





1 Department of Ophthalmology, Indiana University School of Medicine, Indianapolis, IN 46202-5209, USA
2 Stark Neuroscience Research Institute, Indiana University School of Medicine, Indianapolis, IN 46202-5209, USA
Abstract
The Rho GTPase and Rho kinase (ROCK) signaling pathway is essential for cellular mechanics, acting as key regulators of the actin cytoskeleton and actomyosin contractility in various cell types and tissues. Rho GTPases, functioning as molecular switches, and ROCKs, their primary downstream effectors, influence vital cellular processes such as cell shape, movement, growth, and gene regulation. This review explores how this pathway maintains tissue tone, especially its significant role in regulating trabecular meshwork (TM) contractility. It also highlights the critical part of the Rho-ROCK pathway in precisely managing intraocular pressure (IOP). Dysregulation of Rho/ROCK signaling is a known factor in increased aqueous humor (AH) outflow resistance, a major cause of glaucoma, which is a leading cause of irreversible blindness worldwide. The review discusses the molecular mechanisms behind these processes, illustrating how the pathway affects the contractile behavior of tissues in the AH outflow pathway—including the TM and Schlemm’s canal (SC)—by directly impacting actomyosin dynamics and extracellular matrix (ECM) remodeling. It also examines the extensive interaction between Rho/ROCK and other vital signaling pathways such as MAPK/ERK and serum response factor (SRF), emphasizing its integrated role within the complex cellular signaling systems in the AH drainage pathway. This comprehensive discussion concludes by highlighting the promising therapeutic potential of Rho kinase inhibitors (RKIs) as a new class of drugs for glaucoma. These agents not only effectively lower IOP but also show emerging neuroprotective properties, with broader implications for other eye and systemic diseases. Understanding this pathway—from its molecular structure to clinical applications—provides a strong foundation for future research and the development of more precise interventions.
Graphical Abstract
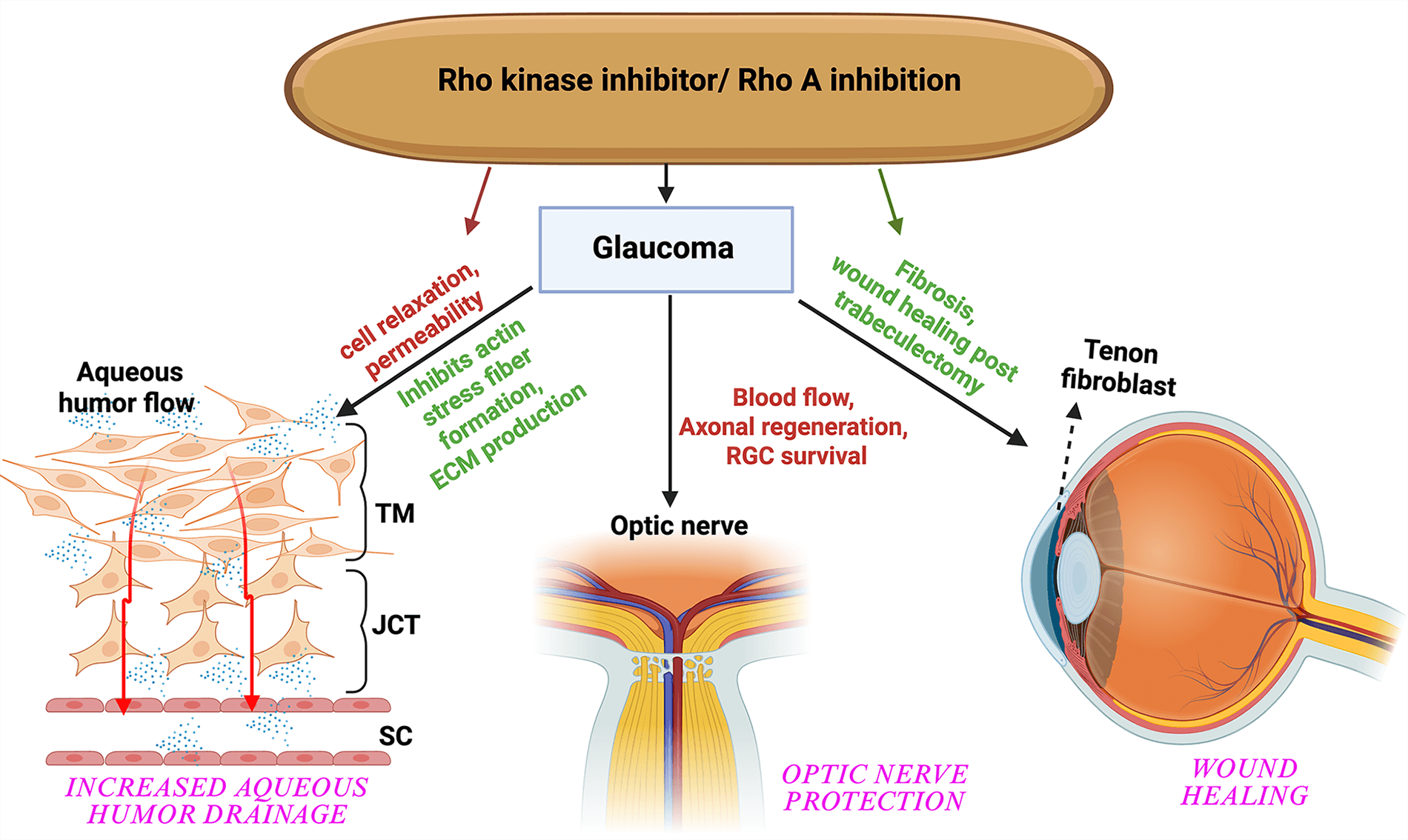
Keywords
- rho GTP-binding proteins
- rho-associated kinases
- actin cytoskeleton
- extracellular matrix
- trabecular meshwork
- glaucoma
- intraocular pressure
The Rho family of proteins constitutes a critical class of small guanosine triphosphate (GTP)-binding proteins, approximately 20–25 kilodaltons (kDa) in size, that are integral members of the broader Ras superfamily [1, 2]. Often referred to as “GTP enzymes” due to their inherent GTPase activity, these proteins function as molecular switches [3]. Their regulatory capacity stems from their dynamic cycling between an active, GTP-bound state, typically localized at the plasma membrane, and an inactive, GDP-bound state, predominantly residing in the cytoplasm. This conformational shift upon GTP binding enables them to interact with and activate a diverse array of downstream effector molecules [4, 5].
