







 , Dae Sung Yoo 2,3,†, Sang Hee Park 2, Dong Seon Kim 1, Si Eun Yoon 2, Ji Hye Yoon 2, Jongsung Lee 1,2, Ji Hye Kim 1,*
, Dae Sung Yoo 2,3,†, Sang Hee Park 2, Dong Seon Kim 1, Si Eun Yoon 2, Ji Hye Yoon 2, Jongsung Lee 1,2, Ji Hye Kim 1,* , Jae Youl Cho 1,2,*
, Jae Youl Cho 1,2,*1 Department of Integrative Biotechnology, Sungkyunkwan University, 16419 Suwon, Gyeonggi-do, Republic of Korea
2 Department of Biocosmetics, Sungkyunkwan University, 16419 Suwon, Gyeonggi-do, Republic of Korea
3 ASK Company Co., Ltd., 41256 Daegu, Republic of Korea
†These authors contributed equally.
Abstract
Araliadiol, a triterpenoid compound isolated from Centella asiatica, exhibits diverse biological activities, including anti-cancer, neuroprotective, and hair growth-promoting properties. However, its protective effects against skin damage caused by environmental pollutants, such as urban particulate matter (UPM), remain unexplored. Given the critical role of oxidative stress in UPM-induced cellular damage, we investigated the potential of araliadiol as a dermoprotective agent and explored its underlying molecular mechanisms.
The stability of araliadiol was evaluated at various temperature conditions and solvent conditions using high-performance liquid chromatography (HPLC). To explore the biological functions and signaling pathways affected by araliadiol, bioinformatic analyses including Gene Ontology (GO) enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, and Monarch phenotype analysis were performed. Cellular responses to araliadiol were assessed in HaCaT and HEK293T cells by measuring reactive oxygen species (ROS) levels and transcription of antioxidant genes. Activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and activator protein-1 (AP-1) signaling pathway was further examined using quantitative polymerase chain reaction (PCR), luciferase assay, western blotting, and immunofluorescence staining. The interaction between araliadiol and mitogen-activated protein kinase kinase 7 (MKK7) was investigated through molecular docking and cellular thermal shift assay (CETSA). DNA damage and apoptosis were examined using the comet assay, γ-H2AX staining, Annexin V/PI flow cytometry, and protein expression analysis.
Araliadiol significantly reduced intracellular levels of ROS by upregulating key antioxidant genes, including HO-1, NQO1, TXNRD1, GCLC, and GCLM. Mechanistically, araliadiol promoted the expression and nuclear translocation of Nrf2, a master transcription factor involved in antioxidant defense. In parallel, araliadiol selectively activates the c-Jun N-terminal kinase (JNK)–AP-1 signaling cascade by directly binding to and activating MKK7, an upstream kinase involved in oxidative stress responses. Given the close association between oxidative stress, DNA damage, and apoptosis, we further investigated the protective capacity of araliadiol in this context. Araliadiol markedly attenuated UPM-induced DNA damage and apoptosis, as evidenced by reduced comet tail formation, decreased γ-H2AX levels, a lower proportion of Annexin V-positive cells, and modulation of apoptosis-related proteins. Meanwhile, although UPM exposure induced the expression of specific antioxidant-associated genes (TXNRD and GCLC), HO-1 protein expression, and AP-1 signaling, it failed to activate Nrf2 transcriptional activity. Instead, UPM exposure resulted in elevated intracellular ROS accumulation and increased DNA damage.
Our findings suggest that UPM exposure alone elicited limited stress-adaptive antioxidant responses without effective cytoprotection. In contrast, araliadiol treatment independently activated robust antioxidant and cytoprotective signaling. Moreover, under UPM exposure, araliadiol further enhanced cellular defense through the activation of the Nrf2 and JNK–AP-1 signaling pathways. These results highlight the therapeutic potential of araliadiol as a dermoprotective agent derived from Centella asiatica, particularly in mitigating pollutant-induced skin damage.
Keywords
- Centella asiatica
- particulate matter
- keratinocytes
- oxidative stress
- nuclear factor erythroid 2-related factor 2
- apoptosis
Urban particulate matter (UPM) is widely recognized as a major environmental factor affecting skin health. Both animal and human studies have demonstrated that exposure to particulate matter (PM) disrupts the skin barrier and elevates oxidative stress through the activation of mitogen-activated protein kinase (MAPK) signaling pathways, leading to increased DNA damage [1, 2]. Furthermore, PM exposure has been shown to upregulate inflammatory mediators and stimulate T helper 17 (TH17) cell differentiation as well as interleukin-7 (IL-7) signaling pathways via activation of the aryl hydrocarbon receptor (AhR) [3, 4]. In keratinocytes, PM induces cell death by modulating the expression and activity of proteins that regulate apoptosis. Collectively, these complex molecular mechanisms link UPM exposure to the exacerbation of inflammatory skin disorders including allergic dermatitis, atopic dermatitis, and psoriasis, as well as to accelerated skin aging [5].
In response to the harmful effects of UPM on the skin, there is increasing
interest in therapeutic approaches utilizing natural compounds with antioxidant
and anti-inflammatory properties. For instance, the citrus flavonoid hesperidin
has been reported to inhibit PM-induced cell death by suppressing oxidative
stress and autophagy [6]. It also confers protection against mitochondrial
damage, cell cycle arrest, and cellular senescence [7]. Similarly, flavonoids
such as catechin, known for their potent antioxidant and anti-inflammatory
activities, have demonstrated protective effects against skin damage caused by
fine dust particles (FDP). In human dermal fibroblasts (HDFs), catechin not only
scavenges reactive oxygen species (ROS) but also maintains collagen synthesis and
inhibits matrix metalloproteinase (MMP) secretion through the regulation of
NF-
Interestingly, signaling pathways activated by UPM exposure appear to exert context-dependent effects in skin cells. Excessive ROS generation induced by UPM primarily activates pro-oxidant and pro-inflammatory signaling cascades, including MAPK-driven AP-1 activation, which is widely associated with the transcriptional induction of MMPs and pro-inflammatory cytokines [5]. In contrast, under certain adaptive conditions, AP-1-MAPK signaling has also been reported to participate in antioxidant defense responses, particularly through interactions with the nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) pathway, leading to the induction of cytoprotective genes [10]. Importantly, AP-1 is a highly context-dependent transcription factor, and its functional outcomes are influenced by factors such as subunit composition, upstream signaling strength, and cellular redox status. Accordingly, while AP-1 activation is classically linked to pro-aging and pro-inflammatory processes, it may also contribute to stress-adaptive responses under specific conditions [11].
Centella asiatica (L.) Urb., an edible and medicinal herb long recognized for its wound-healing and anti-inflammatory properties, has gained renewed attention through a growing interest in plant-derived bioactive compounds for therapeutic use. This herb is rich in phytochemicals, including saponins, flavonoids, and triterpenoids, which contribute to its diverse pharmacological effects [12]. Among these bioactive constituents, araliadiol, a pentacyclic triterpenoid, has emerged as a promising molecule owing to its notable antioxidant and anti-inflammatory activities [13]. However, despite increasing evidence uncovering the deleterious effects of UPM on skin health and the protective potential of various natural compounds, the impact of araliadiol in UPM-induced skin damage remains insufficiently understood.
In our study, bioinformatics analysis revealed that araliadiol treatment is significantly associated with antioxidant signaling pathways, MAPK signaling pathways, and skin-related phenotypes. Based on these findings, we focused our attention on the MAPK and antioxidant signaling pathways. Among antioxidant signaling mechanisms, the Nrf2 pathway is the most well-characterized. Under oxidative stress conditions, cells activate Nrf2-centered antioxidant signaling to enhance the transcriptional regulation of antioxidant-related genes, such as HO-1, NQO1, GCLC, and GCLM [14, 15], thereby facilitating reactive oxygen species ROS clearance and restoration of cellular homeostasis. Notably, previous in vitro studies have demonstrated a functional relationship between MAPK and Nrf2 pathways. For instance, in astrocytes, specific inhibition of MAPK pathways revealed that c-Jun N-terminal kinase (JNK) mediates HO-1 upregulation via Nrf2/ARE [16]. Another study reported that 5-aminolevulinic acid protects murine proximal tubular epithelial cells from cyclosporin A-induced apoptosis by activating the Nrf2 antioxidant pathway through MAPK signaling [17]. Importantly, AP-1 components downstream of MAPK signaling can exert context-dependent roles while AP-1 activation is classically associated with pro-aging and pro-inflammatory outcomes such as MMP induction, emerging evidence suggests that AP-1 may also cooperate with antioxidant signaling under specific stress conditions, thereby contributing to adaptive cellular defense [18]. Therefore, we prioritized investigating the MAPK-Nrf2 axis to evaluate the protective efficacy of araliadiol under UPM-stimulated conditions.
