









 , Liang Zhang 1, Ping Wang 1,*
, Liang Zhang 1, Ping Wang 1,*1 Department of Radiation Oncology, Tianjin Medical University Cancer Institute & Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin’s Clinical Research Center for Cancer, Tianjin Medical University, 300060 Tianjin, China
2 Department of Radiation Oncology, Liaocheng People’s Hospital, 252000 Liaocheng, Shandong, China
Abstract
Lung cancer is the leading cause of cancer-related mortality worldwide, and metastasis is the key factor leading to patient death. Epithelial–mesenchymal transition (EMT), which is crucial to tumor metastasis, is primarily regulated by EMT transcription factors, such as Twist1. As an RNA-binding protein, far upstream element binding protein 3 (FUBP3) shows aberrant expression in various tumors; however, its mechanistic role in lung cancer metastasis remains unclear. This study aims to elucidate the functional role of FUBP3 in lung cancer metastasis and its molecular mechanism in the regulation of Twist1.
Bioinformatics analysis was conducted to examine FUBP3 expression patterns in lung cancer and its association with patient prognosis. The Cancer Genome Atlas database was used, and FUBP3 protein expression levels were detected in clinical lung cancer tissues by immunohistochemical analysis. Lung cancer cell lines with FUBP3 knockdown were established, and the effects of FUBP3 on the metastatic capacity of lung cancer were assessed using Transwell migration and invasion assays, 3D spheroid invasion experiments, and tail vein injection metastasis models. Changes in the expression levels of EMT markers were detected by western blot, quantitative real-time polymerase chain reaction, and immunofluorescence. The interaction between FUBP3 and signal transducer and activator of transcription 3 (STAT3) was verified by co-immunoprecipitation (Co-IP), proximity ligation assay, and immunofluorescence co-localization. The effects of STAT3 inhibitor S3I-201 on FUBP3-mediated pro-metastatic functions were assessed.
Bioinformatics analysis revealed high FUBP3 expression in lung cancer tissues, which correlated with poor patient prognosis. Notably, patients with distant metastasis (M1) stage exhibited higher FUBP3 expression than those at the no distant metastasis (M0) stage. Functional experiments confirmed that FUBP3 silencing inhibited the migration and invasion of lung cancer cells, as well as the formation of pulmonary metastatic foci in vivo. The knockdown of FUBP3 led to an increase in the expression of the epithelial marker E-cadherin and downregulated the expression of the mesenchymal marker vimentin, indicating that FUBP3 promotes lung cancer metastasis by promoting EMT. Subsequent analysis indicated that FUBP3 facilitates lung cancer progression by upregulating Twist1 expression. Both exhibit positive correlations in lung cancer patient tissues. Co-IP and immunofluorescence assays demonstrated a direct interaction between FUBP3 and STAT3 proteins. STAT3 silencing counteracted pro-metastatic effects associated with FUBP3 overexpression in lung cancer metastasis. Treatment with S3I-201 effectively reversed the pro-metastatic phenotype in cells with high FUBP3 expression, restored the typical patterns of EMT marker expression, and reduced the formation of metastatic foci in the in vivo metastasis model.
This study reveals the critical role of FUBP3 in lung cancer metastasis and identifies a new regulatory axis involving FUBP3–STAT3–Twist1. FUBP3 interacts with STAT3, enhancing STAT3-dependent Twist1 expression, which promotes EMT and metastasis. FUBP3 functions as a prognostic biomarker, and STAT3 inhibitors present therapeutic strategies for lung cancer, offering novel insights for precision treatment.
Keywords
- FUBP3
- lung cancer
- metastasis
- epithelial–mesenchymal transition
- Twist1
Epithelial–mesenchymal transition (EMT) is a crucial process enabling tumors to gain invasive and metastatic properties [1, 2]. This process allows epithelial-derived tumor cells to detach from intercellular connections and gain motility akin to mesenchymal cells, facilitating tumor metastasis [3]. EMT is primarily regulated by EMT transcription factors (EMT-TFs), including members of the snail family transcriptional repressor (SNAIL), zinc finger E-box-binding homeobox (ZEB), and twist family bHLH transcription factor (TWIST) families [1, 4]. Twist1, which is a basic helix–loop–helix transcription factor, is pivotal in the EMT of various malignant tumors [5, 6]. Twist1 suppresses the expression of epithelial marker genes, such as E-cadherin, by binding to Enhancer box (E-box) sequences in the promoters of target genes, while concurrently activating the transcription of mesenchymal marker genes, such as vimentin [7]. Clinical studies indicate that Twist1 is overexpressed in lung cancer, and its expression level is closely associated with tumor grading, metastasis, and patient prognosis [8, 9]. However, the transcriptional regulatory mechanisms of Twist1, especially its upstream regulatory networks, are not fully understood.
Recent studies have highlighted the important roles of RNA-binding proteins (RBPs) in tumor development and progression [10, 11]. RBPs regulate gene expression with precision across various levels, including mRNA stability, splicing, localization, and translation. They are integral to processes such as tumor cell proliferation, apoptosis, metastasis, and drug resistance [12]. Traditional concepts indicate that RBPs primarily regulate gene expression at the post-transcriptional level. However, emerging evidence shows that many RBPs also have DNA-binding capabilities and can directly engage in transcriptional regulatory processes, acting as crucial links between transcriptional and post-transcriptional regulation [13, 14].
The far upstream element binding protein (FUBP) family, as a single-strand DNA/RBP family, comprises three members: FUBP1, FUBP2, and FUBP3, which regulate gene transcription through recognition of purine-rich Far Upstream Element sequences (FUSE) [15, 16]. Members of the FUBP family were first identified for their role in regulating c-myc proto-oncogene expression; however, later research has demonstrated that their regulatory networks are significantly more intricate than previously thought [17, 18]. The oncogenic role of FUBP1 in various tumors has been extensively validated, whereas functional studies of FUBP2 and FUBP3 have lagged, especially those about the mechanistic roles of these proteins in tumor metastasis processes [19, 20, 21]. FUBP3, also known as FBP3, is a relatively understudied member of the FUBP family [22]. The FUBP3 protein comprises four K homology domains, can bind single-strand DNA and RNA and is primarily localized in the cell nucleus [22, 23]. Bioinformatics analysis indicates abnormal FUBP3 expression in multiple tumor types, suggesting its potentially essential role in tumor progression [23, 24, 25]. The roles and molecular mechanisms of FUBP3 in tumor biology are not yet comprehensively investigated, in contrast to those of FUBP1, and the role of FUBP3 in the regulation of EMT-related gene expression and its specific functions in tumor metastasis processes remain unclear. This research gap limits our understanding of the complete roles of the FUBP family in tumor development and progression.
Signal transducer and activator of transcription 3 (STAT3) is a key component of the Janus kinase (JAK)–STAT signaling pathway, playing an important role in tumor cell proliferation, angiogenesis, and metastasis [26, 27]. The persistent activation of STAT3 is an important characteristic of various malignant tumors [28, 29]. Gene transcriptional regulation depends on the formation of transcriptional complexes formed by various regulatory molecules, including transcription factors, co-activators, and RBPs [30]. RBPs can serve as “scaffold” proteins, facilitating the recruitment of additional transcriptional regulatory factors and establishing intricate regulatory networks [31, 32, 33]. The mechanism of multiprotein complex formation is crucial in the regulation of genes associated with tumor metastasis. Bioinformatics analysis revealed that FUBP3 is highly expressed in lung cancer tissues and correlates with poor patient prognosis. Further analysis suggests that FUBP3 is involved in the regulation of genes associated with EMT. FUBP3 and STAT3 demonstrate a correlation in expression within tumor samples, indicating a possible functional relationship between them.
We hypothesized that FUBP3 facilitate lung cancer EMT and metastasis by recruiting STAT3 to form transcriptional complexes, thereby activating the transcription of Twist1. This study aims to elucidate the mechanistic role of FUBP3 in lung cancer metastasis, focusing on its interaction with STAT3 and the regulation of Twist1 expression. We validated the effects of FUBP3 on EMT and metastasis in lung cancer through in vitro and in vivo functional experiments. The interactions between FUBP3 and STAT3 and their role in the transcriptional regulation of Twist1 were explored using molecular biology techniques. Additionally, the therapeutic potential of targeting this regulatory axis was evaluated.
Transcriptomic sequencing data for non-small cell lung cancer (NSCLC) were downloaded from The Cancer Genome Atlas (TCGA) database, including tumor tissue samples and paired normal tissue samples from lung adenocarcinoma (LUAD), and lung squamous cell carcinoma. Differential expression analysis was performed using R language (Version 4.2.0, R Core Team, R Foundation for Statistical Computing, Vienna, Austria) and the DESeq2 package (Version 1.36.0, Bioconductor, Fred Hutchinson Cancer Research Center, Seattle, WA, USA) to assess FUBP3 expression levels in tumor and normal tissues. Clinical follow-up data from patients with NSCLC in the TCGA cohort indicated that patients were divided into high- and low-expression groups according to the median FUBP3 expression level. Survival curves were plotted using the Kaplan–Meier method, and overall survival (OS) differences among groups were assessed using the Log-rank test. Functional enrichment analysis using Gene Ontology (GO) and pathway enrichment analysis via the Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed using the Gene Denovo website (https://www.genedenovo.com/). A protein–protein interaction network for FUBP3 was established using the GeneMANIA protein interaction database (https://genemania.org/), and network visualization was performed using Cytoscape software (Version 3.8.2, The Cytoscape Consortium, University of California San Diego, San Diego, CA, USA). Immune score, stromal score, and tumor purity was calculated for each tumor sample with the ESTIMATE algorithm, and the relationship between FUBP3 expression and the tumor microenvironment was analyzed. The relative abundance of immune cell subsets was inferred using the CIBERSORT deconvolution algorithm, and the effects of FUBP3 expression on tumor immune infiltration patterns were explored.
Tissue specimens were collected from 30 patients with NSCLC who underwent
surgical resection in the Department of Thoracic Surgery at our hospital between
January 2025 and August 2025. This cohort included 30 cases of LUAD, which had
paired adjacent normal tissues. Inclusion criteria: (1) pathologically confirmed
primary NSCLC; (2) absence of preoperative chemotherapy, radiotherapy, or
targeted therapy; (3) availability of complete clinicopathological data.
