








1 Department of General Surgery, The Second Affiliated Hospital of Harbin Medical University, 150081 Harbin, Heilongjiang, China
2 School of Basic Medicine, Harbin Medical University, 150081 Harbin, Heilongjiang, China
Abstract
Triple-negative breast cancer (TNBC) is an aggressive malignancy that lacks effective treatment. Immune infiltration plays an important role in anti-tumor responses. Serpin family G1 (SERPING1), a biomarker associated with immune infiltration, has been implicated in multiple cancers, but its role in TNBC remains unclear.
RNA sequencing and clinical data for TNBC were obtained from the Gene Expression Omnibus, the Cancer Genome Atlas, and the Molecular Taxonomy of Breast Cancer International Consortium databases. First, the expression, prognostic value, and biological functions of SERPING1 were analyzed. Then, the tumor microenvironment (TME) was comprehensively characterized, and the relationship between SERPING1 expression and immunotherapy response was assessed. Immunohistochemical staining was performed to confirm SERPING1 expression and the abundance of CD4+ T cells and CD8+ T cells in clinical specimens. Finally, single-cell analysis was conducted to investigate the role of SERPING1 in immune cell activation.
SERPING1 was downregulated in TNBC and was an independent predictor of survival. Functionally, SERPING1 activated the immune response in TNBC patients. Mechanistically, elevated SERPING1 levels lead to increased immune cell infiltration, particularly of CD4+ and CD8+ T cells, in the TME. Moreover, SERPING1 was primarily localized in cancer-associated fibroblasts (CAFs), with SERPING1+ apCAFs exhibiting increased communications with anti-tumor immune cells at the single-cell level.
SERPING1 contributes to enhanced immune cell infiltration, desirable immunotherapy response, and improved prognosis. It thus can be utilized as a promising biomarker for immune infiltration and prognosis. These findings provide novel insights into TME-related immune regulation and may inform strategies to enhance immunotherapy efficacy in TNBC.
Keywords
- triple negative breast neoplasms
- tumor microenvironment
- immunomodulation
- prognosis
- biomarker
- immunotherapy
Breast cancer (BC) is the most common malignancy among women globally, with triple-negative breast cancer (TNBC) accounting for approximately 15–20% of cases. TNBC is associated with a particularly poor prognosis, characterized by its high histological grade and increased rates of metastasis [1]. Due to the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression, TNBC is unresponsive to endocrine and HER2-targeted therapies [2]. As a result, surgery and chemotherapy remained the standard treatments for TNBC patients over the past decades [3].
In recent years, immune checkpoint inhibitors (ICI) have achieved some success across multiple tumor types [4, 5]. As the most immunogenic subtype of BC, TNBC has demonstrated clinical benefit from immunotherapy. The programmed cell death ligand 1 (PD-L1, also known as CD274) blocking antibodies, atezolizumab and pembrolizumab, combined with chemotherapy, have been approved as first-line treatment for PD-L1-positive metastatic TNBC [6, 7, 8]. However, due to limited expression of PD-L1 and substantial heterogeneity of TNBC tumor microenvironment (TME), not all patients benefit from ICI treatment, and approximately 60%–85% of them develop primary resistance to ICI monotherapy [8, 9]. Therefore, there is an urgent need to explore novel therapeutic targets and enhance the efficacy of immunotherapy in TNBC.
Currently, research on solid tumors has shifted from focusing solely on tumor cells to encompassing the TME [10]. Tumor-infiltrating lymphocytes (TILs) play a critical role in treatment response and prognosis, serving as key determinants of immunotherapy efficacy [11]. However, TME comprises not only lymphocytes but also stromal cells, immunosuppressive cells, extracellular matrix (ECM), and other components [12]. Increasing evidence indicates that cancer cells do not act alone but interact closely with different cell populations and ECM [13]. The ECM, as a key structural and supportive component of the TME, constitutes more than 90% of breast tumors. Among its constituents, cancer-associated fibroblasts (CAFs) are the most abundant cell type, characterized by marked heterogeneity and plasticity [14, 15]. CAFs profoundly influence tumor growth, invasion, and immune responses through ECM remodeling, metabolic regulation, and signaling interactions with both tumor and immune cells [16]. With the deepening of research on TME, novel targets correlated with immune infiltration have been identified [17]. These discoveries offer new opportunities to improve cancer treatment, particularly by enhancing immunotherapy and strengthening host anti-tumor immune responses.
SERPING1, located on chromosome 11, encodes the C1-inhibitor, a highly glycosylated plasma protein that regulates the complement cascade, as well as fibrinolytic, clotting, and kinin pathways. It plays a crucial role in controlling vascular permeability and innate immunity [18]. Recently, SERPING1 has gained attention for its involvement in tumor biology and the TME [19]. Evidence indicated that SERPING1 was downregulated in liver cancer cells, with low expression linked to an immunosuppressive TME and poor prognosis [20]. Similarly, the anti-cancer role of SERPING1 was also demonstrated in gastric and prostate cancers [21, 22]. However, limited knowledge exists regarding the distinct roles of SERPING1 in TNBC, particularly in relation to TME and immune status, highlighting the need for further investigation.
Herein, we conducted a comprehensive investigation focusing on the function of SERPING1 in TNBC. We noticed that SERPING1 expression was significantly downregulated in TNBC compared with normal tissue, with low SERPING1 levels associated with poor prognosis and diminished responses to immunotherapy. Mechanistically, SERPING1 regulated the infiltration of various immune cells, especially T lymphocytes. Overall, our findings provided a thorough understanding of the complex roles of SERPING1 in TNBC and highlighted its potential as a predictive biomarker in clinical practice.
Gene expression profiles and clinicopathological information were downloaded from the Cancer Genome Atlas (TCGA) data portal. Single-cell RNA sequencing (scRNA-seq) data (GSE176078) were obtained from the Gene Expression Omnibus (GEO) database. External validation was performed using data from Molecular Taxonomy of Breast Cancer International Consortium (METABRIC), Kaplan-Meier plotter (KM plotter: https://kmplot.com/analysis/) and GEO datasets (GSE45827, GSE103091). Proteomic expression of SERPING1 was analyzed via UALCAN [23]. The immunotherapy response data were derived from GSE91061 dataset and IMvigor210 cohort.
RNA-seq data from 118 TNBC and 99 normal samples from TCGA were selected for
analysis. Using the “DESeq2” package (version 1.40.2), 5239 differentially
expressed genes (DEGs) were identified with
ESTIMATE algorithm was used to calculate the tumor purity and immune scores of TNBC samples in TCGA cohort [24]. xCell, quanTIseq, MCPcounter, CIBERSORT Absolute, and EPIC algorithms were implemented using the “IOBR” R package (version 0.99.8). The relationship between SERPING1 expression and the tumor infiltration status of 29 immune gene sets was assessed using the ssGSEA algorithm [25]. Moreover, the TIMER algorithm(http://cistrome.org/TIMER/) was utilized to investigate the correlations between SERPING1 expression and CD4+ and CD8+ T cell infiltration in basal BC [26].