Our study aims to explore the therapeutic potential of araliadiol against oxidative stress-related skin damage induced by UPM. Using human keratinocyte HaCaT cells exposed to UPM, we systematically investigated the protective effects of araliadiol. Specifically, we evaluated its impact on ROS generation, DNA damage, apoptosis markers, and the modulation of key signaling pathways, including Nrf2 and MAPK. To our knowledge, this is the first comprehensive study to assess araliadiol’s ability to mitigate UPM-induced cellular damage through regulation of oxidative stress, DNA repair, apoptosis, and associated signaling cascades, thus providing new insights into its potential as a skin-protective agent.
The Centella asiatica used in this study was obtained from a registered cultivar, BT-care, cultivated under controlled conditions at the smart farming facilities of ASK Base on Jeju Island, Korea. To preserve its bioactive constituents, freshly harvested Centella asiatica was frozen immediately at –80 °C using a rapid freezer (DF8530; Ilshin Biobase, Seoul, Korea). The frozen plant material was then subjected to freeze-drying with a lyophilizer (FDS 8518; Ilshin Biobase, Seoul, Korea) to remove moisture while maintaining its structural integrity and biochemical properties [19]. The dried samples were then stored under refrigeration until further analysis.
HPLC was used to assess the stability and detection of araliadiol under five different storage conditions: 45 °C, RT, 4 °C, –20 °C, and –80 °C. Analyses were conducted in both the presence and absence of methanol as a solvent. For sample preparation, araliadiol was either dissolved in methanol or analyzed in a solvent-free state, depending on the experimental design. Each sample was filtered through a 0.45-µm syringe filter to eliminate particulates, and a 10 µL volume was injected into the HPLC system.
Separation was performed using a TSKgel ODS-100V column (4.6 mm
The SMILES representation of araliadiol was collected from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and analyzed using Swiss Target Prediction (http://www.swisstargetprediction.ch/result.php?job=1721683916&organism=Homo_sapiens) to identify potential target genes. To identify antioxidant-related genes associated with araliadiol, the predicted targets were compared with a list of known antioxidant-related genes obtained from Gene Cards (https://www.genecards.org/), and a Venn diagram was generated to visualize the overlapping gene sets. Additionally, Gene Ontology (GO) enrichment analysis [20], Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and Monarch phenotype were conducted using R programming to gain insights into the biological processes and signaling pathways associated with predicted targets of araliadiol [21].
To assess the effects of araliadiol on oxidative stress, DNA damage, and apoptosis, we established in vitro models using HaCaT and HEK293T cells. HaCaT cells were selected for their relevance in skin biology, particularly for studying oxidative-stress related skin damage, as UPM induces ROS predominantly in these cells. HaCaT cells provide a suitable model for evaluating the antioxidant and DNA repair properties of bioactive compounds under oxidative stress conditions. HEK293T cells, which have high transfection efficiency, were used to perform a luciferase reporter assay to analyze the regulatory effects of araliadiol on gene expression and related signaling pathways.
HEK293T cells were obtained from the Korean Cell Line Bank (Seoul, Korea), and HaCaT cells were purchased from Cytion (Eppelheim, Germany). Cell line identity was confirmed based on supplier authentication and by reference to the International Cell Line Authentication Committee (ICLAC) database and the ExPASy Cellosaurus, which list no known misidentification or cross-contamination for these cell lines. Both suppliers provide short tandem repeat (STR) profiling data as part of their cell line authentication and quality control procedures. The authenticated cell lines were used directly in this study. All cell lines were routinely tested for mycoplasma contamination and confirmed to be negative.
Both HaCaT and HEK293T cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, Thermo Fisher Scientific, Grand Island, NY, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Gibco, Waltham, MA, USA) and 1% (v/v) penicillin-streptomycin antibiotic solution (Gibco) to maintain sterility and prevent bacterial contamination. To optimize cellular viability and experimental reproducibility, all cultures were inoculated under controlled conditions at 37 °C in a humidified atmosphere with 5% CO2, ensuring a mycoplasma and parasite-free environment.
UPM (SRM 1648a; NIST, Gaithersburg, MD, USA), consisting predominantly of particles within the 1.35 to 30.1 µm size range and a median diameter of 5.85 µm, was used in this study. UPM powder was suspended in PBS at a stock concentration of 100 mg/mL and sonicated for 20 minutes to ensure uniform dispersion. Immediately before use, the stock solution was diluted to a final working concentration of 100 µg/mL and applied to cultured cells, with the volume of culture medium per well specified in the corresponding experimental sections. This concentration was selected based on previous in vitro studies demonstrating reliable induction of oxidative stress responses in skin-related cell models [22].
Cell viability was evaluated using the MTT assay. HaCaT cells (5
Intracellular ROS levels were assessed using the fluorescent probe H2DCFDA
(D6883; Sigma-Aldrich, St. Louis, MO, USA). HaCaT cells were seeded in six-well
plates at a density of 3
Total RNA was extracted from HaCaT cells using TRIzol reagent according to the
manufacturer’s instructions. HaCaT cells were seeded into six-well plates (2 mL
of medium per well) at a density of 3
| Gene Name | Sequence (5′-3′) |
| NQO1 | Forward: TCACCGAGAGCCTAGTTCC |
| Reverse: CTGAGTGAGCCAGTACGATCA | |
| GCLC | Forward: TCGCAAACCATCCTGACTACA |
| Reverse: TCCAAGTAACTCTGGGCATTC | |
| SOD1 | Forward: CCTCTATCCAGAAAACACGG |
| Reverse: CAATGATGCAATGGTCTCCT | |
| HO-1 | Forward: CCAGGCAGAGAATGGTCCTG |
| Reverse: GGCGAAGACTGGGCTCTC | |
| TXNRD | Forward: GCAAGAAGGTGATGGTCCTG |
| Reverse: CTTGCAGGGCTTGTCCTAAC | |
| GCLM | Forward: GGGGAACCTGCTGAACTCT |
| Reverse: TCTGGGTTGATTTGGGAACT | |
| GAPDH | Forward: GACAGTCAGCCGCATCTTCT |
| Reverse: GCGCCCAATACGACCAAATC |
To evaluate the transcriptional activity of antioxidant-related pathways, a
luciferase reporter assay was conducted using HEK293T cells. HEK293T cells were
seeded at a density of 2.5
(1) Araliadiol-only treatment:
Cells were treated with araliadiol (2.5 or 5 µM) for 24 h.
(2) Araliadiol + UPM treatment:
Cells were pretreated with araliadiol (2.5 or 5 µM) for 30 min, followed by exposure to UPM (100 µg/mL) for 24 h.
(3) SP600125 + araliadiol + UPM treatment:
Cells were pretreated with SP 600125 (10 µM) for 30 min, then treated with araliadiol (5 µM) for 30 min, and subsequently exposed to UPM (100 µg/mL) for 24 h.
After treatment, luciferase activity was measured using a luciferase reporter assay system [25]. The relative luciferase activity was calculated to determine the regulatory effects of araliadiol on Nrf2 and AP-1-mediated transcription.
To analyze protein expression, HaCaT cells were cultured in six-well plates (2
mL of medium per well) at a density of 3
To evaluate the cellular localization of key proteins, immunofluorescence
staining was performed on HaCaT cells. Cells were seeded in 12-well plates (1 mL
of medium per well) at a density of 4
The three-dimensional (3D) structure of mitogen-activated protein kinase kinase 7 (MKK7) in its active conformation was obtained from the Protein Data Bank (https://www.rcsb.org/), while the molecular structure of araliadiol was collected from PubChem. The docking process was executed using DiffDock’s transformer-based prediction model, which evaluates potential binding interactions by incorporating protein flexibility and ligand orientation dynamics [28].
HaCaT cells were transfected with Myc-MKK7 for 24 hours, then treated with araliadiol or DMSO as a control for an additional 24 hours, followed by harvesting in ice-cold PBS supplemented with protease and phosphatase inhibitors. The collected cells were subjected to heat shock by gradually increasing temperatures for 3 minutes. The samples were lysed via repeated freeze–thaw cycles, and the soluble protein fractions were isolated by centrifugation. Equal amounts of the supernatant were analyzed by western blotting [29].
HaCaT cells were cultured in six-well plates (2 mL of medium per well) at a
density of 3
Quantitative data were analyzed using GraphPad Prism (version 10; GraphPad
Software, San Diego, CA, USA) and SigmaPlot (Systat Software, San Jose, CA, USA).