Exclusion criteria: (1) concurrent malignancies; (2) severe cardiopulmonary
insufficiency; (3) immunodeficiency diseases. All cases were diagnosed by
qualified pathologists in accordance with the WHO classification criteria. This
study received approval from the Hospital Ethics Committee of Liaocheng People’s
Hospital (approval number: 2025256), and all participants provided informed
consent. Tissue specimens were promptly fixed in 4% paraformaldehyde (P1110,
Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) for 24 hours,
subjected to gradient ethanol dehydration, paraffin embedded, and cut into create
4 µm-thick serial sections. The sections underwent deparaffinization in
xylene (10023418, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China),
rehydration through a gradient of ethanol, and antigen retrieval by microwave
heating in EDTA buffer (pH 8.0, ZLI-9069, Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) for 15 minutes. The sections were washed
with PBS (SH30256.01, HyClone Laboratories Inc., Logan, UT, USA) and treated with
3% H2O2 (H1009, Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) for 10 minutes to block endogenous peroxidase activity. After
blocking with 5% bovine serum albumin (SW3015, Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) at room temperature for 1 hour, the
sections were incubated overnight at 4 °C with the FUBP3 primary
antibody (1:200, ab181111, Abcam plc, Cambridge, UK), washed with PBS, and
incubated with horseradish peroxidase–conjugated goat anti-rabbit secondary
antibody (1:500, PV-6001, Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd., Beijing, China) at room temperature for 30 minutes. Color development was
performed using a DAB chromogen kit (ZLI-9018, Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China), and the nuclei were counterstained with
hematoxylin (G1140, Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China). The sections were dehydrated through with a gradient of ethanol, cleared
in xylene, and subsequently mounted with neutral resin (G8590, Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China). Images were obtained using a
Leica DM4000B microscope (Leica Microsystems GmbH, Wetzlar, Germany) and assessed
independently by two pathologists employing a double-blind approach.
Immunohistochemical staining was evaluated using a semiquantitative scoring
system. The final score was determined by assessing staining intensity (0–3
points) and the percentage of positive cells (0–4 points), and a score of
A549 and H460 cells were underwent various treatments were collected, and total RNA was extracted utilizing TRIzol reagent (15596018, Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA). The concentration and purity of RNA were assessed using a NanoDrop 2000 UV–Vis spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Only samples exhibiting A260/A280 ratios within the range of 1.8–2.0 and A260/A230 ratios exceeding 2.0 were used for subsequent experiments. One microgram of total RNA was reverse-transcribed to synthesize cDNA using a PrimeScript RT reagent kit (RR047A, TaKaRa Bio Inc., Kusatsu, Shiga, Japan). The reaction conditions included incubation at 37 °C for 15 minutes, 85 °C incubation for five seconds, and subsequent storage at 4 °C. cDNA was used as a template for amplification, which was performed using a SYBR Premix Ex Taq kit (RR420A, TaKaRa Bio Inc., Kusatsu, Shiga, Japan) on an ABI 7500 real-time fluorescence quantitative PCR system. The PCR reaction system comprised a total volume of 20 µL, which included 10 µL of SYBR Premix Ex Taq, 0.4 µL of forward primer, 0.4 µL of reverse primer, 2 µL of cDNA template, and 7.2 µL of ddH2O. The amplification program included an initial denaturation step at 95 °C for five minutes and 40 cycles of amplification comprising denaturation at 95 °C for 30 seconds, annealing at 60 °C for 30 seconds, and extension at 72 °C for 30 seconds. Then, product specificity was confirmed through melting curve analysis. Table 1 presents the primer sequences utilized in this study. The relative expression of target genes was calculated using the 2-ΔΔCt method, and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal reference gene. Each sample was analyzed in triplicate.
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
| STAT3 | CAGCAGCTTGACACACGGTA | AAACACCAAAGTGGCATGTGA |
| Twist1 | GGAGTCCGCAGTCTTACGAG | TCTGGAGGACCTGGTAGAGG |
| FUBP3 | GCAAGGTCATCCATGACAACTC | CTCCTTCTGCATCCTGTCAAAC |
GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; STAT3, Signal transducer and activator of transcription 3; Twist1, Twist family BHLH transcription factor 1; FUBP3, Far upstream element binding protein 3.
Cells exposed to different treatments were collected, washed with PBS, and lysed
using prechilled RIPA lysis buffer (R0020, Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China). The samples were then incubated on ice for 30 minutes
with intermittent vortexing. Supernatants were collected following centrifugation
at 12,000 rpm for 15 minutes at 4 °C. Protein concentrations were
determined using the BCA protein assay kit (PC0020, Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). Protein samples were combined with
5
A549 cells were harvested 48 hours after transfection through centrifugation at
800 rpm for 3 minutes at 4 °C. The supernatant was removed, and the
cells were washed twice with prechilled PBS. PBS was completely removed, and the
cell pellets were preserved. Cells were resuspended in prechilled
immunoprecipitation lysis buffer (R0020, Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) and homogenized 30–50 times with a glass homogenizer.
The samples were centrifuged at 12,000 rpm for 5 minutes, and supernatants were
collected as cell lysates. Protein concentrations were determined using a BCA
protein assay kit (Beijing Solarbio, PC0020), and 500 µg of total protein
was used for each reaction. Cell lysates were separated into forward and reverse
co-immunoprecipitation groups. Each group received 1 µg of FUBP3 antibody
(Abcam, ab181111), STAT3 antibody (Cell Signaling Technology, #9139), or a
negative control IgG antibody (#2729, Cell Signaling Technology Inc., Danvers,
MA, USA) overnight and incubated overnight with gentle rocking at 4 °C.
This setup included three conditions: (1) Input (whole cell lysate), (2) IgG
negative control (beads plus nonspecific antibody), and (3) Experimental Groups
(with Anti-FUBP3 or Anti-STAT3 antibodies). Buffer-pretreated protein A/G
magnetic beads (10 µL, SE101, Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China) or agarose beads (10 µL, sc-2003, Santa
Cruz Biotechnology Inc., Dallas, TX, USA) were subsequently added, and the
samples were incubated with gentle rocking at 4 °C for 2–4 hours.
Following immunoprecipitation, samples underwent centrifugation at 12,000 rpm for
30 seconds at 4 °C to pellet the beads. The supernatant was aspirated,
and the beads were washed three or four times with 1 mL of lysis buffer,
centrifuging at 1000 rpm for 1 minute at 4 °C each time. Subsequently,
15–20 µL of 1
After cell digestion, cells were placed onto sterile coverslips that had been
prepositioned in 48-well plates at a density of 5
A549 and H460 human NSCLC cell lines were acquired from the Cell Bank of the Chinese Academy of Sciences and subsequently seeded in culture dishes. All cell lines were validated by STR profiling and were confirmed to be negative for mycoplasma contamination. Cells were cultured in RPMI-1640 complete medium (SH30809.01, HyClone Laboratories Inc., Logan, UT, USA) supplemented with 10% fetal bovine serum (10099-141, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (SV30010, HyClone Laboratories Inc., Logan, UT, USA). They were maintained in a sterile incubator (Thermo Fisher Scientific Inc., Waltham, MA, USA) at 37 °C with 5% CO2 until they reached the logarithmic growth phase. Upon reaching a cell density of approximately 80%, the culture medium was aspirated, and the cells were washed twice with sterile PBS (Hyclone, SH30256.01). One milliliter of trypsin–EDTA digestion solution (25200-072, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) was applied to digest cells for 2–3 minutes until the cells rounded and detached. Digestion was halted by the addition of 2 mL complete medium. Cells were gently pipetted to create a cell suspension, which was then transferred to a 15 mL centrifuge tube and centrifuged at 1000 rpm for 5 minutes. After the removal of the supernatant, cells were resuspended in 2 mL of complete medium and passaged at a 1:3 ratio into new culture dishes. Six milliliters of complete medium were added to each dish, mixed thoroughly, and subsequently returned to the incubator for ongoing culture.
The construction and infection of the FUBP3 shRNA lentiviral vector were conducted in accordance with the specified instructions. The control sequence was a scrambled nonspecific sequence, which was cloned into the pLKO.1 lentiviral vector (#8453, Addgene Inc., Watertown, MA, USA). The vector plasmid was co-transfected with the packaging plasmid psPAX2 (#12260, Addgene Inc., Watertown, MA, USA) and the envelope plasmid pMD2.G (#12259, Addgene Inc., Watertown, MA, USA) in a ratio of 4:3:1 into 293T cells with Lipofectamine 3000 reagent (L3000015, Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA) for transfection. The supernatant containing the virus was collected 48 hours after transfection, filtered using a 0.45 µm membrane, and subsequently stored at –80 °C for future applications. A549, H1299, and H460 cells were cultured in six-well plates and subsequently infected with lentivirus upon achieving a cell density of 50%–60%. Cells were categorized into the NC group (negative control group, infected with scrambled shRNA lentivirus) and the FUBP3-shRNA group (infected with FUBP3 shRNA lentivirus), utilizing a multiplicity of infection of 10. Fresh medium was replaced 24 hours after infection, and following an additional 48 hours of culture, puromycin (2 µg/mL, P8230, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) was introduced for the selection of positive clones. Selection persisted for one to two weeks until stable transfected cell lines were established, and the efficiency of FUBP3 knockdown was confirmed via Western blot analysis. The complete human FUBP3 cDNA was inserted into the pcDNA3.1(+) vector (Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA). Four synonymous nucleotide substitutions were introduced into the shRNA recognition sequence through site-directed mutagenesis to render the construct resistant to shRNA targeting, preserving the original amino acid coding while inhibiting shRNA binding. Cells with FUBP3 knockdown (A549-shFUBP3 and H460-shFUBP3) were transfected with either the shRNA-resistant FUBP3 plasmid or an empty vector control using Lipofectamine 3000 (Invitrogen). This procedure was followed by G418 selection at a concentration of 800 µg/mL for 3 weeks. The successful restoration of FUBP3 expression was confirmed by Western blot analysis. The full-length human Twist1 cDNA was cloned into the pcDNA3.1(+) vector and transfected into A549 and H460 cells utilizing Lipofectamine 3000 (Invitrogen). Stable cell lines were selected using G418 (800 µg/mL), which was administered 3 weeks prior. Twist1 shRNA lentiviral constructs were developed utilizing the pLKO.1 backbone (Addgene, #8453). Lentivirus was generated through the co-transfection of 293T cells with a shRNA plasmid, psPAX2 packaging plasmid, and pMD2.G envelope plasmid in a 4:3:1 ratio. Target cells were infected with the viral supernatant and were subsequently selected using puromycin (2 µg/mL), which was administered for 2 weeks. Knockdown efficiency was confirmed using quantitative real-time polymerase chain reaction. Scrambled shRNA served as the negative control.
Forty-eight hours after lentiviral transduction, A549 and H460 cells were digested into single-cell suspensions and seeded at a density of 1000 cells per well in ultralow attachment six-well plates (3471, Corning Inc., Corning, NY, USA). Cells were cultured in a serum-free sphere culture medium composed of DMEM/F12 (SH30023.01, HyClone Laboratories Inc., Logan, UT, USA). Cultures were incubated at 37 °C with 5% CO2 for 10–14 days, and half of the medium was replaced every 3 days. Following the completion of culture, the cytoskeleton was stained with phalloidin–FITC (1:200, P5282, Sigma-Aldrich LLC, St. Louis, MO, USA) for 30 minutes and washed with PBS, and the nuclei were stained with DAPI (1:1000, C0065, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) for 5 minutes. Images were observed and captured under a laser confocal microscope (Leica TCS SP8, Leica Microsystems GmbH, Wetzlar, Germany).