Major histocompatibility complex (MHC) molecules and immune checkpoints were compared between the high and low SERPING1 expression groups [27]. Immunophenoscore (IPS) scores from The Cancer Immunome Atlas (TCIA) (https://tcia.at/home) and tumor immune exclusion score from TIDE algorithm (http://tide.dfci.harvard.edu/) were assessed to evaluate tumor immunogenicity [28, 29].
Weighted Gene Co-expression Network Analysis (WGCNA) was conducted using the “WGCNA” R package on TNBC samples from the TCGA database [30]. Hierarchical clustering was performed based on gene expression, identifying genes with high similarity within modules using the dynamic cut tree method. The Module Eigengene (ME) value for each module and its correlation coefficients with ESTIMATE scores were calculated. A soft thresholding power of 3 was selected. The minimum module size was set at 30. Then, least absolute shrinkage and selection operator (LASSO) COX regression analysis was used to determine the final hub genes associated with immune infiltration.
To annotate potential biological processes associated with SERPING1, we performed enrichment analysis using Gene Ontology (GO) (http://geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/), with the “ClusterProfiler” R package (version 4.8.2). Multiple testing was corrected using the Benjamini-Hochberg FDR method. GO analysis was conducted based on 3 categories, including biological process (BP), molecular function (MF), and cellular component (CC). Additionally, Gene Set Enrichment Analysis (GSEA) was performed as a secondary step, where it evaluates sets of biologically relevant genes, such as those in specific pathways, and categorizes gene groups based on their enrichment scores.
A total of 18 TNBC samples and 28 normal breast tissue samples were collected from the Second Affiliated Hospital of Harbin Medical University (HMU). Informed consent was obtained from all participants, and the study was approved by the Ethics Committee of HMU. All samples were diagnosed by a minimum of 3 pathologists, and the tissue microarray was conducted following standard procedures. All research was performed following the Declaration of Helsinki.
The tissue microarrays were immunostained as described previously [31]. The
anti-SERPING1 (1:50, 12259-1-AP, Rabbit, Proteintech, Wuhan, China), anti-CD8
(1:200; HPA037756; Merck KGaA, Darmstadt, Germany) and anti-CD4 (1:50; ab231460;
Abcam, Shanghai, China) were used as primary antibodies. Staining intensity was
scored from 0 to 3, with 0 indicating no staining, 1 indicating light brown, 2
indicating brown, and 3 indicating tan. The extent of staining was rated from 0
to 4 based on the percentage of positive cells: 0–25%, 25–50%, 50–75%, and
75–100%. The immunohistochemistry (IHC) score was calculated as the product of
staining intensity and extent scores, yielding a range from 0 to 12. Scores 0–4
were assigned as negative, and scores 5–12 were assigned as positive [32]. The
percentages of CD4+ and CD8+ T cells in each TNBC sample were determined by
analyzing at least five 40
GSE176078 was conducted using the R package “Seurat” according to standard procedures. We used the harmony dimensionality reduction algorithm to de-batch the 9 TNBC samples. We visualized the dimensionality reduction via the “UMAP” function, and all cells were annotated based on reference datasets from the CellMarker 2.0 (http://bio-bigdata.hrbmu.edu.cn/CellMarker/). Intercellular communications were computed by the R package “CellChat”, which leverages a receptor-ligand interaction database.
Statistical analyses were conducted using R software (version 4.3.1; R Foundation for Statistical Computing, Vienna, Austria).
Categorical variables were assessed using the
A total of 5239 DEGs were identified between TNBC and normal tissues in TCGA
database, including 2906 upregulated and 2333 downregulated genes (Fig. 1A).
ESTIMATE algorithm was applied to assess the level of infiltration of immune and
stromal cells in each TNBC sample (Supplementary Table 1). DEGs and
results of ESTIMATE were then incorporated into the WGCNA in search of immune
infiltration-related biomarkers. 8 co-expression modules were recognized with a
power of 3 as the optimal soft threshold (Fig. 1B,C). Based on Pearson’s
correlation coefficient, a heat map illustrating module-trait relationships was
drawn to assess the relationships between modules. The turquoise module exhibited
the highest correlation with immune infiltration scores and was designated as an
“immune infiltration-related DEGs (IRDEGs) module” (Fig. 1D,E). Univariate Cox
regression analysis of the 1239 IRDEGs in turquoise module identified 46 genes
significantly associated with overall survival (OS) (p
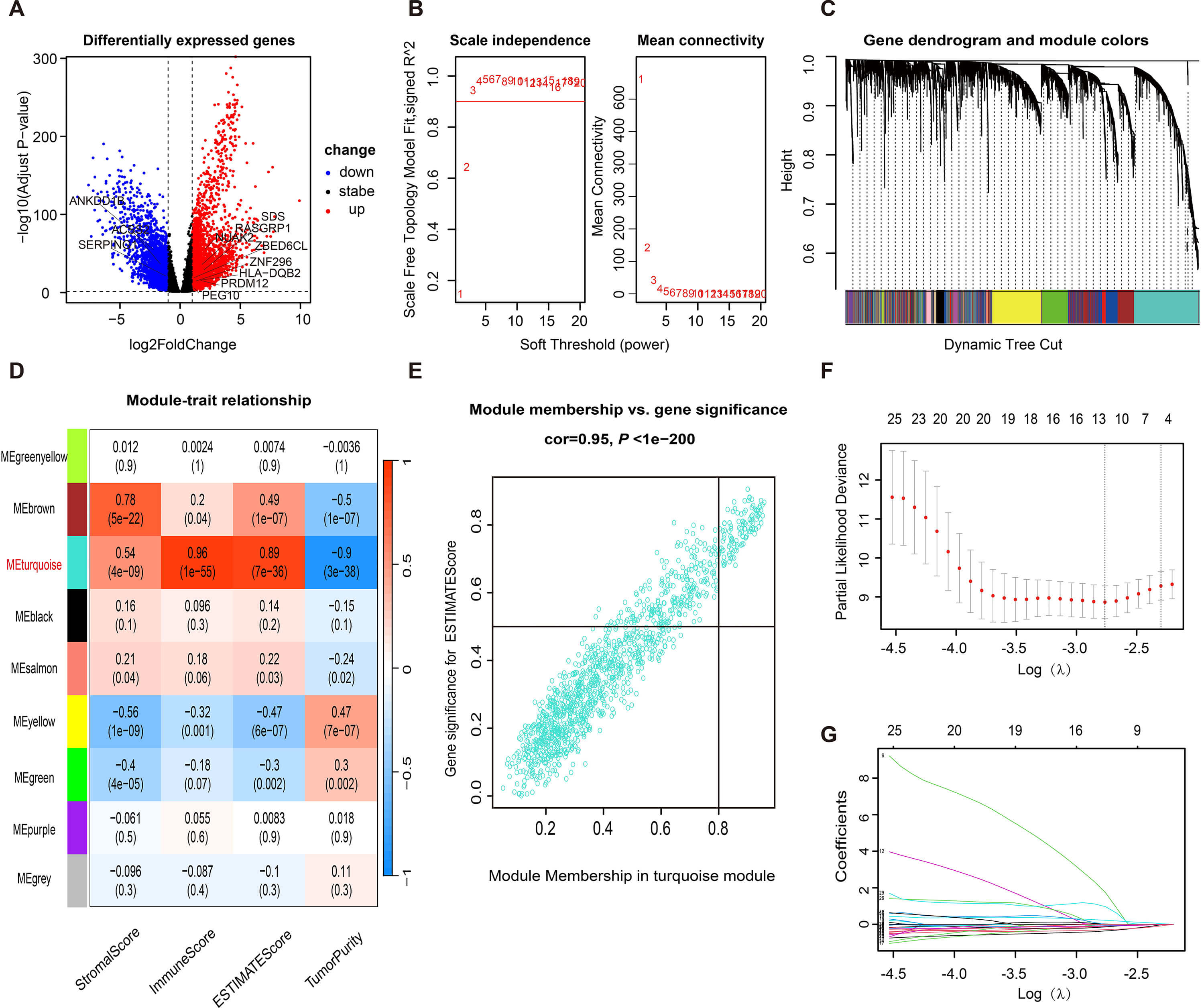 Fig. 1.
