




1 Department of General Surgery, The First Affiliated Hospital of Harbin Medical University, 150001 Harbin, Heilongjiang, China
2 Department of Gastrointestinal Surgery, Yantai Yuhuangding Hospital, Qingdao University, 264000 Yantai, Shandong, China
Abstract
Hepatobiliary malignancies remain a major clinical challenge because they are highly aggressive and resistant to therapy. In eukaryotes, N6-methyladenosine (m6A), the most prevalent internal RNA modification, regulates post-transcriptional gene expression. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3) act as pivotal m6A readers, stabilizing coding and non-coding RNAs to modulate cancer-related signaling networks. In hepatobiliary cancers, dysregulated IGF2BP expression is associated with proliferation, metastasis, metabolic adaptation, and immune evasion, underscoring its potential as a biomarker and therapeutic targets. This review provides a comprehensive overview of IGF2BP-mediated regulatory mechanisms and explores their translational potential in precision diagnostics and targeted interventions.
Keywords
- N6-methyladenosine
- non-coding RNA
- hepatobiliary cancers
- molecular mechanisms
- targeted therapy
- hepatocellular carcinoma
- intrahepatic cholangiocarcinoma
- gallbladder cancer
Hepatobiliary cancers, which include primary liver cancers (PLCs) and biliary tract cancers (BTCs), are prevalent and severe malignancies that pose significant threats to human health. PLCs include hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma (ICC), and combined HCC-cholangiocarcinoma (CC). HCC accounts for approximately 70–85% of PLCs and is the second leading cause of cancer-related mortality after lung cancer [1]. ICC represents approximately 10–12% of PLCs and is the second most common PLC [2]. By contrast, HCC-CC is a notably rare tumour type with an incidence of 1.0–4.7%.
BTCs include gallbladder cancer (GBC), ICC, extrahepatic bile duct cancer, and ampulla of Vater cancer. Interestingly, ICC is fundamentally classified as a BTC because it originates in the bile duct epithelium [3]. GBC is the most prevalent malignant tumour of the biliary tract and originates from the gallbladder epithelium. According to the Surveillance, Epidemiology, and End Results Program, its incidence rate is 2.5 per 100,000 [4, 5, 6]. From 1999 to 2013, extrahepatic bile duct cancer comprised approximately one-third of the cancers of the biliary system, with an annual incidence rate of approximately 1.8%, whereas ICC incidence rates increased to approximately 3.2%. Ampulla of Vater cancer consistently had the lowest incidence among BTCs between 1999 and 2013, with a rate of approximately 7.4 per 1,000,000 [3].
Effectively managing hepatobiliary cancers is a challenge due to the intricate processes governing their growth, metastasis, and therapeutic resistance. Accumulating evidence indicates that genetic mutations [7, 8, 9], epigenetic alterations [10, 11, 12], immune responses [13, 14, 15], and the tumour microenvironment play significant roles in these processes. Given that N6-methyladenosine (m6A) is the most prevalent and abundant epigenetic RNA modification, it has become a major focus in cancer research [16, 17]. As a dynamic and reversible regulatory mechanism, m6A modification significantly influences cancer initiation and progression (Fig. 1). The m6A regulatory system is primarily composed of methyltransferases (writers), demethylases (erasers), and effector proteins (readers). Methyltransferase-like 3 (METTL3) [18, 19, 20], METTL14 [21, 22, 23], METTL16, Wilms’ tumour 1-associating protein (WTAP) [24, 25, 26], vir like m6A methyltransferase associated [27], zinc finger CCCH domain-containing protein 13, and RNA-binding motif protein 15 (RBM15) function as m6A writers [28], facilitating the addition of m6A marks to RNA molecules. Conversely, human AlkB homolog H5 (ALKBH5) [29, 30], fat mass and obesity-associated protein act as m6A erasers [31], removing methylation marks from target mRNAs. YTH domain-containing proteins (YTHDC1/2 and YTHDF1/2/3) [32, 33, 34, 35, 36, 37, 38], heterogeneous nuclear ribonucleoproteins (hnRNPs: hnRNPC and hnRNPA2B1), and insulin-like growth factor 2 mRNA-binding protein (IGF2BP) proteins (IGF2BP1/2/3) function as m6A readers, recognizing m6A-modified transcripts and regulating their fate [39, 40]. Notably, IGF2BPs are frequently overexpressed in hepatobiliary cancers and are associated with a poor patient prognosis, suggesting their important roles as regulators that deserve further investigation.
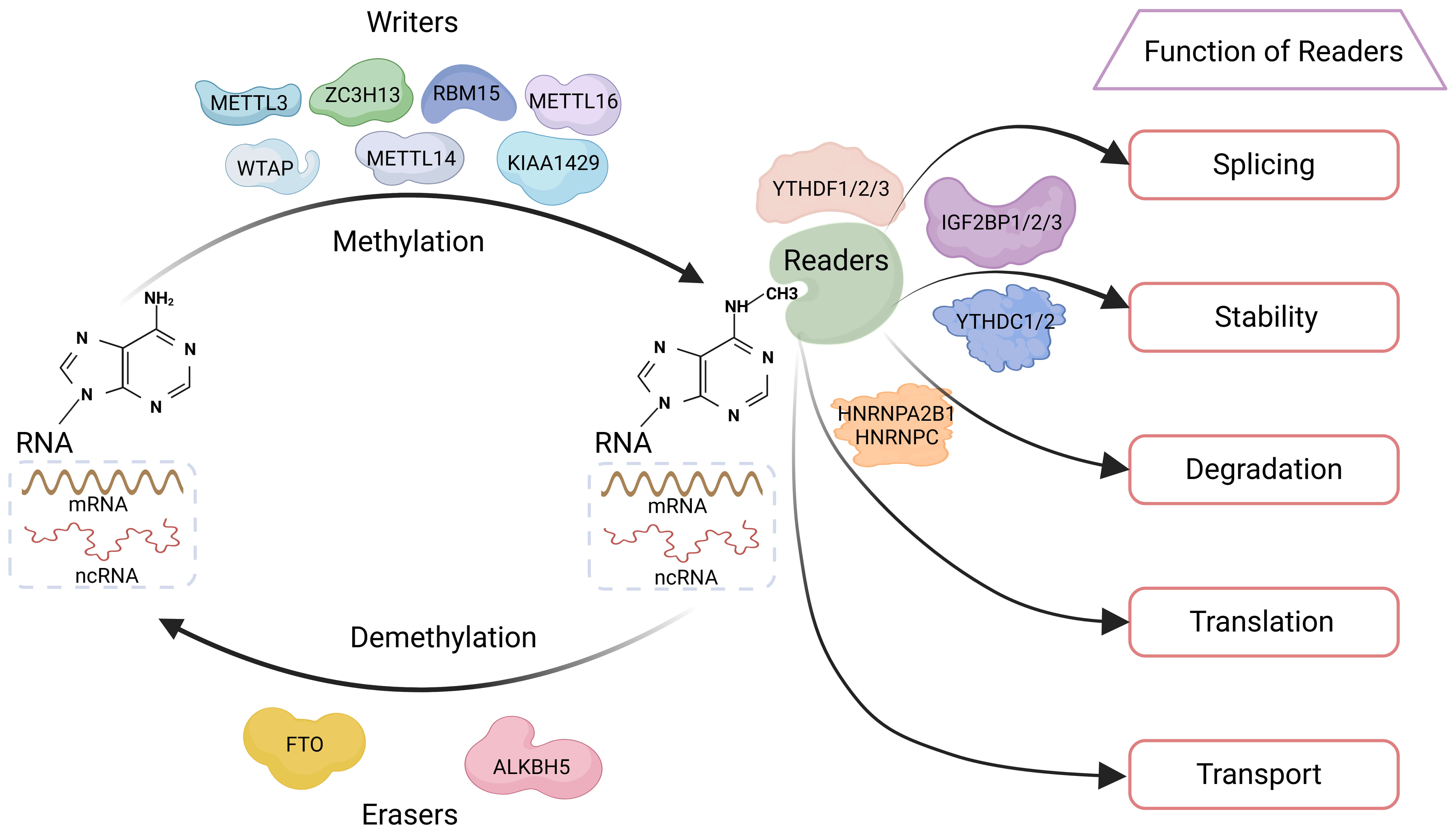 Fig. 1.