These Rho proteins include about 20 members, as shown in Table 1 (Ref. [6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23]). Among them, RhoA, Rac1, and Cdc42 are the most studied, each affecting the actin cytoskeleton [2, 24, 25, 26]. RhoA primarily promotes the assembly of actin stress fibers and focal adhesions, which are crucial for cell adhesion and tension generation [27]. In contrast, Rac1 controls the formation of lamellipodia and membrane ruffles, which are essential for cell migration [28, 29]. Cdc42 mainly governs the formation of actin microspikes and filopodia, structures important for cell sensing and exploration [30, 31, 32].
| Rho GTPase family | Rho GTPases subfamilies | Functions | Reference | ||
| 1 | Rho subfamily | RhoA | Actin myosin contraction, stress and fiber formation | [6] | |
| 2 | RhoB | Cell adhesion and migration | [7] | ||
| 3 | RhoC | Migration and invasion | [8] | ||
| 4 | RhoH subfamily | RhoH | TCR activation | [9] | |
| 5 | Rac subfamily | Rac1 | JNK activation, actin filament stabilization, PIP3 level | [10] | |
| 6 | Rac1B | Cellular transformation | [11] | ||
| 7 | Rac2 | B cell adhesion | [12] | ||
| 8 | Rac3 | Regulation of cell adhesion and differentiation | [13] | ||
| 9 | RhoG | Microtubule dependent transport | [14] | ||
| 10 | Cdc42 subfamily | Cdc42 | Actin organization | [15] | |
| 11 | RhoQ (TC10) | Actin organization, vesicular trafficking | [16] | ||
| 12 | RhoJ (TCL) | Stabilization of focal adhesion | [17] | ||
| 13 | RhoD/F subfamily | RhoD | Growth cone formation | [18] | |
| 14 | RhoF (Rif) | Actin organization | [18] | ||
| 15 | RhoU/V subfamily | RhoU (Wrch) | Cell shape and cell adhesion | [19] | |
| 16 | RhoV (Chp) | Cell shape and cell adhesion | [19] | ||
| 17 | Rnd subfamily | Rnd1 | Microtubule depolymerization | [20] | |
| 18 | Rnd2 | Neurite branching | [21] | ||
| 19 | Rnd3 (RhoE) | Loss of stress fibers | [22] | ||
| 20 | Rho BTB subfamily | RhoBTB1 | Not well characterized but like RhoBTB2 | [23] | |
| 21 | RhoBTB2 | Tumor suppresor | [23] | ||
| 22 | RhoBTB3 | Vesicle trafficking, tumor suppresor | [23] | ||
Beyond their well-documented roles in cytoskeletal reorganization, Rho GTPases are essential to various fundamental cellular processes. These include establishing and maintaining cell polarity, regulating cell adhesion, orchestrating cell motility and migration, facilitating vesicle transport, developing and maintaining synaptic structures, supporting wound healing, and executing cytokinesis [33, 34, 35, 36, 37, 38, 39]. Their widespread involvement emphasizes their critical role in maintaining cellular integrity and function across diverse biological contexts.
Rho-associated protein kinases (ROCKs), specifically ROCK1 and ROCK2, are the best-known and most studied downstream effectors of the small GTP-binding protein Rho, especially RhoA [40]. The ROCKs are serine/threonine kinases with a molecular mass of about 160 kDa, and they play a crucial role in mediating RhoA induced reorganization of the actin cytoskeleton [40]. The functions of ROCKs are diverse, affecting many cellular processes such as contraction, motility, proliferation, apoptosis, cell shape, secretion, and gene expression [41, 42, 43, 44]. The RhoA/ROCK pathway is one of the two main pathways controlling smooth muscle contraction, working alongside myosin light chain kinase activated by calmodulin. They work together by affecting the phosphorylation state of the myosin light chain (MLC) [45, 46]. It is a key regulator of actomyosin contractility, which is essential for force generation in processes like cell movement and muscle contraction.
Rho GTPase and ROCK signaling are crucial and found throughout various biological systems. By establishing their role as fundamental regulators of cellular mechanics, particularly actomyosin contractility, we will provide details on their role in the mechanism of tissue contractility, the regulation of intraocular pressure (IOP), and the implications for ocular diseases, notably glaucoma.
RhoA, Rac1, and Cdc42 are the most thoroughly studied members of the Rho GTPase family, each playing unique yet interconnected roles in organizing the actin cytoskeleton and related cellular processes [47]. Their specific functions are summarized in Table 2.
| Rho GTPase member | Primary cellular functions | Major effectors |
| RhoA | Formation of actin stress fibers and focal adhesions, actomyosin contractility, cell motility, cell proliferation, apoptosis, regulation of vascular tone, glucose uptake (via GLUT4 translocation) | Rho-associated protein kinases (ROCK1, ROCK2), mDia (formin family) |
| Rac1 | Formation of lamellipodia and membrane ruffles, cell migration, cell polarity, synaptic development and maintenance, force-dependent growth of adherens junctions | WAVE, Arp2/3 complex |
| Cdc42 | Formation of actin microspikes and filopodia, cell polarity, synaptic development and maintenance, cytokinesis, cell migration | N-WASP, Arp2/3 complex |
ROCK1 and ROCK2, the two isoforms of Rho kinase, share significant structural homology, with about 65% overall amino acid identity [48]. However, they differ in their cellular localization and activation mechanisms. For example, ROCK1 is mainly found in the cytosol, while ROCK2 can be located in both the cytoplasm and nucleus, often co-localizing with actin and vimentin filaments [43]. Despite their structural similarities and often shared downstream substrates, ROCK1 and ROCK2 do not fully compensate for each other’s loss. This indicates they have distinct or non-redundant functions in certain developmental or disease contexts, such as the known role of ROCK2 in blastocyst development [49, 50]. Their different expression patterns and activation processes contribute to the complex regulation of cellular functions [48].
The structure and regulation of Rho GTPases and Rho kinases enable them to control cellular processes precisely. Understanding these structural and functional details is essential for appreciating their roles as molecular switches and effectors.
Rho GTPases are monomeric proteins (~20 kDa) characterized by a
conserved core G domain [51, 52, 53]. This G domain is a defining feature of the
Ras-like GTPase superfamily and contains five conserved sequence motifs (G1–G5)
that are essential for binding and hydrolysis of guanine nucleotides. The G1
motif, or the P-loop, is crucial because it coordinates the
The isoforms RhoA, RhoB, and RhoC also share identical sequences in their switch
I (residues 27–43 in RhoA) and switch II regions (residues 57–68), with only
minor variations at positions 29 and 43 within switch I [52, 59, 60, 61]. These
subtle differences can significantly influence the binding affinity of RhoGEFs
and downstream effectors. These switch regions undergo substantial conformational
changes upon GTP binding, which is the molecular basis for the active Rho GTPase
to engage with its downstream effectors. A highly conserved glutamine residue
(Gln63 in RhoA/B/C) located within the switch II region is vital for coordinating
the nucleophilic water molecule relative to the GTP
Three main classes of proteins orchestrate the dynamic regulation of Rho GTPase activity.
