxidation in ACH tibiae. Intense cytoplasmic and extracellular 4-HNE
staining was observed in ACH mice, which was substantially reduced in METTL16 OE
and Fer-1-treated groups. Scale bar = 100 µm. (C) Biochemical assays were
used to measure MDA (malondialdehyde) and the Fe2+/total iron ratio, as well
as GSH (glutathione) content. Both METTL16 OE and Fer-1 restored MDA,
Fe2+/total iron levels, and GSH content to near normal levels, whereas
Casp/Nec-1 treatment had little effect. (D) Western blot analysis of
ferroptosis-related proteins in ACH tibiae. GPX4, SLC7A11, and ferritin (FTH1)
were downregulated in ACH mice and restored by METTL16 OE and Fer-1, whereas
ACSL4 and transferrin receptor (TfR1) were upregulated in ACH mice and reduced by
METTL16 OE and Fer-1 treatment. (E) Serum and tissue inflammatory cytokine levels
(TNF-
We further investigated the regulatory role of METTL16/GPX4 in ACH development. The results from H&E staining and bone structure parameters indicated that suppression of GPX4 abolished METTL16-promoted cartilage growth and bone formation (Fig. 6A,B). The results from in vitro experiments also revealed that GPX4 knockdown reversed the METTL-16-elevated viability and proliferation of chondrocytes (Fig. 6C,D). The decreased level of GPX4 protein in chondrocytes following siGPX4 transfection was verified by Western blotting (Fig. 6E).
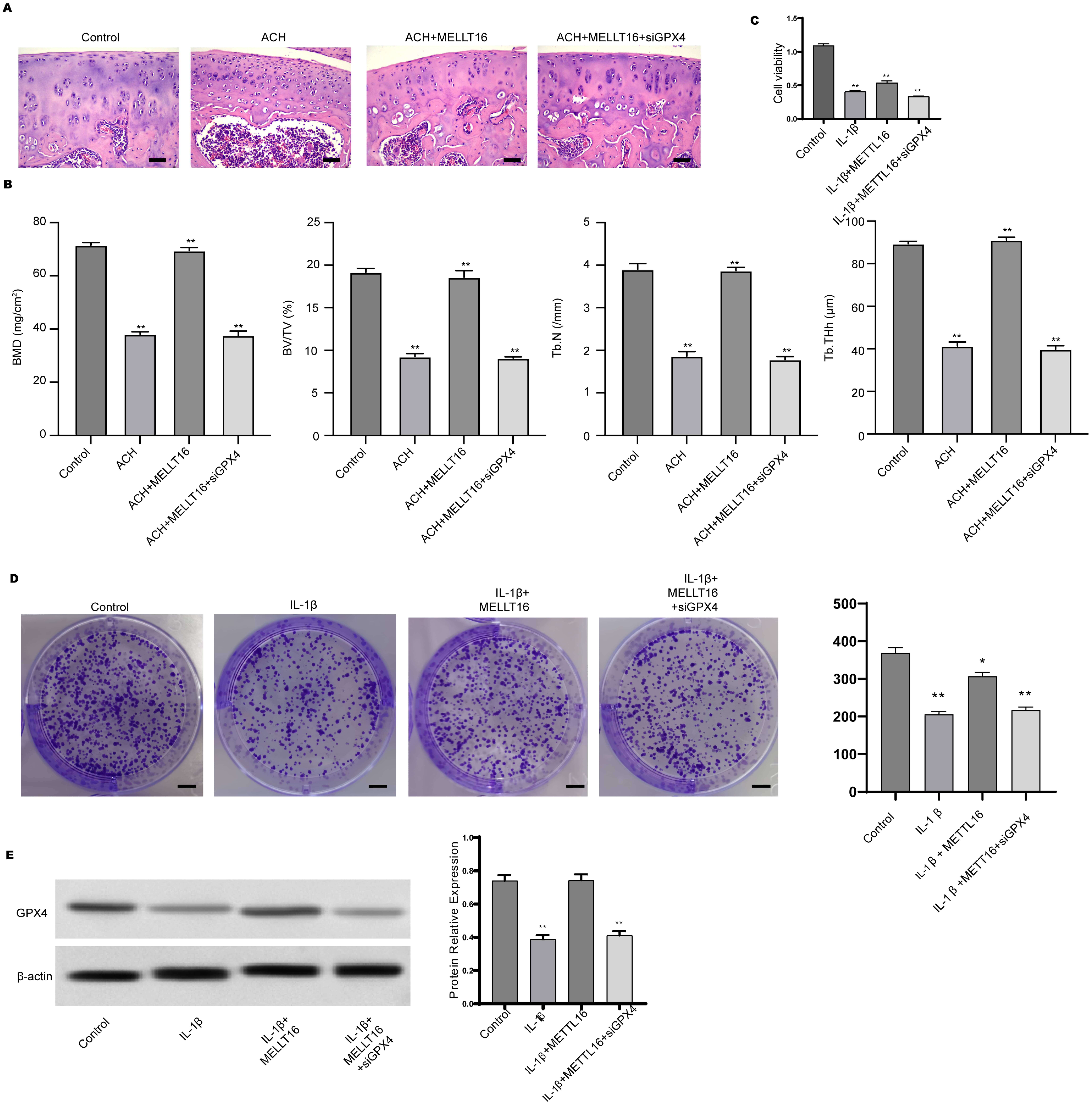 Fig. 6.