Serum-free RPMI-1640 medium was introduced into the upper chamber of Transwell
inserts for cell migration assays. Co-cultured A549 cells were suspended in a
serum-free medium at a concentration of 5
After cell treatment, 100 µL of EdU medium (50 µmol/L) was added to
each well and incubated for 2 hours. Cells underwent three washes with phosphate
buffer, fixed in 4% paraformaldehyde at room temperature for 30 minutes, and
treated with 0.5% Triton X-100 for 10 minutes. After washing with phosphate
buffer, Apollo staining solution was introduced and incubated with agitation at
room temperature in the dark for 30 minutes. Nuclei were stained using a DAPI
solution (5 µg/mL). Immediately after staining, the cells were observed
under a microscope, and five random fields of view selected for counting. The
EdU-positive cell rate for each group was determined using ImageJ software
(Version 1.8, National Institutes of Health, Bethesda, MD, USA). The rate of
EdU-positive cells was calculated as follows: (number of EdU-positive cells/total
number of cells)
Following the transfection treatment of each cell group, cells were fixed using
4% paraformaldehyde fixative (Beijing Solarbio, P1110) at room temperature for a
duration of 30 minutes. After PBS washing, a permeabilization solution (0.1%
Triton X-100, Beijing Solarbio, T8200) was introduced to resuspend the cells and
incubated at room temperature for 5 minutes. The samples were then washed with
PBS three times, and 50 µL of the TUNEL reaction mixture was added to each
sample. The mixture was supplied by the TUNEL assay kit (Roche, 11684795910) and
included TdT enzyme and fluorescently labeled dUTP. After incubation at 37
°C in the dark for 60 minutes, the cells were washed three times with
PBS. Coverslips were mounted with antifade mounting medium (Beijing Solarbio,
S2100) and examined with a fluorescence microscope (Olympus IX73). TUNEL-positive
cells exhibited red fluorescence (excitation wavelength 550 nm, emission
wavelength 570 nm), while DAPI-stained nuclei displayed blue fluorescence. The
rate of TUNEL-positive expression was determined by the formula (number of
TUNEL-positive cells/total number of DAPI-positive cells
A549 and H460 cells were cultured in 24-well plates containing prepositioned sterile coverslips and subsequently stained upon reaching 70% confluence. Cells were fixed using 4% paraformaldehyde (Beijing Solarbio, P1110) at room temperature for 30 minutes, permeabilized with 0.1% Triton X-100 (Beijing Solarbio, T8200) for 30 minutes, and subsequently blocked with 5% BSA (A8010, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) at room temperature for 1 hour to minimize nonspecific binding. Following the removal of the blocking solution, phalloidin–FITC (Sigma-Aldrich, P5282) was applied at a dilution of 1:1000 in 1% BSA–PBS and incubated overnight at 4 °C in the dark within a humidified chamber. Nuclei were stained using a DAPI staining solution (Beijing Solarbio, C0065, 1:1000) at room temperature in the dark for a duration of 10 minutes. Coverslips were mounted using anti-fade mounting medium (Beijing Solarbio, S2100) and examined with a laser confocal microscope (Leica TCS SP8). The excitation wavelength for FITC was 488 nm, indicating green fluorescence associated with F-actin, and DAPI was excited at 358 nm, revealing blue fluorescence corresponding to nuclei.
Sterile coverslips were initially positioned in culture flasks, followed by the seeding and culturing of cells until monolayer confluence was achieved, at which point the coverslips were removed. Cells were fixed with 2.5% glutaraldehyde (Beijing Solarbio, G1102) at 4 °C for 2 hours, followed by secondary fixation with 1% osmium tetroxide (Sigma-Aldrich, 75632) at room temperature for 1 hour. Cells underwent sequential dehydration in 30%, 50%, 70%, 80%, 90%, 95%, and 100% ethanol for 15 minutes at each concentration, and 100% ethanol dehydration was performed twice. After dehydration, CO2 critical point drying was conducted using the Leica EM CPD300, and subsequently, gold sputter coating was applied in a vacuum coating unit, achieving a thickness of approximately 10–15 nm. Samples were examined for cell surface morphology using a HITACHI S-570 scanning electron microscope, operating at an accelerating voltage of 15 kV and a working distance of 10–15 mm.
This research received approval from the Institutional Animal Care and Use
Committee of Liaocheng People’s Hospital (approval number: 2025257). Female
BALB/c nude mice (SPF grade; 5–6 weeks old; 20–22 g body weight) were obtained
from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Laboratory
Animal Production License No. SCXK(Jing)2016-0011). Nude mice were maintained in
a specific pathogen-free barrier system at an environmental temperature of 25
Lung tissues were fixed in 10% neutral formalin for 24 hours, followed by routine dehydration and paraffin embedding to prepare 4 µm serial sections. The steps for HE staining were as follows: Sections were deparaffinized to water, stained with hematoxylin for 5 minutes, rinsed with tap water to eliminate excess dye, differentiated with 1% hydrochloric acid–alcohol solution for several seconds until nuclei became clear, blued with ammonia water for 10 seconds, and rinsed with running water for 10 minutes. After staining with 1% eosin for 3 minutes, the sections underwent sequential dehydration in 85%, 95%, and 100% ethanol for 2 minutes each, were cleared in xylene for two 5-minute intervals, and were subsequently mounted with neutral resin. The morphological characteristics of the pulmonary metastatic foci were examined using an optical microscope (Olympus BX53, Olympus Corporation, Tokyo, Japan), and metastatic foci were counted.
The proximity ligation assay (PLA) was performed using the Duolink PLA
Technology (Sigma-Aldrich, DUO92101) in accordance with the manufacturer’s
guidelines. A549 cells were plated on sterile coverslips in 48-well plates at a
density of 5
Experimental data were analyzed utilizing SPSS 26.0 (IBM Corporation, Armonk,
NY, USA) and GraphPad Prism 10.0 (GraphPad Software LLC, Boston, MA, USA)
statistical software. Results are presented as mean
To identify key regulators of lung cancer metastasis, we focused on FUBP3
through bioinformatics screening (Fig. 1A). Single-cell analysis revealed FUBP3
upregulation in mesenchymal-type lung cancer cells, suggesting its involvement in
mesenchymal transformation (Fig. 1B). TCGA database analysis confirmed elevated
FUBP3 transcription in lung cancer versus normal tissues (p
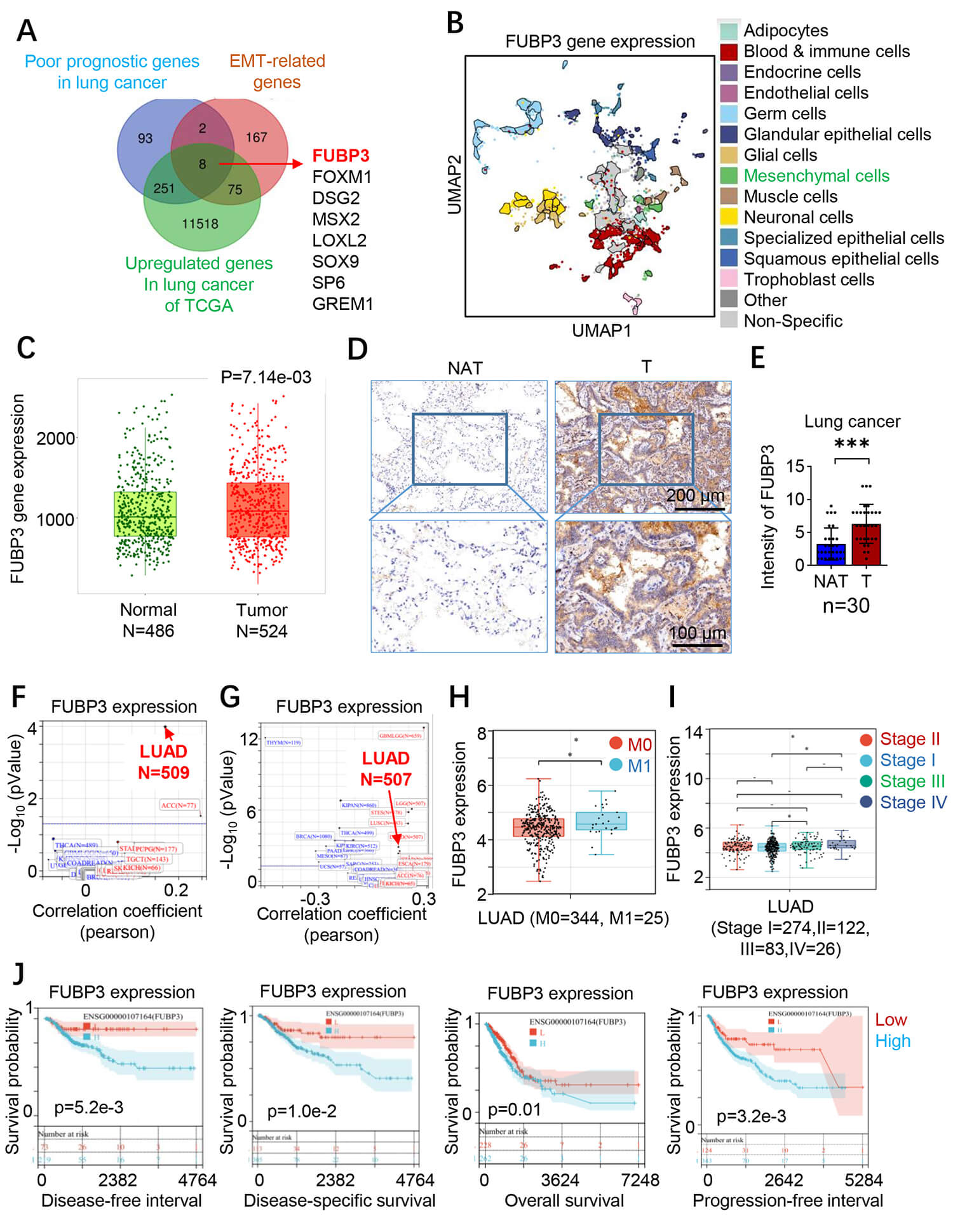 Fig. 1.
Fig. 1.
