1. Introduction
According to the World Health Organization, heart disease is one of the leading
causes of global mortality and morbidity, accounting for approximately 17.9
million deaths annually. Heart diseases such as left ventricular hypertrophy
(LVH) and arrhythmia are particularly common, which poses a significant health
burden [1]. These diseases not only lead to acute illnesses such as heart attacks
and strokes but also contribute to long-term complications such as heart failure
(HF), placing a heavy burden on global healthcare systems. LVH is a pathological
state of cardiac structural remodeling, characterized by hypertrophy and
hyperplasia of the left ventricular (LV) myocardium. It typically occurs due to
prolonged pressure or volume overload (such as hypertension, valve disease, or
heart failure) as a compensatory response, leading to LV wall thickening,
narrowing or dilation of the cavity, and eventual loss of compliance [2, 3]. This
pathologic change not only indicates target organ damage in patients with
hypertension but is also an important risk factor for congestive heart failure
(CHF), arrhythmia, and stroke [4, 5]. Arrhythmia, characterized by an irregular
heart rhythm due to abnormal electrical activity, usually manifests as
tachycardia, bradycardia, or atrial fibrillation (AF), all of which increase the
risk of stroke and CHF [6, 7]. An increasing number of studies indicate that the
wingless-int1 (Wnt)/-catenin signaling pathway plays a key role in their
development and progression.
The canonical Wnt/-catenin pathway comprises four essential elements:
① Wnt proteins (ligands), ② Receptor complex, Frizzled (primary
receptor), LRP5/6 (co-receptors), ③ Dishevelled (Dvl) (scaffold protein
for signal transduction), ④ -catenin (nuclear transcriptional
effector). In the absence of Wnt ligands, -catenin is phosphorylated and
degraded by the “destruction complex” (Axin/APC/CK1/GSK-3).
Upon Wnt activation, the ligand binds to the Frizzled-LRP5/6 receptor complex,
leading to the recruitment of Dvl protein and the disassembly of the
-catenin destruction complex. This results in the stabilization and
nuclear accumulation of -catenin. Subsequently, -catenin forms
a transcriptional complex with TCF/LEF factors to activate target genes [8] (Fig. 1). The Wnt/-catenin pathway critically regulates embryonic development
through cell proliferation, differentiation and migration [9, 10]. Its
dysregulation contributes to various diseases including cancer, stroke,
myocardial infarction (MI), LVH and arrhythmias by modulating multiple cellular
processes [11, 12, 13, 14]. Aberrant activation exacerbates oxidative stress, inflammation
and cell death [15, 16].
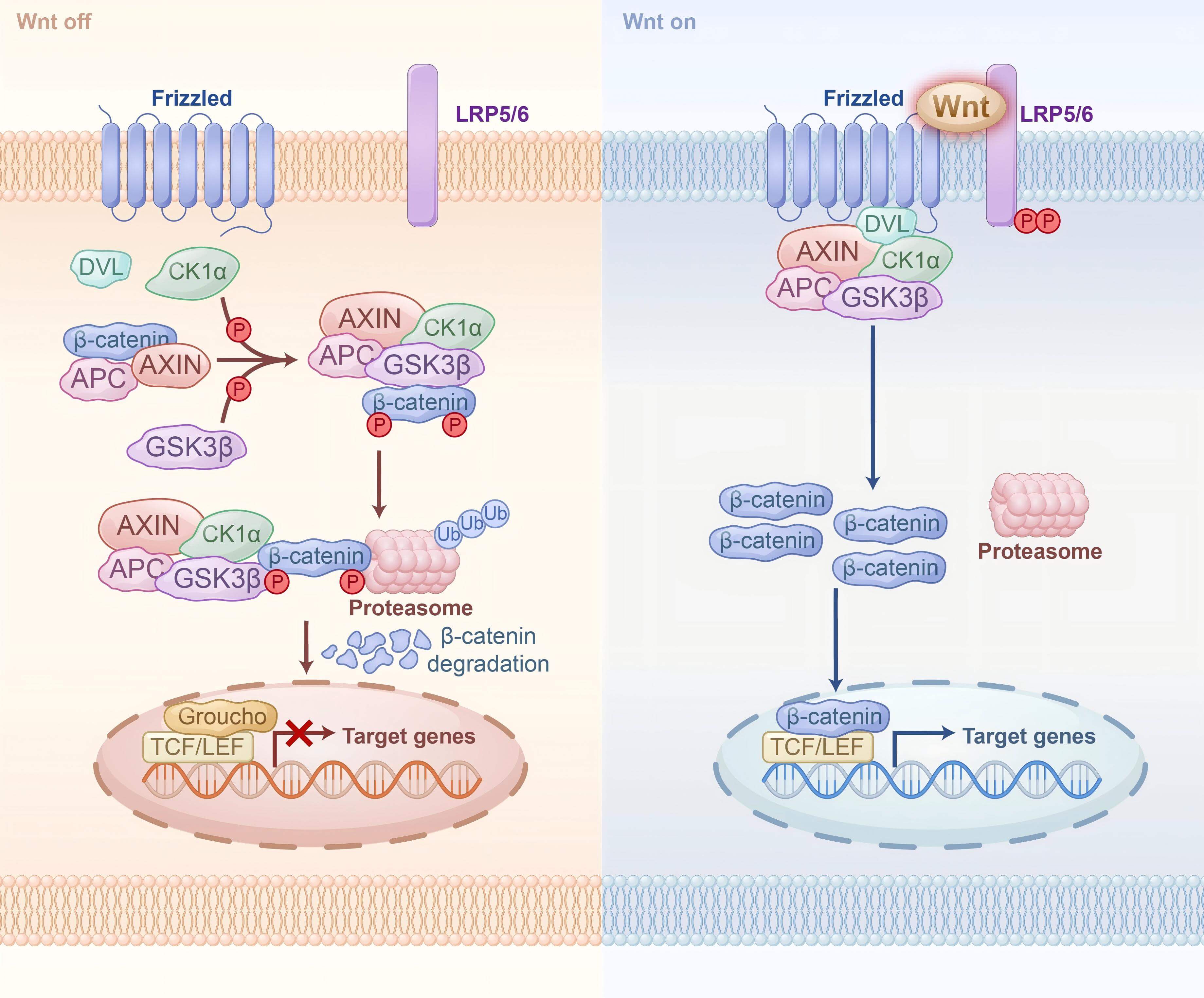 Fig. 1.
Fig. 1.
Wnt/-catenin signaling pathway. (Left) In the absence
of Wnt ligands, -catenin binds to AXIN and APC and is phosphorylated by
GSK-3 and CK1. Once the complex is formed, phosphorylated
-catenin binds to the Proteasome and is degraded, and gene transcription
cannot be interrupted in the nucleus. (Right) In the present of Wnt ligands. Upon
binding of Wnt ligands to LRP5/6 and Frizzled ligands, LRP5/6 phosphorylates and
recruits Dvl proteins to the plasma membrane. Subsequently, Dvl recruits the
destruction complex simultaneously to the cell membrane, and -catenin
dissociates in the cytoplasm and enters the nucleus, where it binds to the
TCF/LEF complex and initiates gene transcription. Dvl, dishevelled; APC,
adenomatous polyposis coli protein; CK1, casein kinase 1;
GSK-3, glycogen synthase kinase 3; TCF, T cell factor; LEF,
lymphocyte enhancer factor-1.
Significant crosstalk exists between Wnt/-catenin signaling and other
key cellular signaling pathways, such as Notch, transforming growth factor beta
(TGF-), mitogen-activated protein kinase (MAPK), nuclear factor-kappa B
(NF-B), extracellular signal-regulated kinase (ERK), and
phosphoinositide 3-kinase/Akt (PI3K/Akt), further complicating its role in
regulating LVH and arrhythmia. Therefore, an in-depth study of the specific
mechanisms of the Wnt/-catenin signaling pathway in LVH and arrhythmia
can help reveal its key role in disease development and provide a theoretical
basis for the development of multi-target therapeutic strategies, which may open
up new avenues for the precision treatment of related cardiovascular diseases.
2. Wnt/-catenin Signaling Pathway in the Regulation of LVH
LVH is an adaptive structural change caused by prolonged pressure overload,
characterized by ventricular wall thickening and changes in the ventricular
cavity size [17, 18]. As the condition progresses, LVH can lead to impaired
cardiac function and HF [19]. The Wnt/-catenin signaling pathway plays a
critical regulatory role in the onset and progression of LVH, significantly
accelerating the process through its involvement in pathological myocardial
hypertrophy, fibrosis, and metabolic reprogramming [20].