These proteins act as positive regulators, facilitating the activation of Rho GTPases by catalyzing the exchange of their bound GDP for GTP. The Dbl homology (DH) domain within GEFs is specifically responsible for this guanine nucleotide exchange activity [70, 71].
Acting as negative regulators, GAPs speed up the natural GTPase activity of Rho GTPases, aiding in the breakdown of bound GTP into GDP. This results in the inactivation of the protein and the ending the signal transduction. GAPs have a conserved catalytic Rho GAP domain [72].
GDIs play a crucial role by inhibiting the dissociation of GDP from Rho GTPases, thereby stabilizing their inactive GDP-bound state and sequestering them in the cytoplasm [73, 74]. They also regulate Rho GTPases by binding to their isoprenyl groups, facilitating their extraction from membranes and thus controlling their localization and activity [75, 76].
Beyond these core regulatory proteins, post-translational modifications (PTMs) significantly increase the complexity of Rho GTPase signaling. While prenylation is essential for plasma membrane localization, other PTMs such as phosphorylation, ubiquitination, and palmitoylation affect the stability and spatial distribution of Rho GTPases [47, 77, 78]. For ROCKs, autophosphorylation of ROCK1 at Ser1333 and ROCK2 at Ser1366 indicates their activation status, and phosphorylation at other specific sites, including ROCK2 Thr967, can further enhance their activity [79, 80].
Spatiotemporal precision is crucial for regulating Rho GTPase [81, 82]. Carefully adjusting Rho GTPase activity across different cellular locations is vital for achieving specific biological effects. This dynamic spatiotemporal activation is often managed by GEF and GAP complexes that interact with various proteins, including components of the cytoskeleton, focal adhesion proteins, adaptors, and Rho GTPase effectors [83]. Such flexible control allows for the diverse and context-specific cellular responses observed, ensuring Rho GTPase activity matches cellular needs. Furthermore, Rho-independent activation mechanisms for ROCKs, like caspase cleavage, add an extra layer of complexity, signaling alternative pathways that could be targeted therapeutically or may contribute to disease independently of Rho GTPase activity. Understanding these mechanisms is essential for developing precise interventions.
The ROCKs, ROCK1 and ROCK2, are serine/threonine kinases that belong to the AGC family of kinases [43, 48, 84, 85]. Structurally, both ROCK1 and ROCK2 consist of an N-terminal kinase domain, a central coiled-coil domain containing the Rho-binding domain (RBD), and a C-terminal auto-inhibitory region [86]. Although their kinase domains are highly similar (92% amino acid identity), their coiled-coil domains are more different (52% homology), which may explain their distinct functional roles [48].
The C-terminal region of ROCKs functions as an auto-inhibitory domain by directly interacting with the kinase interface [87]. Removing this inhibitory part results in constant kinase activation both in vitro and in vivo [88, 89]. The main way ROCK gets activated involves the binding of active GTP-bound Rho (such as RhoA) to the RBD [90]. This binding breaks the auto-inhibition, causing a shape change that leads to an active, “open” form of the kinase domain [86]. Besides Rho-GTP binding, ROCKs can also be activated through mechanisms that do not depend on Rho. ROCK1 can be activated by cleavage through caspase-3 [91], while ROCK2 can be activated by granzyme B and caspase-2 cleavage [92], both of which produce always-active kinase fragments.
The Rho/ROCK signaling pathway is a key regulator of cell and tissue contractility, orchestrating the dynamic interaction of the actomyosin cytoskeleton across various cell types, as shown in Fig. 1. Its role spans from essential cellular processes to the overall function of tissues.
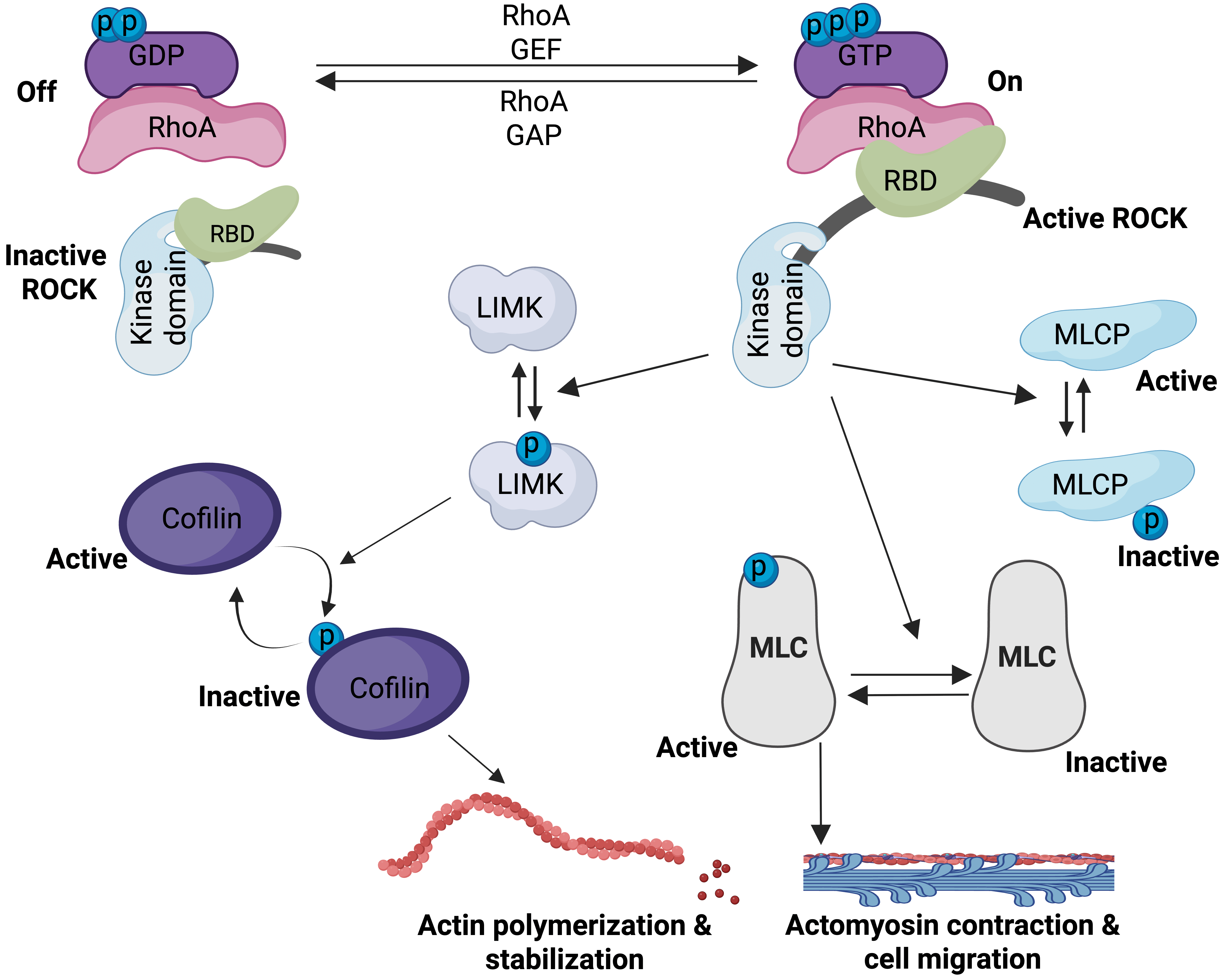 Fig. 1.