Elevated FUBP3 expression correlates with poor survival outcomes
in lung cancer patients. (A) Venn diagram showing the bioinformatics screening
strategy for identifying poor prognostic genes and EMT-related genes in lung
cancer, with FUBP3 among the overlapping candidates. (B) UMAP plot from
single-cell RNA sequencing analysis demonstrating FUBP3 upregulation in
mesenchymal cell populations within lung cancer tissues (p = 7.14
To investigate the functional role of FUBP3 in lung cancer cells, we first examined FUBP3 expression levels in different lung cancer cell lines. Western blot analysis showed that compared to human bronchial epithelial cells BEAS-2B, FUBP3 was universally highly expressed in lung cancer cell lines, with the highest expression in A549 cells (approximately 4-fold), followed by H460 cells (approximately 3-fold), and relatively lower expression in PC-9 cells (approximately 1.2-fold) (Fig. 2A). Based on these results, we selected FUBP3 high-expressing A549 and H460 cell lines for subsequent functional studies. To explore the functional role of FUBP3 in lung cancer progression, we constructed stable FUBP3 knockdown A549 and H460 cell lines and established rescue cell lines to verify phenotype specificity. Western blot analysis confirmed transfection efficiency, with FUBP3 protein levels in the sh-FUBP3 group decreased compared to the control group, while FUBP3 expression was effectively restored in the shRNA-rescue group (Fig. 2B). Clonogenic sphere formation assays demonstrated that FUBP3 knockdown suppressed the proliferative potential and sphere-forming capacity of A549 and H460 cells. Compared to the control group, the sh-FUBP3 group formed significantly fewer clonogenic spheres with reduced sphere volume, while cell sphere-forming capacity was partially restored after rescuing FUBP3 expression (Fig. 2C). These results indicate that FUBP3 plays an important role in maintaining lung cancer cell proliferative capacity and stem cell-like characteristics. To evaluate the effect of FUBP3 on lung cancer cell metastatic capacity, we performed Transwell migration and invasion assays. Migration assay results showed that FUBP3 knockdown inhibited the migration capacity of A549 and H460 cells, with fewer cells migrating through the Transwell membrane compared to the control group, while cell migration capacity was significantly restored after rescuing FUBP3 expression (Fig. 2D). Similarly, invasion assays demonstrated that FUBP3 knockdown weakened lung cancer cells’ invasive capacity to penetrate Matrigel, with invasive cell numbers reduced by approximately 70–80% compared to the control group, and rescue experiments confirmed the FUBP3-dependence of this phenotype (Fig. 2E). To further elucidate the effects of FUBP3 knockdown on cellular biological behaviors, we examined cell apoptosis and proliferation status. TUNEL staining results showed that FUBP3 knockdown induced lung cancer cell apoptosis, with the proportion of TUNEL-positive cells increased approximately 3-fold compared to the control group, while cell apoptosis levels were significantly reduced after rescuing FUBP3 expression (Fig. 2F). These results indicate that FUBP3 possesses anti-apoptotic functions and contributes to lung cancer cell survival. EdU proliferation assays further confirmed FUBP3’s regulatory role in cell proliferation. Results showed that FUBP3 knockdown inhibited DNA synthesis capacity in A549 and H460 cells, with the proportion of EdU-positive cells reduced by approximately 60% compared to the control group, while cell proliferative capacity was restored after rescuing FUBP3 expression (Fig. 2G). Combined with TUNEL and EdU experimental results, we confirmed that FUBP3 maintains survival advantages in lung cancer cells by promoting cell proliferation and inhibiting cell apoptosis.
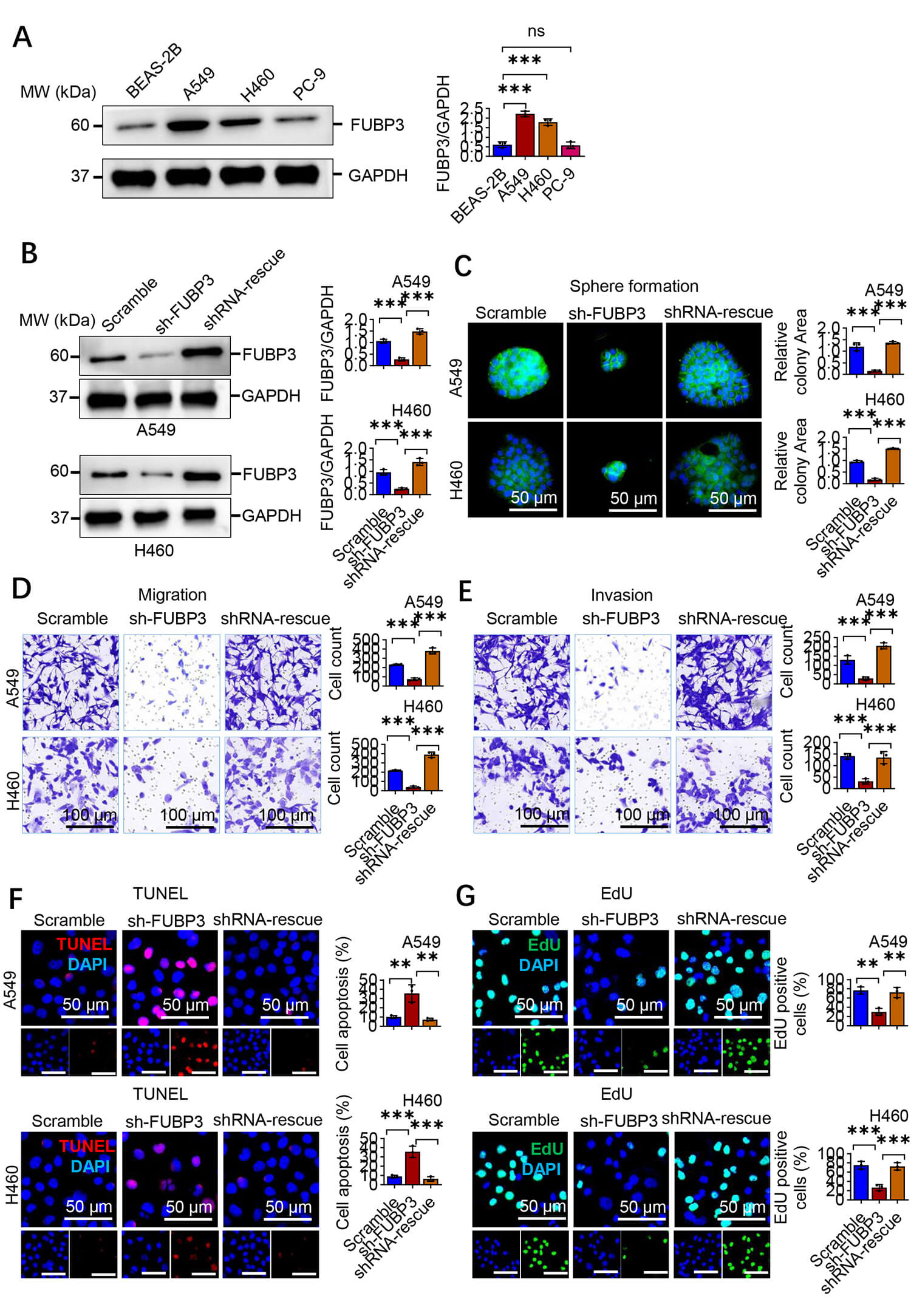 Fig. 2.
Fig. 2.
FUBP3 drives lung cancer progression by promoting metastatic
potential. (A) Western blot analysis of FUBP3 protein levels in lung cancer cell
lines (A549, H1299, H460) compared to normal lung epithelial cells (BEAS-2B).
GAPDH served as loading control. (B) Western blot confirmation of FUBP3 knockdown
and rescue efficiency in A549 and H460 cell lines. (C) Sphere formation assay
evaluating self-renewal capacity in A549 and H460 cells following FUBP3
manipulation. Scale bar: 50 µm. (D) Transwell migration assay assessing
migratory capacity of FUBP3-modified A549 and H460 cells. Scale bar: 100
µm. (E) Transwell invasion assay evaluating invasive potential of A549 and
H460 cells with FUBP3 knockdown and rescue. Scale bar: 100 µm. (F) TUNEL
staining analysis of apoptotic cell death in FUBP3-modified A549 and H460 cells.
TUNEL-positive cells (red), nuclei (blue, DAPI). Scale bar: 50 µm. (G) EdU
incorporation assay measuring cell proliferation in A549 and H460 cells following
FUBP3 manipulation. EdU-positive cells (green), nuclei (blue, DAPI). Scale bar:
50 µm. Data are presented as mean
To validate the functional role of FUBP3 in lung cancer metastasis in
vivo, we established a nude mouse tail vein injection metastasis model. Stably
transfected A549 cells (Scramble control group, sh-FUBP3 knockdown group, and
shRNA-rescue group) were injected into nude mice via tail vein, and mice were
sacrificed after 6 weeks to collect lung tissues for pathological analysis. HE
staining results showed that compared to the control group, the number and size
of pulmonary metastatic foci in the FUBP3 knockdown group were reduced, with
metastatic foci exhibiting smaller and more dispersed distribution patterns,
while metastatic foci formation capacity was significantly restored after
rescuing FUBP3 expression (Fig. 3A). Quantitative analysis results demonstrated
that FUBP3 knockdown inhibited the in vivo metastatic capacity of lung
cancer cells. Control group mice formed approximately 15–20 metastatic foci per
lung on average, while the sh-FUBP3 group showed reduced metastatic foci numbers
to approximately 5–8, with a metastatic inhibition rate of 60–70%. After
rescuing FUBP3 expression, metastatic foci numbers recovered to approximately
12–16, confirming the FUBP3-dependence of the metastatic inhibition effect (n =
6, p
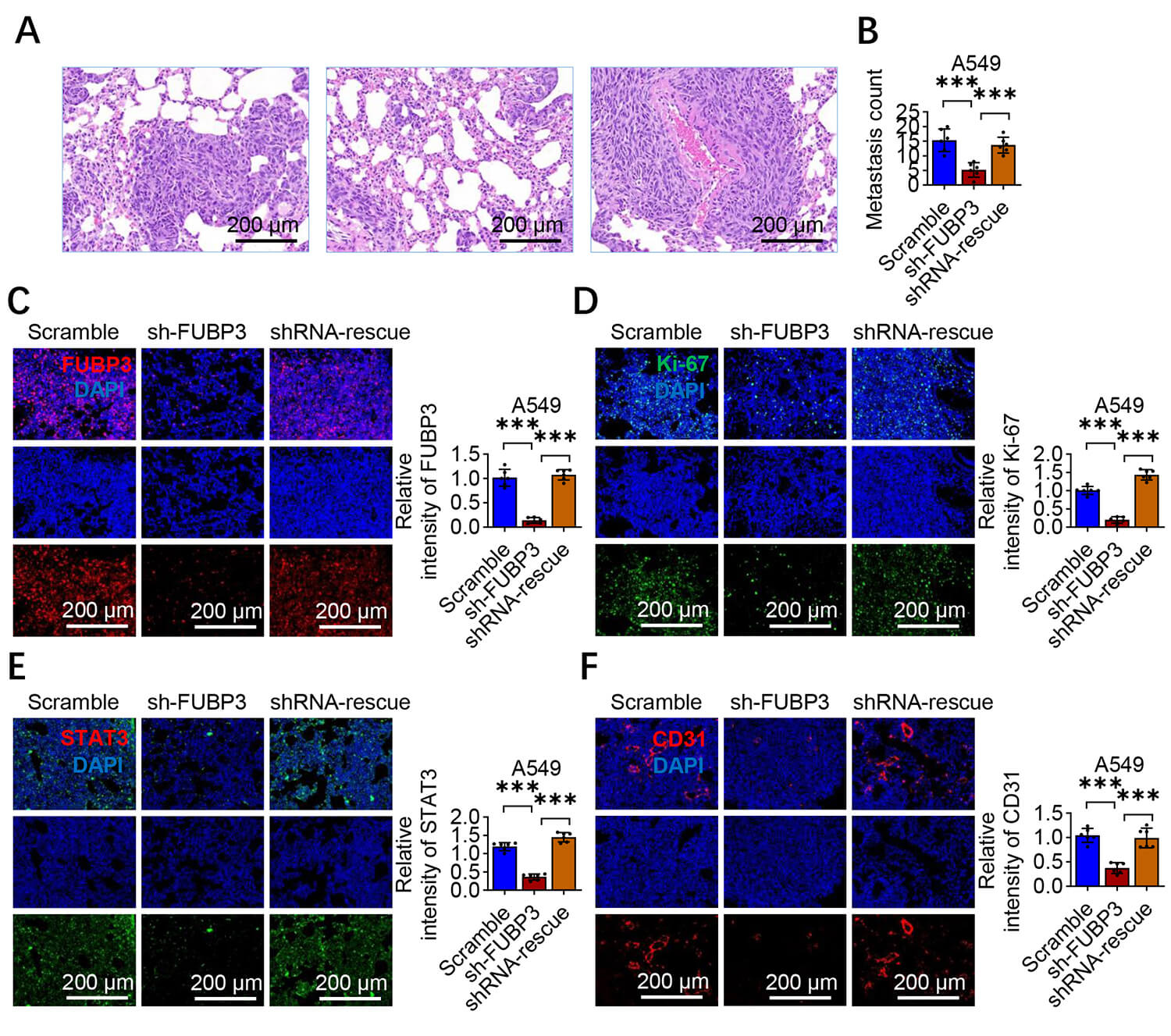 Fig. 3.