Fig. 1.
Identification of hub genes associated with immune infiltration for TNBC. (A) Volcano plot of DEGs of normal and TNBC samples in TCGA cohort. Weighted gene co-expression network analysis: (B) Analysis of network topology for soft powers; (C) Cluster dendrogram developed by the weighted correlation coefficients, genes with similar expression patterns were clustered into co-expression modules, and each color represents a module; (D) The heatmap of module-trait relationships; (E) Correlation Curve of turquoise module. (F,G) LASSO COX regression of eleven immune infiltration-related DEGs. TNBC, triple-negative breast cancer; DEGs, differentially expressed genes; TCGA, the Cancer Genome Atlas; LASSO, least absolute shrinkage and selection operator.
Unpaired and paired analyses of TCGA cohort revealed that SERPING1 was significantly downregulated in cancer tissues compared to normal tissues (Fig. 2A). Consistent results were observed in the GSE45827 dataset (Fig. 2B). To validate the expression of SERPING1, we constructed a tissue array consisting of 18 TNBC samples and 28 normal samples from patients in our department and performed IHC staining. The IHC scores confirmed significantly lower expression of SERPING1 in TNBC compared to normal tissues (Fig. 2C). KM survival analysis from TCGA cohort demonstrated that low SERPING1 group showed inferior OS than high SERPING1 group (Fig. 2D). Both univariate and multivariate Cox regression analyses confirmed that SERPING1 served as an independent prognostic factor for OS in TNBC patients (Table 1). These findings were corroborated by analyses of the METABRIC and GSE103091 cohorts (Fig. 2E and Supplementary Fig. 1). Additionally, among different BC subtypes, the lowest expression of SERPING1 mRNA and protein was observed in TNBC (Fig. 2F and Supplementary Fig. 2). Further validation using KM plotter database indicated that higher SERPING1 expression was associated with improved survival only in basal BC, which includes the majority of TNBC (Fig. 2G–J). Moreover, low SERPING expression was significantly correlated with large tumor size and advanced tumor stage (Fig. 2K–N). These findings highlighted that SERPING1 may serve as a positive prognostic biomarker in TNBC.
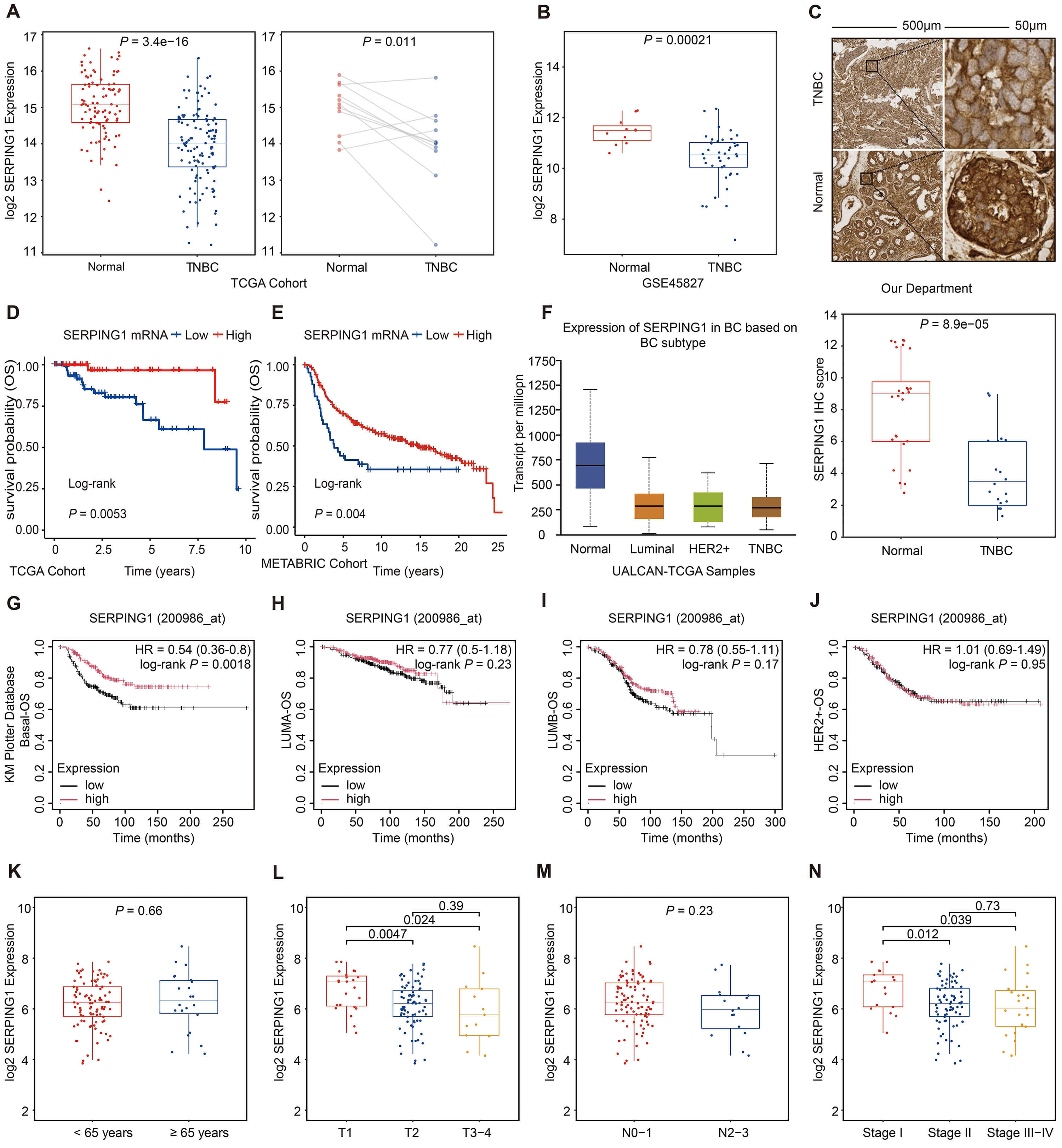 Fig. 2.
Fig. 2.
