



 , Hyun Bon Kang 1,†, Dae Hyun Kim 3,*
, Hyun Bon Kang 1,†, Dae Hyun Kim 3,* , Min Hi Park 1,2,*
, Min Hi Park 1,2,*1 College of Pharmacy, Kyungsung University, 48434 Busan, Republic of Korea
2 Brain Busan 21 Plus Research Project Group, Kyungsung University, 48434 Busan, Republic of Korea
3 Department of Food Science & Technology, College of Natural Resources and Life Science, Pusan National University, 50463 Miryang, Republic of Korea
†These authors contributed equally.
Abstract
Endocrine-disrupting chemicals (EDCs), including bisphenol A (BPA), phthalates, organochlorine pesticides, and heavy metal ions, pose serious threats to reproductive health by interfering with hormonal balance and molecular signaling pathways. Recent research had expanded our understanding of these compounds has beyond their traditional role in hormone receptor interference. EDCs can trigger lasting epigenetic changes, including abnormal DNA methylation, histone modifications, RNA methylation, and altered regulation of non-coding RNA, which can impair reproductive functions such as gametogenesis, folliculogenesis, steroidogenesis, and embryo implantation. Importantly, EDC-mediated epigenetic alterations have been linked to various reproductive disorders, including polycystic ovary syndrome (PCOS), endometriosis, reduced ovarian reserve, and impaired spermatogenesis. For example, BPA exposure alters DNA methylation in estrogen signaling and aromatase gene expression, whereas phthalates disrupt histone acetylation and methylation in hormone synthesis pathways. Similarly, pesticides and heavy metal ions may influence microRNA expression and histone structure, further disrupting endocrine-regulated gene networks. These alterations may occur during sensitive developmental windows and can lead to long-term or transgenerational effects on reproductive health. Understanding how EDCs exert their toxicity through epigenetic mechanisms is essential for early detection of exposure, identification of molecular biomarkers, and development of targeted therapies to reduce reproductive risks. Here, we discuss the emerging molecular evidence that provides a comprehensive overview of how EDCs impair reproductive health through epigenetic pathways, thereby offering a framework for future research and translational applications.
Keywords
- reproductive dysfunction
- endocrine-disrupting chemicals (EDCs)
- histone modification
- DNA methylation
- RNA methylation
- non-coding RNA (ncRNA)
Over the past few decades, increasing evidence has drawn attention to the detrimental effects of environmental pollutants on reproductive health. Among these, endocrine-disrupting chemicals (EDCs), such as bisphenol A (BPA), phthalates, organochlorine pesticides such as dichlorodiphenyltrichloroethane (DDT), and heavy metal ions such as lead ions (Pb2+) and cadmium ions (Cd2+), have gained particular attention because of their widespread use in industrial, agricultural, and consumer products. This leads to chronic, low-level exposure in the general population through ingestion, inhalation, and dermal absorption [1, 2, 3, 4, 5, 6, 7].
Although earlier studies have primarily focused on hormonal disruptions mediated by receptor binding, such as estrogenic or anti-androgenic actions [2, 8], EDCs also act through epigenetic pathways. Epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNA (ncRNA) expression, play pivotal roles in regulating gene activity without altering the nucleotide sequences [9, 10, 11, 12]. These epigenetic processes are essential for maintaining normal reproductive functions, such as gametogenesis, folliculogenesis, ovulation, and steroid hormone biosynthesis [11, 13]. Consequently, when EDCs interfere with these highly coordinated regulatory systems, it can lead to a cascade of reproductive dysfunctions, including infertility, hormonal imbalances, and developmental abnormalities in the reproductive tract.
EDC exposure during critical developmental periods causes abnormal epigenetic reprogramming in reproductive tissues. For instance, BPA exposure hypermethylates Estrogen receptor 1 (ESR1) and Cytochrome P450 family 19 subfamily A member 1 (CYP19A1), reduces histone acetylation (Histone H3 lysine 9 acetylation (H3K9ac)), and upregulates repressive markers such as histone H3 lysine 27 trimethylation (H3K27me3), ultimately impairing ovarian and testicular function [14, 15, 16, 17, 18]. Phthalates such as di(2-ethylhexyl) phthalate (DEHP) and its active metabolite mono(2-ethylhexyl) phthalate (MEHP) disrupt histone-modifying enzyme and DNA methyltransferase (DNMT) activity, reducing the transcription of essential steroidogenic genes such as steroidogenic acute regulatory (STAR) and cholesterol side-chain cleavage enzyme (CYP11A1) [19, 20, 21, 22]. Pesticides such as DDT and chlorpyrifos disrupt the expression of microRNAs (miRNA), such as miR-21 and miR-137, and induce abnormal DNA methylation at hormone receptor gene promoters, thereby impairing reproductive hormone signaling and development [23, 24, 25, 26]. In addition, heavy metal ions, including Pb2+ and Cd2+, promote both global hypomethylation and site-specific hypermethylation of reproductive gene promoters, along with altered histone modification patterns, such as H3K9me2 and H3K27me3 [27, 28, 29].
Recognizing the epigenetic basis of EDC-induced reproductive toxicity is essential for advancing scientific understanding and clinical practice. First, epigenetic alterations are promising biomarkers for the early detection of chemical exposure and reproductive risk, often before visible symptoms appear. Second, elucidating these molecular mechanisms provides insights into how even short-term environmental exposure can result in enduring or even transgenerational reproductive abnormalities owing to the stable inheritance of epigenetic marks. Third, mechanistic knowledge can inform the design of targeted therapeutic interventions, such as epigenetic modulators or antioxidants, and support evidence-based public health regulations aimed at minimizing human exposure to harmful EDCs. These considerations emphasize the importance of multidisciplinary research integrating toxicology, molecular biology, epidemiology, and policy.