Data are presented as the mean
The aerial parts of Centella asiatica (2 kg) were extracted twice with 70% aqueous methanol, yielding a crude extract that was partitioned with hexane. The hexane-soluble fraction was subjected to silica gel column chromatography. Among the fractions collected, H-3 displayed the most prominent bioactivity. Further purification of H-3 using preparative reversed-phase MPLC and Sephadex LH-20 column chromatography led to the isolation of a single compound (Fig. 1A). A final MPLC purification step yielded 28 mg of a triterpenoid compound. This compound was identified as araliadiol based on its spectroscopic characteristics, which were consistent with previously reported values [19].
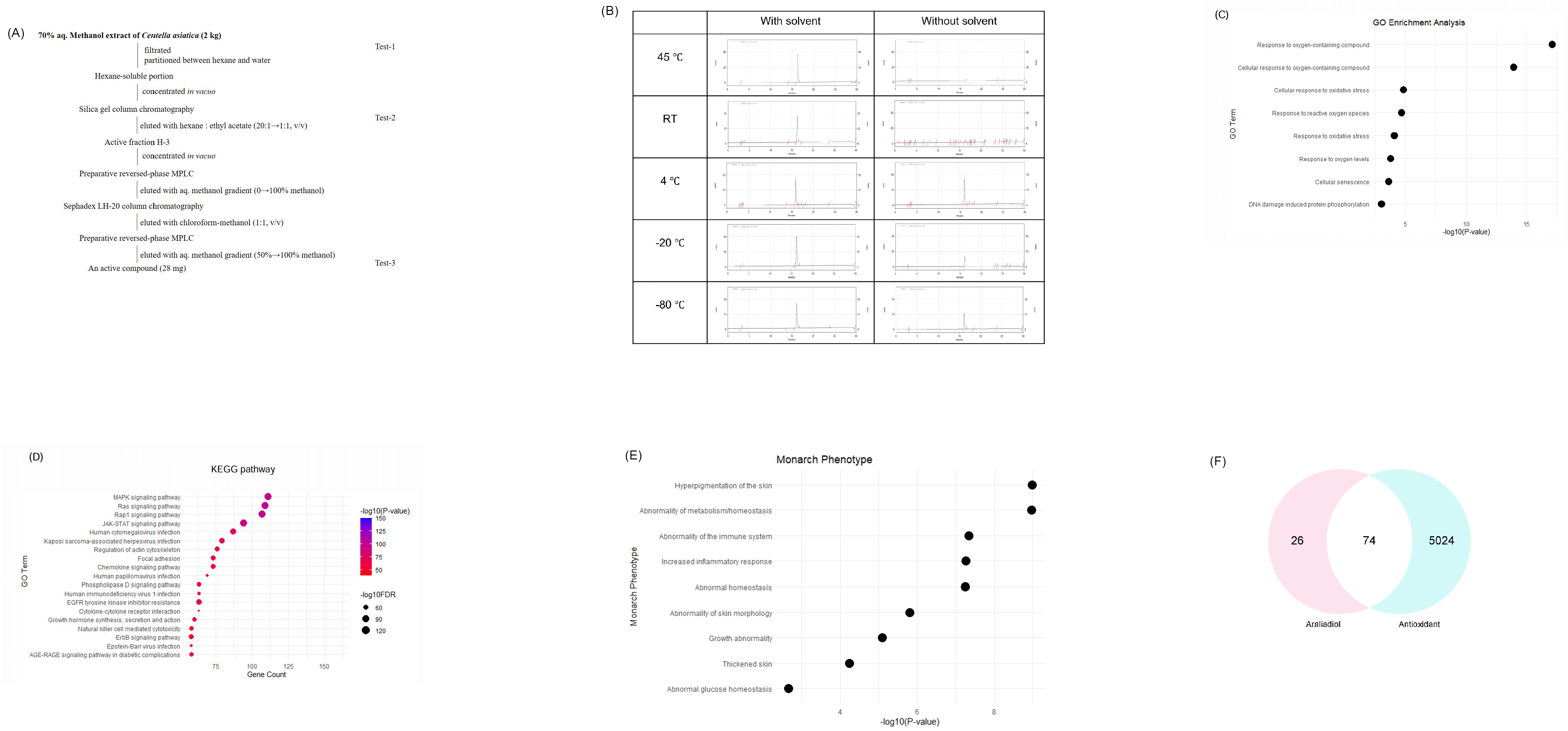 Fig. 1.
Fig. 1.
Extraction and bioinformatic characterization of araliadiol isolated from Centella asiatica. (A) Schematic diagram illustrating the process used to isolate araliadiol from Centella asiatica. (B) Stability profile of araliadiol under diverse temperatures (45 °C, RT, 4 °C, –20 °C, and –80 °C) with or without methanol. HPLC chromatograms (x-axis: minutes; y-axis: mAU) were used to evaluate the chemical stability and quantify the remaining compound over time. (C) GO enrichment analysis of araliadiol-regulated genes. The dot plot illustrates the top significantly enriched GO terms. The x-axis represents –log10 (adjusted p-value) and the y-axis lists the significantly enriched GO term. (D) KEGG pathway analysis of differentially expressed genes following araliadiol treatment. The dot plot presents the top enriched pathways, ranked by statistical significance. (E) Monarch phenotype analysis of araliadiol-associated genes. The dot plot illustrates the top phenotypic terms significantly associated with the gene set, with enrichment ranked by adjusted p-value. (F) Venn diagram illustrating the intersection between predicted araliadiol target genes and established antioxidant-related genes, highlighting 74 shared gene targets. HPLC, high-performance liquid chromatography; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
To assess the stability of araliadiol, HPLC analysis was further conducted under five different storage conditions: 45 °C, room temperature (RT), 4 °C, –20 °C, and –80 °C. Samples were analyzed in both methanol and solvent-free states. HPLC was performed using a TSK gel ODS-100V column with a methanol–water gradient at 25 °C. The retention times and peak intensities of araliadiol in methanol-stored samples remained consistent across all temperature conditions (Fig. 1B). This indicates chemical stability over a wide temperature range when stored in methanol. In contrast, solvent-free samples retained normal retention times and peak intensities only when stored at 4 °C, suggesting limited stability in the absence of solvent (Fig. 1B).
The potential molecular mechanisms underlying the biological activity of araliadiol were explored through bioinformatics analyses, including GO, KEGG, and phenotype enrichment. GO enrichment analysis revealed that araliadiol may significantly influence oxidative stress-related biological processes. The top-enriched GO terms included response to oxygen-containing compound, cellular response to oxidative stress, response to reactive oxygen species, and response to oxidative stress, suggesting that araliadiol may be involved in the activation of antioxidant defense mechanisms (Fig. 1C). Additionally, GO terms such as response to oxygen levels, cellular senescence, and DNA damage-induced protein phosphorylation were also enriched, although with relatively lower statistical significance, implying that araliadiol may be associated with a broader cellular stress response, including ROS-mediated DNA damage (Fig. 1C). KEGG pathway analysis discovered that araliadiol was associated with multiple signaling pathways, notably MAPK and Ras signaling pathways (Fig. 1D). These pathways play critical roles in cellular processes, such as survival and stress adaptation [31]. Monarch phenotype analysis indicated that araliadiol-regulated genes are associated with skin-related phenotypic traits, including hyperpigmentation, abnormal skin morphology, and increased inflammatory response, thereby underscoring the potential dermatological relevance of araliadiol (Fig. 1E). Additionally, Venn diagram analysis comparing araliadiol-regulated genes with known antioxidant-associated genes identified 74 overlapping targets (Fig. 1F). This suggests that araliadiol may exert its antioxidant effects by acting on convergent molecular targets involved in oxidative stress regulation. Collectively, these findings highlight the therapeutic potential of araliadiol in alleviating oxidative stress-induced damage and preserving skin homeostasis.