Downregulated SERPING1 in TNBC leads to
advanced tumor stage and poor prognosis. (A) Box plots of SERPING1
expression in normal tissues and TNBC (left); Dot plots of paired normal and TNBC
(right) based on TCGA database. (B) Box plots of SERPING1 expression in
normal tissues and TNBC according to GSE45827. (C) Representative images of
SERPING1 stained by IHC in TNBC (top) and normal mammary (bottom) were shown on
the top, and Box plots of the distribution of IHC score were shown on the bottom. Scale bar = 500 μm (Left); Scale bar = 50 μm (Right).
(D–F) Kaplan-Meier survival curve of OS based on (D) TCGA and (E) METABRIC
database. (F) Box plots of SERPING1 expression in normal tissues and BC
subtype using the UALCAN tool. (G–J) Kaplan-Meier survival curve of OS based on
Kaplan-Meier plotter database for (G) Basal BC, (H) LUMA BC, (I) LUMB BC and (J)
HER2+ BC. (K–N) Box plots demonstrating the relationship of
SERPING1 expression with (K) patient age, (L) tumor size, (M) lymph node
involvement, and (N) cancer staging. Paired t test was used for paired
normal-TNBC analysis. Other data were analyzed for statistical significance using
Wilcox test. Two-sided p-values
| Variables | Overall survival | ||||
| Univariate | Multivariate | ||||
| HR (95% CI) | p-value | HR (95% CI) | p-value | ||
| Age at diagnosis (%) | |||||
| ref | |||||
| 1.75 (0.56, 5.43) | 0.332 | ||||
| T | |||||
| T1 | ref | ref | |||
| T2 | 2.47 (0.53, 11.38) | 0.247 | 1.55 (0.32, 7.56) | 0.589 | |
| T3–4 | 5.88 (1.07, 32.28) | 0.041 | 1.51 (0.24, 9.43) | 0.661 | |
| N | |||||
| N0 | ref | ref | |||
| N+ | 4.17 (1.47, 11.86) | 0.007 | 4.52 (1.48, 13.77) | 0.008 | |
| Stage* | |||||
| Stage I–II | ref | ||||
| Stage III–IV | 5.30 (2.01, 14.01) | 0.001 | |||
| Histology type | |||||
| IDC | ref | ||||
| ILC | 5.87 (0.74, 46.30) | 0.093 | |||
| Other | 1.29 (0.33, 5.00) | 0.710 | |||
| SERPING1 expression | 0.50 (0.30, 0.83) | 0.008 | 0.47 (0.27, 0.84) | 0.010 | |
OS, Overall survival; HR, Hazard ratio; CI, Confidence interval; ref, Reference; IDC, Invasive ductal carcinoma; ILC, Invasive lobular carcinoma. p-value was considered statistically significant. * Stage was not enrolled in multivariate analysis because it is related to T and N.
Limited evidence exists regarding the biological characteristics of SERPING1 in malignancies, particularly in TNBC. Therefore, GO and KEGG enrichment analyses using the DEGs between low and high SERPING1 groups were conducted to annotate their functions and investigate the role of SERPING1. The results highlighted the crucial role of SERPING1 in immune regulation. GO enrichment analysis indicated that SERPING1 was associated with essential BP such as leukocyte-mediated immunity, lymphocyte-mediated immunity and activation of immune response. Moreover, SERPING1 was linked to specific CC, such as the immunological synapse and MHC protein complex, both crucial for T cell activation. MF analysis revealed enrichment in receptor ligand activity, cytokine activity, immune receptor activity, and cytokine binding, underscoring its functional contribution to immune regulation (Fig. 3A). KEGG pathway analysis also revealed the involvement of SERPING1 in immune signaling, such as cytokine-cytokine receptor interaction pathway and T cell receptor signaling pathway (Fig. 3B). Consistently, GSEA demonstrated significant enrichment of immune response-related pathways in the high SERPING1 group (Fig. 3C).
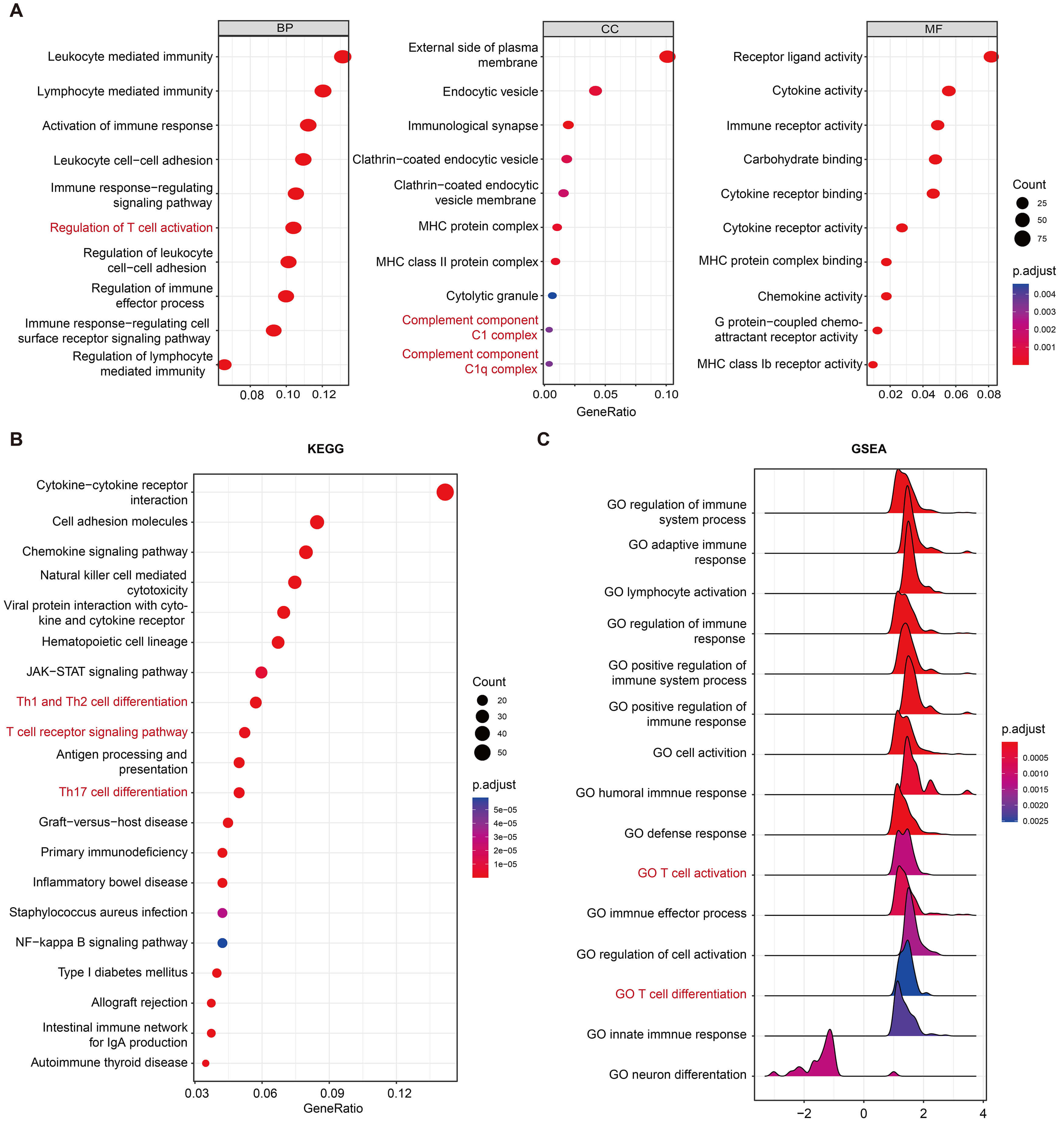 Fig. 3.