Fig. 1.
Activation and functional role of RhoA in actin polymerization and actomyosin contraction. Created in BioRender™. GEF, Guanine Nucleotide Exchange Factor; GAP, GTPase-activating Protein; RBD, Rho-binding domain; LIMK, LIM Kinase; MLCP, Myosin Light Chain Phosphatase; MLC, Myosin Light Chain.
ROCKs are essential in promoting actomyosin contractile force generation, mainly by increasing the phosphorylation of the regulatory light chain of myosin II (MLC2) (Fig. 1). This occurs through a dual mechanism.
Activated ROCKs phosphorylate the PPP1R12A/MYPT1 subunit of myosin light chain phosphatase (MLCP). This phosphorylation inhibits MLCP activity, thereby reducing the dephosphorylation of MLC and leading to sustained, elevated MLC phosphorylation and prolonged contraction [93, 94, 95]. ROCKs can also phosphorylate CPI-17 (Protein Kinase C Potentiated Phosphoprotein Phosphatase 1 Inhibitor), which further contributes to MLCP inhibition and enhances Ca2+ sensitization of smooth muscle contraction [1, 2, 66, 96, 97, 98].
In addition to inhibiting MLCP, ROCKs can directly phosphorylate MLC. This direct phosphorylation stimulates actomyosin ATPase activity, which is a key step in initiating contraction, and contributes to Ca2+-sensitization of smooth muscle contraction and stress fiber formation [99, 100, 101, 102, 103, 104].
Beyond direct myosin regulation, ROCKs also influence actin filament dynamics through the LIM Kinase (LIMK) and Cofilin pathway [105, 106]. ROCKs phosphorylate and activate LIM kinases (LIMK1/2). In response, activated LIMK phosphorylates and inhibits the actin-severing protein cofilin. By preventing cofilin from severing actin filaments, this pathway promotes actin polymerization and increases the stability of actin filaments, thereby contributing to stress fiber formation. Additionally, active RhoA facilitates actin filament polymerization by binding to and activating mDia1, a member of the formin family of actin nucleating factors [107]. The combined effects of these mechanisms—MLC phosphorylation, MLCP inhibition, the LIMK/cofilin pathway, and mDia activation—lead to a substantial increase in the contractile force exerted by myosin II on actin filaments. This results in enhanced stress fiber formation, robust actin filament growth, and stabilization of the actin cytoskeleton. Collectively, these molecular events support the cell and tissue’s ability to generate mechanical force and maintain structural integrity [27, 108].
The diverse array of direct and indirect targets of ROCK moving beyond just the core contractile machinery to include proteins involved in actin dynamics, intermediate filament organization, and even signaling proteins is shown in Table 3 (Ref. [46, 103, 104, 105, 107, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125]). This comprehensive overview demonstrates the pleiotropic nature of ROCK signaling and how it orchestrates a wide range of cellular functions.
| Effector/Target | Role in actomyosin contractility/cellular function | Associated Rho GTPase/ROCK action | References |
| Myosin light chain (MLC) | Increases actomyosin contractility and stress fiber formation | Direct phosphorylation by ROCK; stimulates actomyosin ATPase activity | [104, 109] |
| Myosin phosphatase target subunit 1 (MYPT1) of myosin light chain phosphatase (MLCP) | Sustains MLC phosphorylation, increases Ca2+ sensitization of smooth muscle contraction | Phosphorylated by ROCK, inhibiting MLCP activity | [103, 110] |
| LIM kinases (LIMK1/2) | Leads to actin filament stabilization and polymerization; coordinates microtubule destabilization and actin formation | Phosphorylated and activated by ROCK; phosphorylates and inhibit cofilin | [105, 111] |
| CPI-17 | Enhances Ca2+ sensitization of smooth muscle contraction | Phosphorylated by ROCK; inhibits MLCP activity | [46, 112] |
| mDia1 | Promotes actin filament growth and polymerization | Activated by RhoA; nucleates actin polymerization | [107, 113] |
| ERM proteins (Ezrin, Radixin, Moesin) | Regulates actin filament/membrane interaction; involved in microvilli formation | Phosphorylated by ROCK; decreases intra- or intermolecular head-to-tail association | [114] |
| Adducin | Involved in spectrin/F-actin network assembly; increases cell motility | Phosphorylated by ROCK; increases adducin/F-actin interaction | [115] |
| NHE1 (Na+-H+ exchanger 1) | Promotes actin stress fiber formation | Stimulation of its Na+-H+ exchanger activity by ROCK | [116, 117] |
| Intermediate filaments (GFAP, NF-L, Desmin, Vimentin) | Involved in regulation of cytokinesis | Phosphorylated by ROCK; inhibition of filament formation | [118, 119, 120, 121] |
| Tau, MAP2, CRMP-2 | Involved in microtubule dynamics and growth cone collapse | Phosphorylated by ROCK; reduction of Tau activity, unknown effect on MAP2, CRMP-2 | [122, 123] |
| PTEN | Decreases intracellular PtdIns(3,4,5)P3 level; tumor suppression | Stimulation of phosphatase activity by ROCK; promotes anti-survival | [124, 125] |
The Rho/ROCK pathway is a key, calcium-independent regulator of smooth muscle contraction, playing an important role in various systems. Its role in the cardiovascular system has been studied extensively [126, 127]. It modulates MLC phosphorylation, thus contributing to agonist-induced Ca2+-sensitization in smooth muscle contraction. This means that even at constant intracellular calcium levels, activating the Rho/ROCK pathway can enhance the contractile response. Excessive or prolonged activation of RhoA and/or ROCKs can cause hypercontraction. This pathological state leads to different vascular problems, including age-related hypertension, arterial stiffening, and vasospasm in coronary and cerebral arteries [127]. ROCK activity is also essential for maintaining myogenic tone, the inherent contractile activity of blood vessels, and the tonic component of vascular smooth muscle cell contraction in various vascular beds, ensuring proper regulation of blood flow [128, 129, 130].