This review aimed to clarify how epigenetic mechanisms contribute to the reproductive toxicity of EDCs. We present an integrative analysis of recent molecular studies involving DNA and RNA methylation, histone modifications, and ncRNA dysregulation, focusing on their functional impacts on the male and female reproductive systems. By emphasizing on the key molecular targets and pathways, we also outline future directions for mechanistic research, biomarker discovery, and regulatory action. Although EDCs differ in structure and origin, they ultimately converge on shared molecular pathways that mediate their reproductive toxicity. At the cellular level, they affect reproductive tissues, including granulosa cells, Leydig cells, Sertoli cells, and oocytes, by interfering with essential processes, such as steroid hormone synthesis, gametogenesis, follicular maturation, and embryo implantation.
At the epigenetic level, EDCs alter the activity and expression of enzymes such as DNMT1, DNMT3a, histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone methyltransferases, such as enhancer of zeste homolog 2 (EZH2) and G9a. These disruptions lead to abnormal DNA methylation (such as at ESR1 and CYP19A1), altered histone modification patterns (such as increased H3K27me3 and decreased H3K9ac), and changes in miRNA expression (such as miR-21 and miR-146a), ultimately silencing genes critical for hormonal balance and reproductive cell viability. Functionally, these molecular changes result in decreased estradiol and testosterone production, anovulation, impaired spermatogenesis, reduced oocyte quality, and increased risk of infertility or miscarriage. Understanding these converging epigenetic pathways is essential for developing targeted therapies and identifying early biomarkers of reproductive toxicity.
Epigenetic mechanisms are essential processes that regulate gene expression patterns without altering the underlying DNA sequence [30, 31]. The key epigenetic mechanisms include histone modifications, DNA methylation, RNA methylation, and non-coding RNAs (ncRNAs) such as microRNAs (miRNAs) (Fig. 1) [31, 32, 33].
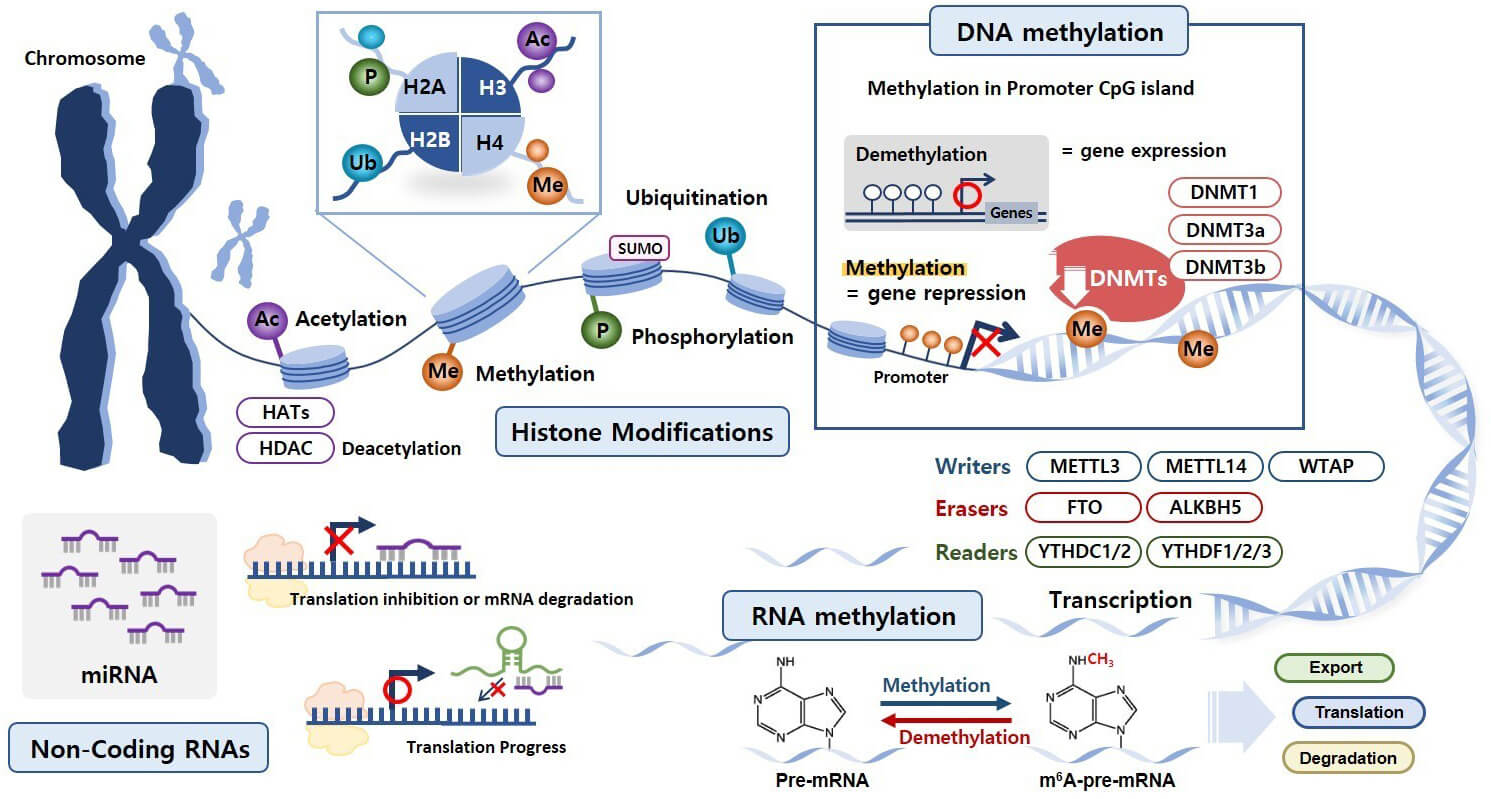 Fig. 1.
Fig. 1.