Fig. 3.
FUBP3 silencing suppresses lung cancer metastatic capacity
in vivo. (A) Experimental scheme of tail vein injection metastasis
model using A549 cells with FUBP3 modifications. Scale bar: 200 µm. (B)
Quantitative analysis of pulmonary metastatic nodules in nude mice (n = 6 per
group). (C) Representative immunofluorescence staining of FUBP3 (green) in lung
metastatic lesions. DAPI (blue) indicates nuclei. Scale bar: 200 µm. (D)
Ki-67 immunofluorescence staining (green) showing proliferation in lung
metastatic tissues. Scale bar: 200 µm. (E) STAT3 (green) and (F) CD31 (red)
immunofluorescence staining in lung metastatic lesions demonstrating reduced
expression in FUBP3 knockdown groups. Scale bar: 200 µm. Data are presented
as mean
To further elucidate FUBP3’s functional pathways, we performed functional enrichment analysis on genes positively co-expressed with FUBP3. Results showed that genes co-expressed with FUBP3 were enriched in multiple tumor-related functional pathways, including focal adhesion, regulation of actin cytoskeleton, non-small cell lung cancer, pathways in cancer, JAK–STAT signaling pathway, vascular endothelial growth factor (VEGF) signaling pathway, and Extracellular Matrix (ECM)-receptor interaction (Fig. 4A). Enrichment of these pathways further supports FUBP3’s important role in regulating cell motility, invasion, and EMT processes, while enrichment of the JAK–STAT signaling pathway suggests that STAT3 may be an important downstream molecule of FUBP3. Cytoskeletal staining showed control A549 and H460 cells exhibited spindle-shaped mesenchymal morphology with pseudopodia. FUBP3 knockdown induced rounded epithelial-like morphology with tighter intercellular connections, which was reversed upon FUBP3 rescue (Fig. 4B). Scanning electron microscopy confirmed that sh-FUBP3 cells had smoother surfaces with reduced pseudopodia, while control and rescue groups displayed abundant membrane protrusions characteristic of motile cells (Fig. 4C). These morphological changes are highly consistent with characteristics of cellular transformation from epithelial to mesenchymal phenotype during EMT processes. To validate EMT marker changes at the molecular level, we performed immunofluorescence staining analysis. E-cadherin immunofluorescence staining showed that FUBP3 knockdown upregulated expression of the epithelial marker E-cadherin, with E-cadherin primarily localized at cell membranes, forming continuous staining patterns at intercellular junctions. In contrast, E-cadherin expression was relatively low in control and rescue groups, with more dispersed distribution (Fig. 4D). Conversely, immunofluorescence staining of the mesenchymal marker Vimentin showed opposite changes. In control and rescue group cells, Vimentin exhibited typical filamentous distribution in the cytoplasm with high expression levels. In the sh-FUBP3 group, Vimentin expression was decreased with significantly reduced filamentous structures, indicating weakened cellular mesenchymal characteristics (Fig. 4E). Western blot analysis further quantitatively confirmed EMT marker expression changes. In both A549 and H460 cell lines, FUBP3 knockdown resulted in upregulated E-cadherin protein levels and downregulated Vimentin protein levels. After rescuing FUBP3 expression, E-cadherin expression was downregulated while Vimentin expression was upregulated, reversing the EMT marker expression pattern (Fig. 4F). This result was consistent across two independent lung cancer cell lines, further confirming FUBP3’s important role in regulating EMT processes.
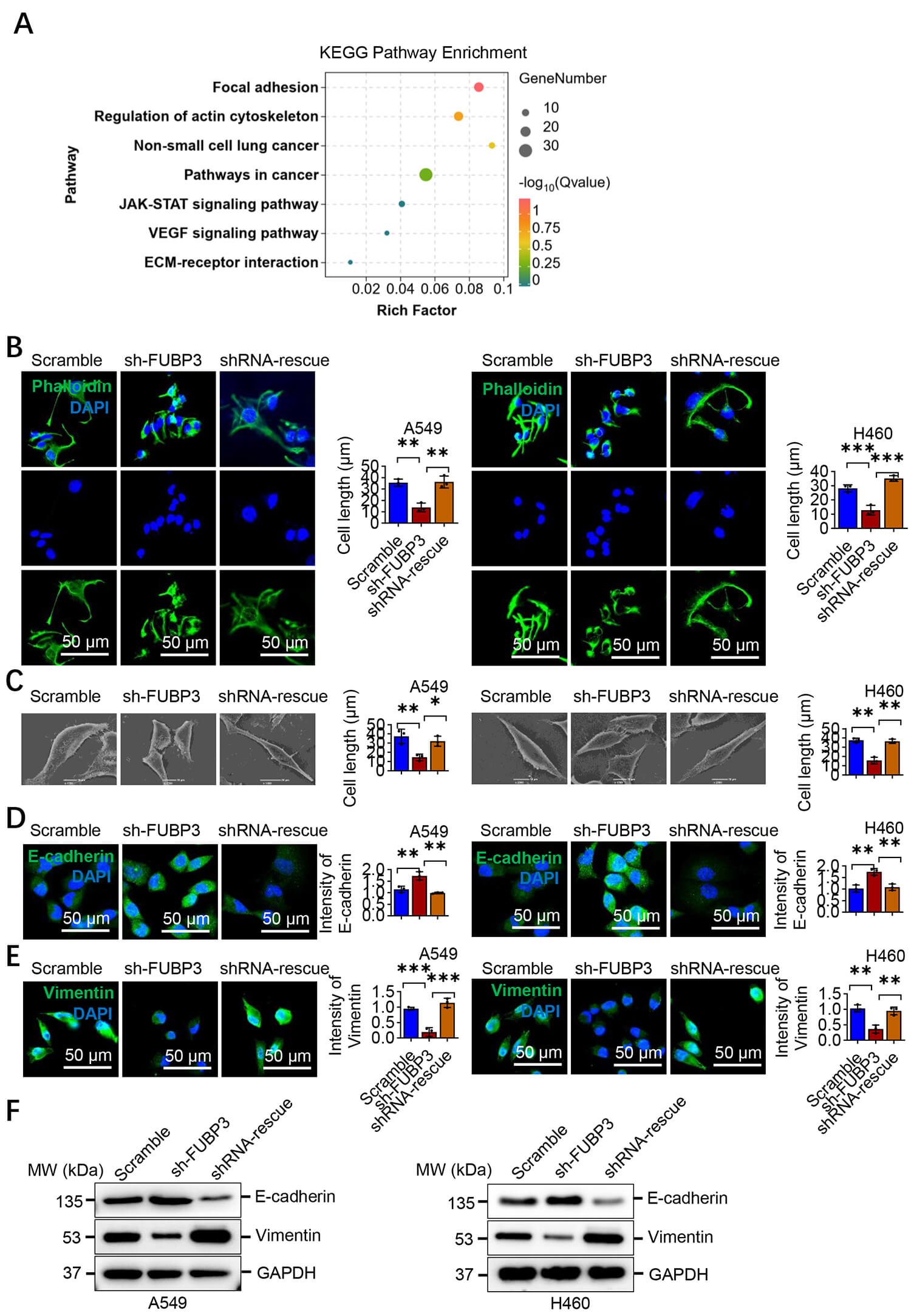 Fig. 4.
Fig. 4.
FUBP3 drives lung cancer metastasis by promoting EMT process.
(A) KEGG pathway enrichment analysis of FUBP3 positively co-expressed genes
showing significant enrichment in focal adhesion, actin cytoskeleton regulation,
JAK–STAT pathway and cancer-related pathways. Analysis was performed using TCGA
lung adenocarcinoma dataset. The bubble size represents gene count and color
indicates adjusted p-value. (B) Phalloidin staining (green) showing
cytoskeleton organization in A549 and H460 cells following FUBP3 manipulation.
DAPI (blue) indicates nuclei. Scale bar: 50 µm. (C) Scanning electron
microscopy analysis revealing morphological changes in A549 and H460 cells with
FUBP3 knockdown and rescue. Scale bar: 10 µm. (D) Immunofluorescence
staining for epithelial marker E-cadherin (green) in FUBP3-modified A549 and H460
cells. Scale bar: 50 µm. (E) Immunofluorescence analysis of mesenchymal
marker Vimentin (red) expression in A549 and H460 cells. Scale bar: 50 µm.
