1. Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder currently
characterized by alterations in social interaction and communication, concurrent
to restricted interests and verbal or behavioral stereotypies. The presence of
raised circulating serotonin levels has long been appreciated to be evident in
ASD [1] in many but not all people classed on this spectrum [2]. Although this
may arise from increased antibodies against monoamine oxidase A (MAO-A) [3], a
number of investigators have proposed that raised circulatory serotonin levels
may occur due to a decreased capacity to use serotonin as a necessary precursor
to initiate the melatonergic pathway, for example from a decrease in chaperone
protein, 14-3-3, stabilization of the first melatonergic pathway enzyme,
Aralkylamine N-acetyltransferase (AANAT) [4]. Decreased 14-3-3 availability can
arise from increased microRNAs (miRNAs) such as miR-451 [5] and miR-375 [6]. This
is supported by data showing decreased circadian/pineal [7] and systemic
melatonergic pathway induction in ASD [5] as well as the clinical utility of
night-time melatonin treatment in management of sleep and wider ASD
symptomatology [8]. Recent work indicates that suppressed pineal and local
melatonergic pathway induction may be a core aspect of ASD, as with many other
diverse medical conditions [9, 10]. Suppressed mitochondrial melatonin may
therefore be intimately linked data showing suboptimal mitochondrial function in
ASD [11, 12] with wider downstream developmental and ongoing consequences.
Numerous factors and processes are associated with ASD pathoetiology and
pathophysiology, including increased phosphorylation and activation of signal
transducer and activator of transcription 3 (STAT3) [13, 14, 15] and nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-B) [16, 17, 18] as well
as related increases in interleukin (IL)-6 [19, 20, 21]. Recent work indicates that
the IL-6/Janus Kinase (JAK)/STAT3 pathway interacts with the specific dimer
composition of NF-B in the nucleus to either upregulate or
down-regulate the mitochondrial melatonergic pathway across diverse cell types,
with NF-B dimer component effects specific to particular cells [22].
These authors showed that the anti-inflammatory effect of IL-10 in pineal, bone
marrow, spleen and peritoneal cells is determined by the interactions of STAT3
and NF-B dimer composition in the regulation of the melatonergic
pathway [22]. As the suppression of the melatonergic pathway across CNS and
systemic cells has long been recognized as an aspect of ASD pathophysiology
[5, 23, 24] the regulation of STAT3 interactions with NF-B dimer
composition in the modulation of the melatonergic pathway is likely to constitute
core aspects of ASD pathoetiology and pathophysiology, including via alterations
in mitochondrial function that are evident in ASD [25].
Alterations in the circadian rhythm [26, 27, 28] and cortisol activation of the
glucocorticoid receptor (GR)- in the course of circadian regulation and
hypothalamic-pituitary-adrenal (HPA) axis activation during stress have long been
linked to ASD pathophysiology [29, 30, 31]. The suppression of pineal melatonin [7],
and systemic melatonin [5] as well as gut microbiome-derived short-chain fatty
acid, butyrate [32] in ASD therefore disinhibits the GR-, thereby
modulating the circadian rhythm and stress response effects of cortisol. This has
relevance across a range of diverse medical conditions linked to decreased pineal
melatonin and increased gut dysbiosis, including Alzheimer’s disease [33], cancer
[34], and diabetes associated conditions, including ASD [35] as well as for ASD
pathoetiology driven by gestational diabetes [36]. Circadian and stress/cortisol
dysregulation arises from a decrease in both pineal melatonin and gut derived
butyrate. Both melatonin and butyrate inhibit GR- nuclear translocation
from the cytoplasm to the nucleus [37, 38], leading to a dysregulated circadian
and stress linked HPA axis activation in ASD pathoetiology and pathophysiology
[39, 40]. As melatonin can inhibit STAT3 and NF-B activation, the
suppression of pineal melatonin in ASD contributes to alterations in STAT3
interactions with NF-B dimer composition, thereby altering the
modulation of the systemic melatonergic pathway in ASD. As local melatonin
upregulation is a key aspect of the resolution of local inflammation, including
as mediated by vagal nerve activation [41], the suppression of the local
melatonergic pathway in ASD has significant implications for attaining resolution
of inflammation systemically. Decreased vagal nerve activation is common in ASD
[42], which may therefore be confounded by a decreased capacity to induce local
melatonin in ASD across body organs/tissues [5]. Similarly, the suppression of
pineal melatonin increases gut dysbiosis and gut permeability [43], which are
typically associated with decreased gut microbiome-derived butyrate, linking the
classical gut associated changes in ASD with alterations in circadian (pineal
melatonin) and systemic (vagal) processes associated with inflammation
resolution.
This article reviews data on circadian and systemic changes in the pathoetiology
and pathophysiology of ASD. It is proposed that alterations in circadian and
systemic processes are strongly determined by variations in the regulation of the
local mitochondrial melatonergic pathway. The mitochondrial melatonergic pathway
is regulated by alterations in the canonical and non-canonical STAT3 interactions
with NF-B dimer composition [22]. This has prevention, treatment and
future research implications including by integrating data showing increased
hyperglycemia inducing methylglyoxal and advanced glycation end-products in ASD
pathophysiology [44], thereby providing a context for the association of
diabetes/hyperglycemia with ASD [35].
The next two sections briefly review the alterations in circadian and local
melatonin regulation. The first section highlights the interactions of pineal
melatonin and cortisol in the course of night-time dampening and resetting in
preparation for the coming day.
2. Night-Time Dampening and Resetting
Altered night-time dampening and resetting may be an aspect of the pathoetiology
and pathophysiology of an array of diverse medical conditions, including
Alzheimer’s disease [33] and cancer [45, 46]. Changes in night-time melatonin and
cortisol interactions may also be core aspects of conditions driving accelerated
aging, such as type 2 diabetes mellitus (T2DM) [47]. T2DM is more common in ASD
and is proposed to contribute to ASD pathophysiology [48]. The overlaps of ASD
and T2DM may therefore arise from suppressed pineal melatonin in ASD [49] and in
T2DM [50]. Suppressed pineal melatonin may arise from a number of factors and
processes that act to increase STAT3 and thereby attenuate AANAT enzymatic
activity that initiates the melatonergic pathway [49, 51]. Approximately 65% of
people with ASD, vs controls, have less than 50% of pineal melatonin levels
[52], highlighting the importance of incorporating pineal melatonin in ASD
pathophysiology. The role of the melatonergic pathway in the pathophysiological
overlaps of ASD and T2DM is highlighted by data showing melatonin and melatonin
receptor levels and allele variants to modulate T2DM [50]. Consequently, any
suppression of pineal and/or local melatonin in ASD [5] would be expected to
increase T2DM risk/symptomatology, with T2DM then contributing to the circadian
and systemic underpinnings of ASD via the attenuation of the capacity of pineal
and systemic cells to induce the melatonergic pathway. Night-time changes in
pineal melatonin and cortisol in ASD, T2DM and aging are shown in Fig. 1 (Ref.
[22, 53, 54, 55, 56, 57]).
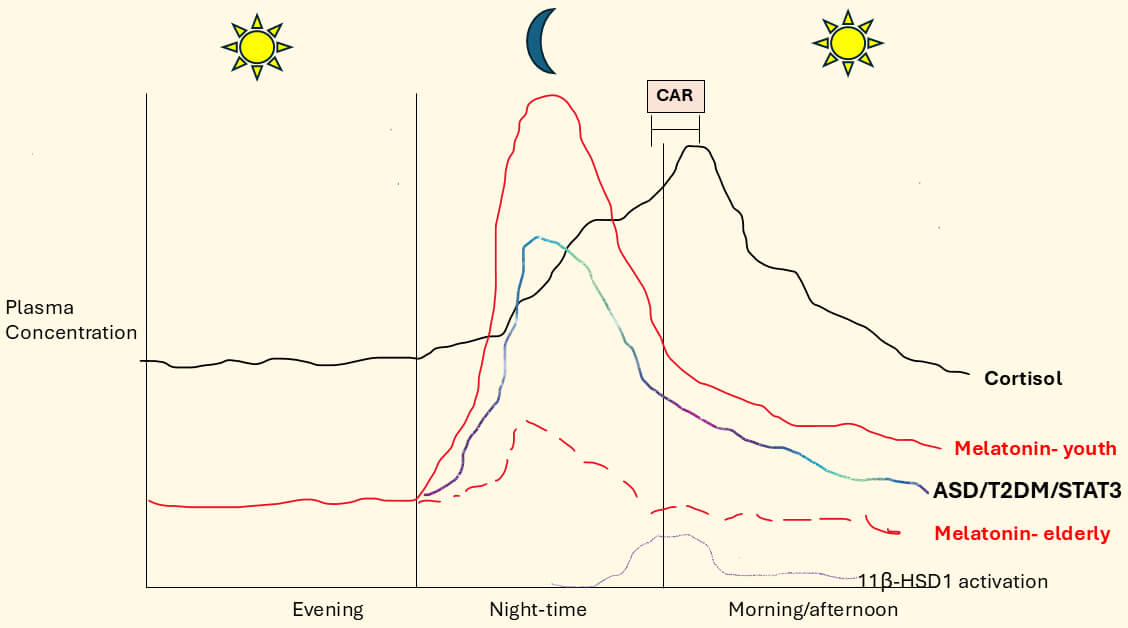 Fig. 1.