Fig. 3.
SERPING1 is involved in regulating multiple immune-related signaling pathways. (A) GO, (B) KEGG and (C) GSEA enrichment of SERPING1-related genes. GO, gene ontology; BP, biological processes; CC, cellular component; MF, molecular function; KEGG, kyoto encyclopedia of genes and genomes; GSEA, gene set enrichment analysis.
To better understand the impact of SERPING1 on immune cells within TME, immune cell infiltration analysis was conducted using the “Xcell” algorithm. High SERPING1 group presented lower tumor purity and higher percentage of anti-tumor immune cells, including CD8+ T cells, CD4+ memory T cells, B cells, macrophages M1 and activated dendritic cells (DC) (Fig. 4A). These findings were validated using other reliable algorithms (Fig. 4B and Supplementary Fig. 3A–C). Moreover, ssGSEA analysis of immune function scores revealed an activated immune status in the high SERPING1 group (Fig. 4C). Given that the enrichment analysis predominantly highlighted T cell-related pathways (regulation of T cell activation, T cell receptor signaling pathway, and Th1 and Th2 cell differentiation) and that SERPING1 demonstrates a positive correlation with CD4+ T cells and CD8+ T cells, we hypothesize that SERPING1 may inhibit tumorigenesis by promoting the infiltration of protective immune cells, particularly T cells (Fig. 4D). Tissue microarrays were prepared from the same batch in our department, ensuring that samples sharing the same ID number had similar background tissue characteristics. Two well-prepared tissue microarrays were subjected to IHC staining for CD4 and CD8. As shown in Fig. 4E, the high SERPING1 expression context exhibited a greater abundance of infiltrating CD4+ T cells and CD8+ T cells. These observations indicated that SERPING1 overexpression was associated with an inflamed TME in TNBC.
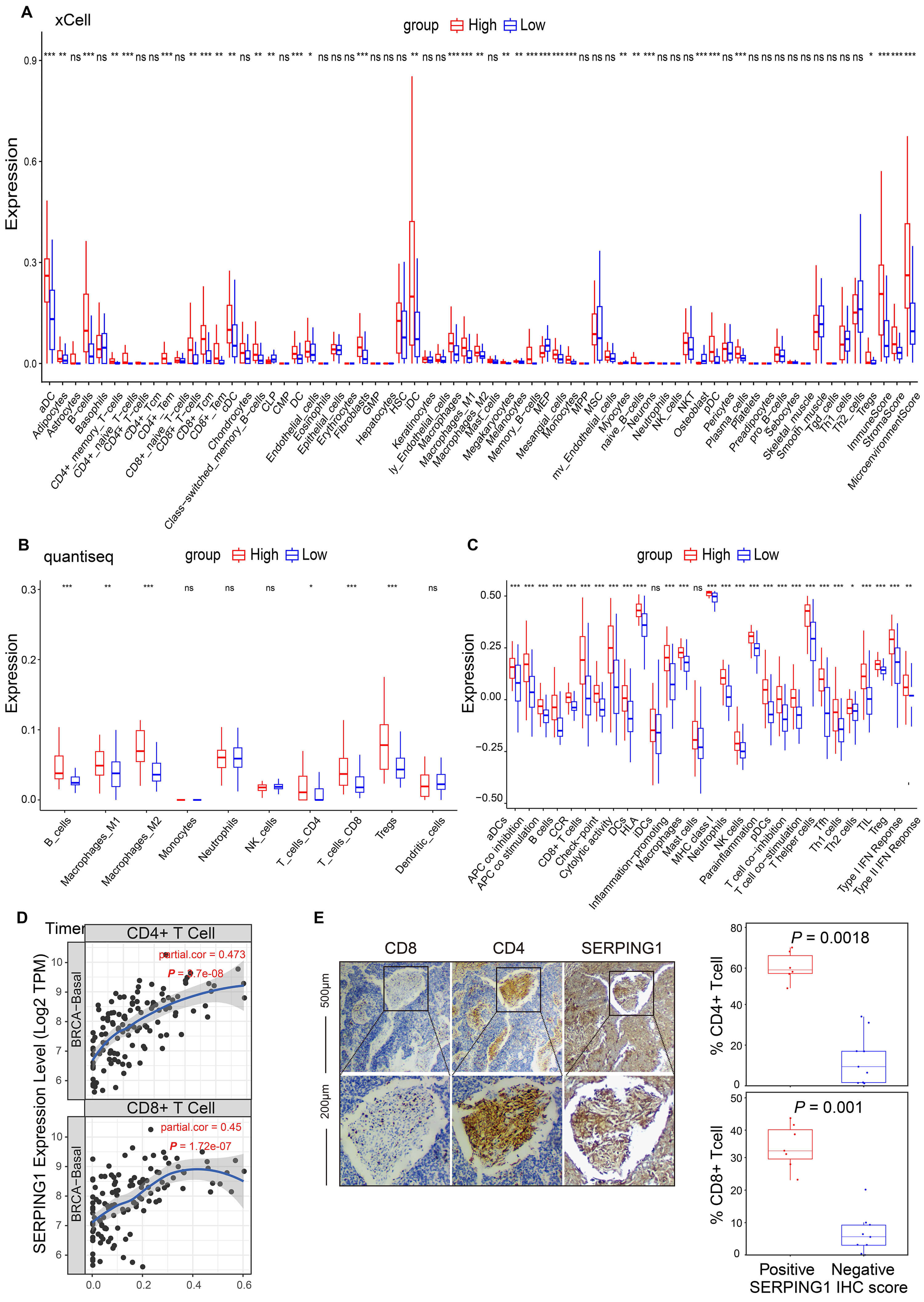 Fig. 4.
Fig. 4.
SERPING1 is markedly associated with the
infiltration of anticancer immune cells in tumors. (A–C) Comparison of immune
infiltration between high and low SERPING1 expression groups was
performed via (A) xCell and (B) quantiseq mode. (C) ssGSEA analysis based on 29
immune signatures. (D) Correlation between SERPING1 expression and
infiltration of CD4+ (top) and CD8+ (bottom) T cells in basal BRCA. (E)
Representative IHC staining of CD8 (left), CD4 (middle), and SERPING1 (right) in
TNBC samples was shown on the left, and Box plots display the percentage of CD4+
(top) or CD8+ (bottom) T cells in TNBC tumor tissue sections were shown on the
right. The difference between groups was analyzed using the Wilcox test. Scale bar = 50 μm (Up); Scale bar = 200 μm (Down).