Before analyzing the efficacy of araliadiol, we evaluated its cytotoxicity in both HaCaT and HEK293T cells. In HaCaT cells, araliadiol exhibited no cytotoxic effects up to a concentration of 40 µM (Fig. 2A), while in HEK293T cells, no cytotoxicity was observed up to 10 µM (Fig. 2B). To investigate whether araliadiol attenuates oxidative stress, intracellular ROS levels were measured using a 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) assay in HaCaT cells. UPM, a major component of air pollutants, has been reported to cause oxidative stress-mediated skin damage [32]. Consistent with previous reports, UPM exposure markedly increased intracellular ROS levels (Fig. 2C), confirming its role as an environmental inducer of oxidative stress. However, under UPM-induced oxidative stress conditions, treatment with araliadiol at 2.5 µM and 5 µM effectively suppressed ROS production, demonstrating its strong antioxidative capacity (Fig. 2C). Additionally, fluorescence-activated cell sorting (FACS) analysis following H2DCFDA staining in UPM-exposed cells demonstrated that araliadiol also effectively attenuated UPM-induced intracellular ROS accumulation (Fig. 2D). H2DCFDA-positive cell levels were normalized to the UPM-treated group, which was defined as 100%. Relative to this group, araliadiol reduced the proportion of H2DCFDA-positive cells to 89% at 2.5 µM and to 48% at 5 µM (Fig. 2D). To validate the reliability of the assay system, ascorbic acid (AA), a well-established antioxidant, was included as a reference compound. In both confocal microscopy and flow cytometry assays following H2DCFDA staining, ascorbic acid (5 and 50 µM) significantly suppressed UPM-induced intracellular ROS levels, confirming the robustness of the assay conditions (Fig. 2E,F). Under the same UPM-challenged conditions, araliadiol at 5 µM also lowered ROS levels to a similar extent as ascorbic acid at the same concentration, further supporting the reproducibility and potential antioxidant activity of araliadiol (Fig. 2E,F). Subsequently, we examined the effect of araliadiol on the expression of antioxidant-related genes, including HO-1, NQO1, SOD1, TXNRD, GCLC, and GCLM. Araliadiol treatment alone resulted in a dose-dependent upregulation of all the genes examined (Fig. 2G). UPM exposure alone also increased the expression of certain antioxidant genes, such as TXNRD and GCLC (Fig. 2H), likely reflecting a compensatory cellular defense response to oxidative stress. Under UPM-stimulated conditions, araliadiol treatment robustly induced the expression of multiple antioxidant-genes, including HO-1, NQO1, TXNRD, GCLC, and GCLM (Fig. 2H). Together, these data suggest that araliadiol consistently induces antioxidant-related gene expression across both basal and UPM-stimulated conditions.
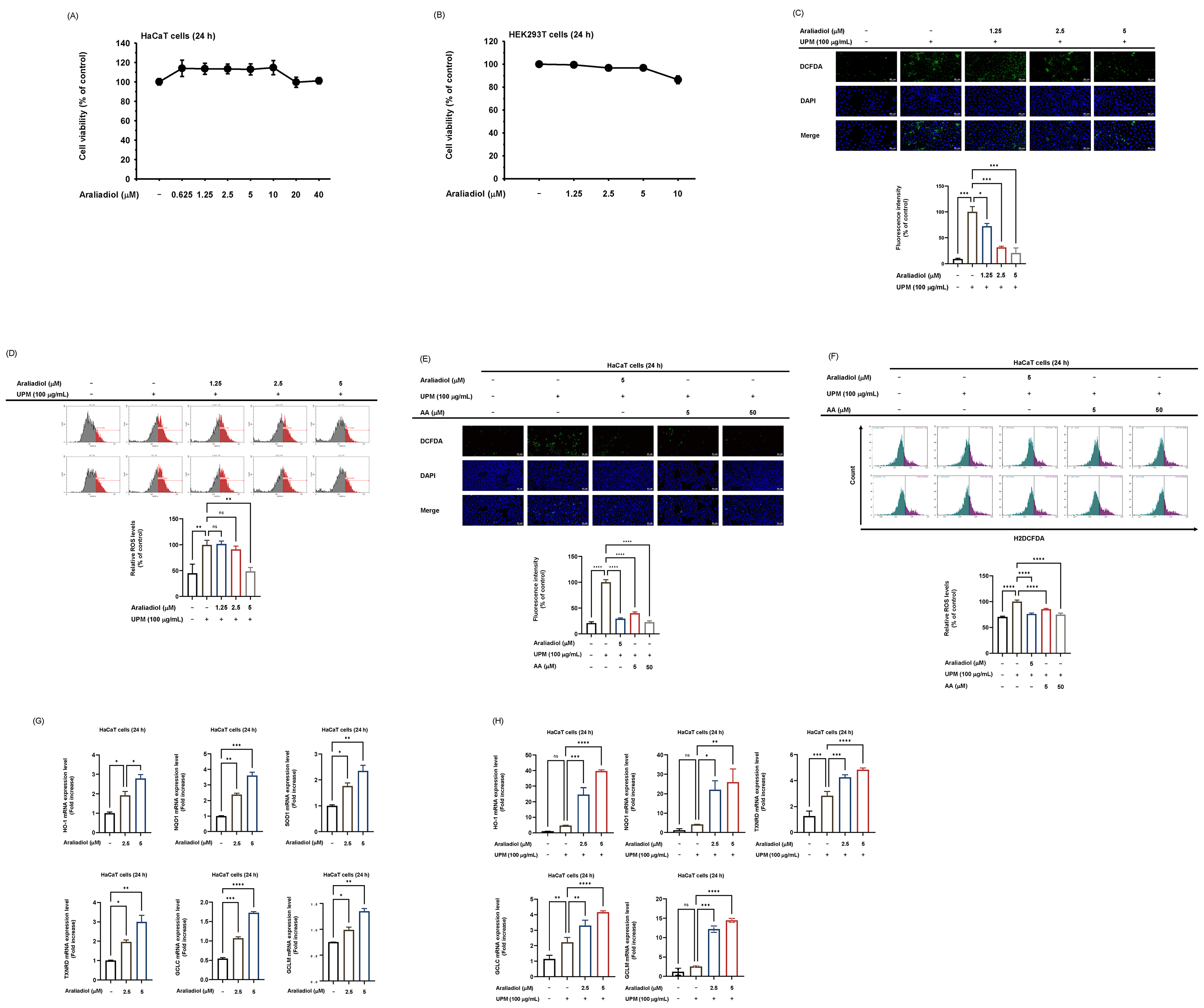 Fig. 2.
Fig. 2.
Antioxidant potential of araliadiol in HaCaT cells exposed to
UPM. (A,B) Cell viability analysis in HaCaT (A) and HEK293T cells (B). Cells
were treated with araliadiol for 24 hours at concentrations ranging from 0 to 40
µM and 0 to 10 µM, respectively. (C,E) Intracellular ROS levels in
HaCaT cells pretreated with araliadiol (0, 1.25, 2.5, and 5 µM) or ascorbic
acid (5, 50 µM) for 30 minutes before exposure to UPM, as assessed by
H2DCFDA staining. Representative fluorescence images and quantitative analyses of
fluorescence intensity are shown. Scale bars = 20 µm (C) and 50 µm
(E). (D,F) Flow cytometry analysis of ROS levels in HaCaT cells with
quantification of H2DCFDA-positive cells. (G,H) (G) qRT-PCR analysis of
antioxidant-related gene expression in HaCaT cells treated with araliadiol (0–5
µM) for 24 hours in the absence of UPM. (H) qRT-PCR analysis of
antioxidant-related gene expression in HaCaT cells pretreated with araliadiol
(0–5 µM) for 30 minutes, followed by UPM exposure for 24 hours. Data
represent means
To elucidate the molecular mechanisms underlying the antioxidant effects of araliadiol, its impact on Nrf2 was evaluated under both basal conditions and UPM-induced oxidative stress. Araliadiol alone significantly enhanced Nrf2-driven luciferase activity at concentrations of 2.5 µM and 5 µM (Fig. 3A). In contrast, UPM treatment alone did not significantly alter Nrf2 luciferase activity, whereas treatment with araliadiol (2.5 µM and 5 µM) under UPM-stimulated conditions led to a marked increase in reporter activity (Fig. 3B).