Epigenetic mechanisms involved in reproductive physiology. Histone modifications, DNA methylation, RNA methylation, and non-coding RNAs (ncRNAs) represent essential epigenetic mechanisms. Four prevalent histone modifications include acetylation (Ac), methylation (Me), phosphorylation (P), and ubiquitination (Ub). Additionally, histone SUMOylation involves the covalent attachment of small ubiquitin-like modifier (SUMO) proteins to specific histone residues. DNA methylation primarily occurs at cytosine residues within cytosine–phosphate–guanine (CpG) dinucleotide regions, commonly referred to as CpG islands. RNA methylation is a reversible epigenetic modification that is tightly regulated by specialized enzymes known as “writers”, “erasers”, and “readers”. Among various ncRNAs, miRNAs regulate gene expression at the post-transcriptional level by binding complementary messenger RNA (mRNA) sequences, resulting in mRNA degradation or translation suppression. HATs, histone acetyltransferases; HDACs, histone deacetylases; DNMT, DNA methyltransferase; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; WTAP, Wilms tumor 1-associated protein; FTO, fat mass and obesity-associated protein; ALKBH5, AlkB homolog 5; YTHDC1, YTH domain containing 1.
Histone acetylation involves HAT-mediated acetyl group addition to lysine residues on histone tails, which neutralizes the positive charge on lysine, thereby reducing interactions between histones and DNA. Therefore, the chromatin becomes less compact, creating an open and transcriptionally active state that promotes gene expression [34, 35].
Histone deacetylation is mediated by histone deacetylases (HDACs), which remove acetyl groups from histone tails to restore the positive charge on lysine residues, strengthening the interaction between histones and DNA. The resulting tighter chromatin structure represses gene transcription by reducing the accessibility of transcription factors to the DNA [36, 37].
Histone methylation is a complex process that mainly occurs at the lysine and arginine residues. The effect of methylation depends on both the specific modified residues, such as H3K4, H3K9, H3K27, and H3K36, and methylation degree (mono-, di-, or tri-methylation). For example, trimethylation at H3K4 (H3K4me3) is typically associated with active transcription, whereas H3K27me3 and H3K9me3 are associated with gene repression. These modifications are tightly controlled by specific histone methyltransferases and demethylases that modulate gene expression during gametogenesis, embryogenesis, and hormone-responsive processes in reproduction [38, 39, 40].
Histone phosphorylation involves adding phosphate groups to the serine, threonine, or tyrosine residues on histones. This modification plays a crucial role in regulating chromatin dynamics during various biological processes. For example, H3 phosphorylation at serine 10 (H3S10ph) and serine 28 (H3S28ph) is closely linked to chromosome condensation during mitosis and meiosis, ensuring proper chromosome alignment and segregation [41, 42].
Histone ubiquitination typically occurs on lysine residues such as H2AK119 and H2BK120 [43]. This modification influences chromatin configuration and gene expression by modulating transcriptional activation, DNA repair, and histone turnover. H2B mono-ubiquitination is associated with active transcription, whereas H2A ubiquitination is often linked to transcriptional repression and DNA damage response [30, 44].
Histone SUMOylation involves the attachment of small ubiquitin-like modifier (SUMO) proteins to histone residues. This modification typically represses gene expression by altering chromatin structure, affecting transcription factor interactions, and promoting the recruitment of repressive protein complexes involved in transcriptional silencing and DNA repair [45, 46, 47, 48].
DNA methylation is an epigenetic modification that primarily occurs at cytosine residues within cytosine–phosphate–guanine (CpG) dinucleotides, known as CpG islands, which are typically located in gene promoter regions. This process involves the covalent addition of methyl groups by DNMTs, primarily DNMT1, DNMT3a, and DNMT3b. DNMT1 is mainly responsible for maintaining DNA methylation patterns during replication, thereby ensuring that these patterns are epigenetically inherited. DNMT3a and DNMT3b are involved in establishing new methylation patterns (de novo methylation) during development and cellular differentiation [49, 50].
DNA methylation generally leads to transcriptional repression by preventing transcription factors from binding to DNA and facilitating methyl-CpG binding domain (MBD) protein recruitment. These proteins in turn recruit histone-modifying enzymes that further enhance gene silencing. However, in some cases, DNA methylation can activate gene expression depending on the specific genomic context and the involvement of certain transcription factors that are sensitive to alterations in methylation [51].
RNA methylation is an important epigenetic modification that primarily affects mRNA expression. The most common and widely studied form of RNA methylation is N6-methyladenosine (m6A), where a methyl group is added to the N-6 position of adenosine. This process is carefully controlled by specialized enzymes known as “writers”, “erasers”, and “readers”.
Writers are enzymes, such as methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and the Wilms tumor 1-associated protein (WTAP), which add methyl groups to RNA and play crucial roles in RNA modification. METTL3 acts as the main catalytic unit of the methyltransferase complex, transferring methyl groups from S-adenosylmethionine (SAM) to adenosine residues in RNA. METTL14 provides structural support to METTL3, stabilizing the complex and improving its efficiency. WTAP regulates this process by guiding the METTL3–METTL14 complex to specific RNA targets and ensuring that it is correctly positioned within the cell [52, 53].
In contrast, erasers such as fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) remove these methyl groups, effectively reversing RNA methylation. FTO was originally linked to obesity but later found to demethylate m6A residues in RNA, impacting RNA stability, translation, and alternative splicing [54]. ALKBH5 specifically removes methyl groups from m6A residues in nuclear RNA, influencing RNA export from the nucleus and affecting overall RNA molecule lifespan and function. This makes ALKBH5 a key player in regulating RNA metabolism and gene expression [52, 55].