In non-muscle cells, ROCKs control a diverse range of cellular processes that are intimately dependent on actin cytoskeleton organization and cell contractility [126]. These include cell-matrix and cell-cell adhesion, cell migration, neurite retraction and outgrowth, and cytokinesis. Actomyosin contractility, driven by Rho/ROCK signaling, is a key determinant for various forms of cell migration and invasion, including cancer cell metastasis [131]. High levels of RhoA/RhoC or ROCK-driven actomyosin contractility can promote amoeboid motility, a rapid mode of cell movement. Rho/ROCK signaling also plays important roles in tissue morphogenesis during development, as evidenced by its influence on eye and wing development in Drosophila [132]. In the context of wound healing, Rho and Cdc42 are essential for the stabilization of the actomyosin ring, while Rac is required for actin mobilization towards the wound site, highlighting the cooperative nature of Rho GTPases in this process [133]. Beyond contractility, Rho/ROCK signaling influences other fundamental cellular functions such as cell proliferation, differentiation, and apoptosis [134, 135].
Cells are constantly exposed to and respond to various mechanical forces, which can originate externally, including fluid shear stress on endothelial cells, compression on skeletal cells, or internally generated by the contractile actin cytoskeleton [136, 137, 138]. These mechanical forces trigger multiple signaling pathways, many of which converge to activate RhoA. The mechanical activation of Rac1 is required for the force-dependent growth of adherens junctions, demonstrating how physical stimuli can directly engage Rho GTPase activity [139, 140].
This highlights a crucial aspect of Rho/ROCK signaling it acts not only as a responder to chemical stimuli but also as a sensor and transducer of physical forces. This pathway dynamically influences cell shape, adhesion, and contractile properties in direct response to the mechanical environment. This is not merely a passive response but a dynamic, bidirectional interplay where mechanical forces activate RhoA, and activated RhoA then generates intracellular tension [141]. This feedback loop is fundamental to understanding tissue homeostasis and disease, particularly in mechanically sensitive tissues like the trabecular meshwork (TM).
The regulation of IOP is a finely tuned physiological process essential for maintaining ocular health [142]. Dysregulation of ocular pressure is the primary modifiable risk factor for glaucoma, a leading cause of irreversible blindness worldwide [143]. The Rho/ROCK signaling pathway plays an indispensable role in the delicate balance of AH drainage and IOP [144].
IOP is maintained by a precise equilibrium between the production of AH by the ciliary epithelium and its drainage from the eye [145, 146, 147]. The primary route for AH outflow, known as the conventional or TM outflow pathway (Fig. 2), accounts for most (up to 90%) of AH drainage. This pathway is anatomically complex, comprising the TM, the juxtacanalicular tissue (JCT), and the endothelial lining of Schlemm’s canal (SC) [146, 147, 148, 149, 150].
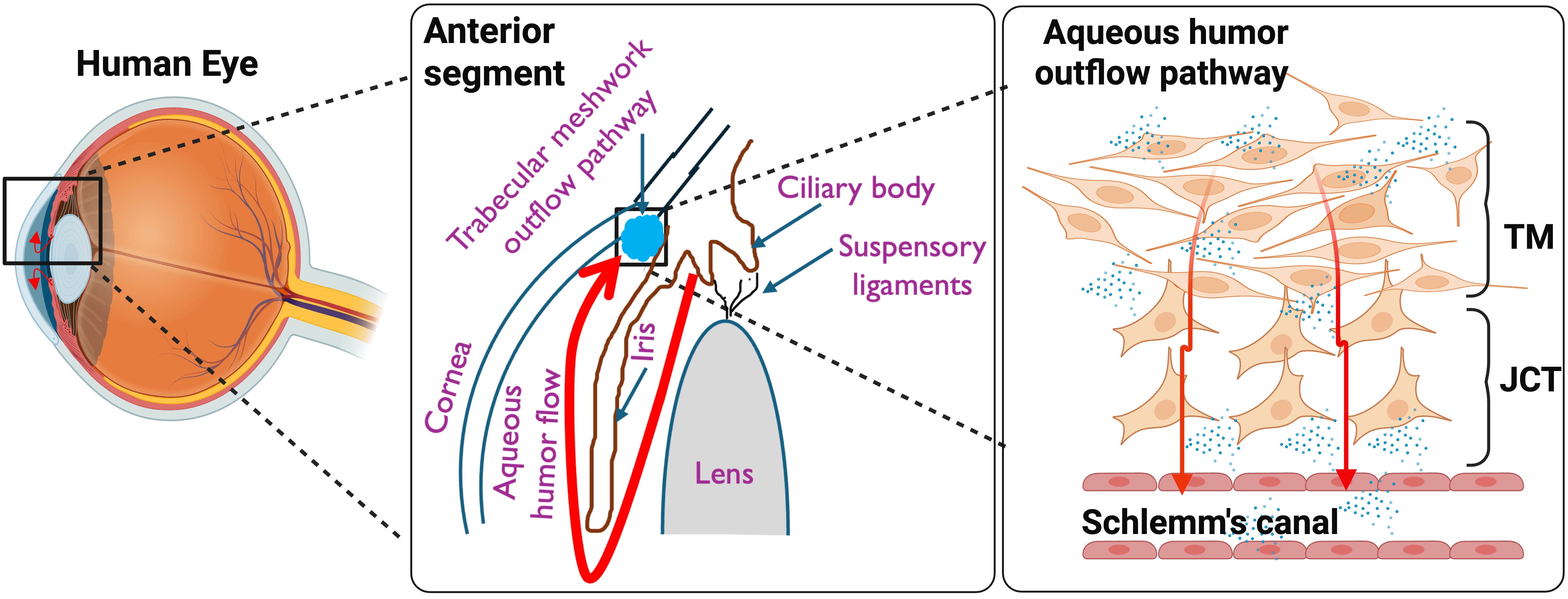 Fig. 2.
Fig. 2.
Overview of a healthy aqueous humor flow pathway. The aqueous humor produced by the ciliary body flows between the lens and iris to enter the anterior chamber. From here, it drains through different layers of trabecular meshwork (TM), juxtacanalicular (JCT), and reaches Schlemm’s canal (SC), thus maintaining the intraocular pressure (IOP) homeostasis. Created in BioRender™.
In primary open-angle glaucoma (POAG), the most prevalent form of the disease, elevated IOP arises from a pathologically increased resistance to AH drainage through this conventional outflow pathway [151, 152]. This sustained elevation in IOP exerts mechanical stress on the optic nerve head, leading to structural changes in the lamina cribrosa. These changes, in turn, impair retinal ganglion cells (RGCs) and their axons, ultimately resulting in progressive vision loss. The intricate interplay between cellular contractility, extracellular matrix (ECM) dynamics, and mechanotransduction within the outflow pathway is critical for understanding IOP homeostasis and the pathogenesis of glaucoma.
The cells within the TM and SC exhibit characteristics akin to smooth muscle
cells, including the expression of key contractile proteins like
Activation of the Rho GTPase/Rho kinase signaling mechanism in the trabecular outflow pathway leads to an increase in IOP by altering the contractile, cell adhesive, and permeability barrier characteristics of the TM and SC tissues [156, 157, 158]. This pathway promotes myosin II activity primarily by inhibiting MLCP and directly phosphorylating MLC, which drives the assembly of contractile actomyosin bundles that generate strong tensile forces within the cells. This activation significantly increases cell stiffness through the formation of actin stress fibers [96, 159, 160].