Fig. 1.
Melatonin and cortisol circadian variations in ASD,
T2DM and over age. Pineal melatonin dramatically decreases over aging as
indicated by ‘elderly’ vs ‘youth’ comparison. ASD, including as influenced by
increased T2DM, suppresses pineal melatonin levels, thereby enhancing the
likelihood of accelerated aging driven changes. Pineal melatonin suppression in
ASD may be mediated by increased pSTAT3 in pinealocytes thereby suppressing AANAT
activation and consequent induction of the melatonergic pathway. Pineal melatonin
suppression in ASD can be driven by the same processes that suppress local
melatonin production across body cells and systems [22], namely the interactions
of heightened levels and activation STAT3 and NF-B, which are
determined by the specific NF-B dimer composition. Night-time and
morning cortisol awakening response (CAR) cortisol levels tend to remain stable
over aging, although in some conditions cortisol levels may remain enhanced
during the day following their morning CAR peak. Melatonin and cortisol are
highly interactive. Melatonin acts on the adrenal cortex to decrease cortisol
release [53, 54] whilst melatonin also suppresses glucocorticoid receptor
(GR)- nuclear translocation from its complex in the cytoplasm [55].
Although other GR exist, including GR-, and GR locations can be plasma
membrane, mitochondrial membrane and mitochondrial matrix [56], most data on
cortisol effects have been restricted to the cytoplasmic GR-. The
suppression of pineal melatonin in ASD, T2DM and over aging may therefore
disinhibit night-time cortisol influence across body cells and systems and
therefore alter how cells, their microenvironments and body systems are prepared
for the coming day. Enhanced GR- activation, as with raised
pro-inflammatory cytokines, increases local cellular cortisol production by 11
beta hydroxysteroid dehydrogenase 1 (11-HSD1) [57], thereby increasing
local cortisol’s influence on cell function and intercellular, homeostatic
interactions in the microenvironment in which all cells exist. Other factors
pertinent to ASD (and T2DM), including gut microbiome-derived butyrate and B cell
lymphoma-2 (Bcl-2)-associated athanogene 1 (BAG-1), which also inhibit
GR- nuclear translocation but are not included for clarity.
Abbreviations: 11-HSD1, 11 beta hydroxysteroid dehydrogenase; BAG-1,
bcl2-associated athanogene 1; CAR, cortisol awakening response; GR,
glucocorticoid receptor; T2DM, type 2 diabetes mellitus; ASD, autism spectrum
disorders; STAT3, signal transducer and activator of transcription 3; AANAT,
Aralkylamine N-acetyltransferase; NF-B, nuclear factor
kappa-light-chain-enhancer of activated B cells.
Night-Time Melatonin and Cortisol Modulation of Oxytocin and Vagal
Nerve
The loss of pineal and local melatonin is typically modelled as a loss of
melatonin’s antioxidant and anti-inflammatory capacity. However, pineal melatonin
can act on a number of systemic processes and body systems to influence processes
of dampening and resolution of inflammation. For example, melatonin directly, and
via oxytocin upregulation [58, 59, 60], can activate the vagal nerve, which dampens
inflammatory activity across different organs and tissues via the release of
acetylcholine (ACh) that activates a number of ACh receptors, especially the
alpha 7 nicotinic acetylcholine receptor (7nAChR), to suppress immune
driven inflammation. This seems mediated via specialized proresolving mediators
(SPMs) upregulation [61], which changes the NF-B dimer composition
allowing different NF-B dimer composition to interact with nuclear
pSTAT3 to upregulate (or down regulate) local melatonin production [22, 62]. Pineal
melatonin also interacts with this set of processes by increasing
7nAChR levels at night [63], thereby upregulating the capacity of ACh
and vagal nerve activation to dampen inflammatory activity. See Fig. 2 (Ref.
[5, 63]).
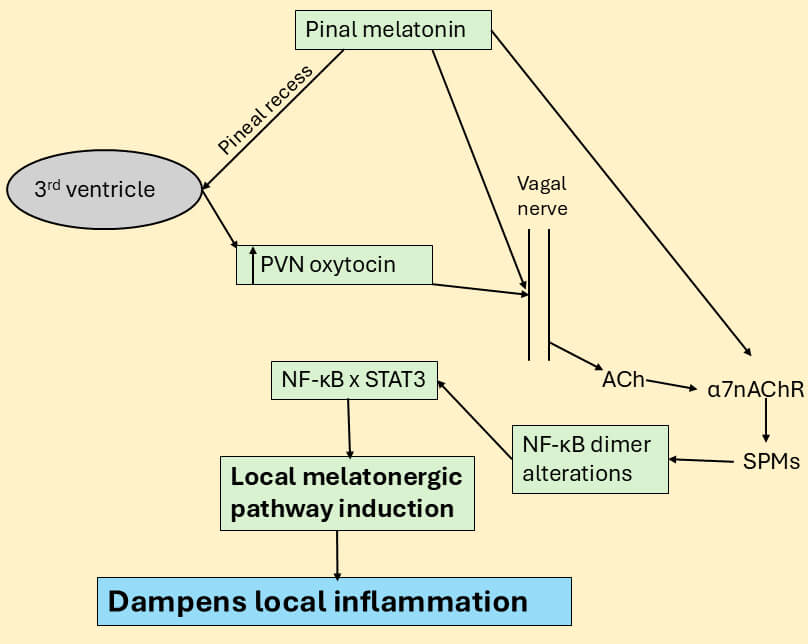 Fig. 2.
Fig. 2.
Pineal melatonin, including via oxytocin, regulates the vagal
nerve. Pineal melatonin directly and via oxytocin induction can activate the
vagal nerve to release acetylcholine (ACh) on to the 7nAChR, which
induces specialized proresolving mediators (SPMs). As pineal melatonin increases
the 7nAChR [63], this may be another route whereby suppressed pineal
melatonin modulates wider processes of dampening and resetting, including by the
vagal nerve. SPMs can alter the NF-B dimer composition by switching
from a pro-inflammatory dimer composition (typically p65/p50) to a resolution
inducing NF-B composition (typically c-Rel/p50) via the upregulation of
the local melatonergic pathway. The suppressed vagal activity and decreased
oxytocin in ASD may therefore be intimately linked to alterations in the
circadian rhythm and the attenuated capacity to upregulate the local melatonergic
pathway in any given organ/tissue [5]. The suppression of pineal and local
melatonin production may therefore be core aspects of ASD pathophysiology,
including from decreased pineal melatonin induction of hypothalamic
paraventricular nucleus (PVN) oxytocin. Abbreviations: 7nAChR, alpha 7
nicotinic acetylcholine receptor; Ach, acetylcholine; NF-B, nuclear
factor kappa-light-chain-enhancer of activated B cells; PVN, paraventricular
nucleus; SPMs, specialized pro-resolving mediators; STAT3, signal transducer and
activator of transcription 3.
In contrast to the effects of melatonin and oxytocin, GR- activation
by cortisol has complex effects on the vagal nerve, including its suppression
[64]. Heightened cortisol effects in ASD are likely to be confounded by
disinhibited GR- activation and consequent alterations in the levels of
GR- and the GR localization site (cytoplasm, plasma membrane,
mitochondrial membrane and mitochondrial matrix), and 11-HSD1 induction
[65, 66]. Consequently, cortisol may have heightened and differential effects in
the absence of raised cortisol levels per se that will be importantly determined
by suppressed pineal and/or local melatonin production. This also applies to the
interactions of cortisol with oxytocin, with cortisol having a rapid negative
feedback on oxytocin induction of adrenocorticotropic hormone (ACTH) and the HPA
axis [67], whilst electroacupuncture suppresses enhanced HPA axis activity via
oxytocin upregulation [68]. The suppression of pineal melatonin and melatonin’s
induction of oxytocin is therefore a significant contributor to alterations in
circadian and stress induced HPA axis activation and regulation. Early life
stressors epigenetically regulate the methylation of the GR and oxytocin
receptors to alter the nature of social interactions, as shown in preclinical
models [69]. The capacity of pineal melatonin to upregulate oxytocin as well as
suppress GR- nuclear translocation and adrenal cortex cortisol
production would indicate that suppressed pineal (and possibly local) melatonin
in ASD will modulate the interactions of the HPA axis with oxytocin and therefore
vagal nerve activation, and that this will interact with early stress induced
epigenetic changes in the GR and oxytocin receptors.