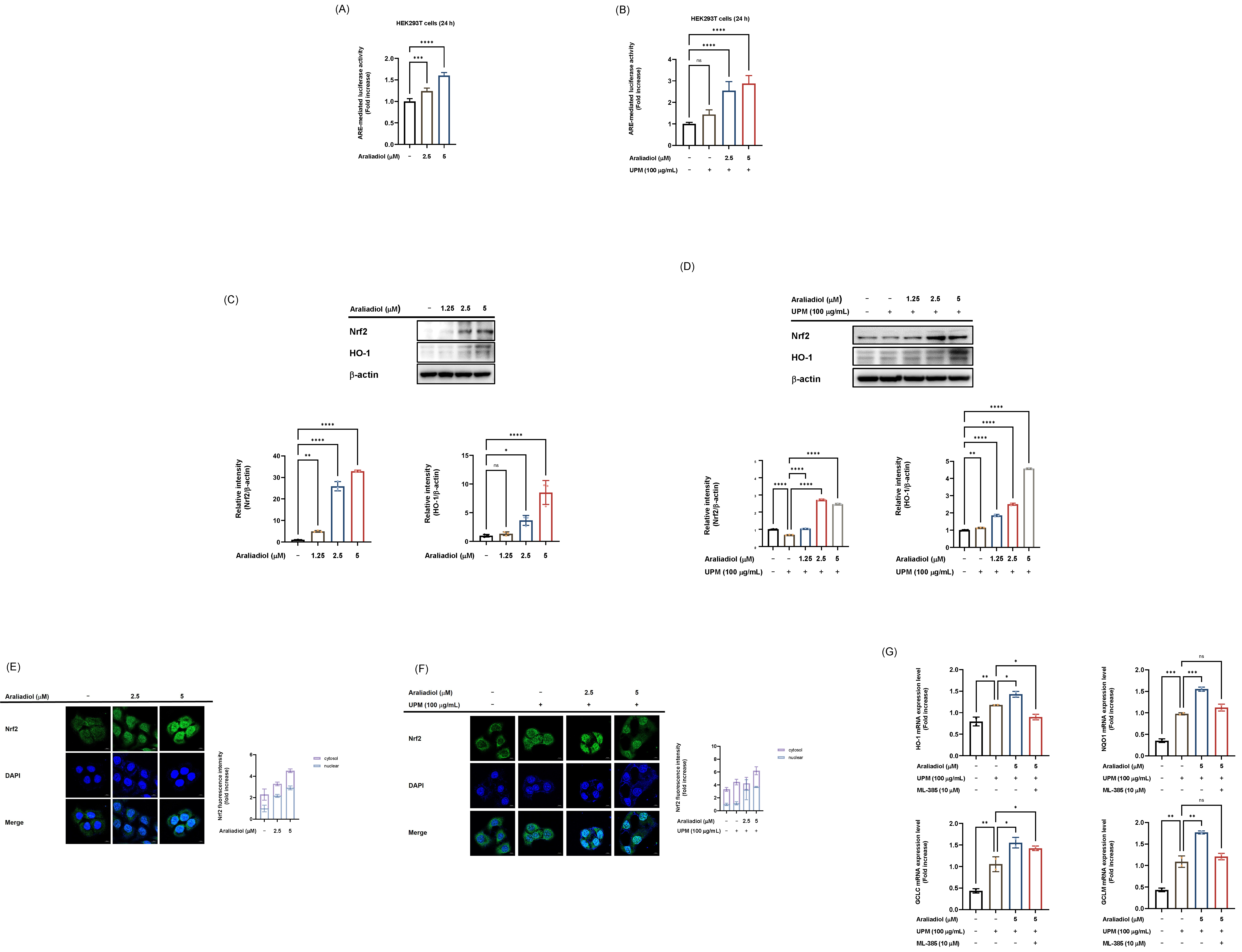 Fig. 3.
Fig. 3.
Activation of the Nrf2 signaling pathway by araliadiol under
basal and UPM-induced oxidative stress conditions. (A) Luciferase reporter assay
measuring Nrf2 transcriptional activity in HEK293T cells treated with araliadiol
(0–5 µM) for 24 hours alone. (B) Luciferase reporter assay measuring Nrf2
transcriptional activity in HEK293T cells pretreated with araliadiol (0–5
µM) for 30 minutes prior to 24 hours UPM exposure. For both assays (A and
B), cells were transfected with a reporter plasmid containing Nrf2 binding sites
upstream of a luciferase gene (Nrf2-luc), along with a
To further investigate Nrf2 activation, western blot analysis was performed to assess the expression of Nrf2 and its downstream target, HO-1. Araliadiol treatment at 2.5 µM and 5 µM significantly elevated the protein levels of both Nrf2 and HO-1, and this effect was maintained under UPM exposure (Fig. 3C,D). In addition, confocal microscopy revealed that araliadiol (2.5 and 5 µM) enhanced the nuclear accumulation of Nrf2 in dose-dependent manner (Fig. 3E). Consistently, under the UPM-stimulated condition, araliadiol treatment (2.5 and 5 µM) further promoted nuclear translocation of Nrf2 (Fig. 3F).
Next, to verify whether araliadiol regulates antioxidant-related genes through the Nrf2 pathway, we employed ML-385, a specific inhibitor of Nrf2. As expected, araliadiol significantly upregulated the expression of antioxidant-related genes, including HO-1, NQO1, GCLC, and GCLM, under UPM exposure; however, this effect was abolished by ML-385 treatment (Fig. 3G), indicating that Nrf2 serves as a critical mediator of araliadiol-induced antioxidant gene expression. Collectively, these findings suggest that araliadiol potentiates Nrf2 activation by upregulating its expression and facilitating nuclear translocation. Collectively, these findings indicate that araliadiol enhances Nrf2 expression and nuclear translocation, and that this Nrf2 activation is preserved under UPM-induced oxidative stress conditions.
Given that Nrf2 activation can be modulated by redox-sensitive transcription factors, including AP-1, we next examined the involvement of AP-1 signaling in the antioxidant effects of araliadiol under basal and UPM-stimulated conditions. Luciferase reporter assays demonstrated that araliadiol at concentrations of 2.5 and 5 µM significantly increased AP-1 transcriptional activity (Fig. 4A). Although UPM stimulation alone could elevate AP-1 activity, co-treatment with araliadiol further enhanced this effect (Fig. 4B). In addition, we assessed the phosphorylation status of AP-1 signaling molecules, including MAPKs (extracellular signal-regulated kinase (ERK), JNK, and p38) as well as AP-1 subunits (c-Jun and c-Fos). Araliadiol (1.25, 2.5, and 5 µM) markedly increased the phosphorylation of JNK and c-Jun, but not of ERK, p38, or c-Fos, under UPM-stimulated conditions (Fig. 4C). Consistently, under UPM exposure, araliadiol markedly enhanced AP-1 luciferase activity, whereas this effect was abolished by the JNK inhibitor SP600125, indicating that araliadiol modulates AP-1 activity via the JNK signaling pathway (Fig. 4D). We also examined the activation of MKK7 and MKK4 as they are well-established upstream regulators of JNK. Araliadiol selectively increased the phosphorylation of MKK7, with no significant change in p-MKK4 levels, suggesting that MKK7 might be a specific upstream target of araliadiol (Fig. 4E). Finally, we verified that araliadiol modulates Nrf2-driven luciferase activity under SP600125 treatment. Consequently, araliadiol completely failed to enhance Nrf2 transcriptional activity when JNK was pharmacologically inhibited, indicating that araliadiol-induced Nrf2 activation is, at least in part, mediated through the JNK pathway (Fig. 4F). Together, these results suggest that araliadiol selectively activates the MKK7–JNK–c-Jun axis to regulate AP-1 activity, and that this signaling cascade contributes to Nrf2 activation particularly under UPM-induced oxidative stress.
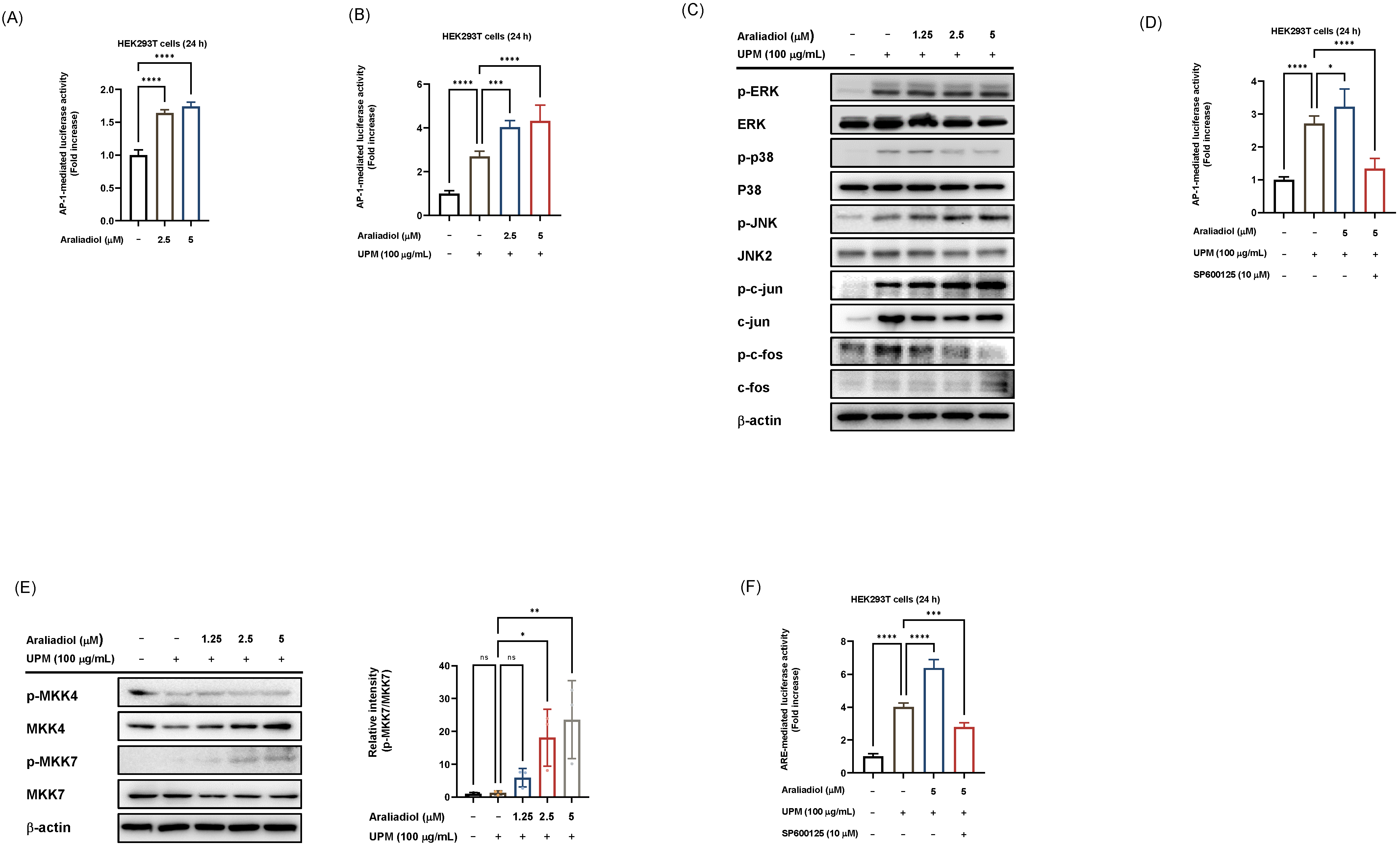 Fig. 4.