Fig. 1.
Dynamic m6A RNA modification and its regulation by writers, erasers, and readers. m6A is a reversible RNA modification that adds a methyl group to the nitrogen-6 position of adenine. This modification is catalyzed mainly by the METTL3-METTL14-WTAP complex, or by METTL16 in certain contexts. Once deposited, m6A is recognized by reader proteins, which in turn influence RNA splicing, nuclear export, stability, targeted degradation, and translation. Removal of m6A by demethylases such as FTO or ALKBH5 allows the modification to be dynamically regulated, highlighting its reversible nature and central role in controlling RNA function. Abbreviations: METTL3/14/16, methyltransferase-like 3, 14, and 16; WTAP, Wilms’ tumour 1-associating protein; ZC3H13, zinc finger CCCH domain-containing protein 13; KIAA1429, vir like m6A methyltransferase associated; RBM15, RNA-binding motif protein 15; FTO, fat mass and obesity-associated protein; ALKBH5, human AlkB homolog H5; IGF2BP1/2/3, insulin-like growth factor 2 mRNA-binding proteins 1, 2, and 3; YTHDF1/2/3, YTH N6-methyladenosine RNA-binding proteins 1, 2, and 3; YTHDC1/2, YTH domain-containing proteins 1 and 2; HNRNPA2B1, Heterogeneous nuclear ribonucleoprotein A2/B1; HNRNPC, heterogeneous nuclear ribonucleoprotein C. Created with http://www.biorender.com/.
IGF2BPs, also known as IGF2 mRNA-binding proteins (IMPs) or Vg1 RBP/IMP/CRD-BP/KSRP/ZBP (VICKZ) family of RNA-binding proteins (VICKZs), is a highly conserved group of RNA-binding proteins comprising IGF2BP1, IGF2BP2, and IGF2BP3 [41, 42]. IGF2BPs are primarily expressed in embryonic tissues, with comparatively lower levels in adult tissues [43]. IGF2BP2 is the predominant isoform expressed in adult tissues. IGF2BPs are expressed in close proximity to the nucleus in nearly all cell types except spermatogenic cells [43, 44, 45]. All three IGF2BP isoforms share a high degree of sequence homology (at least 56%) and each isoform contains six conserved RNA-binding domains: two N-terminal RNA recognition motifs and four C-terminal hnRNP K homology (KH) domains [46, 47]. Unlike YTHDF2, which mediates mRNA degradation, IGF2BPs promote the stability and storage of their target mRNAs in an m6A-dependent manner under both normal and stress conditions. Importantly, KH domains are essential for m6A recognition and may contribute to the tumour-promoting functions of IGF2BPs. Based on these characteristics, this review focuses on the regulatory network of IGF2BPs in hepatobiliary cancers, aiming to elucidate their complex mechanisms and provide insights for therapeutic development.
Initially named and characterized in 1999, IGF2BPs constitute a class of RNA-binding proteins that interact with multiple sites within the 5′ untranslated region (5′ UTR) of IGF2 mRNA [48]. Hence, they are also referred to as IMPs (IGF2 mRNA-binding proteins). The nomenclature of IGF2BPs reflects their regulatory effect on IGF2 expression, with IGF2 being a key ligand in the IGF system.
The IGF system comprises IGF1, IGF2, insulin, and their corresponding receptors (IGF1 receptor [IGF1R], insulin receptor [IR], and hybrid IR/IGF1R), as well as mannose-6-phosphate/IGF2R, at least six IGF-binding proteins (IGFBP1–6), acid-labile subunit, and associated proteases [49]. This network is extensively involved in organismal development, metabolism, and tumorigenesis. Studies have demonstrated that IGF signaling is significantly implicated in hepatobiliary malignancies. For example, analyses of 228 HCC samples, 168 paired cirrhotic liver tissues, and 10 post-surgical non-tumour liver specimens revealed that approximately 15% of HCC tissues exhibited more than 20-fold higher IGF2 mRNA and protein expression compared to non-tumour tissues, suggesting a potential tumour-promoting role in HCC [50].
Elevated IGF2 expression is closely associated with hepatic stellate cell activation, Notch signaling pathway activation, and aggressive tumour phenotypes [50]. Moreover, IGF2 can promote hepatocarcinogenesis through IGF1R signaling and participate in a positive feedback loop involving IGF1R and the transcription factor BTB and CNC homology 1 [51], which supports HCC cell growth and metastasis. Non-coding RNAs (ncRNAs) also modulate IGF2 expression; for example, long non-coding RNA (lncRNA) 91H and microRNA 483-5p (miR-483-5p) can upregulate IGF2 fetal transcripts via the P3/P4 promoters, facilitating HCC progression [52, 53]. In cholangiocarcinoma, tyrosine kinase inhibitor (TKI)-resistant cell lines display markedly elevated expression of IGF2, IGF1R, and IRs, implicating IGF signaling in drug resistance development. Currently, co-administration of IGF pathway inhibitors with TKIs has been proposed as a potential therapeutic strategy [54]. Notably, although IGF2 can bind to IGF2R, this receptor lacks intracellular signaling capability; hence, IGF2 predominantly acts through IGF1R to activate downstream pathways in hepatobiliary cancers [49].