The amygdala [6, 70], hippocampus [71] and ventral tegmental area (VTA)/nucleus
accumbens (N.Acc) [72] show alterations in ASD linked to affect, cognition and
motivation, respectively. Cortisol significantly modulates these three sites and
their associated functions, exemplified by cortisol activation of the
GR- in the central amygdala (CeA), which increases local corticotropin
releasing hormone (CRH) that upregulates the -opioid receptor and its
ligand, dynorphin, in the basolateral amygdala (BLA), leading to feelings of
dysphoria, as shown in preclinical models [73]. This change in affective state
can be prevented by PVN oxytocin projections to CeA astrocytes that suppress CRH
induction by cortisol at the CeA GR- [74]. Such data indicate that the
suppression of pineal and local melatonin induction of oxytocin may allow
cortisol/stress to induce a dysregulated affective state (dysphoria) that is not
uncommon in ASD [75]. Similar factors and processes also regulate hippocampal
cognition and VTA/N.Acc motivation. As indicated above, the suppression of pineal
and local melatonin as well as oxytocin in ASD will modulate the influence of the
vagal nerve and cortisol/stress on affect, cognition and motivation, as shown in
Fig. 3.
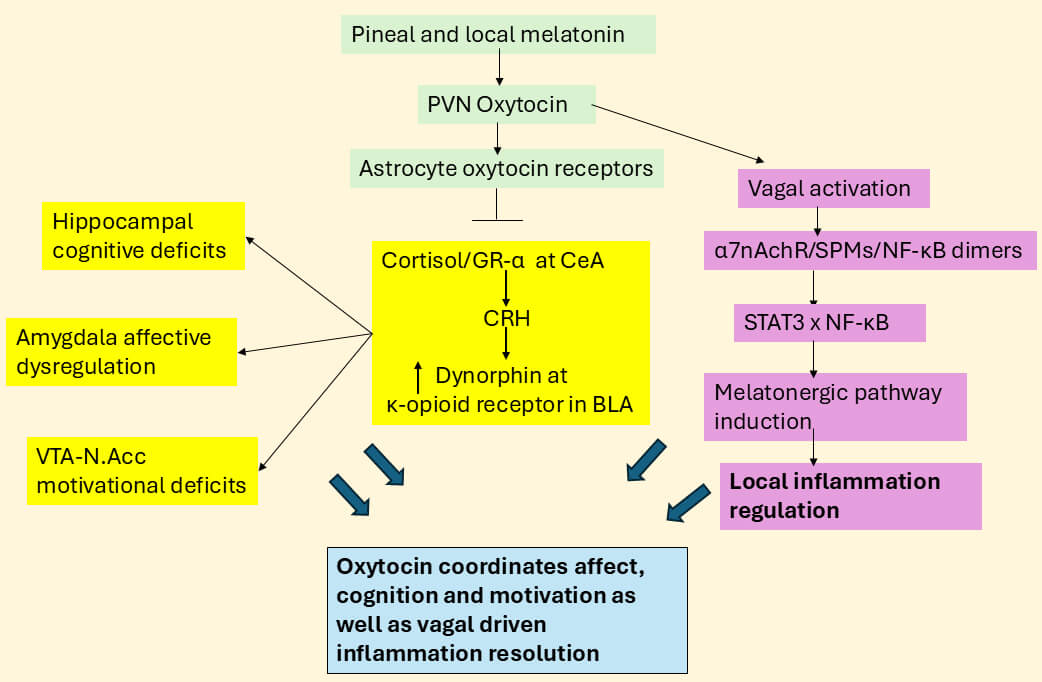 Fig. 3.
Fig. 3.
Melatonin, oxytocin and vagal nerve modulate cognition, affect
and motivation. Pineal and local melatonin may increase oxytocin activation of
the vagal nerve, with vagal ACh driving the 7nAChR/SPMs/NF-B
dimers pathway (purple shading) whilst also coordinating the effects of cortisol
by inhibiting GR- induced CRH in the central amygdala (CeA) thereby
suppressing dynorphin and -opioid receptor activation in the
basolateral amygdala (BLA) with parallel effects in the hippocampus and VTA/N.Acc
in the regulation of cognition and motivation, respectively (mechanisms not shown
for clarity). The changes in the BLA and CeA will also modulate hippocampal and
VTA/N.Acc function, with consequences for wider brain interarea connectivity. The
suppression of pineal and local melatonin in ASD, including by attenuating
oxytocin effects, will therefore have a wide range of CNS and systemic
consequences relevant to classical ASD pathoetiology and ongoing pathophysiology.
Abbreviations: 7nAChR, alpha 7 nicotinic acetylcholine receptor; BLA,
basolateral amygdala; CeA, central amygdala; CRH, corticotrophin releasing
hormone; GR, glucocorticoid receptor; N.Acc, nucleus accumbens; NF-B,
nuclear factor kappa-light-chain-enhancer of activated B cells; PVN,
paraventricular nucleus; SPMs, specialized pro-resolving mediators; STAT3, signal
transducer and activator of transcription 3; VTA, ventral tegmental area; CNS,
central nervous system.
3. Autism, the Opioidergic System, Borderline Personality and Perceived
Social Rejection
There is a growing appreciation of the pathophysiological overlaps of autism
with borderline personality disorder (BPD) [76, 77], with both showing significant
alterations in the opioidergic system. Perceived rejection sensitivity is a key
aspect of BPD pathophysiology [73] and may be an unrecognized aspect of ASD
affective dysregulation [78]. BPD [79], like ASD [80, 81], is associated with very
high levels of non-suicidal self-injury, which may arise from alexithymia and
difficulties in emotion recognition/expression [82] or from white matter
disruption [83] and/or from perceived social rejection induced dysphoria [73]
driven by increased dynorphin in the CeA arising from suppressed oxytocin, as
shown in Fig. 3.
Alterations in the opioidergic system have long been associated with ASD
pathophysiology, with µ-opioid receptor knockout rodents being a
preclinical ASD model that shows improved social interactions following
intranasal oxytocin administration [84]. BPD pathophysiology is intimately
associated with decreased µ-opioid receptor activation coupled with
increased -opioid receptor activation, with some treatment efficacy
being derived from ultra-low dose buprenorphine, a partial µ-opioid
receptor agonist and -opioid receptor antagonist [85, 86]. In different
preclinical ASD models (prenatal valproate and Fmr1-knockout) buprenorphine
increased social behaviors that correlated with increased neuronal activity in
the VTA/N.Acc and medial prefrontal cortex (PFC) [87], with medial PFC activation
negatively feeding back on amygdala activity [88]. Alterations in the
µ-/-opioid receptor ratio across brain regions can therefore have
significant impacts on social behaviors and interarea brain patterned activity,
as indicated in preclinical models, with links to how early developmental
trauma/stress may modulate the pathophysiological overlaps of ASD and BPD
[89, 90, 91].
The regulation of the opioidergic system may be intimately linked to alterations
in pineal melatonin, with melatonin acting on the pituitary to increase the
µ-opioid receptor endogenous ligand, beta-endorphin [92], whilst a specific
fragment of beta-endorphin, called des-tyrosine-gamma-endorphin (DTE),
dramatically increases pineal melatonin, as shown in rodents [93]. In contrast,
melatonin inhibits the sleep disturbing effect of the -opioid receptor
[94, 95]. Acute stress induced CRH increases dynorphin that activates the
-opioid receptor to suppress dopamine release [96, 97], which is
proposed to suppress the reward system and increase anhedonia, whilst chronic
stress can drive dysphoria and low mood via dynorphin activation of the
-opioid receptor [98]. Alterations in nociception are common in ASD,
including hypersensitivity and hyposensitivity [99], with affective aspects of
nociception significantly regulated by right amygdala -opioid receptor
activation [100] and the alterations in the µ-, -and
-opioidergic receptors arising from early developmental stress [101].
This also has pathophysiological relevance in BPD [73] and may be significantly
modulated by suppression of pineal and/or local melatonin availability [102].
Decreased melatonergic pathway availability may therefore regulate the
opioidergic system to modulate diverse aspects of symptomatology in ASD and BPD.
This may have relevance to wider bodies of data showing increased amygdala
-opioid receptor to correlate with poor self-reported social status
[103], indicating that the amygdala µ/-opioid receptor ratio may
regulate our perceived connectedness to others more widely, implicating roles for
pineal and/or local melatonergic pathway regulation in the modulation of wider
affective aspects of social connectedness. Current classification of ASD, in the
absence of any physiological indices, highlights the importance of social
interaction and connectedness, suggesting a significant role for alterations in
the opioidergic system and its regulation in the defining characteristics of ASD.
Alterations in the opioidergic system in ASD and BPD may be partly mediated by
increased gut permeability and gut dysbiosis in ASD [104] as well as in BPD
[105]. Gut permeability/dysbiosis are typically associated with decreases in the
short-chain fatty acid, butyrate. Butyrate is a histone deacetylase inhibitor
(HDACi) and epigenetic regulator that is also used as a metabolic substrate to
increase the melatonergic pathway, as shown in intestinal epithelial cells [106].