(F) Western blot analysis of EMT markers E-cadherin and Vimentin in FUBP3
knockdown and rescue cell lines. Data are presented as mean
To further elucidate the molecular mechanisms by which FUBP3 regulates EMT, we
examined the effects of FUBP3 on major EMT transcription factors. qRT-PCR
analysis showed that FUBP3 knockdown reduced mRNA expression levels of Twist1 and
snail family transcriptional repressor 1 (SNALl), while having no effect on Zinc
finger E-box-binding homeobox 1 (ZEB1) expression. Among these, Twist1 expression
downregulation was most significant, suggesting it may be a key downstream
molecule in FUBP3-mediated EMT regulation (Fig. 5A). Immunofluorescence staining
further confirmed that FUBP3 knockdown led to reduced Twist1 protein expression
in the cell nucleus, with nuclear fluorescence intensity decreased by
approximately 70% compared to the control group (Fig. 5B). Clinical sample
correlation analysis showed that FUBP3 and Twist1 expression levels were
positively correlated in lung cancer patient tissues (p
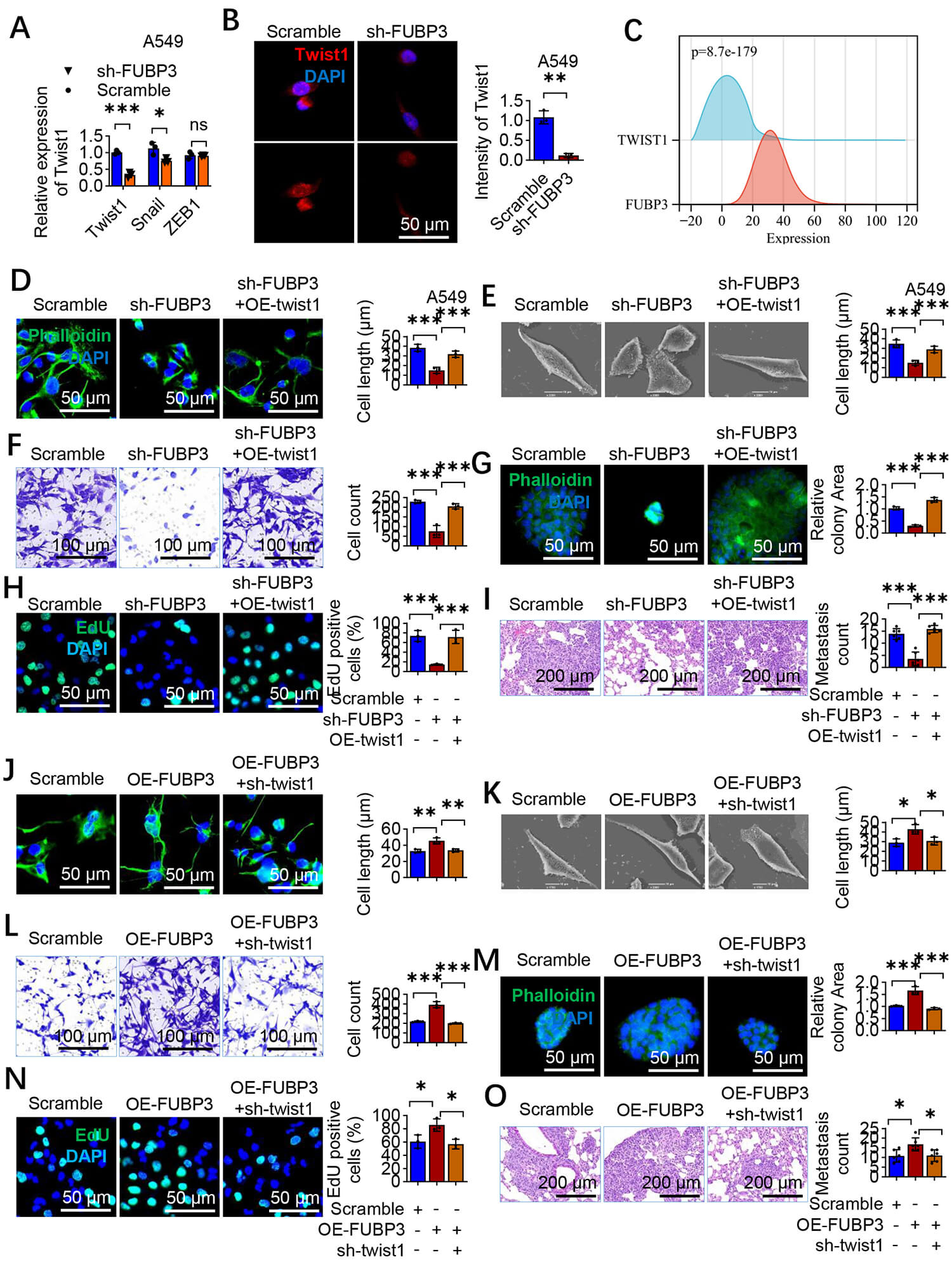 Fig. 5.
Fig. 5.
FUBP3 promotes lung cancer progression through upregulation of
Twist1 expression. (A) qRT-PCR analysis of EMT transcription factors in FUBP3
knockdown A549 cells showing significant reduction in Twist1 expression. Gene
expression was normalized to GAPDH. (B) Immunofluorescence staining confirming
reduced Twist1 protein (green) in FUBP3 knockdown A549 cells. Representative
images (left) and quantification of fluorescence intensity (right) are shown.
DAPI (blue) indicates nuclei. Scale bar: 50 µm. (C) Correlation analysis
showing positive association between FUBP3 and Twist1 expression in lung cancer
patients (p = 8.7
To explore the molecular mechanisms by which FUBP3 regulates Twist1, based on
the enrichment of JAK–STAT signaling pathway in previous functional enrichment
analysis, we focused on investigating the potential interaction between FUBP3 and
STAT3. Protein–protein interaction network analysis revealed a direct protein
interaction relationship between FUBP3 and STAT3, suggesting they may form
functional complexes to co-regulate downstream gene expression (Fig. 6A).
Clinical sample analysis showed that FUBP3 and STAT3 expression levels were
positively correlated in lung cancer tissues (p
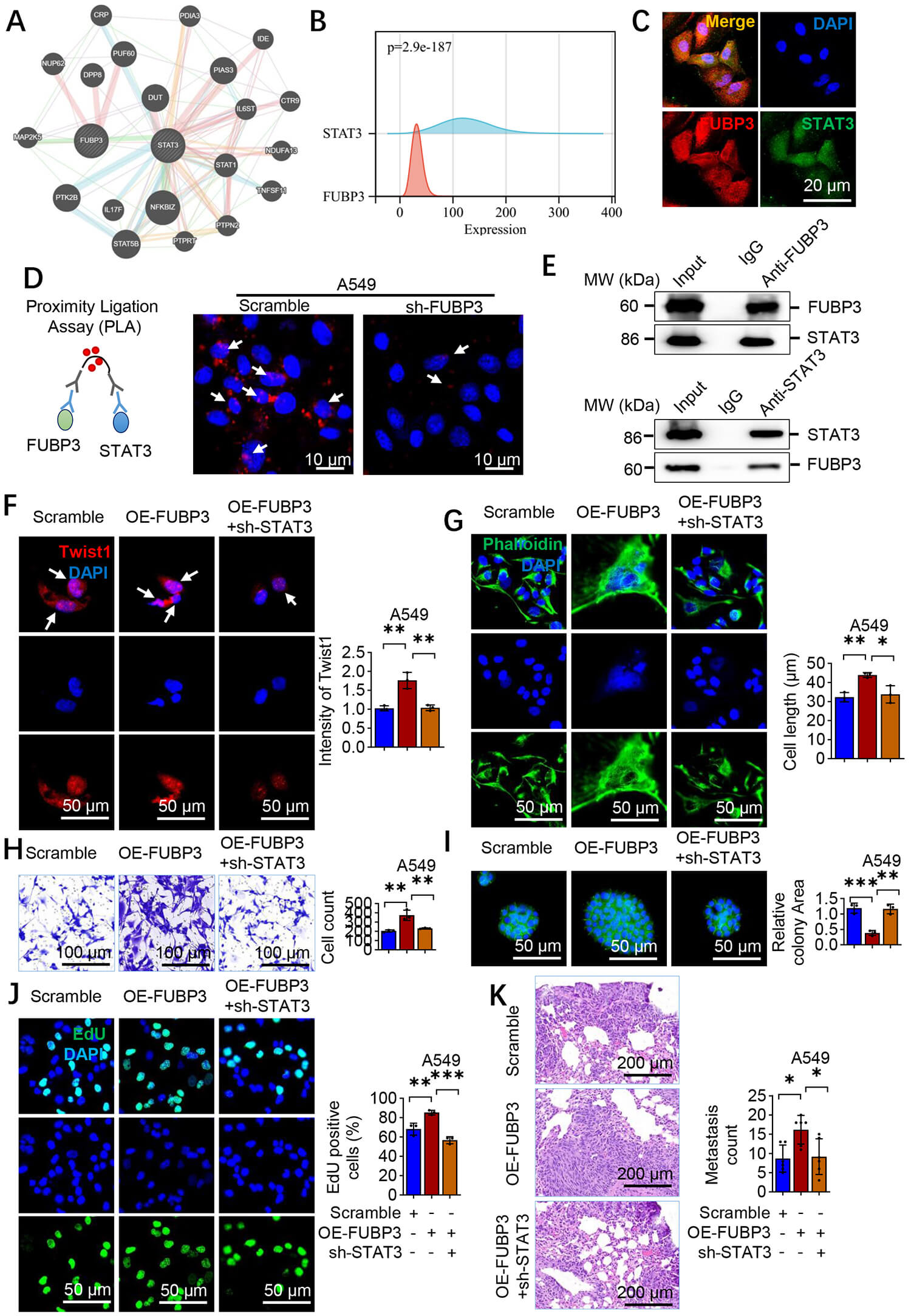 Fig. 6.
Fig. 6.