IGF2BPs act as post-transcriptional regulators of IGF2 by enhancing mRNA stability, preventing degradation, and promoting translation, collectively increasing IGF2 bioavailability. This regulation facilitates IGF1R-mediated signaling, influencing HCC cell growth, proliferation, and differentiation. Beyond IGF2, IGF2BPs can also interact with other tumour-associated targets, such as the 3′ UTR of cellular myelocytomatosis oncogene (MYC), modulating multiple signaling pathways and thereby exerting broader effects on hepatobiliary tumour progression [40].
Given that the IGF2BP nomenclature originates from their initial identification based on IGF2 regulation, it is sometimes mistakenly assumed that they are merely derivatives or downstream effectors of IGF2. To avoid this misconception, using the original term “IMPs” when referring to both IGF2 and IGF2BPs may provide clearer communication, preserving the functional link while enhancing accuracy and clarity.
Since 2018, studies have increasingly established IGF2BPs as critical regulators of tumour initiation and progression. They regulate malignant processes by modulating RNA splicing, transport, stability, degradation, and translation. In hepatobiliary cancers, IGF2BPs regulate diverse biological processes by binding to specific sites on target mRNAs or ncRNAs. The ncRNAs most closely associated with IGF2BP regulation can be broadly classified into three major types: miRNAs, lncRNAs, and circular RNAs (circRNAs).
MiRNAs are short endogenous ncRNAs, approximately 20–24 nucleotides in length, that modulate gene expression post-transcriptionally by binding to the 3′ UTR of target mRNAs. This interaction typically results in mRNA degradation and inhibition of protein translation. Because the protein expression of target genes is critical for regulating fundamental cellular functions, miRNAs can profoundly influence cellular phenotypes [55]. miRNAs are widely expressed across various human tumours, predominantly functioning as oncogenic or tumour-suppressive regulators throughout cancer initiation, progression, and evolution.
LncRNAs, transcripts exceeding 200 nucleotides that lack protein-coding potential, have emerged as crucial regulators of a multitude of oncogenic processes via interactions with proteins, DNA, and RNA. They can act as molecular signals, decoys, guides, scaffolds, or enhancers, thereby modulating gene transcription and functional activity [56, 57, 58]. LncRNAs can also direct proteins and transcription factors to specific genomic loci, influencing gene expression. Additionally, they can counteract miRNA activity by providing specific decoy-binding sites.
CircRNAs are defined by their covalently closed circular structure formed through pre-mRNA back-splicing and are widely expressed in human cells, where they perform key regulatory functions. Established roles of circRNAs include modulating cancer metabolism, metastasis, and apoptosis. While circRNAs were initially considered non-coding due to their lack of traditional cap-dependent translation elements, accumulating evidence indicates that some circRNAs can independently encode polypeptides via cap-independent mechanisms. As key endogenous RNAs, disruptions in circRNA homeostasis can affect cancer cell proliferation and therapeutic resistance by altering gene expression at both the pre- and post-transcriptional levels [59].
Systematic elucidation of IGF2BP-mediated regulatory mechanisms in hepatobiliary cancers will enhance our understanding of their roles in tumour initiation and progression and provide a solid foundation for subsequent mechanistic studies. Fig. 2 illustrates the four major m6A-dependent functions of IGF2BPs in hepatobiliary cancers.
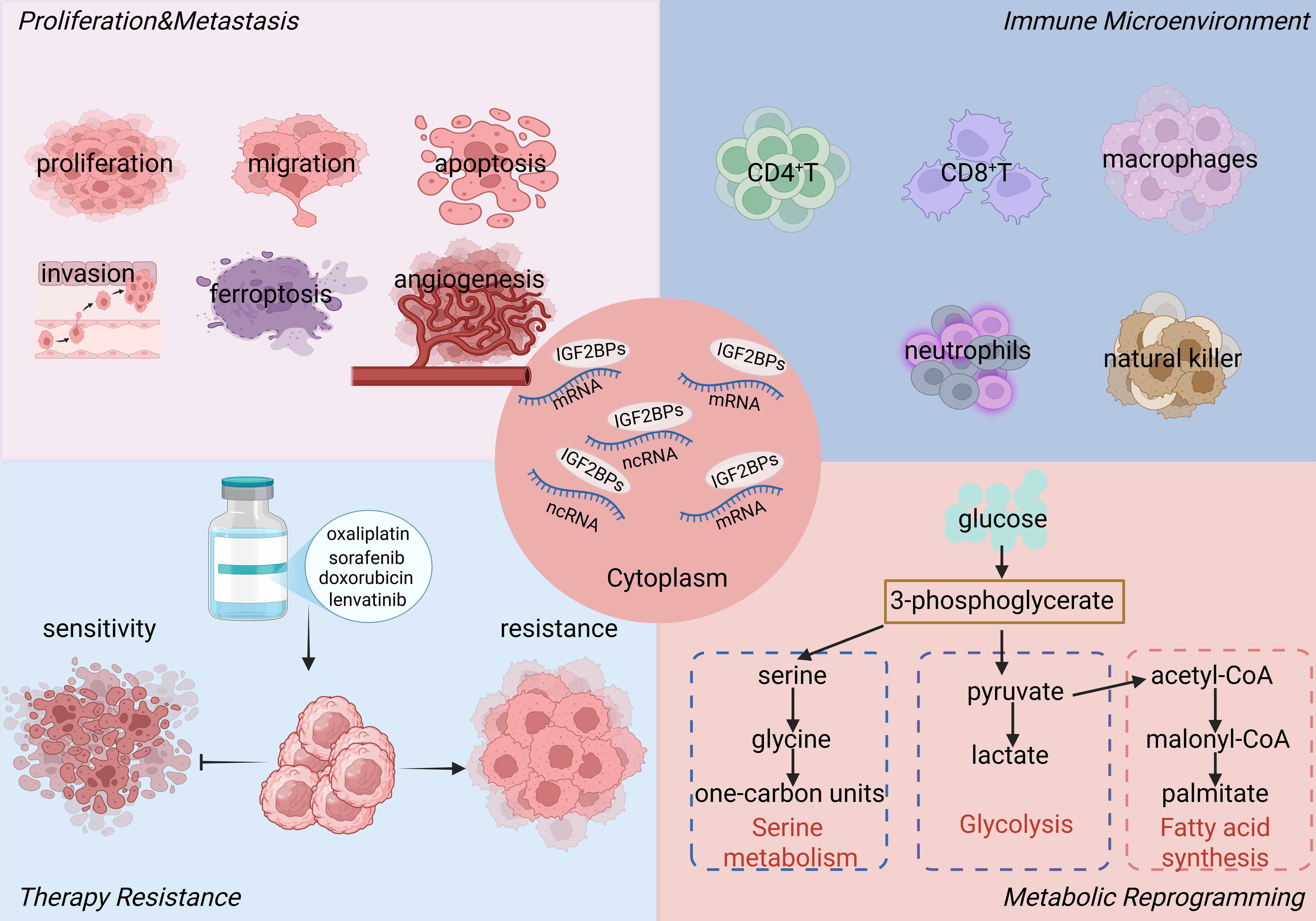 Fig. 2.