Butyrate also epigenetically regulates the µ- and -opioid
receptors [107, 108]. Some of the consequences of stress/cortisol induced gut
permeability/dysbiosis on the opioidergic system may therefore be mediated via
decreased butyrate and its regulation of the melatonergic pathway and/or
opioidergic system. Factors influencing the availability of tryptophan for the
initiation of the tryptophan-serotonin-N-acetylserotonin-melatonin pathway will
also have consequences for butyrate’s effects. As well as increasing gut
dysbiosis/permeability, chronic stress increases -opioid receptor
levels, which are major contributors to sleep disruption, indicating that
suppressed pineal and local melatonin production in ASD will alter the
consequences of chronic stress, including decreasing the
µ-/-opioid receptor ratio that will negatively regulate sleep to
further contribute to circadian and pineal melatonin dysregulation [94].
Suppressed pineal and local melatonin in ASD may therefore be intimately linked
to alterations in the opioidergic system, with significant consequences for
development of interarea brain connectivity, affective regulation, perceived
social rejection and non-suicidal self-injury, with significant overlaps to BPD
symptomatology and pathophysiology.
This begs the question as to how the opioidergic system interacts with canonical
and non-canonical pSTAT3 and NF-B dimer composition in the modulation
of the mitochondrial melatonergic pathway.
Opioidergic System Interactions With STAT3, NF-B and
miRNAs
Activation of the -opioid receptor increases STAT3, with diverse
effects across different body cells and organs [109], including upregulating
mitochondrial function in challenged cardiomyocytes [110]. In other cell types,
-opioid receptor decreases pSTAT3 by sequestering pSTAT3 to the plasma
membrane, as shown in chondrocytes [111]. It is unknown as to how
-opioid receptor activation modulates amygdala pSTAT3, including
whether it differentially impacts on the canonical, nuclear translocating
STAT3Tyr705 and/or non-canonical, mitochondria translocating
STAT3Ser727. Canonical, nuclear translocating pSTAT3 is mediated by
Tyrosine705 phosphorylation of STAT3, whilst non-canonical, mitochondria
translocating STAT3 is mediated by Serine727 phosphorylation. How these different
sites of STAT3 phosphorylation interact with NF-B dimer composition in
regulating the melatonergic pathway and how this then acts to modulate the
opioidergic system requires investigation, see Fig. 4 (Ref. [22, 112]). This will be
important to clarify in future research, especially as -opioid receptor
activation in the basolateral amygdala drives dysphoria that is commonly evident
in ASD [75]. The capacity of melatonin to increase the µ-opioid receptor
ligand, beta endorphin, indicates that pineal and/or local melatonin will
increase the µ-/-opioid receptor ratio, thereby paralleling the
beneficial effects of buprenorphine on social processes in ASD preclinical models
[85, 86, 87, 88, 89, 90, 91].
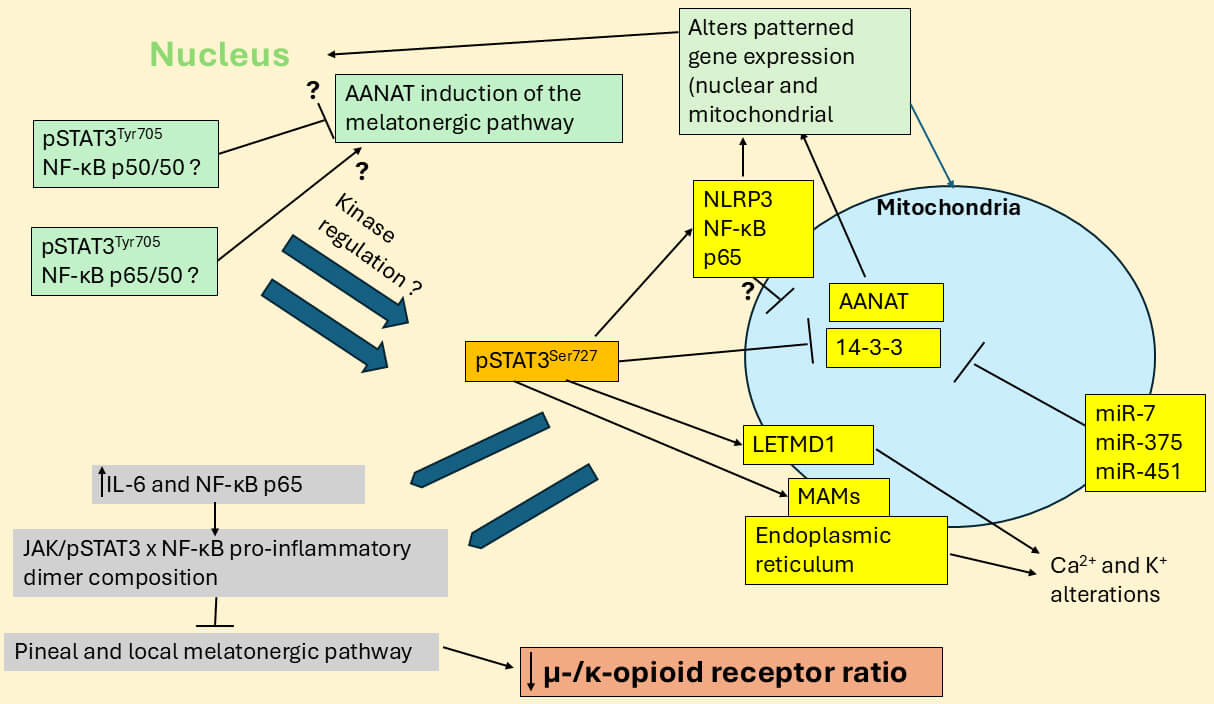 Fig. 4.
Fig. 4.
STAT3 interaction with NF-B dimers modulates
melatonergic and opioidergic pathways. Canonical pSTAT3Tyr705 interacts
with NF-B dimer composition in the nucleus to modulate the cellular
melatonergic pathway, which may be present in the nucleus although more likely in
mitochondria. Nuclear (green shade) translocated STAT3Tyr705 interacts with
NF-B dimer components (such as p65/50 and p50/p50) to stimulate or
inhibit the melatonergic pathway, with specific effects partly dependent upon
cell type [22]. Nuclear STAT3Tyr705 interactions with NF-B dimer
components may also modulate non-canonical, mitochondria translocating
pSTAT3Ser727, including from alterations in specific kinases that
phosphorylate and activate pSTAT3Ser727. At mitochondria, pSTAT3Ser727 can regulate many core aspects of mitochondrial function, including: (1)
regulates mitochondria-associated membranes (MAMs), thereby modulating
endoplasmic reticulum Ca2+ mitochondrial influx, a key driver of alterations
in mitochondrial function; (2) pSTAT3Ser727 can bind and regulate
mitochondrial 14-3-3 availability. As 14-3-3 is required to stabilize AANAT
stabilization to initiate the melatonergic pathway any suppression of 14-3-3
availability, including by miR-7, miR-375 and miR-451, will attenuate
melatonergic pathway availability; (3) In some cells, mitochondrial
pSTAT3Ser727 can form a positive reciprocal feedback loop with LETM1
domain-containing protein 1 (LETMD1), thereby regulating mitochondrial Ca2+
and K+ flux; and (4) Mitochondrial translocation of pSTAT3Ser727 enhances the mitochondrial translocation of the NACHT, LRR and PYD
domains-containing protein 3 (NLRP3) inflammasome, NF-B and p65 with
consequences for patterned gene expression in both the nucleus and mitochondria,
as shown in different cell types. Interestingly, LETM1/LETMD1 has a 14-3-3 like
matrix motif [112] that may bind AANAT and/or form a ‘dimer’ with 14-3-3,
indicating a possibly wider complexity to mitochondrial melatonergic pathway
regulation. Importantly, pro-inflammatory processes (IL-6, NF-B p65,
NLRP3) in a given cell will have consequences for adjacent cells of the local
microenvironment, via increased IL-6 and NLRP3 inflammasome induced IL-1
and IL-18 release driving inflammatory processes in neighboring cells, including
via released IL-6 activating JAK/pSTAT3/NF-B to stimulate or suppress
the melatonergic pathway in cells of the local microenvironment. The suppressed
capacity to induce the melatonergic pathway in a given cell therefore has
implication for the regulation of the melatonergic pathway in neighboring cells
and inflammatory responses within its local microenvironment. The suppression of
pineal and/or local melatonin will have consequences for
µ-/-opioid receptor ratio and therefore the role of the
opioidergic system in ASD, including in the regulation of affect, cognition and
motivation, as highlighted in Fig. 3. The specifics of pSTAT3 interactions with
NF-B dimer composition in ASD cells over the course of development will
be important to determine. Abbreviations: AANAT, aralkylamine
N-acetyltransferase; JAK, Janus kinase; LETM1, Leucine Zipper EF-hand containing
Transmembrane protein 1; MAMs, mitochondria-associated membranes; miR, microRNA;
NF-B, nuclear factor kappa-light-chain-enhancer of activated B cells;
NLRP3: NACHT, LRR and PYD domains-containing protein 3; STAT3, signal transducer
and activator of transcription 3; LETMD1: LETM1 domain-containing protein 1; IL,
interleukin.