2.1 Activation of the Wnt/-catenin Signaling Pathway
Promotes Cardiomyocyte Hypertrophy and Apoptosis
Moderate activation of the Wnt/-catenin signaling pathway plays a
crucial physiological protective role in cardiac repair and regeneration. In
hemodialysis patients, lower serum levels of sclerostin and Dickkopf-related
protein-1 (Dkk-1) are negatively correlated with LVH severity, with Dkk-1
independently predicting left ventricular mass (LVM) and LVM index (LVMI) [21].
This suggests that reduced inhibition of the Wnt/-catenin pathway may
drive cardiac remodeling, highlighting sclerostin and Dkk-1 as potential
therapeutic targets. In human acute infarction tissues and rat hypertension heart
tissues, activation of the Wnt/-catenin signaling pathway triggers MAPK
signaling, including extracellular signal-regulated kinase 1 and 2 (ERK1/2),
c-Jun N-terminal kinase (JNK), and p38, leading to the upregulation of
hypertrophic markers such as atrial natriuretic peptide (ANP), brain natriuretic
peptide (BNP), nuclear factor of activated T cells 3 (NFATc3), and phosphorylated
GATA-binding protein 4 (GATA4), thereby promoting cardiomyocyte hypertrophy and
pathological remodeling, which may ultimately result in LVH [22]. In non-ischemic
transmural samples from failing human left ventricles, increased expression of
the Wnt signaling antagonists secreted frizzled-related protein (sFRP) 3 and 4
(sFRP3 and sFRP4) suppresses the Wnt/-catenin pathway, accompanied by an
elevated Fas/FasExo6Del ratio and downregulation of bcl-xL expression, promoting
a proapoptotic cardiomyocyte phenotype. These changes may drive cardiac
remodeling and compensatory hypertrophy, ultimately contributing to the
development and progression of LVH [23]. In Angiotensin II (Ang II)-induced
ventricular hypertrophy models in mice and rats, downregulation of protein
arginine methyltransferase 7 (PRMT7) activates the Wnt/-catenin pathway,
leading to upregulation of hypertrophic markers such as atrial natriuretic
peptide (ANP), brain natriuretic peptide (BNP), and collagen
type I alpha 1 chain (COL1A1), thereby promoting cardiomyocyte
hypertrophy and collagen deposition [24]. Such activation contributes to adaptive
changes in LVH and cardiac function, highlighting the protective role of this
pathway under stress conditions.
However, excessive activation of this pathway can trigger pathological
myocardial hypertrophy, cardiac remodeling, and HF development. In Ang II-induced
neonatal rat cardiomyocytes and C57BL/6J mouse models, increased expression of
methyltransferase-like 3 (METTL3) enhances m6A methylation, promoting
pri-miR-221/222 expression, and activates the Wnt/-catenin signaling
pathway by inhibiting Dickkopf2 (DKK2), thereby promoting myocardial hypertrophy
[25]. In the mouse LVH model induced by transverse aortic constriction (TAC),
Wnt/-catenin signaling is activated, upregulating nuclear factor-kappa B
(NF-B), -myosin heavy chain (-MHC), TNF-,
fibronectin (FN), and collagen type I (Col I), leading to cardiomyocyte
hypertrophy and fibrosis. As the disease progresses, it further upregulates
angiotensin-converting enzyme (ACE), renin, and Ang II type 1 receptor (AT1),
activates the renin–angiotensin–aldosterone system (RAAS), induces myocardial
cell apoptosis, and exacerbates LVH [26].
Integrin beta-like 1 (ITGBL1) is an extracellular matrix protein associated with
-integrins that can activate the Wnt/-catenin signaling pathway
[27, 28]. In TAC-induced mice, elevated ITGBL1 activates Wnt/-catenin
signaling, mediating fibroblast–cardiomyocyte crosstalk. In cardiomyocytes, this
pathway upregulates -MHC and FN, promoting hypertrophy, while in
fibroblasts, it enhances TGF- expression and interacts with the
TGF-/Smad2/3 pathway, accelerating collagen deposition and fibrosis
[29]. In the isoproterenol (ISO)-induced mouse model of myocardial hypertrophy,
the activation of the Wnt/-catenin signaling pathway promotes
hypertrophy by upregulating cell cycle related protein (Cyclin D1) and
c-Myc. Concurrently, sodium/calcium exchanger-1 (NCX1) overexpression
triggers Ca2+ overload, activating calcium–calmodulin-dependent protein
kinase II (CaMKII) and calcineurin (CaN), which induces apoptosis and activates
MAPK signaling via the nuclear factor of activated T-cells (NFAT)/ETS
transcription factor 2 (ETS2) complex, exacerbating hypertrophy and remodeling
[30, 31]. Collectively, these findings highlight the dual role of
Wnt/-catenin signaling in cardiac physiology and pathology. The
differences in experimental models and activation levels are likely the key
factors underlying the inconsistent findings regarding the role of this pathway
in cardiac function observed in previous studies.
2.2 Activation of the Wnt/-catenin Signaling Pathway
Promotes Fibroblast Fibrosis
Myocardial fibrosis (MF) is one of the main histological features of LVH and
often leads to severe cardiac insufficiency [32, 33]. The Wnt/-catenin
signaling pathway participates in regulating the pathological process of LVH
through crosstalk with other signaling pathways such as NF-B,
TGF-, and ERK, playing a crucial role, particularly in
fibroblast-mediated fibrosis.
The synergistic interaction between Wnt/-catenin and TGF-
signaling significantly exacerbates MF. In patients with chronic kidney disease
(CKD), elevated levels of TGF-1 suppress the cardiac expression of
endogenous Klotho, leading to activation of the Wnt/-catenin signaling
pathway. This, in turn, upregulates the expression of profibrotic markers such as
fibronectin, type I collagen, PAI-1, and MMP-2/9, thereby promoting cardiac
fibroblast-mediated fibrosis [34]. In TGF--stimulated human cardiac
fibroblasts, activation of the Wnt/-catenin signaling pathway is
enhanced synergistically by exogenous WNT3a and the GSK-3 inhibitor
CHIR99021, leading to increased interleukin (IL)-11 production and secretion.
Concurrently, TGF- promotes phosphorylation of TGF--activated
kinase 1 (TAK1), which further stimulates IL-11 expression and upregulates
fibrosis-related genes such as COL1A1 and FN1, thereby
accelerating fibroblast activation, cardiac fibrosis, and contributing to the
progression of LVH [35]. In an acute myocardial infarction (AMI) rat model, Wnt2
and Wnt4 activate -catenin by interacting with Fzd2/4 and LRP6, further
activating the NF-B signaling pathway, which upregulates
fibrosis-related genes such as COL1A1 and FN1, ultimately
worsening cardiac fibrosis and cardiac dysfunction [36]. sFRPs, by antagonizing
the Wnt/-catenin pathway, inhibit fibroblast activation and collagen
synthesis, thus slowing the progression of cardiac fibrosis [37]. In sFRP1
knockout mice, the excessive activation of the Wnt/-catenin pathway
promotes fibroblast proliferation, alpha-smooth muscle actin (-SMA)
expression, and collagen synthesis, ultimately leading to MF and LVH [38]. In a
type 1 diabetes mellitus rat model induced by streptozotocin, NF-B
cooperates with the Wnt/-catenin/GSK-3 pathway to activate the
expression of pro-inflammatory cytokines tumor necrosis factor (TNF)-
and IL-2, thereby inducing myocardial hypertrophy and interstitial fibrosis [39].
In an ISO-induced MF rat model, activation of the Wnt/-catenin pathway
upregulates -catenin, c-Myc, and Cyclin D1 expression, enhancing
fibroblast proliferation and differentiation, thereby exacerbating MF and cardiac
dysfunction [40]. In a high-fat diet-induced hyperlipidemia mouse model,
obesity-induced hypertrophy activates the TGF-/Wnt/-catenin
pathway, promoting -SMA and TGF- expression and inducing MF.
Additionally, the activation of mast cells induced by obesity leads to elevated
expression of serine proteases, such as tryptase and chymase, which are closely
associated with cardiac fibrosis primarily by indirectly activating the
TGF- and Wnt/-catenin signaling pathways, thereby promoting
cardiac collagen deposition and myocardial fibrosis, resulting in cardiac
dysfunction [41].
2.3 Activation of the Wnt/-catenin Signaling Pathway
Promotes Metabolic Reprogramming
The activation of the Wnt/-catenin signaling pathway contributes to the
development of LVH by modulating mitochondrial dynamics, lipid metabolism,
glucose metabolism, and other aspects of metabolic reprogramming.