FUBP3–STAT3 interaction upregulates Twist1 to promote lung
cancer progression. (A) Protein–protein interaction network analysis revealing
direct interaction between FUBP3 and STAT3. (B) Positive correlation between
FUBP3 and STAT3 expression in lung cancer datasets (p = 2.9
To further validate STAT3’s critical role in FUBP3-mediated regulation of lung cancer metastasis and explore potential therapeutic targets, we treated FUBP3-overexpressing A549 cells with the STAT3 small molecule inhibitor S3I-201 and systematically evaluated its antagonistic effects on FUBP3 function. Twist1 immunofluorescence staining analysis showed that FUBP3 overexpression upregulated Twist1 expression in the cell nucleus, with nuclear fluorescence intensity increased approximately 1.5-fold compared to the control group. S3I-201 treatment alone reduced basal Twist1 expression levels. Importantly, S3I-201 effectively reversed the Twist1 upregulation induced by FUBP3 overexpression, reducing its expression levels (Fig. 7A). This result confirms the necessity of STAT3 activity for FUBP3-mediated Twist1 regulation. To comprehensively evaluate S3I-201’s effects on EMT markers, we examined transcriptional level changes in epithelial and mesenchymal markers. qRT-PCR analysis showed that S3I-201 treatment upregulated mRNA expression of the epithelial marker E-cadherin, while FUBP3 overexpression reduced E-cadherin expression. S3I-201 effectively reversed the E-cadherin downregulation induced by FUBP3 overexpression (Fig. 7B). Conversely, expression changes of the mesenchymal marker Vimentin showed opposite trends: S3I-201 treatment reduced basal Vimentin expression, while FUBP3 overexpression upregulated Vimentin expression, and S3I-201 effectively blocked this upregulation effect (Fig. 7C). Cell morphological analysis confirmed S3I-201’s antagonistic effects on EMT processes. Cytoskeletal staining showed that FUBP3 overexpression promoted A549 cell transformation toward typical spindle-shaped mesenchymal-like morphology, with cells becoming longer and thinner. Addition of S3I-201 to FUBP3-overexpressing cells effectively reversed mesenchymal-like morphological changes, with cells re-exhibiting relatively rounded and tightly connected epithelial-like characteristics (Fig. 7D). Functional experiments further confirmed S3I-201’s antagonistic effects on FUBP3’s pro-oncogenic functions. Transwell invasion assays showed that S3I-201 treatment inhibited basal cellular invasive capacity, while FUBP3 overexpression enhanced invasive capacity. Importantly, S3I-201 effectively reversed the invasion-promoting effects induced by FUBP3 overexpression, reducing invasive cell numbers (Fig. 7E). Clonogenic sphere formation assays showed similar result patterns. S3I-201 treatment inhibited cellular sphere formation capacity, while FUBP3 overexpression promoted sphere formation. S3I-201 effectively blocked the promoting effects of FUBP3 overexpression, with both sphere number and size reduced (Fig. 7F). EdU proliferation assays confirmed that S3I-201 not only inhibited basal cellular proliferative activity but also completely reversed the proliferation-promoting effects induced by FUBP3 overexpression (Fig. 7G). TUNEL apoptosis assays showed that S3I-201 treatment increased cellular apoptosis levels, with the proportion of TUNEL-positive cells increased approximately 2–3-fold compared to the control group. FUBP3 overexpression reduced basal cellular apoptosis levels, while S3I-201 reversed this anti-apoptotic effect, restoring apoptosis levels to or even exceeding control group levels (Fig. 7H). This result indicates that STAT3 activity is necessary for FUBP3’s anti-apoptotic function. Most importantly, in vivo metastasis experiments confirmed S3I-201’s therapeutic potential. In the nude mouse tail vein injection model, S3I-201 treatment reduced pulmonary metastatic foci formation. FUBP3 overexpression increased metastatic foci numbers, while S3I-201 effectively reversed this pro-metastatic effect, reducing metastatic foci numbers to below control group levels (Fig. 7I). This in vivo experimental result provides important pharmacodynamic evidence for S3I-201 as a potential therapeutic agent. In summary, the STAT3 small molecule inhibitor S3I-201 comprehensively antagonizes FUBP3’s pro-oncogenic functions by inhibiting STAT3 activity to block the FUBP3-STAT3-Twist1 regulatory axis, reversing EMT processes, inhibiting cell proliferation, invasion, and metastatic capacity, and promoting cell apoptosis. These results not only further confirm STAT3’s central role in FUBP3 function but also provide important experimental evidence for STAT3 inhibition-based lung cancer therapeutic strategies. The effective antagonistic effects of S3I-201 indicate that targeting the FUBP3-STAT3 regulatory axis may become a novel strategy for precision lung cancer therapy (Fig. 8).
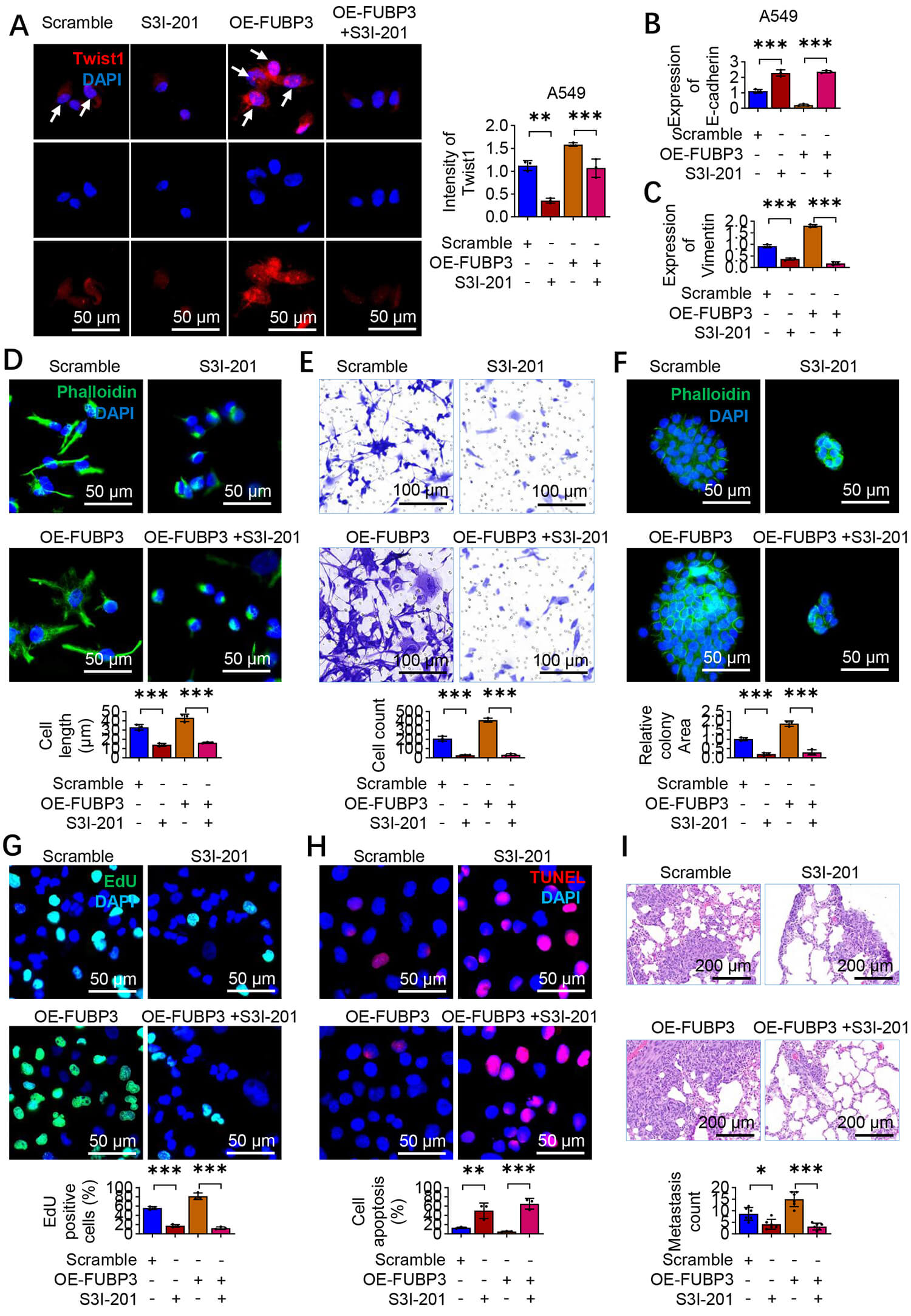 Fig. 7.
Fig. 7.
STAT3 inhibitor S3I-201 reverses FUBP3-induced lung cancer
progression. (A) Immunofluorescence staining showing Twist1 (green) expression
in A549 cells treated with FUBP3 overexpression and STAT3 inhibitor S3I-201.
White arrows indicate positive nuclear expression of Twist1. Scale bar: 50
µm. (B,C) PCR analysis of epithelial marker E-cadherin (B) and mesenchymal
marker Vimentin (C) in A549 cells with FUBP3 overexpression and S3I-201
treatment. (D) Phalloidin staining demonstrating morphological changes in A549
cells with FUBP3 overexpression and S3I-201 treatment. Scale bar: 50 µm.
(E) Transwell invasion assay evaluating the effects of S3I-201 on
FUBP3-overexpressing cells. Scale bar: 100 µm. (F) Sphere formation assay
with FUBP3 overexpression and S3I-201 treatment. Scale bar: 50 µm. (G) EdU
proliferation assay with FUBP3 overexpression and S3I-201 treatment. Scale bar:
50 µm. (H) TUNEL assay assay with FUBP3 overexpression and S3I-201
treatment. Scale bar: 50 µm. (I) In vivo metastasis analysis with
S3I-201 treatment in FUBP3-overexpressing A549 cells. Scale bar: 200 µm.
Data are presented as mean
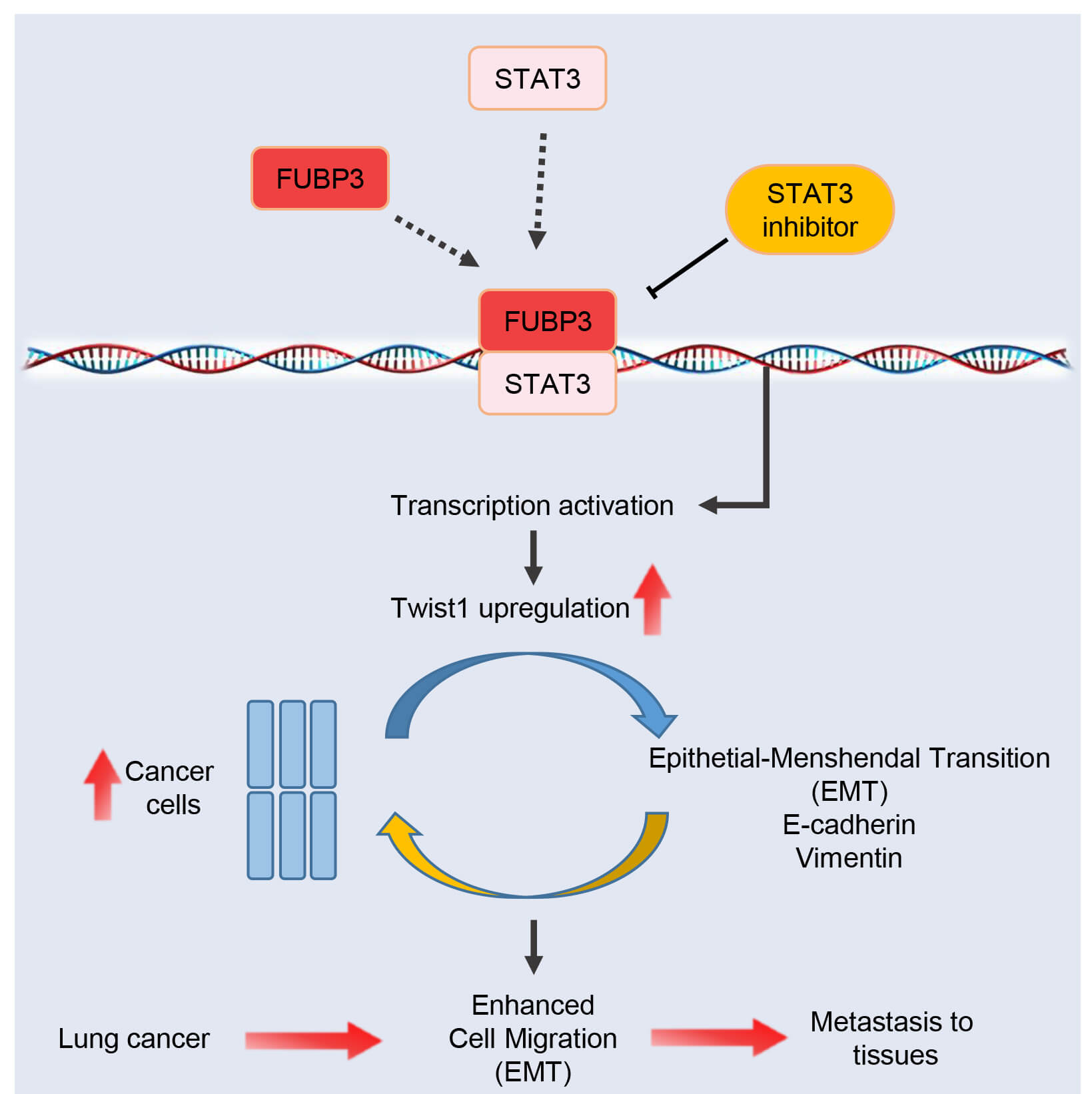 Fig. 8.