Fig. 4.
Effect of araliadiol on AP-1 signal pathways in HaCaT and
HEK293T cells. (A,B,D) Luciferase reporter assays assessing AP-1
transcriptional activity in HEK293T cells transfected with a reporter plasmid
containing AP-1 binding sites upstream of a luciferase gene (AP-1 luc), along
with a
To confirm a direct interaction between araliadiol and MKK7, CETSA was
conducted. The results showed that araliadiol increased the thermal stability of
MKK7 compared to the DMSO control, suggesting direct binding (Fig. 5A). To
further investigate the molecular basis of interactions between araliadiol and
MKK7, we performed molecular docking studies using MKK7 as the target protein and
three compounds: araliadiol, pyrazolopyrimidine (an MKK7 inhibitor), and
anisomycin (an MKK7 activator) [33]. The docking analysis revealed that
araliadiol binds within ATP-binding cleft of MKK7, overlapping with the binding
regions of both pyrazolopyrimidine and anisomycin (Fig. 5B–D). However,
araliadiol and anisomycin were predicted not to directly interact with the key
auto-inhibitory residue Cys218 (C218), but rather to localize near the
glycine-rich loop and
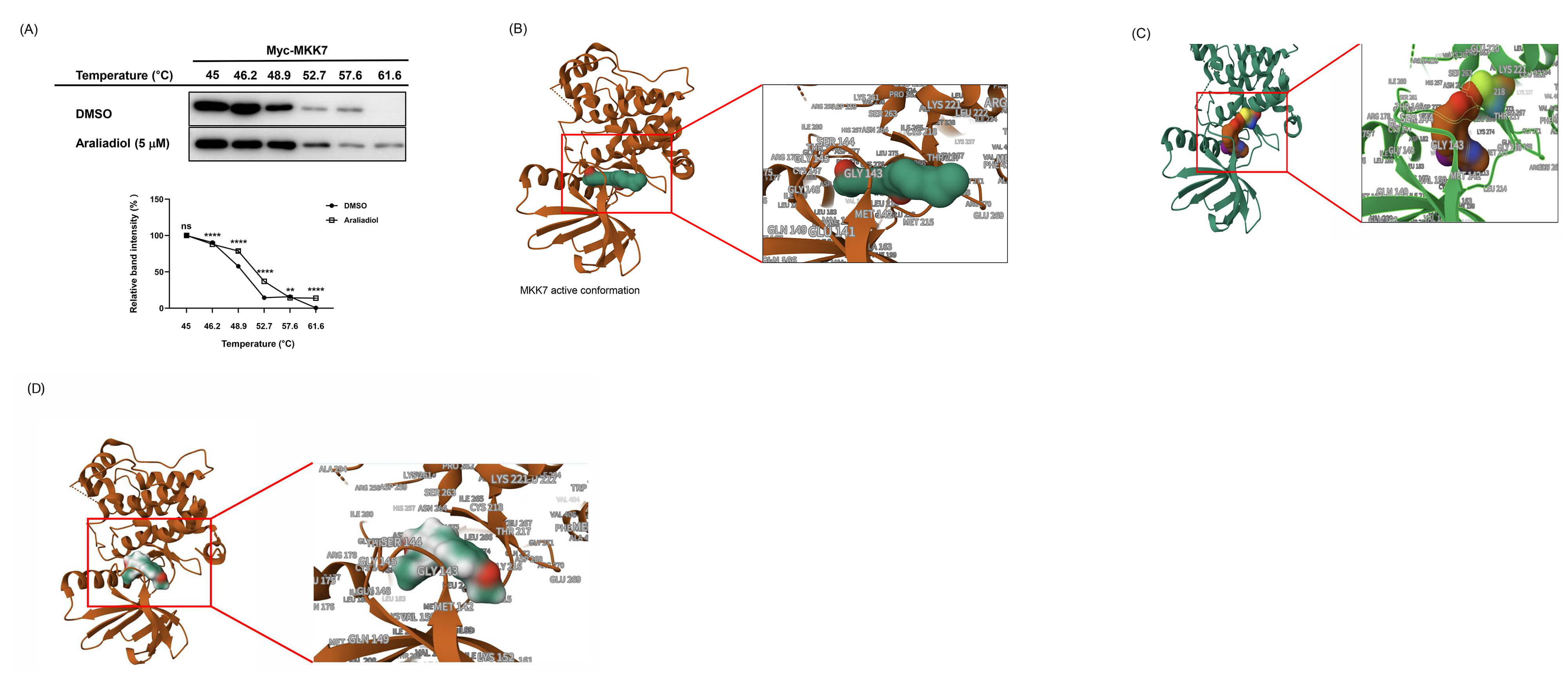 Fig. 5.
Fig. 5.
Araliadiol-MKK7 binding via S263 rather than C218. (A) CETSA is
evaluating the thermal stability of Myc-tagged MKK7 in the presence of araliadiol
or DMSO. HaCaT cells were treated with araliadiol (5 µM) or DMSO and
subjected to a temperature gradient (45–61.6 °C), followed by western
blot analysis. MKK7 band intensity was measured and quantified using ImageJ.
(B–D) Molecular docking analysis showing predicted binding interactions between
araliadiol (green) and the active conformation of MKK7 (brown) (B), between
pyrazolopyrimidine (brown) and the MKK7 (green) (C), and between anisomycin
(green) and MKK7 (brown) (D). Data in (A) represent means
Previous structural studies have shown that C218 contributes to the formation of
a unique auto-inhibitory conformation in MKK7 by forming an n–
DNA damage is one of the major consequences of oxidative stress [35].