Fig. 2.
m6A-dependent functions of IGF2BPs in hepatobiliary cancers. IGF2BPs function as cytoplasmic m6A readers that stabilize and regulate target RNAs. They promote tumour proliferation and metastasis, shape the immune microenvironment, confer therapy resistance, and drive metabolic reprogramming including glycolysis, serine, and fatty acid metabolism. Abbreviations: IGF2BPs, insulin-like growth factor 2 mRNA-binding proteins. Created with http://www.biorender.com/.
IGF2BP1. IGF2BP1 binds to the m6A-modified site within the 3′ UTR of LY6/PLAUR domain-containing 1 (LYPD1), contributing to increased mRNA stability. ALKBH5-mediated m6A demethylation disrupts IGF2BP1 interactions with LYPD1 and progestin and AdipoQ receptor 4, leading to post-transcriptional repression that inhibits cell proliferation, migration, and invasion [60, 61]. IGF2BP1 also stabilizes alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase RNA, which may support tumour initiation and self-renewal [62]. Recognition of m6A sites by IGF2BP1 enhances circMDK expression; circMDK sponges miR-346 and miR-874-3p, leading to increased expression of autophagy related 16 like 1 and activation of the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin pathway, promoting proliferation, migration, and invasion [63]. IGF2BP1 facilitates the translation of circular RNA derived from mitogen-activated protein kinase kinase kinase 4 (circMAP3K4)-455aa by binding m6A sites on circMAP3K4, reducing apoptosis via interaction with apoptosis-inducing factor mitochondria associated 1 and supporting cell survival under cisplatin treatment [64].
Post-translational modifications regulate IGF2BP1 activity. F-box/SPRY domain-containing protein 1 mediates polyubiquitination of IGF2BP1 at lysine residues K190 and K450, enhancing its binding to polo-like kinase 1 and tumorigenic potential [65]. Listerin-induced ubiquitination targets IGF2BP1 for degradation, reducing c-Myc and IGF1R signaling and decreasing proliferation [66]. The METTL3/IGF2BP1 axis influences the sphingomyelin phosphodiesterase acid-like 3A/leucine-rich pentatricopeptide repeat containing pathway, promoting tumour growth and metastasis [67]. IGF2BP1 forms a positive feedback loop with lysine demethylase 5B (KDM5B), which may advance progression; long intergenic non-protein coding RNA 2428 (Linc02428) can antagonize this loop by destabilizing KDM5B RNA [68].
LncRNAs and miRNAs interact with IGF2BP1 to modulate its function. LncRNA interleukin enhancer binding factor 3-antisense RNA 1 (ILF3-AS1) recruits METTL3 to methylate ILF3, enhancing IGF2BP1 interaction and stabilizing ILF3 transcripts, contributing to tumour progression [69]. Linc01134 functions as a competing endogenous RNA by binding miR-324-5p, promoting IGF2BP1 association with Yin Yang-1 RNA and sustaining a feedback loop linked to proliferation, migration, and the epithelial–mesenchymal transition (EMT) [70]. Lin28B-AS1 interacts with IGF2BP1 to enhance m6A recognition, influencing the stability and translation of targets including IGF2, glioma-associated oncogene homolog 1 (GLI1), and MYC [71]. LncRNA neuroblastoma-associated transcript 1 (NBAT1) competes with IGF2BP1 for c-Myc binding, reducing MYC mRNA stability and cell proliferation [72]. LncRNA vimentin (VIM)-AS1 modulates IGF2BP1 binding dynamics, affecting EPH receptor A3 expression and HCC invasiveness [73]. IGF2BP1 upregulates lncRNA MIR4435-2HG, associated with cancer stemness and sphere-forming capacity [74]. RBM15 contributes to cell growth and metastasis by regulating YES proto-oncogene 1 expression through IGF2BP1 [75].
IGF2BP2. IGF2BP2 cooperates with METTL3 to support tumour growth in HCC via m6A-dependent regulation of flap structure-specific endonuclease 1 [76]. METTL16 partners with IGF2BP2 to stabilize SUMO-specific peptidase 3 (SENP3) mRNA, suppressing ferroptosis through SENP3-mediated deSUMOylation of lactotransferrin [77]. IGF2BP2 also regulates multiple lncRNAs involved in tumour progression. Stabilization of glucosylceramidase beta pseudogene 1 activates the bone morphogenetic protein/SMAD signaling pathway, potentially contributing to HCC advancement [78]. LncRNA rhophilin-1-AS1 acts as a competing endogenous RNA by sponging miR-596, relieving repression of IGF2BP2 and promoting proliferation and metastasis [79]. IGF2BP2 stabilizes linc01977, enhancing interaction with RBM39 and inhibiting Notch receptor 2 ubiquitination, contributing to tumour development [80]. METTL14 and IGF2BP2 increase the abundance of lncRNA Rho GTPase activating protein 5 (ARHGAP5)-AS1 by targeting its m6A site. ARHGAP5-AS1 may disrupt the tripartite motif containing 28 (TRIM28)–cold shock domain containing E1 (CSDE1) interaction, prevent CSDE1 degradation, and facilitate translation of VIM and rac family small GTPase 1, potentially accelerating tumour progression [81]. IGF2BP2 also post-transcriptionally upregulates lncRNA transient receptor potential cation channel, subfamily C, member 7-AS1, indirectly elevating high mobility group AT-hook 2 expression and driving HCC progression [82]. LncRNA small nucleolar RNA host gene 1 inhibits miR-7-5p, preventing negative regulation of IGF2BP2 and supporting tumour growth and metastasis [83].
IGF2BP3. IGF2BP3 enhances the mRNA stability of eukaryotic translation initiation factor 5B, which may increase the expression of tumour stem cell marker proteins [84]. It regulates E2F transcription factor 1 expression in an m6A-dependent manner, potentially driving proliferation [85]. Binding to the coding sequence of nuclear factor erythroid 2-related factor 2 (NRF2) mRNA may prolong transcript half-life and suppress ferroptosis [86]. Cyclin D1 (CCND1) expression may be influenced via recognition of m6A sites within the 3′ UTR of CCND1 mRNA and recruitment of the RNA-stabilizing protein poly(A) binding protein cytoplasmic 1 (PABPC1), contributing to cell cycle progression [87]. IGF2BP3 interacts with linc00467 to maintain TNF tumor necrosis factor receptor-associated factor 5 expression, potentially affecting proliferation and metastasis [88]. Binding to the m6A-modified site on exon 3 of diaphanous related formin 3 (DIAPH3) mRNA may increase protein output, whereas linc01089 suppresses DIAPH3 and may promote the EMT through recruitment of hnRNPM to induce exon 3 skipping [89].