Activation of the µ-opioid receptor typically increases pSTAT3 [113] and
has differential effects on NF-B that seem dependent upon cell type and
phenotypic state [114]. NF-B activation also increases the
µ-opioid receptor [115]. Both µ- and -opioid receptor
interact with the chaperone protein, 14-3-3, [116], with 14-3-3
significantly interacting with mitochondrial STAT3Ser727 to regulate
14-3-3 availability [62, 117]. This may be important given the role of
14-3-3 in the stabilization of AANAT in the initiation of the
melatonergic pathway [5, 6]. As indicated, ASD may be associated with an increase
in microRNA (miR)-451 [5], which, like miR-375 and miR-7 can attenuate the
initiation of the melatonergic pathway by AANAT by decreasing 14-3-3
availability [118]. The regulation of 14-3-3 may therefore be of
importance in the coordination of the opioidergic system and mitochondrial
melatonergic pathway, including as arising from mitochondrial, non-canonical
STAT3Ser727 binding and regulating 14-3-3 availability [62]. This
is supported by data showing miR-451 to regulate STAT3 [119, 120] and
NF-B [121, 122], as does miR-375 [123, 124] and miR-7 [125, 126]. Whether
the regulation of 14-3-3 by STAT3Ser727 is coordinated by these
miRNAs with consequences for opioidergic system regulation will be important to
determine. The interactions of canonical and non-canonical pSTAT3 with
NF-B dimer composition in the modulation of the melatonergic pathway
are shown in Fig. 4.
Overall, the data linking canonical and non-canonical pSTAT3 interactions with
NF-B dimer composition in the modulation of the melatonergic pathway
requires extensive further investigation to determine whether this is intimately
linked to alterations in the opioidergic system and opioid receptors at key
sites, as well as the regulation of oxytocin, vagal nerve and gut microbiome.
4. Aryl Hydrocarbon Receptor, STAT3, NF-B, Opioidergic System
and Melatonergic Pathway
The aryl hydrocarbon receptor (AhR) significantly modulates ASD pathophysiology
[127, 128]. The AhR has a number of complex effects that are dependent upon
specific ligands and cell types as well as its site of expression, namely within
the cytoplasm and/or on the mitochondrial membrane [129]. The AhR can be
activated by many ligands including endogenous (FICZ) and induced (kynurenine) as
well as environmental toxins, such as air pollutants and cigarette smoke products
[130]. The AhR also regulates the melatonergic pathway via AhR activation
induction of cytochrome P450 (CYP)1B1 and CYP1A2, which can hydroxylate melatonin
as well as O-demethylate melatonin ‘backwards’ to its immediate precursor,
N-acetylserotonin (NAS) [131]. The association of the AhR with ASD may therefore
be via direct suppression of melatonin availability, whilst the complexity of AhR
effects may arise from cell conditions that determine whether the melatonergic
pathway is available or not. Consequently, AhR effects may be dependent upon
STAT3 interactions with NF-B dimer composition [22].
This is further complicated by the AhR also regulating STAT3 across diverse cell
types and medical conditions, including cancer [132] and cardiovascular diseases
[133]. The raised levels of pro-inflammatory cytokines (IFN-,
IL-1, IL-6, and TNF-) in ASD [134] induce indoleamine
2,3-dioxygenase (IDO) that converts tryptophan to kynurenine to reduce tryptophan
availability for the tryptophan-melatonin pathway, with kynurenine activating the
AhR, to further reduce melatonin availability [135]. Raised kynurenine and
kynurenic acid levels are evident in ASD children, vs controls, with both of
these kynurenine pathway products activating the AhR [136], Such data indicates
an enhanced ligand availability for AhR activation in ASD that concurrently
decreases tryptophan-melatonin pathway availability. Diabetes linked
hyperglycemia increases glucose glycation thereby increasing methylglyoxal
levels, which dramatically suppress tryptophan availability via protein-protein
interactions [137]. This not only decreases tryptophan availability for the
tryptophan-melatonin pathway, thereby limiting tryptophan availability for
conversion to kynurenine and AhR activation. This is likely to contribute to
variations in kynurenine pathway products and therefore AhR activation in ASD
[138]. This requires further investigation as it indicates that AhR complexity
and contrasting effects may arise from an uninvestigated tryptophan-melatonin
pathway availability, including as arising from raised methylglyoxal in
prediabetes, type 1 dianbetes mellitus (T1DM) and T2DM suppressing tryptophan
availability. Such interactions highlight the interconnected nature of ASD with
diabetic pathophysiology and how this may contribute to contrasting results
across studies.
As pro-inflammatory cytokine-induced IDO drives the kynurenine activation of the
AhR, the AhR is intimately associated with a pro-inflammatory NF-B
dimer composition. The AhR and NF-B are classically thought to have
negative reciprocal interactions [139], with the AhR able to bind the
pro-inflammatory component of NF-B, p65, both in the cytoplasm and
nucleus [139]. The AhR can therefore modulate mitochondrial pSTAT3Ser727 effects, given that the mitochondrial translocation of pSTAT3Ser727 also
drives NF-B, p65 and the NLRP3 inflammasome to mitochondria [140] (see
Fig. 4), Mitochondrial NF-B and p65 directly modulate mitochondrial
transcription and function, whilst the NLRP3 inflammasome locates adjacent to the
outer mitochondrial membrane, thereby increasing access to mitochondrial caspases
that cleave pro-IL-1 and pro-IL-18 into their active forms. By
suppressing NF-B and p65 the AhR may therefore change pSTAT3Ser727 regulation of mitochondrial function. Heightened NLRP3 inflammasome and
IL-1 are evident in ASD, as shown in ASD fibroblasts [141], with
mitochondrial ROS driving NLRP3 inflammasome activation. This suggests that the
suppression of the mitochondrial melatonergic pathway in ASD may be a significant
determinant of NLRP3 activation, which may be modulated by AhR suppression of
NF-B and p65 to therefore shape the consequences of pSTAT3Ser727 mitochondrial translocation [142]. As methylglyoxal suppresses tryptophan to
decrease kynurenine availability for AhR activation,
diabetes/hyperglycemia/methylglyoxal and the AhR may therefore interact with
STAT3/NF-B to modulate core aspects of mitochondrial dysfunction,
including the mitochondrial melatonergic pathway, in ASD. Whether the suppression
of the AhR by methylglyoxal decreasing tryptophan availability for conversion to
kynurenine upregulates NF-B p65 induced pro-inflammatory cytokines will
be important to determine. Overall, factors that modulate STAT3 interactions with
NF-B in the modulation of mitochondrial function and the melatonergic
pathway, including genetic, epigenetic and early developmental stressors, as well
as methylglyoxal and the AhR, may be intimate aspects of mitochondrial
dysfunction in ASD. The interactions of the AhR and methylglyoxal with the
tryptophan-melatonin pathway are shown in Fig. 5.
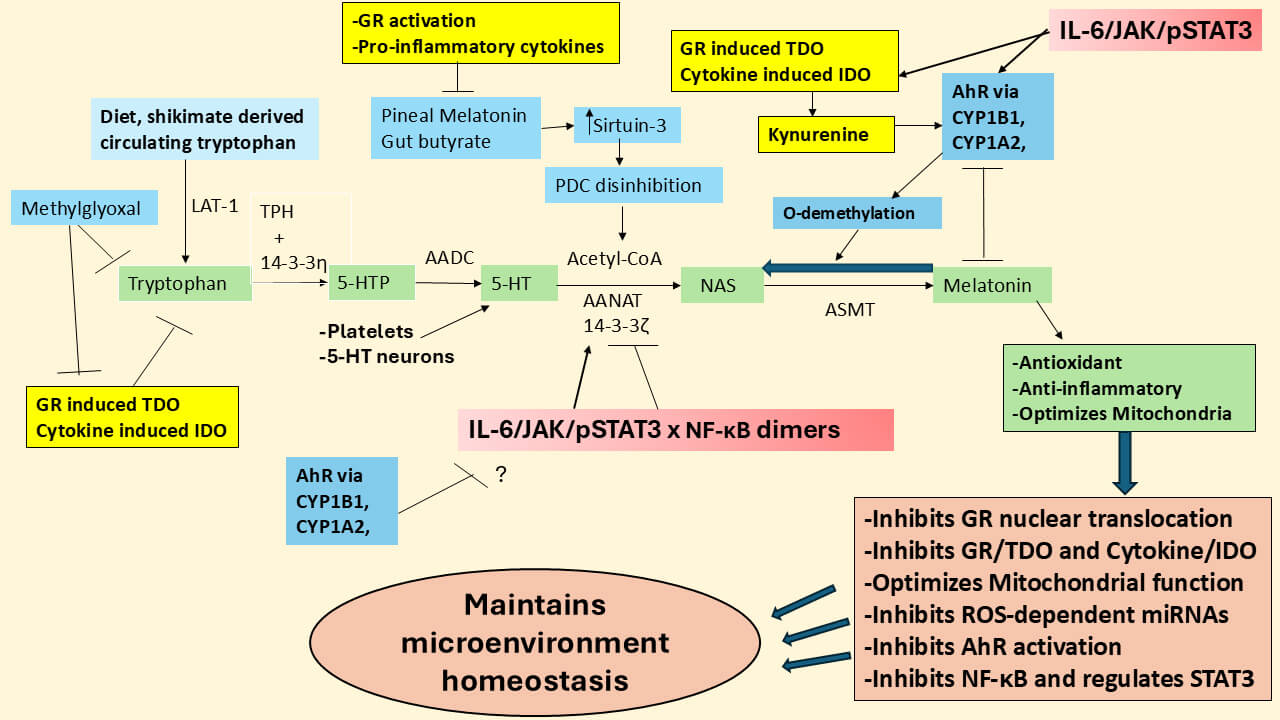 Fig. 5.