Accordingly, to confirm that UPM-induced ROS generation leads to DNA strand
breaks in our model,
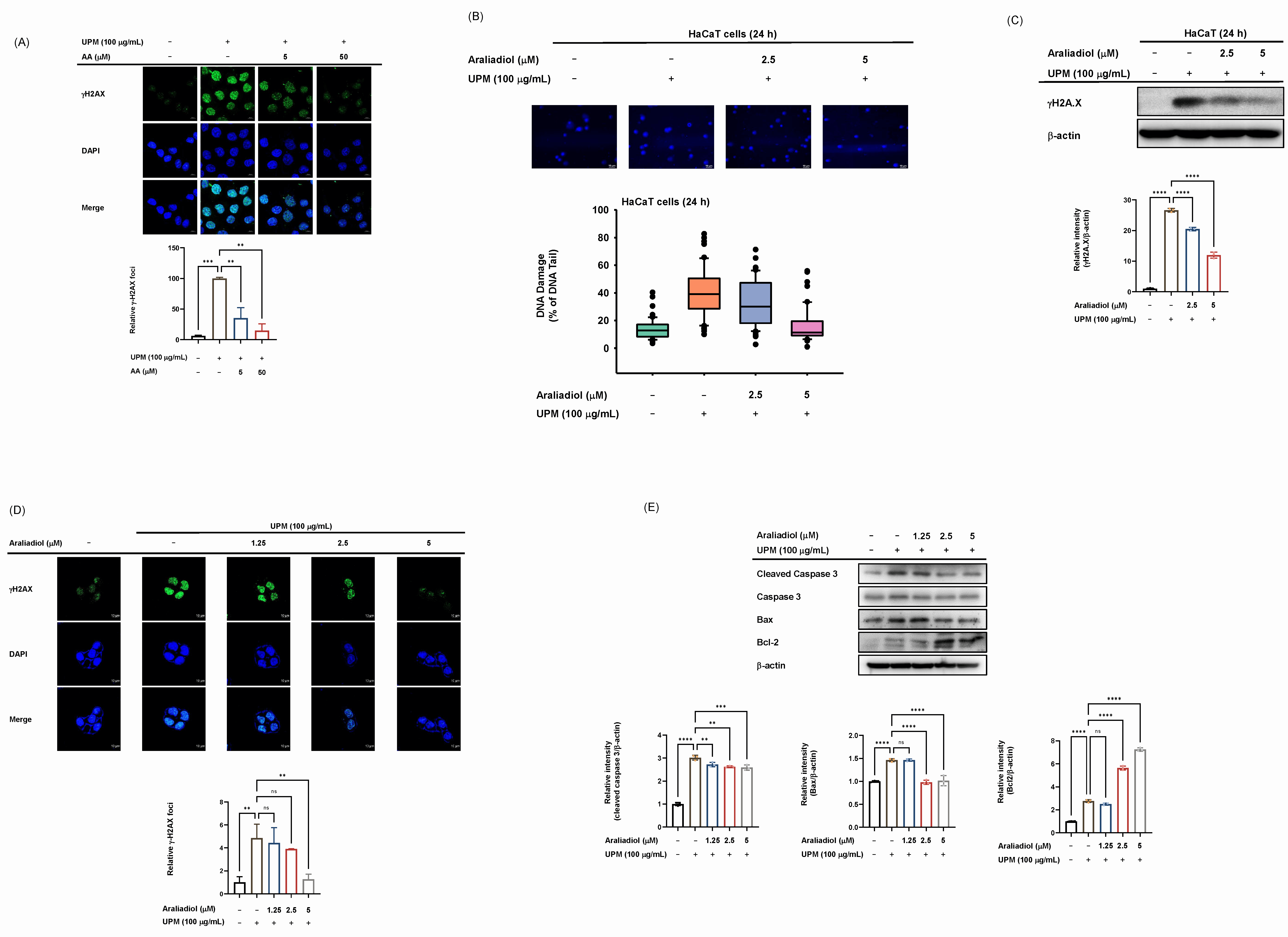 Fig. 6.
Fig. 6.
Effect of araliadiol on DNA damage and apoptosis in UPM-exposed
HaCaT cells. (A,D) Immunofluorescence staining of
This study provides compelling evidence that araliadiol, a pentacyclic triterpenoid derived from Centella asiatica, exerts a significant protective effect against DNA damage and apoptosis through its antioxidant activity in keratinocytes exposed to UPM. UPM has been widely reported to induce oxidative stress in various cell types, including nasal fibroblasts [36], nasal epithelial cells [37], and pulmonary epithelial cells [38]. Consistent with these findings, our data demonstrate that UPM exposure increases intracellular ROS levels in keratinocytes. Given that oxidative stress is closely associated with DNA damage and apoptosis [35, 39], antioxidants serve as a critical defense mechanism against environmentally induced cellular damage. Notably, araliadiol exhibited potent antioxidant effects by attenuating ROS accumulation and upregulating antioxidant gene expression, suggesting that its protective role against UPM-induced cellular damage is primarily mediated through antioxidant mechanisms.
When HaCaT cells were exposed to UPM alone, a modest increase in the expression of certain antioxidant genes, such as TXNRD and GCLC, was observed, likely reflecting a compensatory cellular defense response to oxidative stress. However, this response was insufficient, as Nrf2 activity and expression remained largely unchanged and intracellular ROS levels remained elevated, indicating a limited endogenous antioxidant capacity under UPM exposure. In contrast, araliadiol treatment alone robustly activated the Nrf2 pathway, promoting both Nrf2 expression and nuclear translocation, and leading to the coordinated upregulation of multiple antioxidant genes, including HO-1, NQO1, SOD1, TXNRD, GCLC, and GCLM. Importantly, under UPM-stimulated conditions, araliadiol preserved and reinforced this Nrf2-mediated antioxidant signaling, thereby effectively mitigating ROS accumulation despite ongoing oxidative stress. These findings suggest that while UPM alone elicits only a limited and insufficient antioxidant response, araliadiol confers active and sustained antioxidant protection, both under basal conditions and in the presence of UPM-induced stress.
Air pollution, which is increasing worldwide, contributes to the accumulation of oxidative stress in the body, thereby accelerating aging and increasing the incidence of various skin disorders [40]. Antioxidants, therefore, may be valuable in the treatment and protection against skin diseases induced by particulate matter. Consequently, natural antioxidant compounds that protect skin from damage caused by UPM have been gaining increasing attention in dermatological research. Supporting this, numerous natural antioxidants, including flavonoids, phytosterols, and ginsenosides, have been reported to exert beneficial effects against PM-induced skin damage and aging [40]. Our data likewise suggest the potential utility of araliadiol as a therapeutic candidate for skin protection. In particular, the ability of araliadiol to reduce oxidative stress is expected to provide beneficial effects in skin protection. However, further clinical investigations are warranted to validate the potential of araliadiol as a bioactive material against UPM-induced skin aging and damage.
Nrf2 acts as a master transcription factor orchestrating the expression of a wide array of antioxidant and phase II detoxification enzymes by binding to antioxidant response elements (AREs) within gene promoters. Its cellular activity is tightly controlled not only by proteasomal degradation through its inhibitor Keap1, but also by various post-translational modifications and interactions with upstream signaling cascades [41]. MAPK signaling is one of several upstream modulators of Nrf2 activity, influencing Nrf2 through both direct and indirect mechanisms [42]. For example, JNK1 has been identified as an upstream kinase capable of phosphorylating Nrf2, thereby affecting its nuclear translocation and transcriptional activity [43]. In addition, MAPKs can impact the function of Keap1, indirectly facilitating the stabilization and release of Nrf2, ultimately contributing to the fine-tuning of cellular antioxidant capacity in response to oxidative stress [44]. However, it should be noted that the contribution of MAPKs to Nrf2 activation is context-dependent and may involve complex indirect crosstalk with other pathways. The AP-1 transcription factor, whose activity is regulated by the MAPK pathway, is also implicated in oxidative stress responses and the regulation of Nrf2 signaling. AP-1 and Nrf2 can coordinate gene regulation through adjacent or overlapping binding sites in certain gene promoters, enabling cooperative modulation of target gene expression. Notably, Nrf2 has been shown to form a heterodimer with Jun proteins, thereby enhancing ARE-driven transcriptional activation [45].
Notably, several natural antioxidants, including curcumin, resveratrol, and sulforaphane, have been reported to activate Nrf2 primarily through Keap1-dependent mechanisms and broad redox modulation [46, 47, 48]. While these compounds effectively enhance antioxidant defenses, their regulatory effects on Nrf2 generally do not involve selective engagement of specific MAPK modules. In contrast, our findings demonstrate that araliadiol induces Nrf2 activation through a distinct mechanism that critically depends on the MKK7–JNK–c-Jun axis. KEGG pathway analysis demonstrated that araliadiol robustly engaged the MAPK and Ras signaling pathways. Although the Ras pathway is classically linked to ERK activation, it is also known to activate the JNK and p38 pathways under specific cellular contexts or external stimuli [49, 50]. In line with these predictions, araliadiol treatment selectively enhanced the phosphorylation of MKK7, JNK, and c-Jun in keratinocytes, whereas phosphorylation of ERK, p38, and c-Fos was not observed. These differential effects among MAPK signaling proteins and AP-1 subunits likely reflect the independent and functionally specialized roles of ERK, JNK, and p38 in the activation of each AP-1 subunit [51]. c-Jun is specifically phosphorylated by JNK, and no other MAPK has been identified as being involved in the N-terminal phosphorylation of c-Jun in mammalian cells [52]. In contrast, the activation of c-Fos is facilitated by both p38 and ERK pathways [53, 54]. Taken together with previous reports, our results suggest that araliadiol reinforces cellular defense mechanisms against oxidative stress predominantly via selective activation of the MKK7-JNK-c-Jun axis. Consistent with this hypothesis, pharmacological inhibition of JNK abrogated the araliadiol-induced upregulation of ARE-mediated luciferase activity, further supporting a key role for JNK signaling in mediating the antioxidant effects of araliadiol.
A selective MKK7 inhibitor that covalently binds to C218 has been developed based on the critical role of this residue in maintaining the autoinhibited conformation of MKK7 [33, 34]. A previous study has suggested that MKK7 autoinhibition is maintained through structural features involving C218 and its neighboring residues, including S263, which together restrict ATP access [34]. Molecular docking analysis revealed that araliadiol, anisomycin, and pyrazolopyrimidine all bind near the ATP-binding pocket of MKK7, encompassing the C218 and S263 residues. This observation led us to hypothesize that the interaction between araliadiol and MKK7 may also be influenced by the structural elements involving C218 and its neighboring residue, S263. We expected that selective engagement of araliadiol with S263, in the absence of C218 binding, might destabilize the autoinhibited conformation and thereby promote MKK7 activation. However, these possibilities remain speculative and require further investigation. Collectively, these findings provide new insights into the development of MKK7 activators, highlighting S263 as a promising allosteric site. Targeting the S263 region may represent a novel and effective strategy for the discovery of small-molecule MKK7 activators.