In spontaneously hypertensive rats, Wnt/-catenin activation enhances
sterol regulatory element-binding protein 1 (SREBP1), upregulates fatty acid (FA)
synthesis genes (e.g., stearoyl-CoA desaturase 1 (SCD1) and acetyl-CoA
carboxylase (ACC)), reduces FA transport proteins (CD36, FATP1),
suppresses AMP-activated protein kinase (AMPK) and carnitine palmitoyltransferase
1 (CPT1), thereby promoting FA accumulation and impairing -oxidation,
contributing to left ventricular hypertrophy LVH [42]. In a -catenin
haploinsufficient (WT/CKO) mouse model, suppression of the Wnt/-catenin
signaling pathway reduces adipose triglyceride lipase (ATGL) and
hormone-sensitive lipase (HSL) activity, leading to triglyceride (TAG)
accumulation and limited fatty acid -oxidation. Meanwhile, upregulation
of glucose transporter 4 (GLUT4) and downregulation of pyruvate dehydrogenase
kinase 1 (PDK1) enhance glucose utilization, elevate the NADH/NAD⁺ ratio, and
impair oxidative phosphorylation (OXPHOS) complex I, disrupting mitochondrial
metabolism [43]. These de novo metabolic disturbances occur in the
absence of spontaneous LVH, but directly blunt physiological cardiomyocyte growth
and limit training-induced adaptive cardiac hypertrophy. This finding suggests
that while Wnt/-catenin activation is known to promote pathological
cardiac remodeling, its suppression may conversely constrain the heart’s adaptive
growth capacity under physiological conditions and potentially restrain
pathological remodeling under stress, thus affecting LVH development. In the
volume overload-induced HF model, elevated TNF- and IL-6 activate
Wnt/-catenin signaling, downregulating proliferator-activated receptor
alpha (PPAR) and PPAR-gamma coactivator 1 alpha (PGC-1),
reducing CPT1B and ACADM expression, impairing FA oxidation. Simultaneously,
upregulation of c-Myc enhances the activity of glycolytic enzymes hexokinase 2
(HK2) and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3, disturbing the
glucose and lipid metabolism. Furthermore, Wnt/-catenin signaling
activates the mammalian target of rapamycin (mTOR) pathway, inhibits mitophagy,
promotes reactive oxygen species (ROS) production, and aggravates metabolic
dysfunction and cardiomyocyte apoptosis, ultimately leading to energy imbalance
and worsening cardiac function [44, 45, 46]. In hypoxia/reoxygenation rats,
upregulated miR-423-5p inhibits Myb-related protein B (MYBL2), activates
Wnt/-catenin signaling, enhances caspase 3/7 activity and Bax/cleaved
caspase-3 (c-casp-3) expression, while promoting Drp1-mediated mitochondrial
fission, causing mitochondrial membrane potential (MMP) loss, ROS overproduction,
ATP suppression, and cardiomyocyte apoptosis [47]. Therefore, Drp1 acetylation
may be an early key event in LVH. In TAC-induced heart–kidney syndrome type 2
mice, Wnt/-catenin activation inhibits antioxidant enzymes superoxide
dismutase (SOD) and catalase, and activates NADPH oxidase (NOX), causing ROS
accumulation, cytochrome C release, and apoptosis. ROS suppress Bcl-2/Bcl-xL and
activate Bax/Bad, aggravating mitochondrial permeability transition and promoting
cardiomyocyte apoptosis [26].
The evidence suggests that activation of the Wnt/-catenin signaling
pathway contributes to ventricular hypertrophy by upregulating
hypertrophy-related genes and exacerbating pathological myocardial hypertrophy
through crosstalk with the MAPK and NF-B pathways. This pathway also
promotes fibrosis through interaction with TGF- signaling, with
GSK-3 acting as a key regulator (Fig. 2). Moreover, it drives metabolic
reprogramming, regulating lipid and glucose metabolism as well as mitochondrial
function, all of which contribute to cardiac hypertrophy and functional
impairment (Fig. 3).
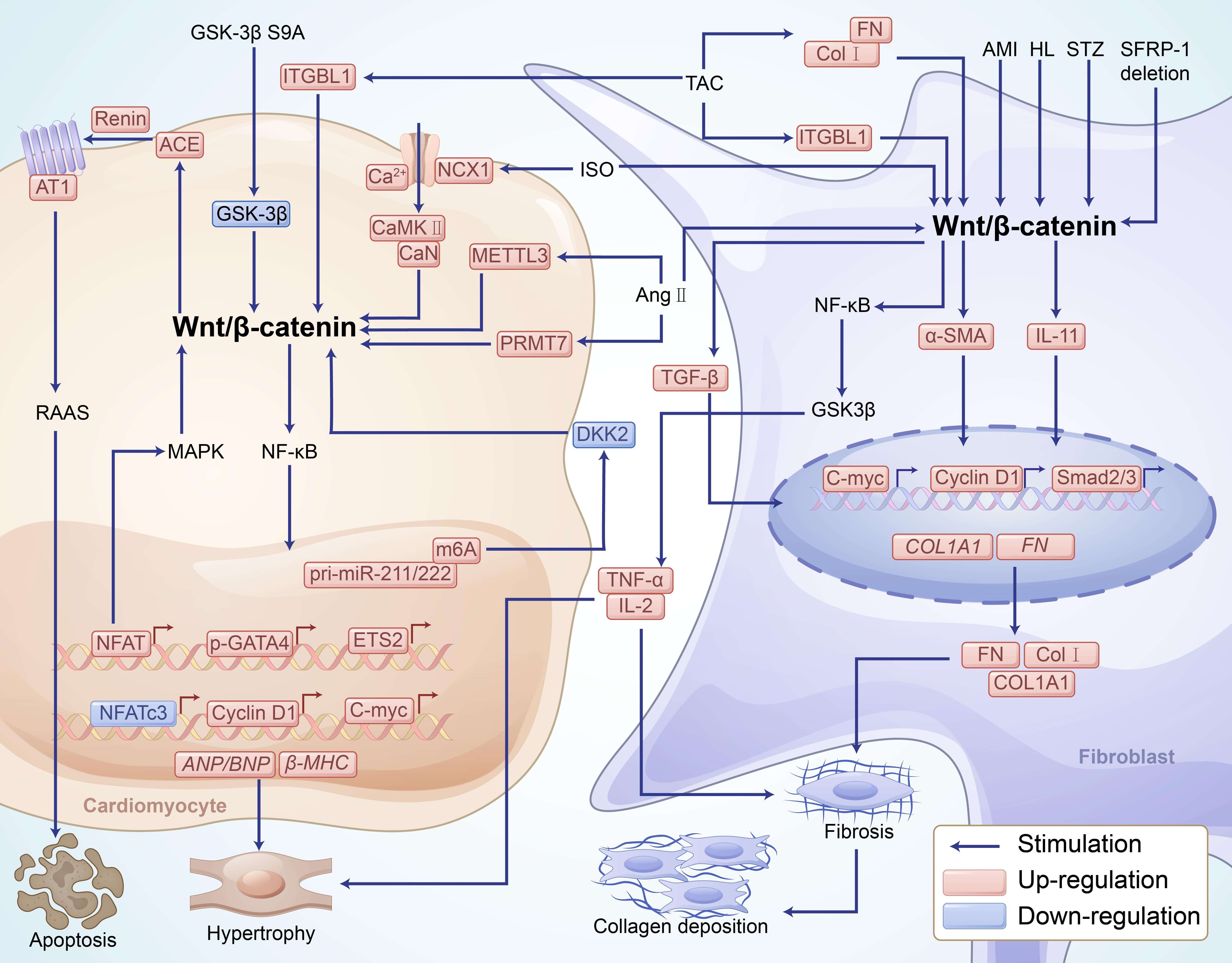 Fig. 2.
Fig. 2.
Activation of the Wnt/-catenin signaling pathway
influences cardiomyocyte hypertrophy and fibroblast fibrosis through various
mechanisms, thereby promoting the development of LVH. Activation of the
Wnt/-catenin signaling pathway in both cardiomyocytes and fibroblasts
collectively promotes the progression of LVH. In cardiomyocytes, this pathway
upregulates transcription factors such as NFAT, p-GATA4, c-Myc, NFATc3, and
Cyclin D1, which promote the expression of hypertrophic markers including
ANP/BNP and -MHC, inducing myocardial hypertrophy and
apoptosis. GSK-3 S9A, TAC, ISO, and Ang II activate the
Wnt/-catenin signaling pathway by inhibiting GSK-3, METTL3, and
DKK2 while upregulating NCX1, PRMT7, ITGBL1, ACE, renin, and AT1. Additionally,
crosstalk between this pathway and the MAPK and NF-B signaling pathways
further amplifies the pathological process. Meanwhile, RAAS activation
exacerbates cardiomyocyte apoptosis, ultimately leading to cardiac dysfunction.