Two-sided p-values
We next investigated whether SERPING1 could predict patient response to ICI therapy. ICI response biomarkers, including MHC molecules and immune checkpoints, were all elevated in the high SERPING1 group (Fig. 5A). In addition, patients with high SERPING1 expression exhibited a significantly higher IPS score and a lower exclusion score, indicating a stronger likelihood of response to immunotherapy (Fig. 5B,C). To further validate the immune association of SERPING1, we analyzed data from a melanoma immunotherapy cohort (GSE91061), which revealed significantly higher SERPING1 expression in responders compared with non-responders (Fig. 5D). Consistently, analysis of the IMvigor210 cohort, a widely used benchmark dataset for immunotherapy studies, showed that high SERPING1 expression was associated with favorable prognosis and improved immunotherapy response (Fig. 5E,F).
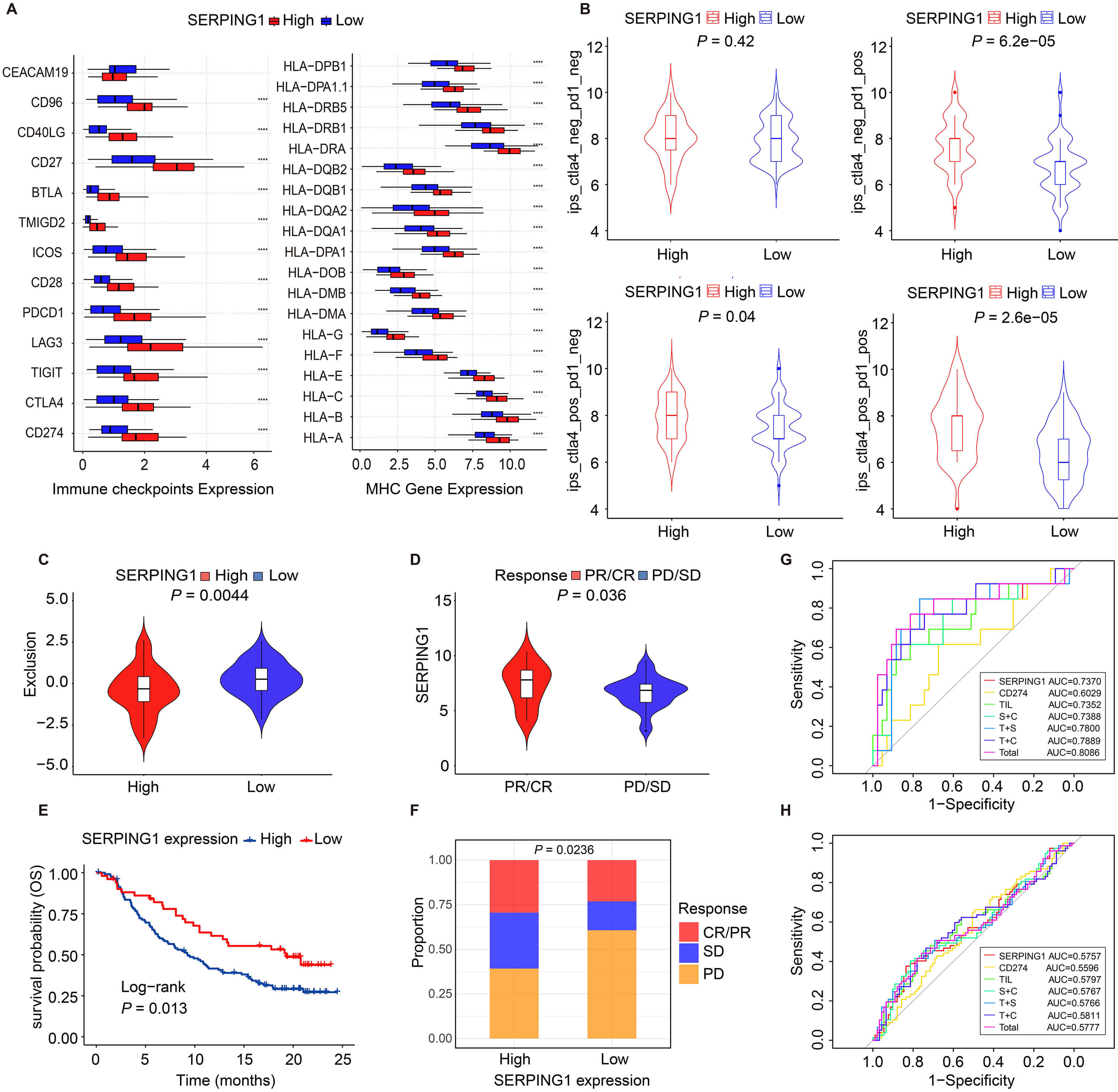 Fig. 5.
Fig. 5.
High SERPING1 expression is sensitive to tumor
immunotherapy. (A) Box plots demonstrating the abundance of immune checkpoints
and MHC molecules between high and low SERPING1 expression groups. (B,C)
Violin plots of (B) IPS scores and (C) exclusion scores between high and low
SERPING1 expression groups. (D) Violin plots of SERPING1
expression across different immunotherapy response groups in GSE91061 dataset.
(E) Kaplan-Meier survival curve of OS based on IMvigor210 cohort. (F) Stacked bar
plots of immunotherapy reactivity between high and low SERPING1
expression groups in IMvigor210 cohort. (G,H) ROC curves of the predictive
performance of SERPING1, CD274, TILs, SERPING1+CD274
(S+C), TILs+SERPING1 (T+S), TILs+CD274 (T+C) and
TILs+SERPING1+CD274 (Total) based on (G) GSE91061 and (H)
IMvigor210 cohort. The difference between groups was analyzed using the Wilcox
test. Two-sided p-values
Subsequently, we compared the predictive performance of SERPING1 with established biomarkers, including CD274 and TILs. In both the GSE91061 and IMvigor210 cohorts, SERPING1 achieved AUC values comparable to those of CD274 and TILs. Furthermore, incorporating SERPING1 into combined models with CD274 and/or TILs modestly improved predictive performance (Fig. 5G,H).
To examine the regulatory role of SERPING1 on tumor immunity at single-cell resolution, we performed dimensionality reduction analysis on scRNA-seq data containing 9 TNBC samples from GSE176078. 15 cell clusters were annotated and assigned to 9 cell types, namely B cells, CD4+ T cells, CD8+ T cells, DC, endothelial cells, epithelial cells, CAFs, macrophages and natural killer (NK) cells (Fig. 6A and Supplementary Fig. 4A). SERPING1 was mainly expressed in CAFs, and it was less expressed in macrophages, endothelial cells and DC, which was consistent with the result of higher immune scores and higher proportions of these cells in high SERPING1 group (Fig. 6B).