Fig. 5.
A diversity of factors can modulate the
tryptophan-melatonin pathway. The mitochondrial melatonergic pathway of the
tryptophan-melatonin pathway (green shade) is evident in all cells where it has
been investigated. Methylglyoxal, via protein-protein interactions with
tryptophan, not only suppresses tryptophan but also tryptophan derived kynurenine
pathway products that can activate the AhR, thereby changing the consequence of
AhR activation as well as GR-induced TDO and cytokine induced IDO by limiting
tryptophan availability. As the AhR can modulate STAT3 and inhibit NF-B
p65, the suppression of tryptophan conversion to kynurenine can change the
influence of the AhR on the regulation of the melatonergic pathway. The AhR
induction of CYP1B1 and CYP1A2 leads to the hydroxylation and/or
‘O-demethylation’ of melatonin, with both processes decreasing melatonin
availability and effects. As IL-6 not only induces the JAK/pSTAT3 pathway but
also IDO, IL-6 may therefore initiate the IDO/kynurenine/AhR/CYP1B1/CYP1A2 to
suppress the tryptophan-melatonin pathway, although this would be dependent upon
tryptophan availability for conversion to kynurenine, and therefore subject to
suppression by methylglyoxal. Alterations in T2DM/hyperglycemia/methylglyoxal and
AhR activation may therefore act on core aspects of ASD pathophysiology by
modulating mitochondrial function, including the mitochondrial melatonergic
pathway, with consequences for cellular function and homeostatic intercellular
interactions. Abbreviations: 5-HT, serotonin; 5-HTP, 5-hydroxytryptophan; AADC,
aromatic-L-amino acid decarboxylase; AANAT, aralkylamine N-acetyltransferase;
AhR, aryl hydrocarbon receptor; ASMT, acetylserotonin methyltransferase; CYP,
cytochrome P450; GR, glucocorticoid receptor; IDO, indoleamine 2,3-dioxygenase;
JAK, Janus kinase; LAT-1, large amino acid transporter 1; NAS, N-acetylserotonin;
NF-B, nuclear factor kappa-light-chain-enhancer of activated B cells;
PDC, pyruvate dehydrogenase complex; ROS, reactive oxygen species; STAT3, signal
transducer and activator of transcription 3; TDO, tryptophan 2,3-dioxygenase;
TPH, tryptophan hydroxylase.
Overall, the AhR has complex effects on many aspects of ASD pathophysiology,
including interactions of STAT3 and NF-B, with consequences for
mitochondrial function and NLRP3 inflammasome activation. These effects are
intimately intertwined with modulation of the mitochondrial melatonergic pathway,
as shown in Figs. 4,5.
5. Autism Pathoetiology Implications
As indicated above the relative suppression of the melatonergic pathway across
CNS and systemic cells may be an important aspect of ASD pathophysiology in all
its manifestations [5], which include a range of severe learning difficulties to
high-functioning ASD. Melatonergic pathway suppression is intimately associated
with STAT3 and its interactions with NF-B dimer composition, as well as
other regulatory factors such as diabetes/methylglyoxal, the AhR and miRNAs, as
indicated above. As ASD is classically conceptualized as a neurodevelopmental
disorder [143], when and how does such mitochondrial melatonergic pathway
dysregulation occur?
Early developmental risk factors for ASD include preeclampsia [144], which is
associated with a decrease in placental melatonin production [145] and may
exemplify the importance of prenatal melatonergic pathway modulation in ASD
pathoetiology. Many of the melatonergic pathway regulatory factors highlighted
above are also important to placental regulation, including miRNAs [146, 147, 148],
STAT3 [149] and NF-B [150]. Preeclampsia increases cortisol transfer
over the placenta via 11-HSD2 suppression [151]. This suggests parallels
to the alterations in night-time dampening and resetting arising from suppressed
pineal melatonin and associated disinhibition of the wider cortisol system, as
indicated in Fig. 1. Do such placental alterations establish an early
developmental pattern of microenvironment interactions in the developing fetus
leading to a subtle change in optimal homeostatic interactions occurring, with
consequences for differential stress responses, arising from a decreased
melatonin/cortisol ratio prenatally? Another prenatal conditions, intrauterine
growth restriction (IUGR), is also associated with increased cortisol transfer
over the placenta [152] and enhanced ASD risk in the offspring [153]. It should
be noted that this does not necessarily indicate that placental melatonin
replicates circadian, pineal melatonin suppression in ASD as the placental
release of melatonin is not circadian [154]. However, a decrease in the placental
melatonin/cortisol ratio may change the nature of cellular and intercellular
homeostasis in the developing fetus within a crucial temporal window.
The AhR is highly expressed in the placenta and modulates many aspects of
placental function, including trophoblast cell proliferation, migration and
apoptosis, as well as energy metabolism [155]. As noted, the AhR via CYP1B1 and
CYP1A2 can hydroxylate melatonin and ‘backward’ convert melatonin to
N-acetylserotonin (NAS) via O-demethylation [131], with NAS being a brain-derived
neurotrophic factor (BDNF) mimic via its activation of the BDNF receptor,
tyrosine receptor kinase (Trk)B [156]. This may suggest an increase in placental
and fetal NAS that not only suppresses melatonin availability but increases TrkB
activation, which in ASD models is associated with ASD pathophysiology via
alterations in -amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
(AMPA) receptor activation [157]. As NAS can also increase hippocampal BDNF
[158], alterations in the placental NAS/melatonin pathway may contribute to the
diverse effects of BDNF and TrkB in ASD pathoetiology, as shown in diverse
preclinical models [159, 160], with relevance to brain overgrowth (macrocephaly)
that is evident in a subset of people with ASD [161].
At least 8 isoforms of the GR- are evident in the placenta and can
vary by fetal sex and birthweight [162], being proposed as an important interface
with the maternal environment and fetal growth [163]. GR- isoforms also
influence nutrient regulation [164]. Preclinical data indicates that prenatal
stress leads to hypermethylation of glucocorticoid-related genes that disrupts
the placental glucocorticoid barrier, with significant consequences for fetal
development [165]. The GR- is present in the human placenta and is
classically modelled as a dominant negative regulator of GR-, although
recent work shows the GR- to have transcriptional consequences that are
independent of its inhibition of GR- [166]. The raised pro-inflammatory
cytokines evident in ASD increase the GR-/GR- ratio to
suppress the capacity of cortisol and corticosteroids to dampen inflammatory
activity [167], including by GR- attenuating the capacity of
GR- to suppress NF-B [166]. How the
GR-/GR- ratio modulates NF-B dimer components and
their interactions with pSTAT3 in the regulation of the placental, fetal and
post-natal melatonergic pathway will be important to determine, including as to
the consequences that this has for night-time dampening and resetting mediated by
the interactions of pineal melatonin and cortisol following the establishment of
the circadian rhythm in the developing infant. As melatonin attenuates
GR- nuclear translocation, the suppression of melatonin will decrease
the threshold for GR- induction and alterations in NF-B
regulation and therefore in the regulation of the melatonergic pathway. This is
one route whereby alterations in the placenta may shift the influence of
melatonin and cortisol in the developing fetus and infant.
The plasma membrane GR is also evident in the placenta, perhaps especially in
syncytiotrophoblasts [168]. However, the presence and regulation of the
mitochondrial membrane GR and mitochondrial matrix GR in the placenta awaits
investigation and the role of placental bcl2-associaited athanogene (BAG)-1 [169]
in the transport of GR to the mitochondrial matrix, as in other cell types [170],
requires further investigation. As melatonin, like gut microbiome derived
butyrate, suppresses GR- nuclear translocation, presumably the
suppression of placental melatonin in ASD prenatal risk conditions (e.g.,
preeclampsia and IUGR) will have consequences for wider cortisol receptors and
their effects in both the placenta and developing fetus, with later developmental
consequences.