Physiological agonists known to activate Rho/ROCK signaling, such as
transforming growth factor-
The consistent observation that activation and the inhibition of the Rho/ROCK pathway increase and decrease the outflow resistance, respectively, is the basis for providing Rho/ROCK as a prime therapeutic target to treat ocular hypertension and slow the progression of glaucoma.
Sustained activation of Rho GTPase/Rho kinase signaling in the AH outflow pathway consistently increases resistance to AH outflow. This leads to the stiffened and contractile morphology of TM cells, reducing the permeability of the outflow pathway [156, 176, 177, 178]. Additionally, increased Rho and activation of Rho have been documented in the optic nerve head (ONH) [179, 180, 181].
Conversely, inhibition of Rho kinase with specific inhibitors like Y-27632
effectively lowers IOP by inducing relaxation of the trabecular meshwork, thereby
enhancing AH outflow [156, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 193, 194]. The relaxation is achieved by reducing MLC
phosphorylation and disorganizing the actomyosin cytoskeleton, leading to
cellular relaxation and a decrease in cell-substratum adhesions in TM and SC
cells. The result is an increase in the size of intercellular spaces within the
TM, which directly facilitates increased aqueous outflow. Furthermore, ROCK
inhibitors reduce the density of actin stress fibers in TM and SC cells,
contributing to eased AH outflow [144, 194, 195]. ROCK inhibitors have also been
shown to reverse TGF-
Rho GTPases operate within a highly complex molecular network characterized by extensive crosstalk, where individual members and their downstream pathways frequently cooperate or antagonize each other to fine-tune cellular responses [203]. Such an intricate molecular signaling cross-talk can manifest at multiple levels: through the regulation of Rho GTPase activity itself via shared or specific GEFs and GAPs, through the modulation of protein expression and stability, and the direct or indirect regulation of downstream signaling pathways [25, 204]. Interestingly, the activation of RhoA and Rac1, two Rho family members, exhibits temporal or spatial separation [205], or one may actively inhibit the other, demonstrating a sophisticated balance in cellular control [160, 206] as shown in Fig. 3 (Ref. [130]). This intricate integration of Rho/ROCK signaling with other major pathways is a crucial aspect of cellular regulation.
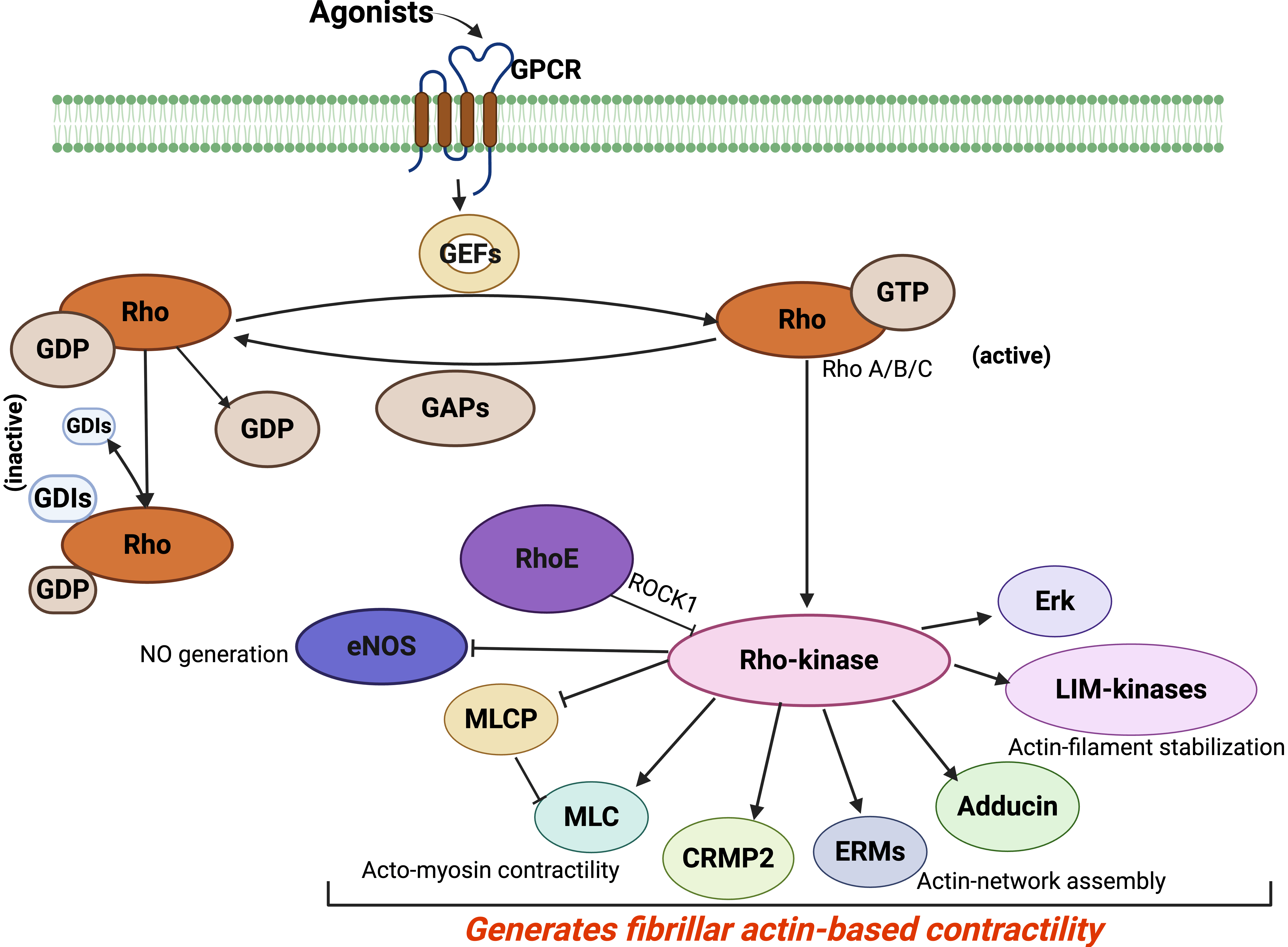 Fig. 3.
Fig. 3.
Rho-kinase activation of various cytoskeleton pathways. Rho GTPases, including RhoA, get activated through a sequential process of guanine nucleotide exchange factors (GEFs) that catalyze the exchange of GDP for GTP, followed by the inactivation of GTPase-activating proteins (GAPs). Rho-kinase is an effector form of Rho. Many other substrates of Rho-kinase include myosin light chain (MLC), MLC phosphatase (MLCP), ezrin/radixin/moesin (ERM), adducin, and LIM-kinases. Endothelial NO synthase (eNOS), guanine nucleotide (GDI), G-protein-coupled receptor (GPCR), collapsing response mediator protein2 (CRMP2), and ezrin/radixin/moesin (ERM). Re-created from [130]. Created in BioRender™.