IGF2BPs. IGF2BPs cooperate with METTL3 to stabilize BMI1 proto-oncogene (BMI1) and ring finger protein 2 mRNAs, promoting HCC initiation and progression [90]. IGF2BP1 and IGF2BP3 enhance lnc-CTHCC abundance, facilitating hnRNP K binding to the Yes1 associated transcriptional regulator promoter, contributing to increased YAP expression and influencing HCC proliferation, angiogenesis, and metastasis [91].
IGF2BP1. Knockdown of IGF2BP1 in HCC cells leads to enhanced apoptosis and reduced programmed death-ligand 1 (PD-L1) expression, potentially relieving immune suppression and modulating the tumour immune microenvironment. This knockdown also correlates with increased infiltration of CD4+ T cells, CD8+ T cells, CD56+ natural killer cells, and F4/80+ macrophages, suggesting a broader impact on immune surveillance [92].
IGF2BP2. Sequencing data analysis indicates that IGF2BP2 regulates its downstream target IGF2 in an m6A-dependent manner, which may influence the recruitment and activity of immune cells, including CD4+ T cells, CD8+ T cells, macrophages, and neutrophils. Further experimental validation is required to confirm these effects [93].
IGF2BP3. Forkhead box M1-mediated upregulation of IGF2BP3 stabilizes ribonucleotide reductase regulatory subunit M2 (RRM2) mRNA through its m6A reader function, correlating with increased RRM2 protein levels. This stabilization may suppress ferroptosis and support macrophage M2 polarization [94]. In cooperation with WTAP, IGF2BP3 enhances circular cell cycle and apoptosis regulator 1 stability, which in turn stabilizes programmed cell death protein 1 (PD-1) protein and potentially contributes to exhaustion of anti-tumour CD8+ T cells [95]. Moreover, IGF2BP3 binds the 3′ UTR of solute carrier family 7 member 11 (SLC7A11) mRNA, facilitating the conversion of naïve CD4+ T cells into induced regulatory T cells and supporting an immunosuppressive microenvironment. Linc00942 recruits IGF2BP3 to maintain these effects [96]. Additionally, IGF2BP3 modulates the tumour immune microenvironment by regulating C-C motif chemokine ligand 5 and transforming growth factor beta 1, which may induce macrophage infiltration and M2 polarization while potentially suppressing CD8+ T-cell activation [97].
IGF2BP1. Fibroblast growth factor 19 (FGF19) binds to FGFR4, forming the FGF19/FGFR4 complex, which strengthens the IGF2BP1 interaction with PD-L1. This association is linked to increased PD-L1 mRNA stability and protein levels, potentially contributing to resistance against immune checkpoint blockade. Experimental evidence shows that FGFR4 knockdown in combination with anti-PD-1 antibody treatment significantly inhibits tumour growth and improves immunotherapy efficacy [98]. Protein arginine methyltransferase 3-mediated methylation of IGF2BP1 may increase heart of glass homolog 1 mRNA stability, potentially promoting oxaliplatin resistance in HCC cells [99].
IGF2BP2. Ferroptosis is correlated with radiosensitivity in HCC and may serve as an indicator of radiotherapy effectiveness. METTL3 and IGF2BP2 cooperatively modulate SLC7A11 mRNA levels, whereasile YTHDF2 potentially influences SLC7A11 protein ubiquitination via suppressor of cytokine signaling 2, collectively impacting radiosensitivity [100]. CircUPF2 binds IGF2BP2 KH3 and KH4 domains, forming a scaffold complex that stabilizes SLC7A11 transcripts and may mediate sorafenib resistance and ferroptosis regulation [101]. Circular eyes absent homology 3 stabilizes deltex E3 ubiquitin ligase 3L mRNA, potentially contributing to radiotherapy resistance [102]. LncRNA OXAR recruits cystatin A to enhance Ku70 stability, which may lead to oxaliplatin resistance in non-alcoholic fatty liver disease-associated HCC; this process is modulated by m6A modification via WTAP and IGF2BP2 [103]. Linc01134 recruits IGF2BP2 to increase mitogen-activated protein kinase 1 (MAPK1) mRNA, potentially activating MAPK signaling and contributing to radiotherapy resistance. It also modulates IGF2BP2 by sponging miR-342-3p, which normally suppresses IGF2BP2 expression [104]. METTL16 and IGF2BP2 enhance m6A modification of laminin subunit alpha 4 and strengthen its interaction with collagen type IV alpha 1 chain, potentially promoting cisplatin resistance [105]. METTL3 and IGF2BP2 increase ww domain-containing E3 ubiquitin protein ligase 2 mRNA abundance, linked to AKT pathway activation and may induce doxorubicin resistance [106]. METTL3 and IGF2BP2 also regulate solute carrier family 50 member 1 to enhance glycolysis, potentially contributing to doxorubicin resistance [107].
IGF2BP3. METTL3 and IGF2BP3 jointly upregulate la-related protein 4B expression in an m6A-dependent manner, activating the serine peptidase inhibitor kazal type 1-mediated epidermal growth factor receptor signaling pathway. This activation increases the expression of stemness-associated transcription factors and facilitates resistance to sorafenib treatment [108].
Metabolic reprogramming is a characteristic of malignancy, enabling tumour cells to meet increased demands for energy and biosynthetic precursors required for proliferation while adapting to environmental stresses such as hypoxia and nutrient deprivation. This involves alterations across multiple metabolic pathways, including glucose, amino acid, lipid, and nucleotide metabolism. Emerging evidence indicates that IGF2BPs, through their m6A reader functions, participate in key metabolic processes in cancer cells, contributing to tumour metabolic plasticity.
IGF2BP1. IGF2BP1 increases the transcript abundance of apoptosis-inducing factor mitochondria-associated 2 in HCC, suppressing ferroptosis and supporting glycolysis [109]. It forms a complex with TRIM71 that binds CCAAT enhancer-binding protein alpha mRNA, influencing serine and glycine metabolism [110]. Cooperation with the nuclear protein non-POU domain-containing octamer-binding protein (NONO) allows IGF2BP1 to regulate ATP citrate lyase mRNA in the cytoplasm, promoting fatty acid biosynthesis [111]. Ring finger protein 5-mediated K63-linked polyubiquitination at specific lysine residues enhances IGF2BP1 m6A recognition, elevating carnitine palmitoyltransferase 1A mRNA levels and facilitating fatty acid oxidation, which affects steatotic HCC progression [112].
IGF2BP2. IGF2BP2 supports glycolytic activity and stemness in HCC by increasing cell division cycle 45 transcript levels via m6A modification [113]. The lncRNA miR4458HG binds IGF2BP2, modulating its regulation of key glycolytic genes, including hexokinase 2 (HK2) and glucose transporter 1, contributing to metabolic reprogramming [114].