In fibroblasts, the Wnt/-catenin signaling pathway interacts with the
TGF--Smad2/3 and NF-B pathways, upregulating the expression of
-SMA, IL-11, COL1A1, and FN1, thereby promoting interstitial fibrosis
and collagen deposition. Additionally, this pathway enhances fibroblast
proliferation and fibrosis through Snail/Twist-mediated
endothelial-to-mesenchymal transition. Ang II, ITGBL1, and ISO activate the
Wnt/-catenin pathway in both cardiomyocytes and fibroblasts, whereas
fibroblast-secreted TGF- further amplifies myocardial hypertrophy and
fibrosis. Meanwhile, NF-B signaling is activated, increasing the
production of pro-inflammatory cytokines such as TNF- and IL-2, which
promote chronic inflammation and exacerbate the progression of LVH. PRMT7,
protein arginine methyltransferase 7; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; COL1A1, collagen type I alpha 1; METTL3,
methyltransferase-like 3; m6A, N6-methyladenosine; DKK2, Dickkopf2;
NF-B, nuclear factor kappa B; -MHC, beta-myosin heavy chain;
ITGBL1, integrin beta-like 1; TGF-, transforming growth factor beta;
Smad2/3, smad family member 2/3; FN, fibronectin; ISO, isoproterenol; Cyclin D1,
cell cycle-related protein D1; c-Myc, cellular Myc; NCX1, sodium/calcium
exchanger 1; CaMKII, calcium–calmodulin-dependent protein kinase II; CaN,
calcineurin; TAK1, TGF--activated kinase 1; sFRP, secreted frizzled
related protein; -SMA, alpha-smooth muscle actin; TNF-, tumor
necrosis factor alpha; IL-2, interleukin 2; T1DM, type 1 diabetes mellitus; STZ,
streptozotocin; GSK-3, glycogen synthase kinase 3 beta.
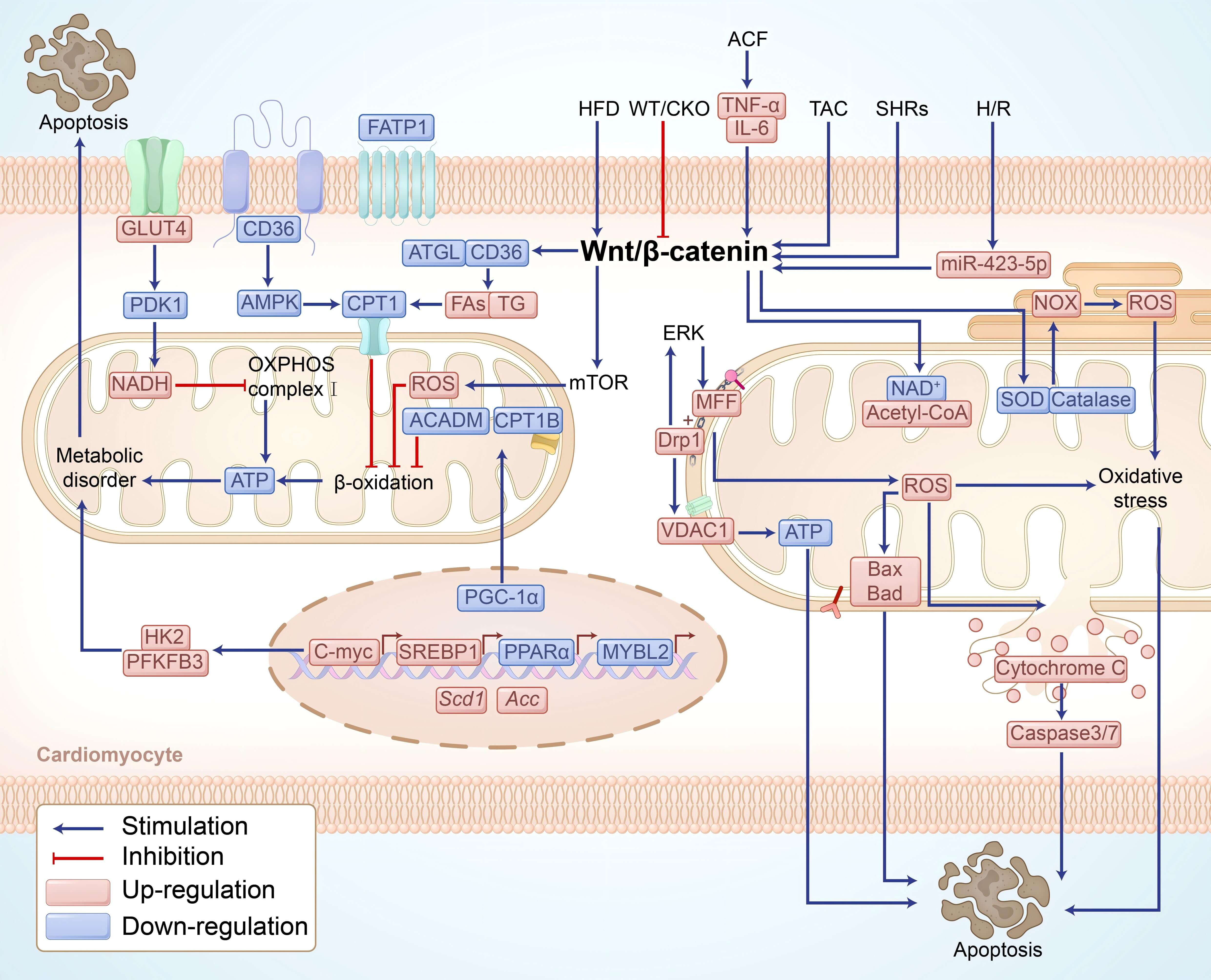 Fig. 3.
Fig. 3.
Activation of the Wnt/-catenin signaling pathway
induces cardiomyocyte apoptosis and promotes the development of LVH by regulating
mitochondrial dynamics, lipid metabolism, and glucose metabolism. Activation of
the Wnt/-catenin signaling pathway reprograms cardiac metabolism by
regulating metabolic transcription factors (SREBP1, c-Myc, PPAR,
PGC-1, and MYBL2), promoting FA synthesis, such as Scd1 and
Acc, while suppressing FA oxidation, including CPT1B and ACADM.
Simultaneously, it downregulates the expression of FA transport proteins (CD36,
FATP1) and lipolytic enzymes (ATGL, HSL), leading to lipid accumulation,
metabolic imbalance, and ultimately, cardiomyocyte apoptosis. In glucose
metabolism, upregulation of GLUT4 and downregulation of PDK1 enhance pyruvate
oxidation, increasing the NADH/NAD⁺ ratio, thereby inhibiting OXPHOS complex I
function and impairing mitochondrial metabolism. Lipid overload induces DRP1
acetylation and promotes its phosphorylation via ERK signaling, strengthening
interactions with MFF and VDAC1, thereby driving mitochondrial fission, reducing
ATP synthesis, and increasing oxidative stress. Additionally,
Wnt/-catenin signaling exacerbates ROS accumulation by inhibiting
antioxidant enzymes (SOD, catalase) and activating NOX, triggering the caspase
3/7 cascade and upregulating pro-apoptotic proteins Bax and Bad, ultimately
leading to cardiomyocyte apoptosis and contributing to LVH progression. LVH, left
ventricular hypertrophy; Wnt/-catenin, Wnt/-catenin signaling
pathway; SREBP1, sterol regulatory element-binding protein 1; c-Myc, cellular
myelocytomatosis; PPAR, peroxisome proliferator-activated receptor
alpha; MYBL2, Myb-related protein B; Scd1, stearoyl-CoA desaturase 1;
Acc, acetyl-CoA carboxylase; FATP1, fatty acid transport protein 1;
AMPK, AMP-activated protein kinase; CPT1, carnitine palmitoyltransferase 1;
GLUT4, glucose transporter 4; PDK1, pyruvate dehydrogenase kinase 1; OXPHOS,
oxidative phosphorylation; Drp1, dynamin-related protein 1; MFF, mitochondrial
fission factor; VDAC1, voltage-dependent anion channel 1; SOD, superoxide
dismutase; NOX, NADPH oxidase; complex I, oxidative phosphorylation complex I;
TG, triglyceride.