Readers are specialized proteins that recognize and bind to methylated RNA, affecting important post-transcriptional processes, such as RNA stability, splicing, export from the nucleus, localization within the cell, and translation. The most well-known readers belong to the YT521-B homology (YTH) family, which includes YTHDF1, YTHDF2, and YTHDF3. YTHDF1 promotes translation by interacting with translation initiation factors. YTHDF2 accelerates m6A-tagged RNA breakdown by directing it to processing bodies (P-bodies) for degradation. YTHDF3 interacts with YTHDF1 and YTHDF2 to coordinate both methylated RNA translation and degradation. Additionally, YTHDC1 is a nuclear reader that influences alternative splicing by helping splicing factors interact with methylated pre-mRNAs, thereby fine-tuning gene expression [52, 56].
In addition to m6A, other RNA methylations occur at different sites and nucleotide positions. For instance, m1A affects RNA structure, stability, and translation by disrupting standard base pairing [57]. 5-methylcytosine (m5C), produced by RNA methyltransferases such as NSUN family proteins and DNMT2, contributes to RNA stability, processing, and nuclear export [13]. N7-methylguanosine (m7G), found at the 5′ cap of mRNA, increases RNA stability and translation efficiency [53]. Meanwhile, 2′-O-methylation (Nm) is common in transfer RNA (tRNA), ribosomal RNA (rRNA), and small nuclear RNA (snRNA), supporting RNA stability and function [58].
ncRNAs are transcribed from DNAs that are not translated into proteins. Instead of functioning as protein synthesis templates, they play an essential regulatory role in gene expression and cellular processes. Among the various ncRNAs, miRNAs, long ncRNAs (lncRNAs), and piwi-interacting RNAs (piRNAs) are particularly important owing to their broad functional impacts [59].
miRNAs are short (approximately 22 nucleotides), single-stranded ncRNAs that
regulate gene expression at the post-transcriptional level. They bind to
complementary sequences in target mRNAs, leading to mRNA degradation or
translation inhibition. miRNAs are involved in various biological processes,
including cell proliferation, differentiation, apoptosis, and metabolism [60].
lncRNAs, which are typically
Together, these different ncRNAs form a complex and highly sophisticated regulatory network that modulates gene expression at both transcriptional and post-transcriptional levels, significantly influencing cellular functions and physiological processes.
Reproductive dysfunction refers to any condition that impairs fertility or reproductive health. The hypothalamic–pituitary–gonadal (HPG) axis plays a central role in regulating reproductive functions, including gonadotropin secretion, steroidogenesis, and gametogenesis. The HPG axis functions through a finely tuned balance of hormonal signaling.
Gonadotropin-releasing hormone (GnRH), secreted by the hypothalamus, stimulates luteinizing hormone (LH) and follicle-stimulating hormone (FSH) release from the pituitary gland. In males, LH primarily acts on Leydig cells to promote testosterone production, whereas in females, it stimulates theca cells to produce androgens. FSH acts on Sertoli cells in males to support spermatogenesis and on granulosa cells in females to promote follicular development and estrogen production [63, 64].
At the cellular level, reproductive dysfunction results from impaired gametogenesis, abnormal follicular development, disrupted embryo implantation, and compromised embryonic growth. For instance, Leydig cell dysfunction can reduce testosterone production, which negatively affects sperm development and libido [65]. In females, disrupted follicular growth can impair ovulation and reduce endometrial receptivity, thereby decreasing the chances of successful implantation and pregnancy.
Male reproductive dysfunction can result from genetic defects, hormonal imbalances, anatomical abnormalities, lifestyle factors, and environmental exposures [66]. Genetic conditions such as Klinefelter syndrome disrupt testicular function, whereas hormonal imbalances within the HPG axis impair gonadotropin production and release. Structural abnormalities, including varicocele and the congenital absence of vas deferens, interfere with sperm production and transport. Lifestyle factors such as obesity, excess alcohol consumption, smoking, and chronic stress significantly reduce fertility [67, 68]. Furthermore, exposure to EDCs, such as phthalates and BPA, exacerbates reproductive dysfunction by disrupting hormonal signaling and increasing oxidative stress [69, 70, 71].
Spermatogenesis is a complex process involving the mitotic proliferation, meiotic division, and post-meiotic differentiation of germ cells [72]. Spermatogonial stem cells undergo mitosis to produce spermatocytes, which subsequently undergo meiosis to generate haploid spermatids. These spermatids mature into spermatozoa through a process called spermiogenesis, which involves chromatin condensation, acrosome and flagellum formation, and cytoplasmic reduction. Oxidative stress caused by increased reactive oxygen species (ROS) production owing to environmental toxins, smoking, and poor dietary habits can severely impair sperm quality [73, 74]. ROS-induced damage, such as lipid peroxidation, mitochondrial dysfunction, and DNA fragmentation, reduces the sperm fertilization potential and embryo viability. Elevated oxidative stress increases sperm apoptosis and decreases sperm motility.
Epigenetic regulation plays a key role in regulating gene expression during spermatogenesis. Aberrant DNA methylation, such as androgen receptor (AR) gene hypermethylation, disrupts testosterone signaling and sperm production [75]. Studies have demonstrated that AR promoter hypermethylation is linked to reduced AR expression in infertile males, which is correlated with low testosterone levels and poor sperm quality [76, 77]. Histone modifications significantly affect chromatin structure and gene expression. Abnormal histone methylation patterns, such as increased H3K9 methylation and decreased H3K4 methylation, have been associated with male infertility by silencing genes critical for spermatogenesis [78]. Furthermore, reduced histone H4 acetylation is associated with decreased sperm motility and poor semen quality [79].
Collectively, these findings highlight that epigenetic dysregulation is a key mechanism underlying male reproductive dysfunction and provides potential targets for diagnostic biomarkers and therapeutic interventions.
Female reproductive dysfunction encompasses various disorders affecting ovarian function, hormone production, and fertility. Clinical manifestations include infertility, ovulatory dysfunction, decreased ovarian reserve, menstrual irregularities, and gynecological disorders such as polycystic ovary syndrome (PCOS) and endometriosis. Female reproductive dysfunction arises from various causes, including genetic predisposition, hormonal imbalances, structural abnormalities of the reproductive tract, lifestyle factors, and environmental exposures [80, 81].