IGF2BP3. Recent studies have identified phosphoglycerate dehydrogenase, phosphoserine aminotransferase 1, and phosphoserine phosphatase as m6A-modified targets of METTL3 and IGF2BP3, with IGF2BP3 stabilizing their transcripts to increase mRNA and protein expression, thereby activating serine metabolism; notably, inhibition of this pathway enhances HCC cell sensitivity to sorafenib and lenvatinib [115]. METTL16 cooperates with IGF2BP3 to stabilize muscle phosphofructokinase mRNA, increasing glycolysis, and its transcription is regulated by POU class 3 homeobox 2 [116]. Eukaryotic cells contain numerous membraneless structures, also referred to as biomolecular condensates, which are formed via liquid–liquid phase separation (LLPS) and have been implicated in transcription, X-chromosome inactivation, DNA damage response, and tumorigenesis [117]. IGF2BP3 interacts with HK2 through LLPS to form stress granules that maintain HK2 mRNA stability, regulating glucose utilization and glycolytic flux under glucose deprivation, whereas Sestrin2 competes for HK2 binding to inhibit stress granule formation and promote cell survival [118]. Enhanced glycolysis leads to lactate accumulation, a hallmark of many solid tumours, and histone lactylation, occurring predominantly on lysine residues, has emerged as a novel post-translational modification that regulates tumour cell function [119]. In lenvatinib-resistant HCC models, lactylation of IGF2BP3 enhances its m6A-binding activity, stabilizing phosphoenolpyruvate carboxykinase 2 and NRF2 mRNAs, supporting gluconeogenesis and maintaining serine and one-carbon metabolism, including critical metabolites such as S-adenosylmethionine and glutathione; these findings suggest that IGF2BP3 lactylation not only augments its m6A reader function but also contributes to resistance against targeted therapies [120]. Moreover, IGF2BP3 enhances minichromosome maintenance 10 mRNA stability in an m6A-dependent manner; although its lactylation status was not examined in this study, it is plausible that post-translational modification contributes to the observed increase in transcript stability, providing a potential mechanistic explanation for its regulatory activity [121].
ICC. In ICC, IGF2BP1 supports cell proliferation and metastasis through
two regulatory axes: c-Myc–cyclin-dependent kinase inhibitor 2A axis and the
zinc finger protein of the cerebellum 2–p21-activated kinase 4–AKT–matrix
metallopeptidase 2 axis [122]. METTL3 and IGF2BP3 contribute to hepatic leukemia
factor (HLF) expression, which regulates downstream targets frizzled class
receptor 4 and forkhead box Q1 (FOXQ1), thereby facilitating self-renewal and
tumorigenic activity via the frizzled class receptor 1–
GBC. In GBC, IGF2BP2 binds lncRNA transient receptor potential cation channel subfamily M member 2-AS at site 271A to stabilize it, supporting activation of the neurogenic locus notch homolog protein 1 (NOTCH1) pathway in a process requiring PABPC1 and NUMB endocytic adaptor protein (NUMB). PABPC1 interacts with argonaute RNA-induced silencing complex (RISC) catalytic component 2 (AGO2) and miRNAs (miR-31-5p, miR-146a-5p) to suppress NUMB, indirectly enhancing NOTCH1 signaling [124]. IGF2BP2 also forms a feedback loop with circEZH2 to regulate the miR-556-5p/stearoyl-CoA desaturase 1 axis, contributing to lipid metabolic reprogramming and tumour growth and metastasis [125]. IGF2BP3 maintains kallikrein-related peptidase 5 transcript stability, promoting protease-activated receptor 2/AKT axis activity and facilitating cell proliferation and migration [126]. Furthermore, IGF2BP3 modulates the interaction between RBM14 and nuclear protein NONO, cooperating with NONO to regulate alternative splicing of discs large MAGUK scaffold protein 1 (DLG1), favoring the DLG1-S isoform associated with oncogenic activity [127].
Overall, IGF2BPs orchestrate coding and non-coding RNA networks to modulate oncogenic signaling, metabolism, and cell fate in hepatobiliary malignancies. These mechanistic roles underscore their multifaceted contribution to tumour initiation, progression, and therapeutic resistance, providing a foundation for further investigation into their biological functions (Table 1, Ref. [60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 75, 76, 77, 78, 80, 81, 82, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 118, 120, 121, 122, 123, 124, 125, 126, 127]).
| Cancer type | Reader | Writer/Eraser | Target | RNA outcome | Cellular phenotypes | Ref |
| HCC | IGF2BP1 | ALKBH5 | LYPD1, PAQR4 | Stability | Proliferation, migration, invasion | [60, 61] |
| HCC | IGF2BP1 | - | MGAT5 | Stability | Initiation, self-renewal | [62] |
| HCC | IGF2BP1 | - | CircMDK | Stability | Proliferation, migration, invasion | [63] |
| HCC | IGF2BP1 | - | CircMAP3K4 | Stability, translation | Apoptosis | [64] |
| HCC | IGF2BP1 | - | PLK1 | Stability | Proliferation | [65] |
| HCC | IGF2BP1 | - | MYC, IGF1R | Stability | Proliferation | [66] |
| HCC | IGF2BP1 | METTL3 | SMPDL3A, LRPPRC | Stability | Growth, metastasis | [67] |
| HCC | IGF2BP1 | - | KDM5B | Stability | Proliferation, metastasis | [68] |
| HCC | IGF2BP1 | METTL3 | ILF3 | Stability | Tumorigenesis, metastasis | [69] |
| HCC | IGF2BP1 | - | YY1 | Stability | Proliferation, migration, EMT | [70] |
| HCC | IGF2BP1 | - | IGF2, GLI1, MYC | Stability, translation | Growth, proliferation, migration, invasion | [71] |
| HCC | IGF2BP1 | - | MYC | Stability | Proliferation | [72] |
| HCC | IGF2BP1 | - | EPHA3 | Stability | Migration, invasion | [73] |
| HCC | IGF2BP1 | RBM15 | YES1 | Stability | Growth, metastasis | [75] |
| HCC | IGF2BP2 | METTL3 | FEN1 | Stability | Proliferation | [76] |
| HCC | IGF2BP2 | METTL16 | SENP3 | Stability | Ferroptosis | [77] |
| HCC | IGF2BP2 | METTL3 | GBAP1 | Stability | Metastasis, growth | [78] |
| HCC | IGF2BP2 | - | Linc01977 | Stability | Growth, metastasis | [80] |
| HCC | IGF2BP2 | METTL14 | LncRNA, ARHGAP5-AS1 | Stability | Invasion | [81] |
| HCC | IGF2BP2 | - | LncRNA TRPC7-AS1 | Stability | Proliferation, invasion | [82] |
| HCC | IGF2BP3 | - | EIF5B | Stability | Proliferation, invasion, EMT | [84] |