3. Wnt/-catenin Signaling Pathway in the Regulation of
Arrhythmia
Arrhythmias are cardiac autonomic disorders caused by abnormal electrical
activity and conduction disorders of cardiomyocytes, typically manifesting as
ectopic beats and impulse reentry. The most common types include AF, atrial
flutter, and ventricular fibrillation [7, 48]. Acute or chronic myocardial injury
often leads to electrical remodeling of the heart, myocardial hypertrophy, and
fibrosis. These pathological changes can interfere with the normal conduction of
cardiac electrical signals and induce arrhythmia [7]. Common symptoms include
sinus arrest, sinus block, bradycardia, and, in severe cases, sudden death [48, 49]. The Wnt/-catenin signaling pathway plays an important regulatory
role in the occurrence and development of arrhythmia, affecting the electrical
activity stability of the heart by regulating oxidative stress, atrial fibrosis,
and metabolic reprogramming.
3.1 Activation of the Wnt/-catenin Signaling Pathway
Promotes Oxidative Stress
Abnormal activation of Wnt/-catenin signaling can promote ROS
production. It has been demonstrated that in the peripheral plasma of patients
with persistent AF, BNP expression and the content of Diacron-reactive oxygen
metabolite (dROM) are increased, and the heart undergoes oxidative stress [50].
Meanwhile, activation of Wnt/-catenin signaling pathway and increased
protein expression of ANP and BNP were found in human cardiomyocytes treated with
ISO in vitro [51]. Therefore, the activation of Wnt/-catenin
signaling pathway in cardiac myocytes induces oxidative stress by up-regulating
the expression of BNP protein, leading to the occurrence of AF. In AngII-treated
rat atrial tissue, SIRT3 protein sulfhydrylation was inhibited,
Wnt/-catenin signaling pathway was activated, ROS production was
increased, MDA expression was increased, while GSH and SOD expressions were
decreased, leading to atrial oxidative stress [52]. Additionally, Wnt and
TGF- signaling pathways contribute to oxidative stress in
alcohol-treated human pluripotent stem cell-derived cardiomyocytes, increasing
susceptibility to AF [53].
3.2 Activation of the Wnt/-catenin Signaling Pathway
Promotes Cardiac Fibrosis
Cardiac fibrosis is an important pathological process of arrhythmia [54].
Previous studies have demonstrated that depolarization of fibroblasts in cardiac
scar tissue can induce arrhythmias through electrical coupling between
fibroblasts and cardiomyocytes [55, 56]. Activation of the Wnt/-catenin
pathway is associated with increased expression of cardiac fibrosis genes [32, 57]. During fibrosis, activation of cardiac fibroblasts promotes excessive
deposition of the extracellular matrix (ECM) and ECM proteins (mainly col I and
col III) [58, 59]. The Wnt/-catenin signaling pathway promotes AF
generation by interacting with miRNA molecules or TGF-, FRAT, and other
signaling pathways.
Dvl-associated antagonist of -catenin 2 (DACT2) expression is decreased
in the right atrial cardiomyocytes of patients with AF. In vitro, Loss
of DACT2 resulted in the accumulation of -catenin in HL-1 cells
and the activation of TGF- in fibroblasts. This cascade resulted in
electrical remodeling of HL-1 cells, as well as increased deposition of col I and
col III in fibroblasts, ultimately contributing to fibrosis. These changes induce
AF [60]. This suggests that DACT2 can regulate the electrical-structural
remodeling between fibroblasts and cardiomyocytes by regulating the
Wnt/-catenin and TGF- signaling pathways and induce AF [61].
Snail1 is a key marker in epithelial-mesenchymal transition (EMT) and
participates in the formation process of cardiac fibrosis [62]. A study has found
that the canonical Wnt signaling pathway is activated in the myocardium of AF
patients, which leads to the up-regulation of Snail1 protein level in endothelial
cells, induces the expansion of cardiomyocytes and the increase of collagen
tissue, and atrial fibrosis induces the occurrence of AF [63]. The expression of
miR-124-3p was increased in plasma exosomes extracted from patients with AF.
Notably, co-culture of these exosomes with rat fibroblasts revealed that
upregulated miR-124-3p inhibited Axin1 expression, activated the
downstream Wnt/-catenin pathway, and stimulated -SMA
expression, promoting fibroblast proliferation [64]. In the rat AF model induced
by acetylcholine–CaCl2, miR-27b-3p expression in the left atrium was
downregulated, leading to Wnt/-catenin pathway activation and
significant upregulation of TGF-1 and fibrotic markers Col I, Col III,
and a-SMA. Furthermore, increased atrial fibrosis and decreased connexin43 (CX43)
expression interfere with the electrical coupling between cardiomyocytes and
promote the occurrence of AF [65]. In the same model, reported the increased
expression of monocyte chemotactic protein-induced protein 1 (MCPIP1) in
cardiomyocytes and decreased expression of miR-26p-5a, which activated the
FRAT/Wnt/-catenin signaling pathway, leading to MF [66]. In mouse
cardiomyocytes with acute MI, LIM kinase 2 (LIMK2) expression is significantly
increased, promoting fibroblast proliferation and activation and ventricular
remodeling through activation of the Wnt/-catenin signaling pathway,
thereby increasing susceptibility to AF [67]. Additionally, in TAC-treated mouse
hearts, the activation of TGF- signaling pathway promoted the activation
of Wnt/-catenin signaling pathway and cell activation in fibroblasts,
increased collagen expression, induced cardiac fibrosis [68].
3.3 Activation of the Wnt/-catenin Signaling Pathway
Regulate Metabolic Reprogramming
Abnormal activation of Wnt/-catenin signaling significantly impacts
arrhythmias through mitochondrial dysfunction [69]. In the atrial tissue of rats
treated with AngII, the sulfhydryl modification of SIRT3 protein was inhibited,
the Wnt/-catenin signaling pathway was activated, the expression of
SLC7A11 and GPX4 decreased, and ferroptosis occurred in the cells, which
increased the expression of fibrosis markers, and incuring atrial fibrosis [52].
At the same time, in the alcohol-treated atrial tissue of mice, decreased SIRT3
inhibited AMPK-PGC-1 signaling, up-regulated DRP1 expression, and
down-regulated MFN2 and MFN1 expression, leading to atrial fibrosis [70]. These
results suggest that Wnt/-catenin signaling pathway can cross-talk with
AMPK-PGC-1 signaling pathway to regulate mitochondrial homeostasis in
atrial tissue. In ISO-treated mouse cardiac fibroblasts, hypermethylation of the
sFRP3 promoter leads to a significant reduction in its expression, which
activates Wnt/-catenin signaling, accompanied by increased DRP1
expression and enhanced mitochondrial fission and migration [71]. The activation
of the Wnt/-catenin signaling pathway may also be involved in AF
occurrence by regulating the abnormal expression of proteins related to the
mitophagy pathway. In rat myocardial fibroblasts treated with ISO, the decreased
expression of sirtuin 1 (Sirt1) and increased phosphorylation of forkhead box
O-3a (FOXO3a) and NF-B activates the Wnt/-catenin signaling
pathway, leading to cell fibrosis [72]. In Ang II-treated mouse fibroblasts,
FOXO3a expression was upregulated, PTEN-induced putative kinase 1 (PINK) and
parkin expression was increased, p62 expression was decreased, mitophagy was
increased, and MMP was decreased, promoting fibroblast proliferation and
increasing -SMA, col I, and III expression, thereby elevating AF
susceptibility [73]. The Wnt/-catenin signaling pathway can activate the
P38 MAPK signaling pathway, inducing the occurrence of myocardial fibrosis [74].
Meanwhile, in Ang-II-treated atrial myocytes of AF rats, it was found that MAPK14
expression was significantly increased, ROS production was elevated, Parkin
protein expression was upregulated, P62 expression was significantly reduced,
mitochondrial quantity decreased, vacuolation increased, mitophagy was
excessively activated, Bcl2 expression was significantly decreased, and apoptosis
occurred, leading to atrial fibrosis and AF [75]. Therefore, the activation of
the Wnt/-catenin signaling pathway may induce excessive mitophagy by
activating the MAPK signaling pathway, thereby promoting AF. In addition,
disturbance of lipid metabolism in the atrial muscle is involved in the
occurrence of AF [76]. The activation of the Wnt/-catenin signaling
pathway can promote the expression of PGC-1 [77]. In high-fat diet
(HFD)-treated mouse cardiomyocytes, AMPK phosphorylation is inhibited, whereas
PGC-1, ANP, and -MHC expression are upregulated, leading to
cardiomyocyte hypertrophy and increased AF susceptibility [78, 79]. Although the
activation of Wnt/-catenin signaling in myocardium and fibroblasts can
cause adverse effects, in epicardial cells, the activation of this signaling
pathway may reduce the adipogenic process of epicardial cells. In boron-treated
mouse preadipocytes, the Wnt/-catenin signaling pathway was activated,
adipogenic-related gene expression Cebp,
Ppar, and fatty acid-binding protein 4
(Fabp4) expression was downregulated, and adipogenesis was inhibited
[80]. In the epicardial preadipocytes of patients undergoing cardiac surgery,
significantly increased sodium-glucose cotransporter 2 (SGLT2) expression and
upregulated expression of FABP4 promote adipogenesis and ROS production in
cardiomyocytes, inducing AF. In the HFD-induced mice heart, ANP secreted by
cardiomyocytes inhibits the Wnt/-catenin signaling pathway, thereby
inducing epicardial cell transformation into adipocytes through
epithelial–mesenchymal transition and fat secretion, inducing AF [81].