Both Ras and Rho proteins function as GTP-regulated molecular switches that govern multiple, often interconnected, signaling pathways in eukaryotic cells. The mitogen-activated protein kinase (MAPK) pathway, particularly the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), is a well-established regulator of cell growth, proliferation, differentiation, and motility. The Rho/ROCK and Ras/MAPK/ERK pathways intersect at numerous points, and their coordinated action is crucial for various cellular processes, including oncogenic transformation and in the TM [165, 171, 207].
Specific mechanisms of crosstalk include:
The phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway is a key intracellular
signaling cascade that regulates cell metabolism, migration, immune function, and
survival [221]. A well-documented point of crosstalk between Rho/ROCK and
PI3K/Akt involves the phosphatase and tensin homolog (PTEN) [125, 222].
ROCK-mediated activation of PTEN can lead to a decrease in nitric oxide (NO)
production, which in turn reduces the survival of endothelial cells [223, 224].
Both Rho/ROCK and PI3K/Akt pathways can be activated by standard upstream signals
and receptors, such as G-protein coupled receptors (GPCRs) and receptor tyrosine
kinases (RTKs), further illustrating their interconnectedness [211, 225, 226].
The complex interplay and crosstalk between pathways like Rho/ROCK,
TGF-
Both ROCK and NF-
Transforming growth factor-beta 2 (TGF-
In the central nervous system (CNS), myelin-associated inhibitory factors such as Nogo, Myelin-Associated Glycoprotein (MAG), Oligodendrocyte-Myelin Glycoprotein (OMgp), and chondroitin sulfate proteoglycans (CSPGs) released from reactive astrocytes in glial scars activate the RhoA/ROCK pathway in neurons [230]. This activation inhibits axon growth and causes growth cone collapse, contributing to the limited regeneration capacity after CNS injury.
Here we present the complex network of cellular signaling, showing that Rho/ROCK signaling does not work alone. It is demonstrated that Rho/ROCK signaling is closely connected with other major pathways. The specific examples of Rho/ROCK affecting MAPK/ERK and, most notably, the PI3K/Akt pathway through PTEN, reveal advanced regulatory loops and overall cellular functions. This detailed mechanism is crucial for understanding the wide-ranging effects of RKI therapies and for predicting potential off-target effects or benefits from targeting this pathway. It also suggests that treatment strategies must consider the whole signaling network to prevent unintended effects and to improve treatment success by affecting multiple pathways.
Interestingly, in the late 1990s, a report was published using a broad spectrum serine-threonine kinase inhibitor, H-7, that demonstrated an increase in outflow facility in monkeys [231]. Although H-7 is a non-selective ROCK inhibitor with low potency against ROCK, this paper mentioned Rho A but did not mention ROCK. Furthermore, extensive research on Rho GTPase and Rho kinase signaling—particularly its role in TM tissue contractility, IOP regulation, and glaucoma—has paved the way for significant translational advancements, especially in ophthalmology [144, 179, 184, 187, 232].
The ROCK inhibitors (RKIs) are an emerging and promising class of anti-glaucoma drugs that specifically target the diseased trabecular outflow pathway, improving AH outflow through the conventional route. Ripasudil (K-115) [233, 234, 235, 236] and Netarsudil (AR-13324) [232, 237, 238, 239, 240, 241, 242] are two leading RKIs that have received clinical approval for glaucoma treatment in Japan and the United States, respectively.
RKIs mainly lower IOP by relaxing the TM and SC cells [156, 165, 184, 187, 243, 244, 245, 246, 247, 248]. This relaxation is achieved by decreasing actin stress fiber density, disassembling focal adhesions, and lowering cell stiffness and tension in TM and SC cells. On a molecular level, this involves directly inhibiting MLC phosphorylation or increasing MLCP activity, leading to MLC dephosphorylation and reduced actomyosin contractility. Most recently, it has been shown that ROCK inhibitors target the ECM by lowering the traction forces sensed by the TM cells [249, 250]. The overall effect is an increase in intercellular space within the TM and improved permeability, which facilitates AH outflow. Netarsudil also inhibits norepinephrine transport (NET), potentially contributing to IOP reduction by decreasing AH production through preventing norepinephrine reuptake at noradrenergic synapses [237, 238]. Another mechanism includes lowering episcleral venous pressure [239, 251].
Clinical trials have demonstrated that RKIs are safe and efficacious, showing non-inferiority when compared to traditional anti-glaucoma medications such as beta-blockers and prostaglandins (Table 4, Ref. [199, 200, 232, 233, 234, 235, 237, 238, 239, 240, 241, 251, 252, 253, 254, 255, 256, 257]). They are effective as monotherapy and, importantly, show enhanced efficacy when combined with other hypotensive medications. The Netarsudil/latanoprost fixed-dose combination has demonstrated superior IOP reduction compared to individual components [252, 258, 259, 260]. Common adverse effects reported include conjunctival hyperemia (the most frequent, affecting ~53% of patients with Netarsudil), corneal verticillata (cornea staining), conjunctival hemorrhage, instillation site pain, conjunctivitis, and blepharitis. Most of these effects are generally mild, self-limiting, and temporary [253, 261, 262, 263, 264]. The very mechanism that makes RKIs effective for glaucoma—by relaxing the TM outflow pathways to lower IOP—also leads to the relaxation of conjunctival blood vessels, causing them to dilate and resulting in conjunctival hyperemia. Corneal verticillata caused by RKIs is typically asymptomatic and does not affect vision. It is also reversible, with the deposits usually resolving over time after the medication is discontinued [253, 261, 262, 263, 264, 265].
| Drug (approval status) | Primary mechanism of IOP reduction | Additional ocular benefits | Common adverse effects | Reference |
| Ripasudil (approved Japan) | Relaxation of trabecular meshwork (TM) and Schlemm’s canal (SC) cells by inhibiting MLC phosphorylation and MLCP inhibition, reducing actomyosin contractility, increasing intercellular spaces | Neuroprotection (RGC survival, axon regeneration), improved ocular blood flow, anti-fibrotic effects | Primarily conjunctival hyperemia (redness), often mild and temporary, along with blepharitis (eyelid inflammation) and allergic conjunctivitis | [199, 200, 233, 234, 235, 251, 253] |
| Netarsudil (approved USA) | Relaxation of TM and SC cells (like Ripasudil); also inhibits norepinephrine transporter (NET) to reduce AH formation | Neuroprotection (RGC survival, axon regeneration), improved ocular blood flow, anti-fibrotic effects | Conjunctival hyperemia ( |
[232, 237, 238, 239, 240, 241, 252, 254, 255, 256, 257] |
This table summarizes key therapeutic agents, their mechanisms, and clinical considerations, making it highly useful for experts who want to quickly understand the practical aspects of Rho/ROCK research in glaucoma management.