Disruptions in the hypothalamic–pituitary–ovarian (HPO) axis due to genetic or hormonal factors can lead to irregular ovulation and impaired hormone production [82]. Structural abnormalities such as uterine fibroids and fallopian tube obstructions interfere with embryo implantation and increase miscarriage risk. Lifestyle factors, including obesity, smoking, poor diet, and chronic stress, significantly reduce ovarian reserve and increase infertility risk [68]. Furthermore, exposure to EDCs and environmental toxins exacerbates reproductive dysfunction by inducing oxidative stress and causing hormonal dysregulation [83].
PCOS is characterized by hyperandrogenism, insulin resistance, and disrupted follicular development [84]. Elevated androgen levels suppress normal follicular development, whereas insulin resistance promotes excess androgen production by ovarian theca cells, further exacerbating anovulation and infertility. Endometriosis involves ectopic growth of endometrial tissue, leading to chronic inflammation, pelvic pain, and reduced fertility [85]. Increased estrogen production and elevated inflammatory cytokine levels in endometriosis contribute to adhesion formation, ovarian dysfunction, and implantation failure [86].
Oxidative stress caused by elevated ROS levels in the ovarian follicles and endometrial tissue significantly impairs oocyte quality, embryo viability, and endometrial receptivity [87]. Elevated ROS levels also disrupt embryonic development, luteal phase function, and progesterone production, thereby reducing implantation success rates and increasing miscarriage risk.
Epigenetic modifications, including DNA methylation and histone modifications, play crucial roles in regulating ovarian function and reproductive hormone signaling. The hypermethylation of specific genes, such as estrogen receptor alpha (ESR1), has been linked to reduced ovarian responsiveness to gonadotropins [88]. Alterations in histone acetylation and methylation impair ovarian steroidogenesis and folliculogenesis. Additionally, miRNA, particularly miR-200 and miR-21, expression is dysregulated in reproductive dysfunction, which negatively affects ovarian steroidogenesis, follicular maturation, and overall fertility. For example, miR-21 overexpression impairs ovarian granulosa cell function, whereas abnormal miR-200 levels have been associated with PCOS pathogenesis [89, 90].
These findings emphasize the critical role of epigenetic dysregulation in female reproductive dysfunction and highlight potential diagnostic biomarkers and therapeutic targets for improving reproductive health.
Endocrine disruptors interfere with normal hormonal functions by mimicking, blocking, or altering hormone signaling pathways (Fig. 2) [91]. Both human epidemiological studies and animal research have consistently linked exposure to EDCs with impaired sperm production, compromised ovarian function, abnormal gonadal development, and reproductive disorders such as endometriosis and PCOS [92, 93, 94, 95, 96]. Among the EDCs, BPA and phthalates have been extensively studied because of their widespread presence and established reproductive toxicity.
 Fig. 2.
Fig. 2.
Mechanisms of reproductive dysfunction induced by endocrine-disrupting chemicals (EDCs). Gonadotropin-releasing hormone (GnRH), released by the hypothalamus, stimulates the anterior pituitary to secrete luteinizing hormone (LH) and follicle-stimulating hormone (FSH). In males, LH acts on Leydig cells to promote testosterone synthesis, while FSH supports spermatogenesis through Sertoli cell regulation. In females, LH and FSH coordinate androgen synthesis in theca cells, follicular maturation, and estrogen production in granulosa cells. Reactive oxygen species (ROS), generated in response to EDCs, contribute to oxidative stress through mechanisms such as lipid peroxidation, mitochondrial dysfunction, and DNA fragmentation, ultimately exacerbating reproductive dysfunction. The upper left panel depicts the steroidogenic pathway in Leydig cells, while the upper right panel illustrates steroidogenesis in theca and granulosa cells. EDC exposure modulates the expression of key steroidogenic enzymes, impairing hormone biosynthesis. Blue and red arrows indicate upregulated and downregulated expression of enzymes involved in steroid hormone synthesis, respectively. DEHP, di(2-ethylhexyl) phthalate; MEHP, metabolite mono(2-ethylhexyl) phthalate; StAR, steroidogenic acute regulatory; CYP11A, cholesterol side-chain cleavage enzyme; HSD3B, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1; DHT, dihydrotestosterone; DBP, dibutyl phthalate; BBP, Benzyl butyl phthalate.
Previous studies have examined the effects of endocrine disruptors on reproductive health, with BPA being one of the most extensively studied compounds. BPA significantly affects reproductive function through epigenetic mechanisms, as demonstrated by the consistent findings of numerous animal and human studies [14]. Exposure to BPA during the prenatal and neonatal periods markedly changes DNA methylation patterns, histone modifications, and miRNA expression, which play crucial roles in regulating ovarian follicle development, steroidogenesis, and the reproductive cycle [11].
Among these epigenetic mechanisms, DNA methylation is strongly associated with the effects of BPA on ovarian gene expression. Animal studies have revealed that BPA exposure hypermethylates the genes essential for ovarian steroidogenesis, such as ESR1, aromatase (CYP19A1), and luteinizing hormone receptor (LHR), thereby downregulating their expression, which impairs ovarian function and reduces reproductive capacity [15, 16]. In addition, BPA alters histone acetylation and methylation patterns at the promoters of critical reproductive genes. For instance, in mouse models, BPA exposure decreased H3K9 acetylation and increased H3K27 trimethylation, modifications that typically repress transcription and significantly disrupt the expression of genes necessary for normal ovarian development and function [17, 18]. These histone alterations are mediated by the BPA-induced dysregulation of chromatin-modifying enzymes. BPA reduces HAT activity and increases HDAC expression, leading to global histone hypoacetylation and the transcriptional repression of reproduction-related genes. Moreover, females with endometriosis exhibit distinct epigenetic profiles, including altered miRNA expression patterns linked to elevated serum and urinary BPA levels [97]. Epigenetic reprogramming is associated with increased disease severity and a high risk of recurrence.