The evidence suggests that activation of the Wnt/-catenin signaling
pathway plays a crucial role in the development of AF by promoting processes such
as oxidative stress and fibrosis in myocardial tissue. This is achieved through
the increase in ROS and MDA levels, as well as interactions with other key
pathways like TGF- and FRAT. The Wnt/-catenin pathway has a
dual role in metabolic reprogramming: in myocardial cells and fibroblasts, its
activation contributes to MF and oxidative damage in myocardial cells and
fibroblasts, but inhibits adipogenesis in epicardial preadipocytes, highlighting
the complexity of its role in arrhythmogenesis. This multifaceted involvement in
AF is summarized in Fig. 4.
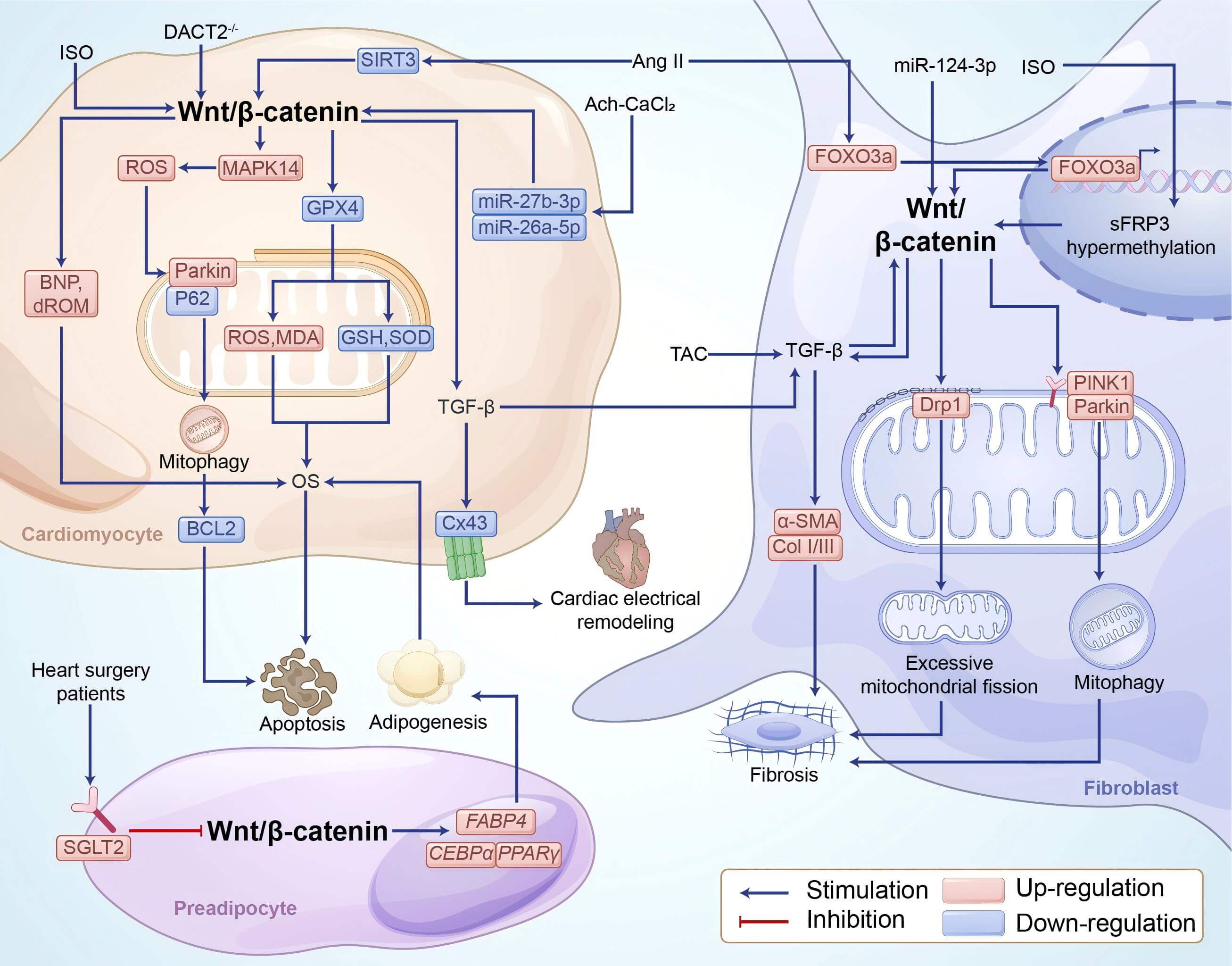 Fig. 4.
Fig. 4.
Mechanisms of Wnt/-catenin in regulating AF. In
cardiomyocytes, ISO can activate Wnt/-catenin signaling directly,
promote the expression of BNP and dROM, ultimately induce OS. At the same time,
Ang-II activates Wnt/-catenin signaling pathway by reducing the
expression of SIRT3, thereby decreases the expression of GPX4 and increases the
expression level of MAPK protein. On the one hand, it promotes the production of
ROS and the expression of MDA, and reduces the expression levels of GSH and SOD,
leading to oxidative stress. On the other hand, up-regulation of Parkin and
down-regulation of P62 expression could promote mitophagy, reduce the level of
BCL2, and eventually lead to cell apoptosis. Absence of DACT2 and Ach–CaCl2 can activate the Wnt/-catenin signaling pathway directly or indirectly
by decreasing the expression of miR-26p-5a and miR-27b-3p, induces the increase
of TGF-. Meanwhile, it decreases CX43 expression and induces cardiac
electrical remodeling. Furthermore, it promotes the activation of the
TGF- signaling pathway in fibroblasts. In fibroblasts, TAC, increased
miR-124-3p expression, ISO-induced hypermethylation of SFRP3, and Ang-II-induced
increase in FOXO3a expression all promote the activation of Wnt/-catenin
signaling. This, in turn, directly promotes TGF- signaling and induces
upregulation of fibrosis-related proteins (-SMA and Col-I/III).
Meanwhile, by promoting the expression of PINK1, parkin, and Drp1, it promotes
mitophagy and excessive mitochondrial fission, inducing fibrosis. Moreover, in
preadipocytes, SGLT2 expression inhibits the Wnt/-catenin signaling
pathway, increases the gene expression of pPAR,
CEBP, and FABP4, and promotes adipogenesis, finally
inducing OS in cardiomyocytes. OS, oxidative stress; ISO, isoproterenol; DACT2,
dishevelled-associated antagonist of beta-catenin homolog 2; SIRT3, Sirtuin 3;
MAPK, mitogen-activated protein kinase; BCL2, B cell lymphoma 2; GPX4,
glutathione peroxidase 4; GSH, glutathione; SOD, superoxide dismutase; ROS,
reactive oxygen species; MDA, malondialdehyde;
Ach–CaCl2, acetylcholine–CaCl2; Ang II, angiotensin II;
TGF-, transforming growth factor-beta; -SMA, alpha-smooth
muscle actin; COL I/III, collagen I/III; CX43, connexin 43; SGLT2, sodium-glucose
cotransporter 2; FABP4, fatty acid binding protein 4; TAC, transverse aortic
constriction; FOXO3a, forkhead box O-3a; sFRP3, secreted frizzled-related protein
3; PINK1, PTEN-induced putative kinase 1.
4. Therapeutic Strategies Targeting Wnt/-catenin Signaling in
LVH and Arrhythmia
Accumulating evidence highlights the involvement of Wnt/-catenin
signaling in the pathological progression of LVH and arrhythmias. A range of
molecular interventions—including small-molecule inhibitors, gene therapies,
and bioactive natural compounds—have demonstrated the ability to modulate this
pathway effectively, offering promising therapeutic avenues for the management of
these cardiac conditions.
4.1 Targeting of Wnt/-catenin Signaling in LVH
Pharmacological and molecular targeting of the Wnt/-catenin pathway has
demonstrated significant potential in alleviating myocardial hypertrophy,
fibrosis, and cardiac remodeling. Given that ventricular remodeling—including
hypertrophy, fibrosis, and structural alterations—is central to LVH
progression, modulating these processes represents a promising therapeutic
strategy.