As in any cell the increased glycolysis in preeclamptic trophoblasts leads to
glycation induced methylglyoxal [171]. These authors showed that preeclamptic
trophoblasts increase methylglyoxal and methylglyoxal induced advanced glycation
end products (N(6)-(carboxymethyl)lysine [CML], and
Nε-(carboxyethyl)lysine [CEL], as well as methylglyoxal-derived
hydroimidazolone [MG-H]), coupled to a decrease in glyoxalase (Glo)1 that
metabolizes methylglyoxal [171]. Maternal plasma concentrations of methylglyoxal,
CML and MG-H1 increase as early as the 12th week of gestation indicating that
these products may be potential early biomarkers of preeclampsia [171]. Notably,
mitoQ (a mitochondrial oxidant quencher) prevented these preeclamptic
methylglyoxal driven changes when the data was replicated in a trophoblast cell
line. This data readily links to the decreased placental melatonin in
preeclampsia and how its loss in mitochondria can prevent melatonin from
offsetting the consequences of suboptimal mitochondrial function as indicated by
raised mitochondria oxidant production and its influence on patterned gene
expression via ROS-dependent miRNAs. This also has implications for intercellular
fluxes and therefore for alterations in the homeostatic interactions of the local
microenvironment.
Melatonin increases mitochondria located sirtuin-3 [172] which suppresses
oxidant production at three points of the electron transport chain [173].
Consequently, the detrimental effects of suppressed placental melatonin may, at
least partly, arise from a decrease in melatonin induction of trophoblast
sirtuin-3. Decreased trophoblast sirtuin-3 and associated increase in the
acetylation, and inhibition, of the antioxidant enzyme, manganese superoxide
dismutase (MnSOD), are evident in preeclampsia and contribute to increased
mitochondrial ROS driven alterations in patterned gene expression [174]. The
suppression of the placental melatonergic pathway therefore modulates
mitochondrial function, at least partly via a decrease in mitochondrial sirtuin-3
and endogenous antioxidants.
As noted above, methylglyoxal can directly downregulate tryptophan availability
by protein-protein interactions [137], indicating that the necessity to
upregulate methylglyoxal in the course of glycolysis may be intimately linked
across diverse cell types to the suppression of the melatonergic pathway. In many
circumstances, this would seem to arise from the increased glycolysis and
methylglyoxal suppression of the tryptophan-melatonin pathway as an indicant of
the need for chemoattracted immune cells to deal with the changes/challenges
occurring, a situation where the local production of melatonin would suppress
immune cell efficacy. Whether this is pertinent in preeclampsia and how it
associates with ASD pathoetiology will be important to determine. As the
glucocorticoid receptor (GR) can be glycated by methylglyoxal to alter its
function [174], the raised levels of methylglyoxal in preeclampsia may not only
suppress melatonin but also alter the nature of the wider cortisol system
response, including GR subtypes and sites of localization. This requires future
investigation.
The above would indicate that the understanding of ASD etiology may require a
fuller investigation of processes and conditions, such as preeclampsia, and how
they may contribute to the alterations in the mitochondrial melatonergic pathway
that are proposed to be a core factor in ASD pathophysiology. Given the
importance of melatonin and cortisol (and their interactions) in the night-time
dampening and resetting of body cells, microenvironments and systems across the
life-span, it would not seem incongruous that factors influencing melatonin and
cortisol levels and effects as well as their interactions will be important in
determining the consequences of environmental sampling that occurs over the
course of pregnancy. This also provides a framework for understanding ASD genetic
susceptibility factors and the processes on which they act in ASD etiology. See
Fig. 6.
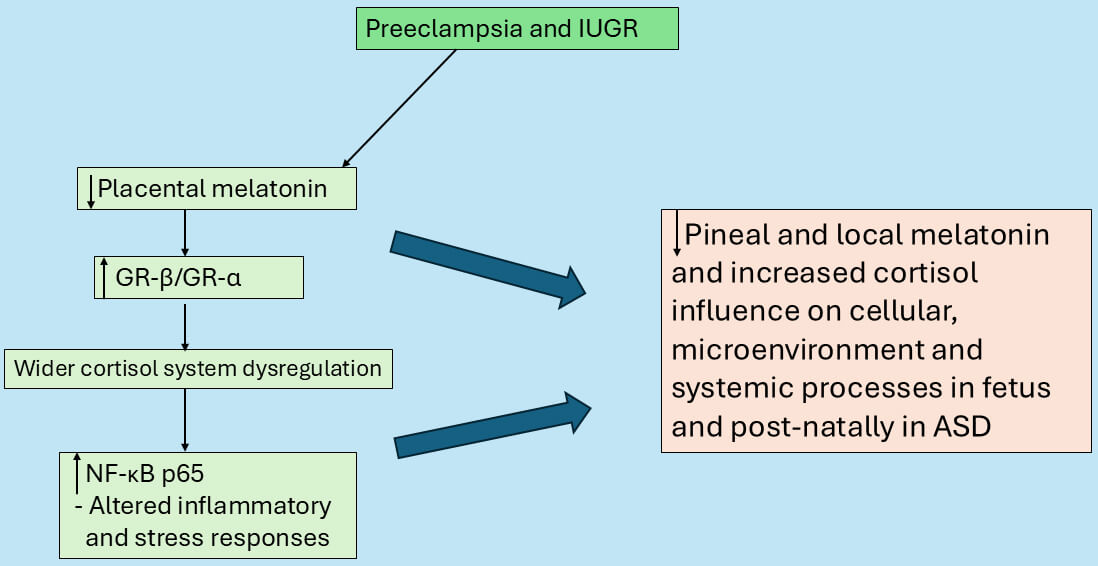 Fig. 6.
Fig. 6.
Preeclampsia and IUGR suppress placental melatonin to
change cortisol fetal effects. Suppressed placental melatonin may disinhibit the
GR-, which increases GR-, thereby enhancing NF-B p65
activation to dysregulate stress responses. These changes in the placenta drive
alterations in the developing fetus in a crucial temporal window that shapes
homeostatic interactions in local microenvironments and later postnatal
development. Abbreviations: GR, glucocorticoid receptor; IUGR, intrauterine
growth restriction; NF-B, nuclear factor kappa-light-chain-enhancer of
activated B cells.
6. Future Research Implications
(1) Does suppressed pineal melatonin in ASD initially disinhibit GR-
activation with consequent alterations in the wider cortisol ‘system’, including
the levels of GR- and the GR localization site (cytoplasm, plasma
membrane, mitochondrial membrane and mitochondrial matrix), as well as
11-HSD1 induction [65, 66]. Would such dysregulation of melatonin and
cortisol at night modulate oxytocin levels as well as the interactions of
cortisol with oxytocin, such as cortisol’s rapid negative feedback on the
oxytocin induction of adrenocorticotropic hormone (ACTH) and the HPA axis [67]?
Does suppressed pineal and/or local (PVN) melatonin decrease oxytocin and
therefore vagal nerve stimulation that dampens local inflammatory activity?
(2) As early life stressors epigenetically regulate the methylation of the GR
and oxytocin receptors to alter the nature of social interactions in preclinical
models [69], would the suppressed capacity to induce pineal and local melatonin
in ASD modulate the impact of early life stressors via melatonin’s capacity to
induce oxytocin and suppress GR- nuclear translocation? Is this also
modulated by the loss of pineal melatonin’s suppression of gut
permeability/dysbiosis and potentiation of butyrate production, given that
butyrate also suppresses GR- nuclear translocation?
(3) Does µ-, vs -, opioid receptor activation differentially
modulate amygdala, especially basolateral amygdala (BLA), pSTAT3 either via
canonical STAT3Tyr705 and/or non-canonical STAT3Ser727, thereby
impacting on the regulation of the local melatonergic pathway in BLA neurons
and/or astrocytes?
(4) Are the interactions of the opioidergic system and receptors with the
melatonergic pathway dependent upon 14-3-3 regulation and availability, including
as a consequence of mitochondria located STAT3Ser727 interacting with, and
regulating, 14-3-3 availability? Is the availability of 14-3-3
also determined by the miRNAs, miR-451, miR-375 and miR-7 [5]?
(5) Is 14-3-3 regulation by STAT3Ser727 coordinated with miR-451,
miR-375 and/or miR-7 levels and their regulation, with consequences for
opioidergic system/receptor levels?
(6) Does the increase in methylglyoxal levels in ASD, by decreasing tryptophan
availability [137], contribute to the variability of increased serotonin and
kynurenine pathway products in ASD. Would methylglyoxal, by decreasing tryptophan
availability for conversion to kynurenine, therefore attenuate AhR activation,
including in the modulation of the melatonergic pathway as well as the AhR
suppression of NF-B? This would indicate specific consequences for AhR
activation in the presence or suppression of the tryptophan-melatonin pathway? Is
this an unrecognized aspect of the complexity and mixed results linked to AhR
activation?
(7) Are the complexity of AhR effects determined by whether the melatonergic
pathway is present or not in a given cell, with consequences not only for a given
cell but for its interactions with other cells in its local microenvironment?
(8) Does the association of preeclampsia and IUGR with ASD risk arise from a
decrease in placental melatonin/cortisol ratio to alter cellular,
microenvironment and systemic melatonergic pathway availability across body
cells? Does this arise from an early developmental ‘crucial window’? Is this
‘crucial window’ determined by alterations in the homeostatic interactions of
cells in their given microenvironment?