BPA disrupts reproductive health by altering miRNA expression. In animal models,
exposure to BPA during critical developmental periods has been associated with
the altered expression of key miRNAs, including miR-146a, miR-21, and miR-200
family members, as well as ovarian cell proliferation, apoptosis,
steroidogenesis, and inflammatory signaling regulators [98, 99, 100, 101]. For instance,
miR-21 upregulation suppresses phosphatase and tensin homolog (PTEN), a negative
regulator of the PI3K/AKT pathway, thereby promoting granulosa cell survival and
potentially contributing to abnormal follicular persistence [102]. Likewise,
miR-146a targets interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF
receptor-associated factor 6 (TRAF6), which are key components of the
NF-
Epidemiological studies in humans have corroborated molecular findings observed in animal models. Clinical studies have identified significant changes in DNA methylation patterns of genes related to steroidogenesis and hormone signaling in women with higher urinary BPA concentrations, which correlate with reproductive disorders such as PCOS, endometriosis, and infertility [104]. For example, elevated BPA levels in females with PCOS have been associated with CYP19A1 and ESR1 hypermethylation, correlating with elevated androgen levels and insulin resistance [105]. Additionally, females with endometriosis display distinct epigenetic profiles, including altered miRNA expression patterns linked to elevated serum and urinary BPA levels, which influence disease severity and recurrence [106]. Beyond BPA, growing attention has been paid to its structural analogs and derivatives such as bisphenol S (BPS) and bisphenol F (BPF), which are commonly used as BPA substitutes in plastics and consumer products. Emerging evidence suggests that BPS and BPF may exert endocrine-disrupting and epigenetic effects similar to or even more potent than those of BPA [107]. Studies have shown that BPS exposure alters the methylation of genes involved in steroidogenesis and increases oxidative stress in ovarian tissues [108, 109]. BPF is associated with disrupted histone acetylation patterns and altered reproductive miRNA expression [110]. Although marketed as safe alternatives, these compounds appear to share similar epigenetic interferences, raising concerns about their reproductive toxicity and necessitating further investigation.
These findings clearly indicate that the reproductive toxicity of BPA is driven primarily by its impact on the epigenetic landscape of reproductive tissues. BPA disrupts hormonal signaling and regulates genes critical for reproductive function by inducing changes in DNA methylation, histone modifications, and non-coding RNA expression. Taken together, these epigenetic alterations provide strong mechanistic evidence linking EDC exposure to reproductive dysfunction.
Phthalates, commonly used as plasticizers in daily products, are increasingly linked to reproductive health through epigenetic mechanisms. Among the most common phthalates, diethylhexyl phthalate (DEHP) induces significant changes in DNA methylation and histone modification, thereby disrupting the transcriptional regulation of genes involved in steroidogenesis and spermatogenesis [4].
DEHP exposure leads to the epigenetic dysregulation of reproductive tissues. Animal studies have demonstrated that exposure to DEHP increases H3 acetylation at the promoters of reproductive genes, leading to abnormal activation [19]. Simultaneously, DEHP reduces DNA methylation at key steroidogenic genes, such as steroidogenic factor-1 (SF-1) and CYP17A1, which are essential for testosterone biosynthesis [20, 21]. Hypomethylation disrupts the hormonal balance required for proper testicular function. Simultaneously, DEHP increases H3 acetylation (H3K9ac) and H3K4 trimethylation (H3K4me3) at these gene loci, while decreasing repressive markers such as H3K9me2 and H3K27me3 [111, 112, 113, 114]. These chromatin changes are mediated by altered expression of epigenetic enzymes, including downregulation of DNMT3a and HDACs and upregulation of HATs and histone methyltransferases (HMTs), such as mixed-lineage leukemia protein 1 (MLL1). This imbalance creates a transcriptionally active chromatin state, leading to aberrant expression of reproductive genes. This impairs hormone synthesis and reduces testosterone production and sperm quality, which has been consistently observed in animal models [113]. Furthermore, these chromatin alterations appear to be long lasting and may have transgenerational consequences [115].
MEHP, a biologically active DEHP metabolite, exerts strong epigenetic effects. Studies using MA-10 mouse Leydig cells have shown that MEHP significantly disrupts the expression of key steroidogenic proteins, particularly STAR protein [22]. This effect is driven by both histone and DNA modifications in the star gene promoter, resulting in transcriptional repression and impaired Leydig cell function. Furthermore, MEHP increases repressive histone markers such as H3K27 trimethylation (H3K27me3) and decreases activating markers such as H3K9 acetylation (H3K9ac), thereby promoting a condensed chromatin structure [114, 116]. It also downregulates coactivator proteins, such as CBP/p300, and reduces the activity of ten-eleven translocation methylcytosine dioxygenase (TET) enzymes, contributing to stable gene silencing. These epigenetic changes are not limited to star and other steroidogenic genes, including Cyp11a1 and Hsd3b1, indicating broad suppression of the steroid biosynthesis cascade. Importantly, the epigenetic effects of MEHP persist even after exposure ends, raising concerns about long-term and potentially heritable reproductive dysfunction [117].