Beyond their IOP-lowering capabilities, RKIs offer significant neuroprotective properties, which is a crucial advantage in the comprehensive management of glaucoma, a neurodegenerative disease. This dual benefit of IOP lowering and neuroprotection addresses both the primary risk factor and the neurodegenerative component of glaucoma, offering a more holistic therapeutic strategy [266, 267, 268]. The mechanisms of neuroprotection are summarized in Sections 7.4.1–7.4.4.
RKIs have been shown to enhance RGC survival and promote RGC axon regeneration in various animal models of optic nerve injury (e.g., optic nerve crush injury, experimental glaucoma). They achieve this by suppressing ROCK signaling in the retina and optic nerve, which is typically activated by myelin-associated axon growth inhibitors (like Nogo, MAG, OMgp) and components of the glial scar (like chondroitin sulfate proteoglycans, CSPGs) that impede axonal regeneration. RKIs can effectively reverse these inhibitory effects, promoting axonal sprouting and functional recovery [269, 270].
RKIs improve ocular blood flow, particularly to the optic nerve head, by promoting vasodilation and reducing vasoconstriction often mediated by endothelin-1. Impaired ocular blood flow is a recognized factor in glaucoma pathogenesis [186, 271, 272].
RKIs protect retinal ganglion cells against neurotoxic injury induced by agents like N-methyl-d-aspartate and mitigate damage from ischemic reperfusion injury in animal models [199, 201, 243].
While not explicitly detailed as a direct mechanism in all studies, the general involvement of ROCK in cell apoptosis and its crosstalk with pro-survival pathways like PI3K/Akt via PTEN suggests an indirect influence on RGC survival by modulating the balance between pro-survival and pro-apoptotic signals [273, 274].
The therapeutic potential of Rho/ROCK modulation extends beyond glaucoma, underscoring its widespread pathophysiological relevance.
RKIs have demonstrated efficacy in improving corneal wound healing and promoting corneal endothelial cell differentiation and regeneration, making them promising therapeutic agents for corneal diseases [135, 275, 276, 277, 278].
Rho/ROCK pathway activation is implicated in the pathophysiology of various retinal and vitreous diseases, including diabetic retinopathy (DR), age-related macular degeneration (AMD), and proliferative vitreoretinopathy (PVR). RKIs are being explored as potential therapeutic targets for these conditions due to their anti-fibrotic and anti-angiogenic effects [279, 280, 281, 282].
RKIs serve as potent anti-scarring agents by inhibiting the transdifferentiation of fibroblasts into myofibroblasts. This property is highly relevant in glaucoma surgery (to prevent bleb scarring, which is a common cause of surgical failure) and in conditions like PVR [283, 284, 285, 286].
Beyond ophthalmology, dysregulation of RhoA/ROCK signaling is linked to a wide range of age-related and smooth muscle-related systemic diseases, including hypertension, atherosclerosis, heart failure, diabetes, and various neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease [287, 288, 289, 290].
This section highlights the significant translational impact of Rho/ROCK research, demonstrating how fundamental scientific understanding has led to tangible clinical advancements. Despite the significant advancements, several challenges and promising future research avenues exist for Rho/ROCK modulation.
A notable challenge in RKI therapy is the current lack of highly selective Rho kinase inhibitors. Given the ubiquitous expression and pleiotropic functions of ROCKs, non-specific inhibition could lead to undesirable systemic consequences. Future research aims to develop more isoform-specific inhibitors to minimize off-target effects.
For neuroprotective effects, particularly targeting RGCs and the optic nerve, traditional topical or oral routes may be insufficient due to anatomical barriers. Novel drug delivery systems, such as sustained-release implants or exosome-based delivery, are being explored to ensure effective drug concentrations at the target site and to improve patient compliance and tolerability.
While initial clinical trials show promise, further independent, large-scale, prospective randomized controlled trials are essential to comprehensively elucidate the long-term therapeutic value and safety profiles of RKIs.
Ongoing research is needed to determine the optimal dosage regimens and identify specific patient populations that would benefit from RKI therapies the most.
The demonstrated success of fixed-dose combinations (e.g., Netarsudil/latanoprost) suggests a future direction in combining RKIs with existing glaucoma treatments to achieve enhanced efficacy and broader therapeutic benefits.
Novel drugs targeting specific pathways relevant to mechanosensing and mechanotransduction, which are directly tied to the Rho/ROCK pathway and play a role in elevating IOP, can be pharmacologically targeted. These can be the focal adhesions, integrins, G-proteins, to name a few.
The Rho GTPase and ROCK signaling pathway is a highly influential regulatory axis in cellular and tissue biology. It fundamentally controls actomyosin contractility and cytoskeletal dynamics, as well as its adaptive response to mechanical forces through mechanotransduction. The success of RKIs in glaucoma therapy highlights the clinical significance of this research. Drugs like Ripasudil and Netarsudil have demonstrated considerable effectiveness in lowering IOP by relaxing the conventional outflow pathway, offering a new approach compared to traditional glaucoma treatments. The combined benefit of IOP reduction and neuroprotection represents a significant advancement, addressing both the primary risk factor and the neurodegenerative aspect of glaucoma, thus providing a comprehensive treatment strategy. The balanced discussion of clinical efficacy alongside common side effects reflects a genuine clinical perspective. Exploring applications beyond glaucoma emphasizes the wide therapeutic potential of modulating this pathway, indicating its broad relevance across various diseases. Finally, recognizing the challenges highlights key areas for future research and innovation with new drug targets. Future efforts will likely focus on refining these targeted approaches and discovering new strategies to leverage this fundamental signaling pathway for improved patient outcomes.
Conceptualization: PPP, SR. Writing—original draft preparation: PPP, SR. Figure preparation: SR, PPP. Writing: review and editing: SR, PPP. Supervision: PPP. Project administration: PPP. Funding acquisition: PPP. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We want to thank all the members of the Pattabiraman lab for their valuable input and thoughtful discussions. The figures were created using BioRender (https://BioRender.com/oytvb7f, https://BioRender.com/j7tv0f4 and https://BioRender.com/rg9wrta).
This work was supported by the National Institutes of Health/National Eye Institute R01EY029320, R01EY035412, and R01EY036107 (PPP); an award from the Ralph W. and Grace M. Showalter Research Trust and the Indiana University School of Medicine (PPP); the RPB Departmental Pilot Grant (PPP), Glick Research Endowment Funds (PPP), and a Challenge grant from Research to Prevent Blindness to IU.
The authors declare that they have no conflicts of interests.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.