Fig. 6.
METTL16 improves chondrogenesis through the regulation of GPX4
expression. (A) H&E staining of proximal tibia tissue. Scale bar = 200 µm.
(B) Results for bone mineral density (BMD), bone structure as analyzed by measuring
the bone volume to total tissue volume ratio (BV/TV), trabecular number (TbN), and trabecular
thickness (TbTh). (C–E) Primary chondrocytes were stimulated with IL-1
RNA epigenetic modification has drawn great attention in recent years and has
been implicated in multiple biological processes during normal tissue development
and disease progression [18, 19]. METTL16 is an important m6A
methyltransferase that has been extensively studied in the context of various
diseases [20, 21, 22]. For example, the level of METTL16 correlates with the
progression of glioma [23] and indicates poor prognosis of melanoma [24]. METTL16
was also reported to promote the proliferation of gastric cancer cells via
regulation of cyclin D1 expression [25]. Mendel et al. [26] reported
that METTL16-mediated methylation of structured RNA is essential for the
development of mouse embryos. Mechanistic studies have also demonstrated that
METTL16 enhances the stability and translational efficiency of PPAR
METTL16 was also recently reported to be an important regulator of RNA translation, variable splicing, and stability [18, 30]. METTL16 mediates the degradation of MAT2A mRNA, which was disrupted by oxidative stress, which consequently aggravates the apoptosis of nucleus pulposus cells and exacerbates the process of intervertebral disc degeneration [31]. Furthermore, METTL16 mediates the translation of CIDEA in a m6A-dependent manner and promotes non-alcoholic fatty liver disease [32]. In the current work, we found that METTL16 upregulated the m6A modification of GPX4 mRNA and suppressed its degradation, thereby increasing the protein level of GPX4. GPX4 is one of the most critical regulators of ferroptosis [33]. As a glutathione peroxidase, GPX4 has been identified to catalyze GSH production and promote ROS clearance [34]. Ferroptosis involves an iron-dependent Fenton reaction that results in the overproduction and buildup of reactive oxygen species (ROS). These ROS can be countered by GPX4, meaning that induction of GPX4 expression is a potential strategy to reduce cell death caused by ferroptosis [35]. Cartilage tissue exists in a hypoxic and avascular microenvironment, with inherently high baseline levels of oxidative stress, making it more prone to rapid accumulation of ROS under pathological conditions [36]. Secondly, chondrocytes have only a weak ability to regulate iron homeostasis. Inflammatory factors can upregulate TfR1, but chondrocytes have only a limited capacity for iron storage and export, resulting in a significantly increased risk of iron overload. Thirdly, the cell membranes of chondrocytes are rich in polyunsaturated fatty acids, providing abundant substrates for lipid peroxidation [37]. Critically, chondrocytes are highly dependent on the GPX4/GSH axis to scavenge lipid peroxides, yet their own capacity for GSH synthesis is limited, rendering this defense system fragile and susceptible to collapse [38]. Consistent with the functions of GPX4 in ferroptosis and cell death, we observed decreased expression of GPX4 in the tibia tissues of ACH transgenic mice. Moreover, we found that suppression of primary chondrocyte ferroptosis by METTL16 was mediated by GPX4. In addition to the GPX4-dependent antioxidant system, ferroptosis can also be regulated through the FSP1–CoQ10 pathway. In the present study, we did not further investigate FSP1 for several reasons. First, accumulating evidence suggests that chondrocytes are highly dependent on the GPX4/GSH axis to counteract lipid peroxidation, whereas FSP1 expression and activity are relatively low or tissue-restricted in cartilage cells. Second, our data showed pronounced alterations in ACSL4, iron metabolism, and GPX4 expression, indicating that lipid remodeling and GPX4 inactivation represent the dominant ferroptotic mechanisms in chondrocytes under inflammatory conditions. Finally, given the limited antioxidant reserve and weak iron-handling capacity of chondrocytes, disruption of the GPX4 pathway is likely sufficient to trigger ferroptosis, making it the primary focus of this study. Nevertheless, future studies exploring potential crosstalk between METTL16 signaling and the FSP1 pathway may further refine our understanding of ferroptosis regulation in cartilage.
In summary, our work demonstrated that METTL16 is downregulated during ACH and that overexpression of METTL16 improved bone growth and alleviated the ferroptosis of chondrocytes. Mechanistically, METTL16 increased the m6A modification of GPX4 mRNA and upregulated its expression in chondrocytes. Our data provided a novel regulatory mechanism for ACH progression.
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
LH and XT performed the research and collected experimental data. MY provided technical assistance for experiments, performed the research and sample processing, and analyzed the data using statistical methods. XMW and XWW analyzed the data using statistical methods. BW designed the study, supervised the research process and provided essential conceptual advice. LH wrote the first draft of the manuscript. All authors contributed to critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Animal Care and Use Committee of Pediatric Orthopaedic Hospital, Honghui Hospital, Xi’an Jiaotong University (No. KT2023-01-05-01) and conducted in accordance with the relevant guidelines and regulations. This study is reported in accordance with the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments). For further details, see: https://arriveguidelines.org.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL45159.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.