| HCC | IGF2BP3 | - | E2F1 | Stability | Proliferation | [85] |
| HCC | IGF2BP3 | - | NRF2 | Stability | Ferroptosis | [86] |
| HCC | IGF2BP3 | - | CCND1 | Stability | G1/S phase transition, proliferation | [87] |
| HCC | IGF2BP3 | - | TRAF5 | Stability | Proliferation, metastasis | [88] |
| HCC | IGF2BP3 | - | DIAPH3 | Alternative splicing | EMT, migration, invasion, metastasis | [89] |
| HCC | IGF2BP1/2/3 | METTL3 | BMI1, RNF2 | Stability | Proliferation, invasion | [90] |
| HCC | IGF2BP1/3 | - | Lnc-CTHCC | Stability | Proliferation, angiogenesis, metastasis | [91] |
| HCC | IGF2BP1 | - | PD-L1 | Stability | Apoptosis, immune cell infiltration | [92] |
| HCC | IGF2BP2 | - | IGF2 | Stability | Immune cell infiltration | [93] |
| HCC | IGF2BP3 | - | RRM2 | Stability | Ferroptosis | [94] |
| HCC | IGF2BP3 | WTAP | CircCCAR1 | Stability | Growth, metastasis | [95] |
| HCC | IGF2BP3 | - | SLC7A11 | Stability | Proliferation, ferroptosis | [96] |
| HCC | IGF2BP3 | - | CCL5, TGF- |
Stability | Macrophage migration, polarization | [97] |
| HCC | IGF2BP1 | - | PD-L1 | Stability | Proliferation, invasion | [98] |
| HCC | IGF2BP1 | - | HEG1 | Stability | Growth | [99] |
| HCC | IGF2BP2 | METTL3, YTHDF2 | SLC7A11 | Stability | Ferroptosis | [100] |
| HCC | IGF2BP2 | - | SLC7A11 | Stability | Proliferation | [101] |
| HCC | IGF2BP2 | - | DTX3L | Stability | Proliferation, apoptosis | [102] |
| NAFLD | IGF2BP2 | WTAP | LncRNA OXAR | Stability | Growth | [103] |
| HCC | ||||||
| HCC | IGF2BP2 | - | MAPK1 | Stability | Proliferation, apoptosis | [104] |
| HCC | IGF2BP2 | METTL16 | LAMA4 | Stability | Proliferation, invasion, migration | [105] |
| HCC | IGF2BP2 | METTL3 | WWP2 | Stability | Proliferation, glycolysis | [106] |
| HCC | IGF2BP2 | METTL3 | SLC50A1 | Stability, translation | Proliferation, glycolysis, apoptosis | [107] |
| HCC | IGF2BP3 | METTL3 | LARP4B | Stability | Proliferation, metastasis, angiogenesis | [108] |
| HCC | IGF2BP1 | - | AIFM2 | Stability | Proliferation, glycolysis, Invasion, apoptosis | [109] |
| HCC | IGF2BP1 | - | CEBPA | Stability | Proliferation | [110] |
| HCC | IGF2BP1 | - | ACLY | Stability | Fatty acid metabolism | [111] |
| Steatotic HCC | IGF2BP1 | - | CPT1A | Stability | Proliferation, migration, invasion | [112] |
| HCC | IGF2BP2 | - | CDC45 | Stability | Proliferation, migration, invasion, EMT, stemness, glycolysis, apoptosis | [113] |
| HCC | IGF2BP2 | - | HK2, GLUT1 | Stability | Proliferation, glycolysis | [114] |
| HCC | IGF2BP3 | METTL3 | PHGDH, PSAT1, PSPH | Stability, translation | Serine metabolism | [115] |
| HCC | IGF2BP3 | METTL16 | PFKM | Stability | Growth, invasiveness, sphere formation, glycolysis | [116] |
| HCC | IGF2BP3 | - | SESN2 | Stability | Proliferation, glycolysis, apoptosis | [118] |
| HCC | IGF2BP3 | - | PCK2, NRF2 | Stability | Serine metabolism | [120] |
| HCC | IGF2BP3 | - | MCM10 | Stability | Proliferation, migration | [121] |
| ICC | IGF2BP1 | METTL3/14 | MYC, ZIC2 | Stability | Growth, metastasis | [122] |
| ICC | IGF2BP3 | METTL3 | HLF | Stability | Self-renewal, tumorigenicity, proliferation, metastasis | [123] |
| GBC | IGF2BP2 | - | LncRNA | Stability | Angiogenesis, proliferation, migration | [124] |
| TRPM2-AS | ||||||
| GBC | IGF2BP2 | - | CircEZH2 | Stability | Lipid metabolism, ferroptosis, proliferation | [125] |
| GBC | IGF2BP3 | - | KLK5 | Stability | Proliferation, migration | [126] |
| GBC | IGF2BP3 | - | DLG1 | Alternative splicing | Proliferation | [127] |
Abbreviations: HCC, hepatocellular carcinoma; ICC, intrahepatic
cholangiocarcinoma; GBC, gallbladder cancer; IGF2BP1/2/3, insulin-like growth
factor 2 mRNA binding proteins 1, 2, and 3; METTL3/14/16, methyltransferase-like
proteins 3, 14, and 16; ALKBH5, human AlkB homolog H5; RBM15, RNA-binding motif
protein 15; WTAP, Wilms’ tumour 1-associating protein; YTHDF2, YTH
N6-methyladenosine RNA binding protein 2; LYPD1, LY6/PLAUR domain-containing 1;
PAQR4, AdipoQ receptor 4; MGAT5, alpha-1,6-mannosylglycoprotein
6-beta-N-acetylglucosaminyltransferase; circMAP3K4, circular RNA derived from
mitogen-activated protein kinase kinase kinase 4; PLK1, polo-like kinase 1; MYC,
cellular myelocytomatosis oncogene; IGF1R, IGF1 receptor; SMPDL3A, sphingomyelin
phosphodiesterase acid-like 3A; LRPPRC, leucine rich pentatricopeptide repeat
containing; KDM5B, lysine demethylase 5B; ILF3, interleukin enhancer binding
factor 3; YY1, Yin Yang-1; EMT, epithelial–mesenchymal transition; GLI1,
glioma-associated oncogene homolog 1; EPHA3, EPH receptor A3; YES1, YES
proto-oncogene 1; FEN1, flap structure-specific endonuclease 1; GBAP1,
glucosylceramidase beta pseudogene 1; LncRNA ARHGAP5-AS1, lncRNA Rho GTPase
activating protein 5-AS1; LncRNA TRPC7-AS1, lncRNA transient receptor potential
cation channel, subfamily C, member 7-AS1; EIF5B, eukaryotic translation
initiation factor 5B; E2F1, E2F transcription factor 1; NRF2, nuclear factor
erythroid 2-related factor 2; CCND1, cyclin D1; TRAF5, TNF receptor-associated
factor 5; DIAPH3, diaphanous related formin 3; BMI1, BMI1 proto-oncogene; RNF2,
ring finger protein 2; PD-L1, programmed death-ligand 1; RRM2, ribonucleotide
reductase regulatory subunit M2; CircCCAR1, circular cell cycle and apoptosis
regulator 1; SLC7A11, solute carrier family 7 member 11; CCL5, C-C motif
chemokine ligand 5; TGF-
Beyond their well-characterized m6A reader activity, IGF2BPs also exhibit m6A-independent RNA-binding functions that contribute to HCC progression. IGF2BP3 binds circ_0098823, supporting its regulatory influence on dynamin 1-like and HCC cell metastasis [128]. Moreover, IGF2BP3 participates in the formation of the miR-191-5p–AGO2 RISC, which suppresses tight junction protein 1 expression and contributes to HCC invasiveness [129]. Additionally, IGF2BP3 interacts with the flanking intronic regions of circCOCH, promoting assembly of the circCOCH–miR-4550a–AGO2 RISC complex, which is associated with HCC cell proliferation and migration [130]. These findings highlight m6A-independent RNA-binding as an additional mechanism by which IGF2BP3 promotes HCC progression.