In TAC and phenylephrine-induced LVH models, the long non-coding RNA taurine
up-regulated gene 1 (TUG1) suppresses miR-34a, upregulating Dickkopf proteins and
thereby inhibiting Wnt/-catenin signaling, leading to reduced expression
of hypertrophy-associated genes [82, 83]. Similarly, overexpression of sFRP2
attenuates pressure overload-induced LVH by inhibiting active -catenin,
reducing fibrosis and apoptosis [84]. Targeting upstream regulators, the
porcupine inhibitor CGX1321 downregulates Wnt/-catenin target genes
(Fzd2, Cyclin D1, c-Myc) in TAC-induced LVH models, while concurrently inhibiting
non-canonical pathways (NFATc3 and c-Jun), thus exerting dual anti-hypertrophic
and anti-fibrotic effects [85, 86]. The small-molecule compound Cardiomogen 1
(CDMG1) selectively inhibits Wnt/-catenin signaling, promoting cardiac
progenitor cell formation, cardiomyocyte differentiation, and cardiac
regeneration in zebrafish models [10, 87]. In embryonic stem cell models, CDMG1
exerts concentration-dependent effects on cardiac lineage commitment, while
minimizing off-target developmental interference [10]. Collectively, these
findings highlight the therapeutic promise of Wnt/-catenin pathway
modulators in treating pathological cardiac remodeling through multi-level
regulation of hypertrophy, fibrosis, and regenerative capacity.
In addition to directly inhibiting hypertrophic responses, Wnt/-catenin
pathway inhibition also ameliorates cardiac fibrosis associated with LVH. In an
ISO-induced myocardial fibrosis rat model, triptolide suppresses
Wnt/-catenin activation, resulting in decreased expression of fibrosis
markers such as Col I and -SMA, thereby alleviating myocardial fibrosis
and improving LV function [40]. In zebrafish heart injury models, activation of
Notch signaling, suppresses Wnt/-catenin signaling by promoting the
expression of Wnt antagonists Wif1 and Notum1b, enhances cardiomyocyte
proliferation, inhibits fibrosis, and facilitates cardiac regeneration,
ultimately counteracting hypertrophy and apoptosis [88]. Similarly, in Ang
II-induced LVH mouse models and H9c2 cardiomyocytes, nuclear protein localization
protein 4 (NPLOC4) suppresses the -catenin/GSK-3 axis, enhances
mitochondrial dynamics and mitophagy through ERO1-mediated modulation
of mitochondria-associated membranes (MAMs), thus alleviating cardiac hypertrophy
and fibrosis [89].
Wnt/-catenin pathway inhibition also contributes to improved metabolic
remodeling. Overexpression of secreted frizzled-related protein 5 (sFRP5) in MI
models inhibits Wnt/-catenin signaling, activates AMPK by enhancing
GSK-3 phosphorylation, promotes mitochondrial fusion (upregulating MFN1,
MFN2) while reducing fission markers (p-Drp1, Mid49, MFF), ultimately improving
mitochondrial integrity, decreasing oxidative stress, and mitigating left
ventricular remodeling [90].
4.2 Targeting Wnt/-catenin Signaling in Arrhythmias
Pharmacological modulation of the Wnt/-catenin signaling pathway shows
therapeutic potential in mitigating oxidative stress, fibrosis, and cardiomyocyte
apoptosis, as well as improving cardiac dysfunction linked to arrhythmias.
Treating healthy individuals deprived of sleep for 48 hours with statins can
inhibit the Wnt/-catenin signaling pathway by suppressing endoplasmic
reticulum stress in myocardial cells, reduce the expression of MDA, inhibit
oxidative stress, and lower the incidence of arrhythmia [91, 92]. Additionally,
in a sunitinib-induced myocardial fibrosis rat model, sacubitril/valsartan
regulates the antioxidant system thioredoxin-interacting protein
(TXNIP)/thioredoxin (TRX) and inhibits the Wnt/-catenin/SOX9 signaling
axis, thereby alleviating oxidative stress and reducing the incidence of AF [16, 93].
Targeting Wnt signaling pathways or their associated proteins has been shown to
reduce atrial fibrosis in arrhythmic conditions. For instance, miR-27b-3p
overexpression in AF rats inhibits the Wnt/-catenin pathway,
downregulates fibrosis markers Col I, Col III, and CX43, and reduces atrial
fibrosis [65]. Angiotensin receptor blockers (ARBs) also mitigate atrial fibrosis
in AF rats, prolong the effective atrial refractory period, and alleviate AF by
blocking the activation of FZD8 and the Wnt5a signaling pathway [94]. However, a
study reports contradictory findings, such as Wnt1 upregulation in 24-month-old
rat LV fibroblasts treated with relaxin, which inhibits the TGF-
pathway, reduces fibrosis markers, and decreases arrhythmia susceptibility [95].
Targeting Wnt/-catenin signaling pathways or associated proteins
through metabolic reprogramming can also help alleviate arrhythmias.
Empagliflozin, an SGLT2 inhibitor, inhibits adipogenesis in preadipocytes by
modulating the Wnt/-catenin pathway which can be regarded as a new
therapeutic strategy for AF patients [69, 96]. In HFD-induced mouse
cardiomyocytes, L-carnitine (LCA) promotes AMPK phosphorylation, suppresses
Wnt/-catenin signaling, increases the expression of fatty acid-related
transmembrane protein CD36 and PGC-1, reduces fat accumulation, and
diminishes inflammatory markers (e.g., IL-1, IL-6, and TNF-).
Additionally, CX43 and CX40 expression is enhanced, which reduces susceptibility
to AF [97, 98].
Therapeutic strategies targeting the Wnt/-catenin signaling pathway,
including GSK-3 inhibitors, Wnt antagonists (such as sFRP2, sFRP4, and
sFRP5), pioglitazone, and small molecules like cardiomogen, have shown promise in
the treatment of LVH and arrhythmias. These interventions have demonstrated
potential in improving mitochondrial function, promoting cardiomyocyte
regeneration, and reducing LV remodeling, as supported by various preclinical
studies [69, 97, 99]. Additionally, agents such as flavonoids, angiotensin
inhibitors, and empagliflozin modulate the Wnt/-catenin pathway,
mitigating AF and reducing fibrosis, further corroborating their therapeutic
efficacy in heart disease management [94, 98]. Collectively, these findings
highlight the therapeutic potential of targeting Wnt/-catenin signaling
in LVH and arrhythmias. An overview of these strategies and their mechanisms of
action is summarized in Table 1 (Ref. [10, 40, 65, 69, 82, 83, 84, 86, 87, 88, 89, 91, 92, 94, 96, 98, 99, 100]).
Table 1.
The therapeutic strategy targeting Wnt/-catenin for
LVH and arrhythmia.
| Treatment |
Target |
Model |
Conclusion |
Reference |
| long non-coding RNA TUG1 |
Inhibit Wnt/-catenin pathway |
TAC and deoxyadrenaline-induced LVH mouse |
Inhibition of miR-34a expression and an increase in DKK protein levels significantly reduced the expression of cardiac hypertrophy-related genes and alleviated cardiac hypertrophy |
[82, 83] |
| sFRP2 |
hypertension induced LVH mouse |
Improvement in cardiomyocyte hypertrophy, interstitial fibrosis, and cardiomyocyte apoptosis |
[84] |
| CGX1321 |
TAC-induced LVH mouse |
Reduced expression of myocardial hypertrophy-related genes (frizzled-2, cyclin-D1, and c-Myc), inhibition of the non-classical Wnt signaling pathway, reduced levels of NFAT and phosphorylated c-Jun, and inhibition of the fibrosis process |
[86] |
| Cardiomogen1 |
Zebrafish model |
Cardiomyocyte proliferation and wound healing accelerated regeneration after heart injury. Simultaneously, it promoted the formation of cardiac progenitor cells and increased the number of cardiomyocytes, thus expanding the size of the embryonic heart |
[10, 87] |
| TP |
ISO-induced myocardial fibrosis rat model |
Reduced expression of fibrosis markers (e.g., COL-I and -SMA), attenuation of myocardial hypertrophy and fibrosis, and improvement of left ventricular function |
[40] |
| Upregulated notch signaling |
Zebrafish heart damage model |
Promoted the expression of Wnt antagonists Wif1 and Notum1b, enhanced cardiomyocyte proliferation, inhibited fibrosis, and improved ability of heart regeneration |
[88] |
| NPLOC4 |
Ang II-induced LVH mouse and H9c2 cardiomyocytes |
Upregulation of ERO1 expression, regulating MAMs, enhancing mitochondrial dynamics and mitophagy, and regulating fibrosis and myocardial hypertrophy |
[89] |
| statins |
|
48-Hour Sleep Deprivation induced Arrhythmia patients |
Suppressing endoplasmic reticulum stress reduce the expression of MDA, inhibit oxidative stress in myocardial cells, and lower the incidence of arrhythmia |
[91, 92] |
| ARB |
AF rats |
Inhibition of FZD8 expression, inhibition of atrial fibrosis in AF rats, and prolonged effective atrial refractory period |
[94] |
| miR-27b-3p |
AF rats |
Downregulation of fibrosis-related proteins Col I, Col III, and CX43 inhibited atrial fibrosis |
[65] |
| LCA |
HFD-induced AF mouse |
Promoted AMPK phosphorylation, elevated the expression of CD36 and PGC-1, alleviated fat accumulation, reduced the production of inflammatory factors (such as IL-1, IL-6, and TNF-), and increased CX43 and CX40 expression |
[98, 100] |
| Empagliflozin |
Activate Wnt/-catenin |
Cardiac surgery patient |
Inhibition of SGLT2 expression, suppression of adipogenesis in preepicardial adipocytes, and alleviation of oxidative stress in cardiomyocytes |
[69, 96, 99] |
Wnt/-catenin, wingless-int1/-catenin; TUG1, taurine
up-regulated gene 1; sFRP2, secreted frizzled-related protein 2; NPLOC4, nuclear
protein localization protein 4; ARBs, angiotensin receptor blockers; LCA,
L-carnitine; TAC, transverse aortic constriction; LVH, left ventricular
hypertrophy; AF, atrial fibrillation; ISO, isoproterenol; HFD, high-fat diet;
Dkk-1, Dickkopf-related protein-1; -SMA, alpha-smooth muscle actin; COL
I/III, collagen I/III; MAMs, mitochondria-associated membranes; CX43, connexin43;
PGC-1, PPAR-gamma coactivator 1 alpha; TNF-, tumor necrosis
factor-; SGLT2, sodium-glucose cotransporter 2.