Given the highly conserved nature of the ATP-binding pocket across MAP2Ks, targeting this site often encounters challenges related to lack of specificity and off-target effects. Accordingly, focusing on an allosteric site such as S263 presents a promising alternative strategy for the selective modulation of MKK7 function. To further advance the development of MKK7-selective activators based on araliadiol, future research should prioritize detailed investigations into the binding dynamics of araliadiol, together with structure-based drug design to optimize affinity and specificity.
Finally, although the present study provides mechanistic insights into the antioxidant and cytoprotective effects of araliadiol, it is subject to the inherent limitations of in vitro keratinocyte models. Such systems do not fully capture the complexity of human skin physiology, including epidermal barrier dynamics, interactions among multiple skin-resident cell types, and systemic immune responses, all of which can influence oxidative stress responses and compound bioavailability. To address these limitations, recent studies have emphasized the value of advanced skin models that enable more physiologically relevant evaluation of cosmetic actives and cosmoceuticals. For example, three-dimensional (3D) Nrf2 reporter epidermis models allow direct monitoring of antioxidant and stress-responsive signaling in stratified human epidermis under conditions closer to native skin physiology [55, 56]. In this context, future studies incorporating 3D reconstructed skin systems and ex vivo–like exposure models will be valuable for validating the efficacy and safety profiles of araliadiol. Furthermore, the incorporation of appropriate in vivo models will be essential to more comprehensively evaluate the feasibility, pharmacodynamics, and translational potential of araliadiol-based interventions.
Collectively, these multidisciplinary approaches may help establish araliadiol and its derivatives as promising modulators of the MKK7-JNK signaling pathway.
This study demonstrates that araliadiol functions as a multifaceted protective agent against environmental pollutants such as UPM, primarily through antioxidant properties. The efficacy and underlying molecular mechanisms involving Nrf2 and AP-1 signal pathways are summarized in Fig. 7. These findings highlight the potential of araliadiol for therapeutic applications in oxidative stress-related skin disorders. Future studies are warranted to further explore its clinical relevance, particularly in the context of skin protection and the prevention of skin aging.
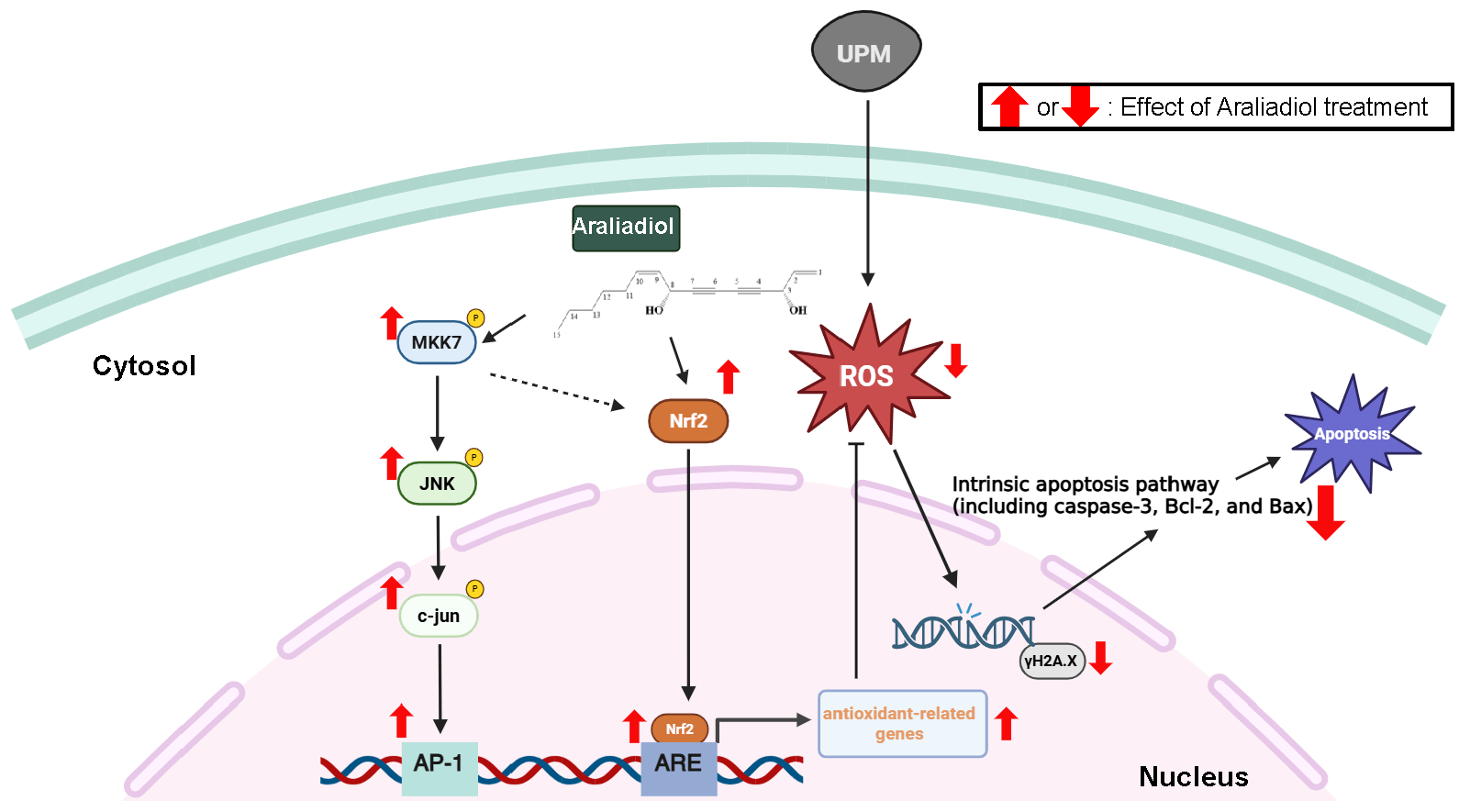 Fig. 7.
Fig. 7.
Schematic summary of the protective effects and underlying molecular mechanisms of araliadiol in UPM-exposed HaCaT cells. Araliadiol exerts cytoprotective effects against UPM-induced oxidative stress, DNA damage, and apoptosis in HaCaT cells. Mechanistically, araliadiol activates the Nrf2 signaling pathway and enhances AP-1 signaling through direct activation of MKK-7. Notably, based on a previous report, it is hypothesized that JNK activation may contribute to Nrf2 pathway activation. This potential crosstalk is represented by a dotted line in the figure, indicating the need for further investigation. The antioxidant activity of araliadiol leads to reduced intracellular ROS levels, thereby mitigating DNA damage and suppressing apoptosis. Collectively, these findings suggest that araliadiol is a promising therapeutic candidate for protecting the skin against damage induced by environmental pollutants such as UPM. The chemical structure of araliadiol was drawn using ChemSketch.
All data generated or analyzed during this study are included in this published article. The raw data supporting the findings of this study are available from the corresponding author upon reasonable request.
ZP and JYC conceptualized and designed the research study. DSY, SHP, DSK, SEY, and JHY performed the experiments and data collection. JL and JHK analyzed and curated the data, and prepared the figures. ZP and DSY drafted the manuscript. JHK and JYC reviewed and edited the manuscript. JYC supervised the overall project administration and acquired funding. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Experimental research involving plants was conducted in compliance with relevant institutional and national guidelines. The plant material used in this study was Centella asiatica, obtained from a registered cultivar (BT-care) cultivated under controlled conditions at the smart farming facilities of ASK Base on (Jeju Island, Korea). The plant material was not collected from wild or endangered species, and no specific permits were required for its use.
Not applicable.
This research was supported by grants provided by the Basic Science Research Program (Grant No.: 2017R1A6A1A03015642) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, and by a Korea Basic Science Institute (National Research Facilities and Equipment Center) funded by the Ministry of Education, Korea (Grant No.: 2020R1A6C101A191).
The authors declare no conflict of interest. Centella asiatica and araliadiol used in this study was provided by ASK Company Co., Ltd., but the company had no involvement in the study design, data analysis, interpretation, or manuscript preparation, the judgments in data interpretation and writing were not influenced by this relationship.
During the preparation of this work, the authors used ChatGPT (OpenAI, San Francisco, CA, USA) to improve the clarity, fluency, and grammar of the text and to refine phrasing for academic style. After using this tool, the authors carefully reviewed and edited the content, and take full responsibility for the final version of the manuscript.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.