IGF2BPs are aberrantly expressed across multiple malignancies, including hepatobiliary cancers, and have been implicated in processes such as cell proliferation, migration, metabolic reprogramming, and therapy resistance, suggesting potential as diagnostic biomarkers and therapeutic targets. Accumulating evidence supports their evaluation as diagnostic or prognostic markers in various cancers. For example, IGF2BP1 and IGF2BP3 have been proposed as biomarkers for high-risk colorectal cancer populations and patient monitoring [131], whereas IGF2BP3 is associated with metastatic risk in renal clear cell carcinoma and has been implicated in triple-negative breast cancer [132, 133]. Additionally, serum antibody detection against IGF2BPs shows promise for cancer screening; IGF2BP2 autoantibodies are detectable in a subset of patients with HCC but not in precancerous conditions such as chronic hepatitis or liver cirrhosis [134], and IGF2BP1 antibodies may assist in the diagnosis of esophageal squamous cell carcinoma and colorectal cancer [135, 136]. Combining IGF2BP antibody assays with conventional tumour markers could improve diagnostic performance.
Therapeutic strategies targeting IGF2BPs are being explored due to their multifaceted oncogenic functions. Current approaches include small-molecule inhibitors, oligonucleotide-based therapies, and natural compounds. Small-molecule inhibitors of IGF2BP1 include compound 2-[(8-bromo-5-methyl-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio]-N-(1-phenylethyl)acetamide (C20H18BrN5OS, ChemBridge ID: 6896009), compound 7773, and the allosteric inhibitor BTYNB. C20H18BrN5OS interferes with IGF2BP1 binding to MYC mRNA, which is associated with reduced downstream signaling [137]; compound 7773 competes for the KH3-4 domains of IGF2BP3, limiting target mRNA interaction and modulating non-small-cell lung cancer and ovarian clear cell carcinoma progression [138]. BTYNB alters IGF2BP1 structural function to limit downstream gene expression, restraining ovarian cancer and melanoma cell proliferation and exhibiting tumour-suppressive activity in ICC patient-derived xenograft models [122, 139]. IGF2BP2 inhibitors include JX5, CWI1-1, CWI1-2, and certain benzoylbenzoic acid and thiourea derivatives (e.g., compounds 4, 6, 9), which target the KH3-4 domains to block mRNA binding and demonstrate antitumour effects in leukemia, colorectal cancer, and HCC models [139, 140, 141]. While no specific IGF2BP3 inhibitors are available yet, BTYNB has been reported to simultaneously downregulate IGF2BP1 and IGF2BP2, suggesting potential cross-family effects that require further investigation [141].
Oligonucleotide-based therapies represent another approach for IGF2BP modulation. Antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), miRNAs, and aptamers can disrupt IGF2BP interactions with mRNA [142]. ASOs complementary to IGF2BP1 recognition sequences interfere with its binding to MYC or cluster of differentiation 44 mRNA, reducing target gene expression [143, 144]. Structured oligonucleotide S1 binds specific protein conformations to interfere with IGF2BP1–GLI1 interactions [145]. siRNAs directly targeting IGF2BP expression have demonstrated antitumour effects across multiple cancer types [142].
Several natural compounds reportedly modulate IGF2BP expression, including isoliquiritigenin, berberine, isocorydine derivative (d-ICD), nitidine chloride, Tamarix articulata, quercetin, and epigallocatechin gallate [146, 147, 148]. Notably, d-ICD downregulates IGF2BP3 to reverse HCC chemoresistance, and nitidine chloride reduces IGF2BP3-mediated m6A modification of metabolism-related targets [149]. Additionally, Tamarix articulata, quercetin, and epigallocatechin gallate inhibit IGF2BP1/3 expression via miR-1275 upregulation, which may suppress tumour progression [150]. Traditional Chinese medicine Cucurbitacin B has also been reported to target IGF2BP1 in PLC treatment [151].
Despite promising preclinical data, clinical translation of IGF2BP-targeted therapies faces challenges. Broad tissue expression complicates the definition of “aberrant expression”, and most inhibitors remain at cellular or animal model stages, necessitating further validation of efficacy and safety in humans. Systematic functional studies and rigorous pharmacological evaluation are therefore required to advance IGF2BPs as therapeutic targets, although their potential for precision diagnostics and therapy in hepatobiliary malignancies is increasingly recognized.
The IGF2BP family (IGF2BP1/2/3) is increasingly recognized for its role in hepatobiliary tumour biology, participating in both m6A-dependent and m6A-independent RNA regulatory networks that influence proliferation, metastasis, immune evasion, metabolic adaptation, and therapy resistance. Through interactions with diverse ncRNAs and stabilization of oncogenic transcripts, IGF2BPs contribute to complex signaling cascades that support tumour progression and modulate the tumour microenvironment.
Despite substantial progress in understanding IGF2BP functions, several important gaps remain. Isoform-specific roles, context-dependent regulation, interactions with other epitranscriptomic regulators, and heterogeneity of IGF2BP expression across tumour subtypes and patient populations are not yet fully characterized, which limits the direct translation of preclinical findings into clinical applications.
Future research should aim to address these gaps by: (i) clarifying upstream signals and structural determinants that influence IGF2BP activity; (ii) integrating multi-omics approaches to map IGF2BP-regulated networks during tumour evolution; (iii) developing selective IGF2BP inhibitors and ncRNA-targeted delivery systems; and (iv) assessing IGF2BPs as biomarkers for patient stratification and therapeutic response in clinical studies.
Overall, IGF2BPs represent functionally significant nodes within the RNA regulatory landscape in hepatobiliary cancers, providing opportunities for more targeted interventions and potentially informing personalized therapeutic strategies.
TP conceived and designed the overall study, oversaw the literature collection and content organization, critically revised the manuscript, and performed the final review. ZY, QL, PX, CJ, and JS collected and analyzed the literature and prepared the figures and tables. ZY, QL, PX, CJ, and JS drafted the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the National Natural Science Foundation of China, grant number 82172792, and the Outstanding Youth Funds of the First Affiliated Hospital of Harbin Medical University, grant number HYD2021J11.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.