Fig. 8.
Proposed mechanistic model. Schematic illustration of the FUBP3-STAT3-Twist1 regulatory axis promoting epithelial-mesenchymal transition (EMT) and lung cancer metastasis. FUBP3 physically interacts with STAT3 to enhance Twist1 transcription, leading to downregulation of epithelial marker E-cadherin and upregulation of mesenchymal marker Vimentin, accompanied by actin cytoskeleton remodeling. This cascade ultimately drives EMT process, enhancing cancer cell migration, invasion, self-renewal capacity, and metastatic potential.
This research highlights the significant function of FUBP3 in lung cancer metastasis via a newly identified FUBP3-STAT3-Twist1 regulatory axis that facilitates the EMT. Our findings indicate that FUBP3 physically interacts with STAT3 to enhance Twist1 transcription, resulting in cytoskeletal remodeling, downregulation of E-cadherin, and upregulation of vimentin, which ultimately facilitates cancer cell invasion and metastasis. Pharmacological inhibition of STAT3 with S3I-201 effectively reverses metastatic phenotypes induced by FUBP3. The findings offer mechanistic insights into lung cancer metastasis and identify FUBP3 as a potential therapeutic target for clinical intervention.
FUBP3, a member of the RBP family, has seen relatively delayed functional studies in tumors [23, 24, 34]. This study demonstrates that FUBP3 upregulation in lung cancer correlates with poor patient prognosis, aligning with existing research indicating that its family member, FUBP1 plays an oncogenic role in multiple tumors. Bioinformatics analysis indicates that lung cancer patients exhibiting elevated FUBP3 expression experience reduced OS. Furthermore, FUBP3 expression levels in M1 stage patients are significantly greater than those in M0 stage patients, implying a direct correlation between FUBP3 and the metastatic potential of lung cancer. This clinical association establishes a significant pathological basis for future functional studies and indicates that FUBP3 may function as a potential biomarker for prognostic evaluation in lung cancer. Functional experiments provide additional evidence for the role of FUBP3 in promoting lung cancer metastasis. Silencing FUBP3 expression via RNA interference technology resulted in decreased migration and invasion capabilities of lung cancer cells. This finding was corroborated across multiple lung cancer cell lines, suggesting a universal regulatory role of FUBP3 in lung cancer metastasis. In vivo metastasis experiments indicate that FUBP3 knockdown leads to a reduction in pulmonary metastatic foci formation, thereby confirming the critical regulatory role of FUBP3 in lung cancer metastasis in the in vivo context. The findings correspond with contemporary research trends concerning the functions of RBPs in tumor metastasis, underscoring their significant role as regulatory factors in this process.
The EMT process is a critical step for tumors to acquire metastatic capacity [35, 36, 37], and our study confirms that FUBP3 promotes lung cancer metastasis through regulating the EMT process. After the knockdown of FUBP3, epithelial marker E-cadherin expression was upregulated, mesenchymal marker vimentin expression was downregulated, and cellular morphology shifted toward epithelial characteristics. The observed changes suggest that FUBP3 is an important regulatory factor in the EMT. Further analysis indicates that FUBP3 knockdown results in the enrichment of EMT-related gene sets, encompassing various metastasis-associated biological processes such as cytoskeletal remodeling and interactions with extracellular matrix receptors. This finding establishes a direct connection between FUBP3 and EMT, a fundamental mechanism underlying tumor metastasis, thereby offering significant insights into the molecular framework of FUBP3-induced metastasis. FUBP3 knockdown resulted in modest apoptosis and impaired cell proliferation, which may partially explain the observed effects on migration, invasion, and metastasis. The considerable decrease in metastatic capacity observed across multiple functional assays seems to exceed what would be expected from altered cell survival alone, suggesting that FUBP3 coordinately regulates multiple cancer hallmarks, including cell survival and metastatic behavior. Future research utilizing anoikis-resistant cell models or apoptosis inhibitors may elucidate the distinct roles of FUBP3 in cell survival and metastatic potential.
Twist1, as a core transcription factor in the EMT process, has been a research hotspot regarding its regulatory mechanisms [6, 38, 39]. Our findings indicate that FUBP3 enhances lung cancer progression through the upregulation of Twist1 expression, thereby uncovering a novel mechanism of Twist1 transcriptional regulation. Correlation analysis indicates a positive correlation between FUBP3 and Twist1 expression in lung cancer patient tissues, thereby confirming their functional association in clinical samples. Rescue experiments provide additional evidence that re-expression of Twist1 can partially counteract the inhibitory effect of FUBP3 knockdown on cellular metastatic capacity, suggesting that Twist1 serves as a significant downstream target for FUBP3’s pro-metastatic function. This finding clarifies the mechanism of FUBP3 action and introduces a new regulatory factor to the upstream regulatory network of Twist1.
Immunofluorescence co-localization and co-immunoprecipitation experiments validated the direct interaction between FUBP3 and STAT3, establishing a molecular foundation for the formation of transcriptional complexes. STAT3, an important transcription factor, demonstrates sustained activation in tumors and is associated with multiple malignant phenotypes. This protein–protein interaction pattern is prevalent in transcriptional regulation and signifies a novel application in the regulation of EMT transcription factors. Silencing of STAT3 can counteract the pro-metastatic effects associated with FUBP3 overexpression in lung cancer metastasis, thereby reinforcing the essential role of STAT3 in the metastatic regulation mediated by FUBP3. Treatment experiments utilizing the STAT3 inhibitor S3I-201 yield significant evidence for the clinical applicability of the findings from this study. The S3I-201 treatment has the capacity to reverse the pro-metastatic phenotype observed in cells with high FUBP3 expression and to restore the typical expression patterns of EMT markers. This finding confirms the essential function of STAT3 within the FUBP3–Twist1 regulatory axis and indicates that targeting STAT3 could be a viable approach for treating lung cancers with high FUBP3 expression. In vivo experiments indicate that S3I-201 treatment decreases the formation of pulmonary metastatic foci, thereby offering proof-of-concept for precision therapy targeting the FUBP3–STAT3–Twist1 axis.
This study establishes the FUBP3–STAT3–Twist1 regulatory axis, offering a new theoretical framework for comprehending the mechanisms of lung cancer metastasis. This regulatory axis encompasses the roles of RBPs, signal transduction molecules, and transcription factors, highlighting the intricate and multifaceted aspects of gene expression regulation. This regulatory pattern elucidates the pro-metastatic function of FUBP3 and serves as a reference model for exploring the mechanistic roles of other RBPs in tumor metastasis. The findings of this study hold significant translational value from a clinical perspective. FUBP3 expression levels may function as biomarkers for prognostic evaluation in lung cancer patients, aiding in the identification of individuals at elevated metastatic risk. The interaction between FUBP3 and STAT3 presents new targets for drug development. Designing specific inhibitors to disrupt these protein–protein interactions may facilitate precision intervention in metastatic processes. The effectiveness of STAT3 inhibitors reinforces the viability of this therapeutic approach. Our findings indicate that FUBP3 physically interacts with STAT3 and is essential for STAT3-mediated upregulation of Twist1 and EMT in NSCLC. The exact molecular mechanism by which FUBP3 facilitates STAT3-dependent transcriptional regulation necessitates additional research. However, our findings align with growing evidence indicating that STAT3 modulates various transcription factors implicated in cancer metastasis. Prior research indicates that STAT3 activation enhances Twist1 expression across multiple cancer types [40, 41, 42, 43]. Furthermore, STAT3 has been shown to influence EMT-related transcription factors via direct DNA binding as well as indirect mechanisms involving protein–protein interactions and transcriptional co-regulation [44, 45, 46]. The physical interaction between FUBP3 and STAT3, as demonstrated by Co-IP and PLA experiments, and the functional dependence of Twist1 expression on both proteins indicate that FUBP3 act as a critical co-factor that enhances the transcriptional activity of STAT3. The mechanisms by which this occurs, whether through direct recruitment to the Twist1 promoter, stabilization of STAT3 transcriptional complexes, or modulation of chromatin accessibility, should be further investigated. Our data identify FUBP3 as a new regulator of the STAT3–Twist1 axis, laying the groundwork for comprehending FUBP3’s involvement in the promotion of NSCLC metastasis.
This study has several limitations. First, the binding pattern and specific regulatory mechanisms of the FUBP3–STAT3 complex on the Twist1 promoter were not comprehensively examined. Second, the consistent functionality of this regulatory axis across various lung cancer subtypes should be validated. Additionally, apart from Twist1, the FUBP3–STAT3 complex may regulate other genes associated with EMT. Comprehensive screening and functional validation of these potential targets will be crucial for future research.
This study elucidates the critical role of FUBP3 in lung cancer metastasis and its underlying molecular mechanisms. Our findings indicate that FUBP3 is highly expressed in lung cancer tissues and is closely associated with poor patient prognosis. Functional studies confirm that FUBP3 enhances the migration and invasion capabilities of lung cancer cells by facilitating EMT processes. Mechanistic investigations indicate that FUBP3 drives lung cancer EMT and metastasis by recruiting STAT3, leading to the formation of transcriptional complexes that specifically activate the expression of the key EMT transcription factor Twist1. The STAT3 inhibitor S3I-201 effectively reverses the pro-metastatic effects mediated by FUBP3, offering evidence for targeted therapeutic approaches. This study establishes the FUBP3–STAT3–Twist1 regulatory axis, offering a novel theoretical framework for elucidating the molecular mechanisms underlying lung cancer metastasis and enhancing the functional understanding of RBPs in tumor metastasis. FUBP3 may function as a biomarker for prognostic evaluation in lung cancer, and its interaction with STAT3 presents new avenues for drug development.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
WW, LZ and PW designed the study. WW and LZ performed the experiments and analyzed the data. WW and PW wrote the initial draft of the paper, with contributions from all authors. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiments met the ethical requirements and were approved by the Institutional Animal Care and Use Committee of Liaocheng People’s Hospital (approval number: 2025257). We confirm that all animal experiments reported in the manuscript were conducted in accordance with the ARRIVE guidelines. The clinical study was approved by the Medical Ethics Committee of Liaocheng People’s Hospital (approval number: 2025256). All procedures were conducted in accordance with the ethical standards of the Declaration of Helsinki, and written informed consent was obtained from all participants.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47541.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.