Phthalate-induced epigenetic disruption has been shown to negatively affect
female reproductive health. Animal studies have shown that exposure to DEHP and
MEHP results in marked alterations in histone acetylation and methylation in the
promoters of key ovarian steroidogenic genes, such as CYP19A1 and
ESR1. These changes impair follicular development and disrupt the
ovulatory cycle [118, 119]. Moreover, MEHP exposure in endometrial cell cultures
increases the secretion of inflammatory cytokines, particularly tumor necrosis
factor-alpha (TNF-
Human clinical studies have supported these findings in animal models. Research has consistently linked elevated urinary levels of phthalate metabolites to menstrual irregularities, reduced ovarian reserve, premature ovarian aging, a high risk of preterm birth, and other adverse reproductive outcomes [121, 122]. These clinical findings provide molecular evidence that phthalates interfere with reproductive function through epigenetic dysregulation, underscoring their relevance as a public health concern. Taken together, phthalate-induced epigenetic modifications disrupt the hormonal balance and transcriptional regulation in reproductive tissues, providing a mechanistic explanation for their roles in both male and female reproductive dysfunctions.
Pesticides, including DDT and chlorpyrifos, are potent endocrine disruptors, with growing evidence that their reproductive toxicity is largely mediated through epigenetic mechanisms [123]. Epidemiological and experimental studies have consistently shown that pesticide exposure negatively affects reproductive health in both males and females by altering DNA methylation, modifying histone structure, and disrupting ncRNA expression [23, 124, 125].
The epigenetic effects of prenatal and developmental exposure to DDT and its persistent metabolite dichlorodiphenyldichloroethylene (DDE) have been extensively studied. Research shows that DDT exposure can lead to abnormal DNA methylation of genes critical for reproductive development and hormone regulation [23, 24, 25]. For example, both DDT and DDE have been associated with hypomethylation at the promoter regions of estrogen-responsive genes such as ESR1, which disrupts estrogen signaling pathways essential for reproductive organ formation and fertility. Furthermore, transgenerational studies have found that maternal exposure to DDT alters the methylation patterns of key reproductive genes, including insulin-like growth factor 2 (IGF2), which persists across multiple generations [24, 26]. These epigenetic changes reduce fertility, delay sexual maturation, and increase the incidence of undescended testes in male offspring.
Similarly, chlorpyrifos, a widely used organophosphate pesticide, exerts reproductive toxicity through comprehensive epigenetic modifications. Exposure to chlorpyrifos alter miRNA expression profiles, which may disrupt critical reproductive signaling pathways. For example, studies on zebrafish embryos have shown that organophosphate compounds can upregulate the expression of miR-137 and miR-141, resulting in developmental abnormalities [126]. Although direct evidence of chlorpyrifos-induced miRNA alterations in mammalian reproductive tissues remains limited, the neurotoxic and reproductive effects consistently observed in animal models suggest that miRNA dysregulation may play a central role in chlorpyrifos toxicity. Recent animal studies have demonstrated that chlorpyrifos exposure leads to abnormal DNA methylation patterns in genes regulating the HPG axis, such as GnRH1, ESR1, and AR [127]. In particular, GnRH1 promoter hypomethylation is associated with early onset of puberty and disrupted reproductive hormone signaling [128]. In addition, chlorpyrifos alters histone modification states in reproductive tissues. In animal models, decreased histone H3K9 acetylation (H3K9ac) and H4 acetylation (H4ac) have been observed, leading to the transcriptional repression of genes involved in steroidogenesis and gametogenesis [129]. These chromatin changes appear to be mediated by the dysregulation of epigenetic enzymes such as DNMT1 and HDAC1, which are downregulated following chlorpyrifos exposure. The resulting imbalance in the chromatin remodeling machinery may contribute to the long-term suppression of reproductive gene expression, with potential long-term consequences for fertility.
The findings of clinical studies align closely with animal study findings, further strengthening the link between pesticide exposure and epigenetic changes in reproductive tissues [16]. Epidemiological studies have reported abnormal DNA methylation patterns at key loci, such as ESR1 and AR genes, in individuals with past pesticide exposure [130]. For instance, ESR1 promoter hypomethylation dysregulates estrogen signaling, which may impair endometrial receptivity and ovulation. Likewise, AR hypermethylation can weaken androgen signaling, contributing to reduced spermatogenesis and impaired Leydig cell function in males [131, 132]. These epigenetic modifications correlate with a range of reproductive issues, including reduced sperm concentration and motility, disrupted menstrual cycles, anovulation, and increased incidence of infertility and miscarriage. Moreover, these methylation changes may persist even after the end of exposure, indicating potential long-term or transgenerational effects. In occupational cohorts, a high pesticide burden has also been linked to altered methylation of imprinted genes such as IGF2 and H19, which play critical roles in embryonic growth and placental development, suggesting possible implications on pregnancy outcomes and offspring health [133, 134]. Taken together, these findings support a mechanistic model in which pesticide-induced epigenetic modifications, such as DNA hypomethylation, histone alterations, and miRNA dysregulation, directly contribute to endocrine disruption and impaired reproductive capacity.
Divalent heavy metal ions such as Pb2+ and Cd2+ are widely recognized as potent endocrine disruptors due to their environmental persistence and strong reproductive toxicity [5, 6, 7]. It is the ionic forms, rather than the elemental metals or their poorly soluble salts, that are primarily responsible for these toxic effects. Although they are not classified as toxins in the classical toxicological sense, these heavy metal ions function as environmental contaminants that exert harmful biological effects through multiple pathways, including oxidative stress, hormonal disruption, and epigenetic alterations. Extensive research has consistently demonstrated the harmful effects of heavy metal exposure on male and female reproductive health [135].
In males, exposure to Pb2+ and Cd2+ has been strongly associated with
impaired spermatogenesis, characterized by reduced sperm count, poor sperm
motility, and abnormal sperm morphology [136, 137]. These disruptions are
primarily attributed to damage to the seminiferous epithelium and dysfunction of
the blood–testis barrier, as observed in rodent models. In particular, Cd2+
induces necrosis of Sertoli and germ cells, whereas Pb2+ tends to affect
Leydig cells and the HPG axis. Animal studies have shown that exposure to these
heavy metal ions impairs testicular function by downregulating the expression of
key enzymes such as STAR, CYP11A1, and
17
Clinical studies support these findings, showing that elevated blood levels of Pb2+ and Cd2+ are associated with reduced testosterone production, increased oxidative stress, and compromised sperm DNA integrity in exposed individuals [141]. Additionally, these metals increase testicular oxidative stress by elevating ROS and depleting antioxidant enzymes, such as superoxide dismutase (SOD) and glutathione peroxidase (GPx), resulting in lipid peroxidation and mitochondrial dysfunction in germ cells. Such oxidative damage contributes to DNA fragmentation in sperm, chromatin condensation defects, and reduced fertilization potential.