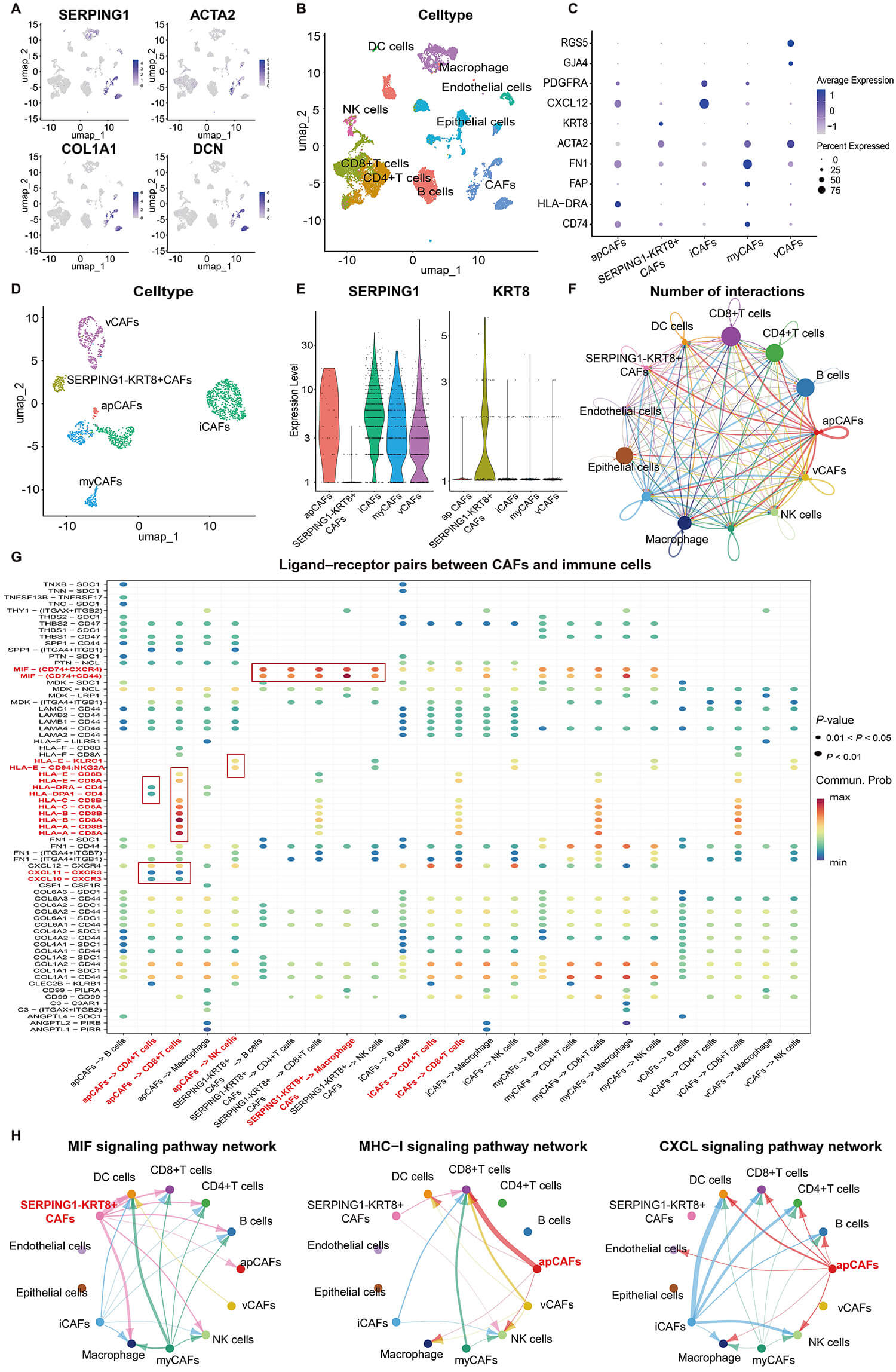 Fig. 6.
Fig. 6.
The contribution of SERPING1 to cellular communication at the single-cell level. (A) The UMAP plots exhibit the expression levels of SERPING1 and CAFs marker genes. (B) UMAP plot for the dimension reduction and visualization of 9 cell types within the TME. (C) Expression levels of marker genes for each CAFs subtype. (D) UMAP plot for the dimension reduction and visualization of 5 CAFs subtypes. (E) Violin plots of SERPING1 and KRT8 in each CAFs subtype. (F) The cellular interaction network between different cell types in TNBC. (G) Bubble plots exhibiting significant interactions between 5 CAFs subtypes with other immune cells by the ligand-receptor pairs. (H) The cellular interaction network between different cell types in various signaling pathways, including MIF (left), MHC-Ⅰ (middle) and CXCL (right) signaling pathways. UMAP, uniform manifold approximation and projection; TNBC, triple-negative breast cancer; TME, tumor microenvironment; MIF, macrophage migration inhibitory factor; MHC-Ⅰ, major histocompatibility complex-Ⅰ; CXCL, chemokine ligand; CAFs, cancer-associated fibroblasts; apCAFs, antigen-presenting CAFs; iCAFs, inflammatory CAFs; myCAFs, myofibroblastic CAFs; vCAFs, vascular CAFs.
CAFs were then isolated and re-clustered into 8 subpopulations (Supplementary Fig. 4B). Based on canonical marker genes, these subpopulations were annotated as antigen-presenting CAFs (apCAFs), inflammatory CAFs (iCAFs), myofibroblastic CAFs (myCAFs), vascular CAFs (vCAFs) and a distinct SERPING1-KRT8+ CAF population (Fig. 6C,D). This SERPING1-KRT8+ subset could not be assigned to any established CAF subtypes. Notably, SERPING1 was expressed in all other CAF subtypes but was absent in the SERPING1-KRT8+ subset (Fig. 6E). Cell communication analysis revealed that SERPING1+ apCAFs exhibited the most interactions with immune cells (Fig. 6F). Ligand-receptor interaction analysis indicated that these apCAFs engaged extensively with T cells via the MHC-I, MHC-Ⅱ, and CXCL signaling pathways, all of which have been previously implicated in immune activation. In contrast, the MIF signaling pathway, associated with immunosuppression, was significantly enriched in the crosstalk of SERPING1-KRT8+ CAFs (Fig. 6G,H). Furthermore, SERPING-KRT8+ CAFs displayed reduced MHC-I expression and significantly fewer interactions with CD8+ T cells compared to all SERPING1+ CAF subtypes (Fig. 6G,H and Supplementary Fig. 4C). These findings indicated that SERPING1 loss in CAFs may impair immune activation with TNBC TME.
TNBC is the most aggressive subtype of BC, with limited treatment strategies and high immunogenicity. Recently, anti-PD-L1 antibodies have been approved for the treatment of both early- and late-TNBC, demonstrating significant benefits. Although the application of molecular targeted therapies and ICIs has extended survival in some patients with advanced TNBC, treatment resistance and relapse remain frequent due to the complexity of the TME. Within the TME, immune cells, remodeled ECM, soluble factors, epigenetic alterations, and reprogrammed fibroblasts interact to shape the anti-tumor immune response, driving tumor initiation, progression, and metastasis. Therefore, an in-depth investigation of the role of TME in TNBC is crucial for optimizing therapeutic strategies and enhancing immunotherapy efficacy.