(9) How do variations in the GR-/GR- ratio modulate placental
NF-B dimer composition and therefore the regulation of the melatonergic
pathway via interactions with STAT3? Does this have consequences for subsequent
post-natal night-time dampening and resetting mediated by the interactions of
pineal melatonin and cortisol, suggesting that non-circadian variations in
placental/fetal melatonin and cortisol modulate their subsequent post-natal
levels and effects? How does an increased GR-/GR- ratio and
decreased 11-HSD2 in the placenta and post-natal cells modulate
pSTAT3Tyr705 and pSTAT3Ser727? Does an increase in the
GR-/GR- ratio, via increased NF-B, drive a
maintained inflammation that enhances immune cell chemoattraction to resolve
inflammation and therefore coupled to suppression of the melatonergic pathway?
This could indicate that ‘glucocorticoid resistance’ acts to signal the necessity
of immune system chemoattraction and activation by minimizing the effects of
cortisol and melatonin.
(10) As the glucocorticoid receptor (GR) can be glycated by methylglyoxal to
alter its function [175], the raised levels of methylglyoxal in preeclampsia may
not only suppress melatonin via protein-protein interactions [137] but also alter
the nature of the wider cortisol system response, including GR subtypes,
GR-/GR- ratio and sites of GR localization. This requires
future investigation.
(11) Do alterations in the regulation of the melatonergic pathway and its
interactions with cortisol occur prior to placenta formation? The melatonergic
pathway is evident in oocytes and the granulosa immune cells that regulate oocyte
selection and development. Would this have relevance to intercellular
interactions in blastocysts and the subsequent interface with the endometrial
wall and maternal immune cells in the course of shallow placentation?
(12) Hyperserotonemia in ASD is associated with learning difficulties [176]. It
requires investigation whether this arises from decreased conversion of
hippocampal serotonin to melatonin given the importance of melatonin in long-term
potentiation (LTP) regulation [177, 178]. Does the wide range of cognitive
capacity in people classed with ASD arise from factors regulating mitochondrial
melatonergic pathway availability in the hippocampus?
(13) ASD is associated with an increased risk of cancer and COVID-19 fatality
[179, 180]. This may be especially evident in people with ASD and learning
difficulties, with ASD linked to a decreased cytotoxicity of natural killer (NK)
cells [181, 182]. Is the suppressed capacity to induce the melatonergic pathway in
ASD across diverse cell types [5] also evident in NK cells? Exogenous melatonin
increases NK cell cytotoxicity, which is also powerfully regulated by melatonin
over the circadian rhythm [183], suggesting that the suppression of endogenous NK
cell tryptophan-melatonin pathway by canonical and noncanonical STAT3
interactions with NF-B dimer composition may modulate the NK cell
melatonergic pathway and associated cytotoxicity. This will be important to
determine in ASD, given the capacity of melatonin to increase NK cell elimination
of tumor cells and viral infected cells. Alternatively, is the increased risk of
cancer and COVID-19 fatality in ASD linked to increased concurrent T2DM and
raised methylglyoxal levels that bind tryptophan to attenuate the initiation of
the tryptophan-melatonin pathway [137]?
7. Treatment Implications
(1) Although the above clearly provides future research that should shape
prevention and treatment, it is clear that the utilization of melatonin in ASD
will provide some circadian and systemic benefits to decrease symptomatology.
(2) Given the overlapping pathophysiology of ASD with Borderline personality,
there may be some utility of ultra-low dose buprenorphine, with possible
particular relevance to stress-induced by social rejection and associated
emotional dysregulation [184]. It is also important to note that low dose
buprenorphine has also shown clinical utility in single case studies of people
with ASD, with improvement in social interaction processes [185].
(3) As hyperglycemia driven methylglyoxal modulates tryptophan availability for
the tryptophan-melatonin pathway, quercetin may have some utility in ASD due to
its quenching of methylglyoxal [186]. Preclinical models would indicate that
quercetin and its derivatives have utility in ASD [187].
(4) Other dietary factors/nutriceuticals, such as the polyphenol,
epigallocatechin gallate (EGCG), have some clinical utility in ASD, which is
typically modelled as being mediated via sealing the gut barrier, decreasing
dysbiosis and increasing butyrate [188]. However, EGCG is also a monoamine
oxidase inhibitor and therefore may increase serotonin availability for the
melatonergic pathway in people with ASD without hyperserotonemia [189]. EGCG also
inhibits the AhR [190], which as indicated above may be intimately linked to the
regulation of core ASD pathophysiology.
(5) Another nutriceutical, resveratrol, which inhibits the AhR and increases
sirtuins [191], is also proposed to have benefits in offsetting the effects of
prenatal stress/valproate induction of ASD-like characteristics in preclinical
models [192]. Whether resveratrol regulates the STAT3 interaction with
NF-B in the modulation of the melatonergic pathway will be important to
determine in regard to its potential clinical efficacy.
(6) Recent work has highlighted the potential of repetitive transcranial
magnetic stimulation (rTMS) in the treatment of neurodevelopmental disorders,
including ASD [193]. Interestingly, rTMS decreases systemic cortisol [194] and
increases pineal melatonin [195] indicating that rTMS will have significant
impacts on how CNS and systemic processes are dampened and reset at night.
Whether the rTMS upregulation of pineal melatonin increases oxytocin and oxytocin
activation of the vagal nerve, as indicated above in Fig. 3, will be important to
determine in clinical investigations. It will be important to clarify whether
rTMS effects, both at the site of direct application and systemically, involve
alterations in canonical and noncanonical STAT3 and its interactions with
NF-B dimer composition, as some data may suggest [196, 197]. The
association of rTMS with the regulation of fear processing and post-traumatic
stress disorder (PTSD) [198, 199] may underpin and reshape the conceptualization
of an altered stress response in ASD, as previously indicated for another
neurodevelopmental disorder [200]. The extent to which the effects of rTMS are
mediated via pineal melatonin, including in the regulation of the gut
barrier/permeability [201] and/or oxytocin stimulation of the vagal nerve having
efficacy as consequence of melatonin availability in gut cells will be
interesting to determine.
8. Conclusions
The above highlights the potential relevance of alterations in the melatonergic
pathway in ASD with pathoetiological and ongoing pathophysiological implications.
It is proposed that the interactions of canonical and non-canonical STAT3 with
NF-B dimer composition may be an important, under-explored aspect of
ASD biological underpinnings. This provides a perspective of core processes on to
which many previously disparate bodies of data on ASD can be incorporated and
integrated. The understanding of the role of the mitochondrial melatonergic
pathway in early developmental processes, as exemplified by preeclampsia, should
provide a body of knowledge that will allow the monitoring and targeting of early
developmental processes in the pathoetiology of ASD.
Abbreviations
11-HSD1, 11-hydroxysteroid dehydrogenase type 1; 5-HT, serotonin; 5-HTTP, 5-hydroxytryptophan; 7nAChR, alpha 7nicotinic acetylcholine receptor; AADC, aromatic-L-amino acid decarboxylase; AANAT, aralkylamine N-acetyltransferase; acetyl-CoA, acetyl-coenzyme A; ACTH, adrenocorticotropic hormone; AhR, aryl hydrocarbon receptor; ASMT, N-acetylserotonin O-methyltransferase; BAG-1, bcl-2 associated athanogene 1; BDNF, brain-derived neurotrophic factor; BLA, basolateral amygdala; CAR, cortisol awakening response; CeA, central amygdala; CRH, corticotrophin releasing hormone; CSF, cerebrospinal fluid; CYP, cytochrome P450; FKBP4, FKBP prolyl isomerase 4; GR, glucocorticoid receptor; GRE, glucocorticoid receptor element; HDAC, histone deacetylase; HPA, hypothalamic-pituitary-adrenal; hsp, heat shock protein; IDO, indoleamine 2,3-dioxygenase; IUGR, intrauterine growth restriction; LETM1, Leucine Zipper EF-hand containing Transmembrane protein 1; lnc, long non-coding; LAT-1, large amino acid transporter 1; MAMs, mitochondria-associated membranes; MHC, major histocompatibility complex; N.Acc, nucleus accumbens; NAS, N acetylserotonin; NF-B, nuclear factor kappa-light-chain-enhancer of activated B cells; NK, natural killer; NLRP3, NACHT, LRR and PYD domains-containing protein 3; OXPHOS, oxidative phosphorylation; PDC, pyruvate dehydrogenase complex; PVN, paraventricular nucleus; SPMs, specialized pro-resolving mediators; STAT3, signal transducer and activator of transcription 3; T2DM, type 2 diabetes mellitus; TCA, tricarboxylic acid; TDO, tryptophan 2,3 dioxygenase; TPH, tryptophan hydroxylase; VTA, ventral tegmental area.
Author Contributions
GA confirms sole responsibility for the following: study conception and design and manuscript writing. GA read and approved the final manuscript.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of Interest
The author declares no conflict of interest.