In females, heavy metal ions exposure is strongly associated with ovarian
dysfunction, disrupted follicular development, and diminished reproductive
capacity [142, 143]. Cd2+ accumulates in ovarian tissue owing to its long
biological half-life and ability to mimic essential divalent cations, such as Ca
and Zn, thereby interfering with cellular signaling and enzyme activity.
Experimental studies have shown that Cd2+ disrupts ovarian steroidogenesis
by downregulating the expression and activity of key enzymes involved in estrogen
and progesterone synthesis, including cytochrome P450 side-chain cleavage enzyme
(CYP11A1), aromatase (CYP19A1), and 3
At the molecular level, heavy metals contribute to reproductive toxicity, primarily through epigenetic dysregulation. Pb2+, Cd2+, and soluble ionic forms of mercury (such as HgCl2) and nickel (such as NiSO4 or NiCl2) alter DNA methylation and histone modification profiles, particularly at promoters of genes essential for hormonal signaling, gametogenesis, and oxidative defense. In contrast, arsenic trioxide (As2O3) does not dissociate in solution but acts as a precursor to toxic trivalent arsenic compounds and organoarsenicals, and should be evaluated separately in toxicological contexts. DNA methylation changes are among the most widely documented epigenetic effects of heavy metal exposure. Pb2+ and Cd2+ induce global hypomethylation, compromising genomic stability, while simultaneously promoting hypermethylation at specific CpG-rich promoters. For example, aberrant methylation of follicle-stimulating hormone receptor (FSHR), LHR, ESR1, and AR has been reported in animal models and human tissues, leading to decreased hormonal sensitivity and disrupted reproductive function [27, 28].
Simultaneously, heavy metal exposure disrupts histone modifications, further contributing to epigenetic dysregulation. Cd2+, for example, has been associated with reduced H3K9 acetylation and increased H3K27 trimethylation at the promoter regions of steroidogenic genes such as CYP11A1, STAR, and HSD17B3, as well as antioxidant genes such as GPX4 and SOD2 [29]. These changes promote chromatin condensation and transcriptional repression, ultimately impairing the expression of genes critical for reproductive functions. Beyond Cd2+ and Pb2+, other heavy metals also exhibit distinct epigenetic signatures in reproductive tissues. For instance, As causes global hypomethylation and gene-specific hypermethylation (e.g., p16, p53), along with inhibition of HAT activity, disrupting oocyte maturation and endometrial receptivity [145]. Hg, particularly methylmercury (MeHg), decreases DNMT expression and alters histone marks (such as H3K9me2 and H4K20me3), impairing spermatogenesis and inducing germ cell apoptosis [146]. Ni2+, typically as NiSO or NiCl2, also exerts notable epigenetic effects. Ni exposure induces global DNA hypomethylation and gene-specific hypermethylation at loci involved in gonadal development and hormone synthesis [147]. It enhances repressive histone marks such as H3K9me2 and H3K27me3 via the upregulation of methyltransferases such as G9a and EZH2, leading to chromatin condensation and transcriptional silencing. These effects are associated with disrupted spermatogenesis and follicular viability in experimental models [148, 149].
These findings suggest that heavy metal ion-induced reproductive toxicity is driven by epigenetic dysregulation. A deep understanding of these molecular alterations could provide essential insights into the mechanisms of heavy-metal-induced reproductive dysfunction and guide the development of targeted prevention and treatment strategies.
Collectively, these epigenetic alterations illustrate how heavy metals act as endocrine disruptors by reprogramming the epigenome of the reproductive tissues. Heavy metal ions contribute to long-term reproductive dysfunction and transgenerational health effects through mechanisms involving aberrant DNA methylation, histone modification, and oxidative stress-linked epigenetic remodeling.
EDCs pose a significant threat to reproductive health by interfering with hormonal regulation and inducing stable epigenetic changes that alter gene expression profiles critical for gametogenesis, steroidogenesis, and follicular development. This review highlights the multifaceted epigenetic mechanisms, including DNA methylation, histone modifications, and ncRNA regulation, through which various EDCs such as BPA, phthalates, pesticides, and heavy metal ions impair reproductive functions in both sexes.
Accumulating evidence indicates that these epigenetic alterations are not only persistent but may also exert transgenerational effects, raising public health concerns regarding long-term reproductive consequences. However, gaps remain in our understanding of compound-specific epigenetic signatures, dose–response relationships, sex-specific effects, and reversibility of these modifications.
Future studies should prioritize the identification of epigenetic biomarkers for the early detection of EDC exposure and susceptibility as well as development of targeted epigenetic therapies. Additionally, integrative approaches combining epigenomics, transcriptomics, and single-cell analysis are essential for elucidating the tissue- and cell-type-specific effects of EDCs. A deep understanding of these mechanisms will guide risk assessment, public policies, and therapeutic intervention strategies for mitigating EDC-induced reproductive dysfunction.
DHK and MHP conceived and designed the study. SH and HBK conducted the literature search, analyzed the data, and prepared the figure visualizations. SH, HBK, and MHP wrote the manuscript. DHK and MHP reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was supported by the Regional Innovation System & Education (RISE) program through the Institute for Regional Innovation System & Education in Busan Metropolitan City, funded by the Ministry of Education (MOE) and the Busan Metropolitan City, Republic of Korea (2025-RISE-02-005-000).
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGPT to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.