In our study, by integrating clinical data, bioinformatics analyses and IHC validation, we identify SERPING1 as a potential biomarker associated with immune infiltration in TNBC. SERPING1 encodes a serine protease inhibitor that inactivates C1r and C1s proteases of the C1 complex in the classical complement pathway, thereby suppressing complement activation. Initially, SERPING1 was primarily studied in the context of hereditary angioedema and partial complement component deficiency. Recently, its role in various tumors has gained increasing attention [20, 33]. In BC, low SERPING1 expression was reported as an early diagnostic marker for bone metastasis and was significantly downregulated in metastatic lymph nodes of hormone receptor-positive BC [34, 35]. However, the specific function and mechanism of SERPING1 in TNBC remain unclear. Our study revealed that SERPING1 expression was decreased in TNBC, with low expression strongly associated with advanced stage and poor patient survival. These findings are consistent with studies in prostate and gastric cancers, indicating that SERPING1 loss may act as an adverse factor in the progression of multiple tumor types [21, 36, 37].
Research by Shen et al. [38] demonstrated that SERPING1 could inhibit lung cancer progression via the TSC2/mTOR signaling pathway and SERPING1 was linked to immunotherapy response. In this study, GO and KEGG functional enrichment analyses indicated that SERPING1-related genes were primarily involved in immune regulatory pathways, suggesting a potential role of SERPING1 in anti-tumor immune regulation in TNBC. In addition, we applied multiple robust bioinformatics approaches to analyze the immune microenvironment of TNBC samples from the TCGA cohort. Our results revealed that high SERPING1 expression was significantly associated with an immune-activated TME, characterized by elevated infiltration of anti-tumor immune cells, including CD8+ T cells, CD4+ T cells, M1 macrophages, and DC. Immunotherapy response analysis showed that the high SERPING1 group exhibited significantly increased levels of MHC molecules, immune checkpoint genes, and IPS, indicating enhanced immune responsiveness. Moreover, immunotherapy efficacy analysis in two independent external cohorts further supported its potential as a biomarker for immunotherapy prediction.
GSEA analysis further indicated that SERPING1-related genes are primarily
enriched in T cell regulatory pathways, thereby enhancing anti-tumor immunity
through activation of T cells within the TME. To validate this hypothesis, IHC
staining was performed on tissue microarrays from our hospital. The results
demonstrated that infiltration levels of CD4+ and CD8+ T cells were significantly
higher in the SERPING1-positive group compared with the SERPING1-negative group.
Subsequently, scRNA-seq analysis revealed that SERPING1 was
predominantly expressed in CAFs. CAFs are the most abundant cellular component of
the TME and constitute a highly heterogeneous population derived from multiple
cell types. They exert complex functions in tumor progression by secreting
soluble factors, releasing exosomes, providing nutrients, remodeling the ECM, and
modulating immune cell activity [15]. Recent studies have clarified the functions
of distinct CAF subpopulations in BC, which can be roughly classified into
myCAFs, iCAFs and apCAFs [39]. MyCAFs are mainly located near tumor cells, highly
express ECM-related proteins, and promote the formation of an immunosuppressive
TME. In contrast, iCAFs are situated farther from tumor cells, exhibit low
Cell communication analysis indicated that SERPING1+ apCAFs exhibit strong interactions with anti-tumor immune cells, including CD8+ T cells, CD4+ T cells, and NK cells. Consistent with our findings, previous studies have demonstrated that SERPING1+ CAFs are closely associated with T cell infiltration in nasopharyngeal carcinoma and melanoma [45, 46, 47]. In contrast, ligand-receptor interaction analysis of SERPING1-KRT8+ CAFs revealed enrichment of the MIF signaling pathway, which has been reported to promote immunosuppression by inhibiting p53 expression and activating multiple oncogenic signaling pathways [48, 49]. Compared with all SERPING1+ CAF subtypes, SERPING1-KRT8+ CAFs exhibited markedly fewer interactions with T cells and lacked enrichment of CXCL signaling pathways. Moreover, previous research reported that KRT8 upregulation in CAFs was associated with enhanced tumor growth and metastasis [50]. Differential expression between SERPING1+ apCAFs and SERPING1-KRT8+ CAFs revealed enrichment in cytokine secretion pathways (Supplementary Fig. 5). Collectively, our findings indicated that the reduced SERPING1 expression in CAFs may diminish cytokine and chemokine production, limit immune cell infiltration, and consequently promote an immunosuppressive microenvironment associated with poor prognosis in TNBC. However, the precise molecular mechanisms of SERPING1 in TNBC warrant further investigation.
To the best of our knowledge, this is the first study to systematically elucidate the role of SERPING1 in TNBC patients. However, there are still some limitations. First, the IHC validation cohort included only 18 TNBC samples due to limited tissue availability and strict inclusion criteria. Second, although our conclusions were supported by multiple public datasets and single-cell analyses, functional experiments are needed to verify the role of SERPING1 in CAFs. Additionally, the molecular mechanisms underlying SERPING1-mediated immune regulation in TNBC remained to be fully elucidated. Further studies with larger clinical cohorts and mechanistic experiments are warranted to validate and extend these findings.
In conclusion, our analysis indicated that SERPING1 is a favorable prognostic factor, with its expression positively correlated with immune activity and response to immunotherapy in TNBC. Mechanically, SERPING1+ apCAFs could promote immune activation through interactions with T cells in the TME. These findings highlighted SERPING1 as a potential biomarker for immune infiltration and prognostic prediction in TNBC. We expected to provide the foundation for further in-depth research on SERPING1 in TNBC.
The paper is listed as, “Integrated single-cell and bulk RNA sequencing identifies SERPING1 as an immune infiltration regulator and therapeutic target in Triple-negative breast cancer ” as a preprint on (SSRN) at: (https://www.researchsquare.com/article/rs-7064973/v1.pdf).
The datasets generated and/or analyzed during the current study are available in the TCGA database on the Xena website (http://xena.ucsc.edu/), the GEO (https://www.ncbi.nlm.nih.gov/geo/), the METABRIC database (https://www.bccrc.ca/dept/mo/), the Kaplan-Meier plotter (KM plotter, https://kmplot.com/analysis/), and the UALCAN tool (https://ualcan.path.uab.edu/). The datasets used and analyzed during the current study are available from the corresponding author on reasonable request. Further inquiries can be directed to the corresponding authors.
YHS and BLG were responsible for conception and design of this study, YHS wrote the main manuscript text, YHJ, JWL, RZG, ABH, YQD and YSL collected and analyzed datasets, XLW, MCL, HYZ, ZYR and YLL prepared all figures. WLC, ZBF, YYJ, YLC and DLC interpreted the data, YYJ prepared Supplementary Materials. All authors reviewed the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the Ethics Committee of HMU (Ethics Code: YJSDW2024-260) on May 29, 2024. All participants provided written informed consent prior to enrolment in the study. This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki.
The authors thank the Department of Pathology at the Second Affiliated Hospital of Harbin Medical University for their professional assistance. We acknowledged the TCGA database, GEO database, Kaplan-Meier Plotter database and UALCAN tool for providing their platforms and contributors for uploading their meaningful datasets.
This study was funded by grants from the National Natural Science Foundation of China (81872135).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47089.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.