5. Clinical Translations and Challenges
Therapeutic strategies targeting Wnt/-catenin pathway—such as PPIs,
sFRP2, Porcupine inhibitors, relaxin, and miRNA modulators—have demonstrated
promising efficacy in ameliorating cardiac hypertrophy and fibrosis in
preclinical models. Currently, Wnt-targeted interventions are in the early stages
of clinical investigation. For instance, statins may indirectly inhibit the
Wnt/-catenin pathway by alleviating endoplasmic reticulum stress, thus
reducing arrhythmia risk in sleep-deprived individuals [91, 92]. Liensinine [101]
and the LncRNA RNA GAS5 [102] have also been identified as potential therapeutic
targets for arrhythmia. GSK-3 inhibitors, including tideglusib [103] and lithium
[104], have shown promise in ameliorating arrhythmic phenotypes in arrhythmogenic
cardiomyopathy (ACM) [105]; however, their clinical application remains limited
due to potential carcinogenicity [106], pro-hypertrophic effects [107, 108],
risks of immunosuppression [109], and off-target activity. In CKD, downregulation
of Klotho induced by TGF-1 activates Wnt/-catenin signaling,
providing a novel therapeutic target [34]. The drug pyrvinium has been shown to
prevent adverse cardiac remodeling and promote cardiomyocyte proliferation,
thereby offering a potential therapeutic benefit for LVH [110]. Moreover, a
variety of emerging Wnt pathway inhibitors—including small molecules (e.g.,
LGK-974 [111], CGX1321 [112], IWR-1 [113], and ICG-001 [114]) and traditional
Chinese medicine formulas (e.g., Linggui Zhugan Decoction formula [115])—have
demonstrated favorable safety profiles and translational potential in preclinical
studies. Several of these agents have already advanced into early-phase clinical
trials. The SIRT2 inhibitor AGK2 holds promise in improving conditions
characterized by cardiac fibrosis [116]. However, most clinical evidence remains
correlative, with a paucity of interventional studies targeting specific patient
subgroups. The heterogeneity in Wnt-related protein expression across disease
subtypes underscores the need for personalized treatment strategies based on
molecular profiling [21].
Despite the clear mechanistic relevance of the Wnt/-catenin pathway in
cardiovascular disease, clinical translation faces substantial challenges. The
structural complexity of the pathway and its involvement in multiple
physiological systems pose risks of off-target effects [117]. Additionally,
current animal and in vitro models fail to fully recapitulate human
cardiac pathology, particularly regarding age-related changes, comorbidities, and
molecular heterogeneity, limiting the extrapolation of preclinical findings
[118]. Furthermore, while many current studies emphasize average therapeutic
outcomes, others are limited to short-term observations, and patient responses to
Wnt pathway inhibitors vary considerably across individuals. Nevertheless,
Wnt-targeted therapeutic strategies remain promising.
Although emerging therapeutic strategies targeting the Wnt/-catenin
pathway have shown promise in basic and preclinical studies, translational
barriers remain due to the pathway’s inherent complexity, disease heterogeneity,
and limitations of current experimental models. The clinical advancement of
Wnt-targeted drugs for malignancies highlights their broader translational
potential in cardiovascular medicine [119]. To realize this potential, future
research should integrate systems biology, big data analytics, and single-cell
technologies to comprehensively dissect the regulatory network of Wnt signaling
and its crosstalk with other pathways [120]. This will enable the design of
mechanism-driven, biomarker-based patient stratification strategies and help
clarify patient-specific molecular signatures. Furthermore, optimizing dosing
regimens to minimize off-target effects and developing companion diagnostics for
precise patient selection will be essential. Robust long-term clinical trials and
real-world studies are also needed to verify sustained therapeutic efficacy and
monitor potential adverse effects, ultimately translating mechanistic insights
into safe and effective personalized therapies for cardiovascular disease.
6. Conclusion
The Wnt/-catenin signaling pathway is a pivotal regulator in the
pathogenesis and progression of LVH and arrhythmias. By modulating cardiomyocyte
hypertrophy, fibroblast-mediated fibrosis, oxidative stress, and metabolic
reprogramming, it contributes to cardiac structural remodeling and
electrophysiological dysfunction. Its extensive crosstalk with key signaling
cascades such as TGF-, NF-B, and MAPK further complicates the
disease landscape and presents additional therapeutic challenges.
Mechanism-guided clinical trial designs and a better understanding of Wnt pathway
interactions with other signaling networks may provide the foundation for
multitargeted therapies. Such approaches could ultimately improve clinical
outcomes and patient prognosis.
Abbreviations
-SMA, alpha-smooth muscle actin; AMPK, AMP-activated protein kinase; Ang II, angiotensin II; AF, atrial fibrillation; ANP, atrial natriuretic peptide; -MHC, beta-myosin heavy chain; CDMG1, cardiomogen 1; Col I, Collagen type I; Dvl, dishevelled; ERK, extracellular signal-regulated kinases; FA, fatty acid; FN, fibronectin; Fzd, Frizzled; GSK-3, Glycogen synthase kinase-3; HF, heart failure; HFD, high-fat diet; IGF-R, insulin like-growth factors receptor; ITGBL1, integrin beta-like 1; IL, interleukin; ISO, isoproterenol; LVH, left ventricular hypertrophic; MDA, malondialdehyde; MFN, mitochondrial fusion protein; MMP, mitochondrial membrane potential; MAPK, mitogen-activated protein kinase; MF, myocardial fibrosis; MI, myocardial infarction; NF-B, nuclear factor-kappa B; PGC-1, PPAR-gamma coactivator 1 alpha; ROS, reactive oxygen species; sFRP2, secreted frizzled related protein 2; SGLT2, sodium-glucose cotransporter 2; TGF-, transforming growth factor beta; TAC, transverse aortic constriction; Wnt/-catenin, wingless-int1/-catenin.
Author Contributions
ZG, JW, LX, XG, XZ, YX and YS collected the literatures and interpreted the data. YX, RT, ZG and JW wrote the original manuscript. LX and XG drew the figures. RT, GZ and JY designed manuscript conception and critically revised manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
We would like to express our gratitude to ChatGPT for their meticulous editing of the English grammar, and we express our gratitude to EditSprings for the expert linguistic services.
Funding
The authors would like to acknowledge the Research Start-up Fund of Jining Medical University (Reference: 600791001.J.y.), the College Students’ Innovation Training Program of Jining Medical University (Reference: 202410443002), the Outstanding Talent Research Funding of Xuzhou Medical University (D2016021), the Natural Science Foundation of Jiangsu Province (BK20160229), and the Postdoctoral Foundation of Xuzhou Medical University (RC5052112).
Conflict of Interest
The authors declare no conflict of interest.
Declaration of AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the authors used ChatGPT in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.







 , Guoan Zhang 7,*
, Guoan Zhang 7,*